

Abstract
Resident alveolar macrophages (AMs) suppress allergic inflammation in murine asthma models. Previously we reported that resident AMs can blunt inflammatory signaling in alveolar epithelial cells (ECs) by transcellular delivery of suppressor of cytokine signaling 3 (SOCS3) within extracellular vesicles (EVs). Here we examined the role of vesicular SOCS3 secretion as a mechanism by which AMs restrain allergic inflammatory responses in airway ECs. Bronchoalveolar lavage fluid (BALF) levels of SOCS3 were reduced in asthmatics and in allergen-challenged mice. Ex vivo SOCS3 secretion was reduced in AMs from challenged mice, and this defect was mimicked by exposing normal AMs to cytokines associated with allergic inflammation. Both AM-derived EVs and synthetic SOCS3 liposomes inhibited activation of STAT3 and STAT6 as well as cytokine gene expression in ECs challenged with IL-4/IL-13 and house dust mite (HDM) extract. This suppressive effect of EVs was lost when they were obtained from AMs exposed to allergic inflammation-associated cytokines. Finally, inflammatory cell recruitment and cytokine generation in the lungs of OVA-challenged mice were attenuated by intrapulmonary pretreatment with SOCS3 liposomes. Overall, AM secretion of SOCS3 within EVs serves as a brake on airway EC responses during allergic inflammation, but is impaired in asthma. Synthetic liposomes encapsulating SOCS3 can rescue this defect, and may serve as a framework for novel therapeutic approaches targeting airway inflammation.
Keywords: Allergic airway inflammation, Alveolar macrophages, Extracellular vesicles, Epithelial cells, Suppressor of cytokine signaling 3, Liposomes
Introduction
Carrying out the lung’s principal physiologic function of gas exchange in the face of a continuous barrage of inhaled allergens, toxins, and microbes requires calibrated or, where necessary, restrained inflammatory responses to these diverse insults. The development of chronic inflammatory processes, such as allergic airway inflammation, implies the dysregulation of these normal homeostatic mechanisms (1). Macrophages are well known for their functional plasticity and their pleiotropic role in orchestrating immune responses (2, 3), and the alveolar macrophage (AM) is the resident immune cell of the pulmonary mucosal surface. Lung macrophages had long been overlooked as cellular participants in the development of allergic airway inflammation (4). However, recent studies in mouse models have revealed an important dichotomy in which resident AMs play largely suppressive roles (5–7) while recruited monocyte-derived macrophages play largely pathogenic roles in allergic airway inflammation (5, 8, 9).
Given their paucity as well as their relative immobility in the normal mammalian lung (10, 11), AMs would be anticipated to employ paracrine means to restrain the inflammatory behavior of the alveolar and airway epithelial cells (ECs) which comprise this mucosal surface. A form of paracrine communication whose importance is increasingly appreciated involves the transfer of extracellular vesicles (EVs) containing various molecular species of cargo from donor to recipient cell (12). We have identified transcellular delivery of EVs containing suppressor of cytokine signaling 3 (SOCS3) from AMs to ECs as a new paradigm for regulating inflammation in the lung (13, 14). SOCS3 serves as the endogenous brake on Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling, which is critical in transducing the effects of numerous cytokines and growth factors (15). We have demonstrated that these AM-derived EVs are rapidly internalized by, and inhibit JAK-STAT signaling and inflammatory gene expression within, ECs (13, 16).
Pharmacologic inhibition of JAK is used clinically in rheumatoid arthritis, and its potential is being widely explored in other inflammatory diseases (17), including allergic asthma (18–22). While SOCS3 has been suggested to promote type 2 immune responses by its actions in lymphocytes and eosinophils (23, 24), its upregulation in bronchial ECs has been associated with anti-inflammatory actions (25, 26). The important role of the epithelium as both a source of and responder to inflammatory mediators in allergic asthma has gained increased recognition (27, 28), and its generation of chemokines is critical in recruitment of a variety of leukocyte lineages. Impaired delivery of SOCS3 from AMs to ECs in the setting of allergic inflammation, then, could promote inflammatory responses. Here we show that vesicular SOCS3 secreted by AMs restrains allergic inflammatory responses in bronchial ECs in vitro, but this brake is compromised in the lungs of asthmatic subjects and in two mouse models of allergic asthma. We also demonstrate that intrapulmonary administration of liposomes with SOCS3 as their sole cargo has the capacity to restrain inflammation in a mouse model of allergen challenge.
Materials and Methods
Human subjects and BALF sample acquisition
Subjects were men and women, ages 18–75, with or without mild to moderate stable asthma. Asthma diagnosis was based on symptoms, methacholine challenge, and/or bronchodilator reversibility. Stability was based on an absence of changes in asthma medications and no exacerbations requiring treatment with steroids within 30 days of bronchoscopy. Exclusion criteria included a smoking history greater than 30 pack-years, history of lung disease other than asthma, and other medical conditions that might increase the risks associated with bronchoscopy. BALF samples were acquired from a total of 16 subjects undergoing research-related bronchoscopies at the University of Michigan Hospital Medical Procedure Unit. BALF samples were collected and treated as protocolized research specimens with a uniform instillation volume (180 mL). Informed consent was obtained from each subject prior to sample collection in accordance with the Declaration of Helsinki and with approval of the University of Michigan Institutional Review Board.
Animals
Pathogen-free male and female C57BL/6 mice aged 6–8 weeks were purchased from The Jackson Laboratory. The mice were housed in groups of 5 and they had ad libitum access to water and food. Mice were treated in accordance to relevant national and local guidelines and regulations regarding the use of experimental animals and with approval of the University of Michigan Committee for the Use and Care of Animals.
Mouse models of allergic airway inflammation
Male (n=10 per group) and female (n=5 per group) mice were sensitized with 20 μg ovalbumin (OVA, Sigma-Aldrich, St. Louis, MO) mixed with 2 mg of alum (Thermo Fisher Scientific, Waltham, MA) in 150 μl PBS through intraperitoneal injection on day 0. On days 7 and 8, mice were challenged with nebulized 1% OVA, as described previously (29). Control groups (males, n=5 per group and females, n=3 per group) were sensitized with PBS and were challenged with nebulized PBS.
Male mice (n=5 per group) were also sensitized and challenged with 100 μg of Dermatophagoides pteronyssinus HDM extract (Greer Laboratories) protein suspended in 50 μl PBS and administered by oropharyngeal (o.p.) administration on days 0, 7 and 8 as described previously (5). Mice exposed to 50 μl PBS on the same days served as controls. For both OVA and HDM models, lung lavage fluid was collected on day 9. The first flush of 600 μl was stored separately for cytokine analysis. Additional flushes were performed to collect all lung cells and total numbers were counted. Approximately 50,000 lung cells per mouse were cytospun onto slides at 800 rpm for 2 min. The percentage of eosinophils and neutrophils among 300 total cells was determined in the cytospins by differential counting after H&E staining of the slides.
Cells
A continuous SV40-transformed line of primary AMs originally obtained from lavage fluid of normal mice (MHS, CRL-2019) (30), which we have utilized previously as a source of EVs (16), and a transformed human bronchial EC line (BEAS-2B) were purchased from American Type Culture Collection. Normal primary mouse AMs were obtained by lung lavage of a male C57BL/6 wild type mouse (The Jackson Laboratory) and subsequently immortalized by infecting with the J2 retrovirus carrying v-raf and v-myc oncogenes as previously described (31). Primary AMs were also obtained by lavage from PBS- and allergen-challenged mice. AMs and AM cell lines were cultured in RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco). However, because serum itself is a source of EVs, AMs were cultured in serum-free RPMI 1640 medium when they were being used as a source of EVs.
Isolation of EVs
Upon reaching confluency, AM medium was replaced with serum-free RPMI 1640 for 90 min (at 37°C, 5% CO2), and AM conditioned medium (CM) was harvested as a source of basally secreted EVs. Cell debris and apoptotic bodies were removed from CM by centrifugation at 4°C at 500 × g and 2500 × g, respectively. Two different methods were used to purify EVs in this study. In initial studies with MHS cells, EVs were pelleted from MHS CM by 17,000 × g ultracentrifugation for 30 min, with quantification of EV numbers performed as described previously (16). During the course of our studies we observed that the yield of EVs by this isolation method was limited due to their rupture owing to the high shear forces from ultracentrifugation. This prompted us to instead employ the gentler method of centrifugal filtration of AM CM through a 100 kDa exclusion filter (MilliporeSigma) (32), and this technique was employed for EV isolation from the J2-immortalized AM cell line. While this approach provided a higher yield of EVs and vesicular SOCS3, EVs isolated using both methods had similar properties and modulatory characteristics.
In vitro challenge of BEAS-2B cells
BEAS-2B ECs were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (Gibco) in six-well tissue-culture plates, and once 80% confluent, they were serum-deprived overnight. The next day, serum-free RPMI medium alone (2 mL), AM CM (2 mL) or AM EVs (at a ratio of 5 EVs per EC in 2 mL RPMI) were added to each well of ECs and incubated for 2 h. ECs were then washed and stimulated with human IL-4 + IL-13 (both 10 ng/mL, Peprotech) or HDM (10 μg/mL, Greer Laboratories) and harvested after 1 h for analysis of transcription factor phosphorylation by western blot or harvested after 6 h for analysis of cytokine mRNA by qPCR.
Western blotting
BEAS-2B ECs were lysed and protein concentrations were determined by the DC protein assay (modified Lowry assay from Bio-Rad Laboratories). Samples containing 40–50 μg EC lysate protein were separated by SDS-PAGE using 8% gels (for phosphorylated proteins), and those containing 10 μg AM lysate protein or 20–30 μg AM CM (or mouse lavage fluid after 100 kD filtration) protein were separated on 12.5% gels (for SOCS3 proteins) and then transferred to nitrocellulose membranes. After blocking with 4% BSA, the membranes were probed overnight with antibodies against phospho- and total STAT3 (both Cell Signaling, 1:1000), phospho-STAT6 (Cell Signaling, 1:500), β-actin (Sigma-Aldrich, 1:10,000) or SOCS3 (Abcam, 1:750). Films were developed using ECL detection (Amersham Biosciences) after incubation with peroxidase-conjugated secondary antibody (Cell Signaling). Relative band densities were determined by densitometric analysis using Image J software.
RNA isolation and qPCR
BEAS-2B ECs and primary AMs were suspended in 700 μl TRIzol reagent (ThermoFischer Scientific) and RNA was extracted using the RNeasy Micro Kit (Qiagen) according to manufacturer’s instructions and converted to cDNA. Levels of mRNA were assessed by qPCR performed with a SYBR green kit (Applied Biosystems) on an ABI Prism 7300 thermocycler (Applied Biosystems). Expression of eotaxin-1, TSLP, IL-33, IL-6, GM-CSF, inducible nitric oxide synthase (iNOS), and found in inflammatory zone 1 (FIZZ1, also termed resistin like alpha) was assessed (sequences of primers used can be found in Supplemental Table 1). Relative gene expression was determined by the ΔCT method, and either GAPDH (for ECs) or β-actin (for AMs) was used as a reference gene.
Synthetic SOCS3 liposomes
To generate synthetic vesicles with SOCS3 as lone cargo molecule, recombinant mouse SOCS3 was cloned, expressed, and purified to homogeneity as described previously (33). Thin phospholipid films were prepared from a 1:1 mixture of dioleoyl phosphocholine and dioleoyl phosphoglycerol and dried. These were mixed with PBS with or without recombinant SOCS3 protein (3.5 μg/mL) and intermittently vortexed to produce multilamellar vesicles, followed by serial extrusion. Resulting liposomes were centrifuged to remove unloaded SOCS3 protein and pellets suspended in PBS and stored at 4 °C until use. Empty (PBS control) and SOCS3 liposomes both had a mean diameter of ~110 nm as determined by dynamic light scattering and comprised a single, homogenous population of liposomes, as determined by polydispersity index (33). U.S. Patent application 16/071,290 describing these liposomes was filed on July 19, 2018.
For in vitro experiments with BEAS-2B ECs we employed a dose of liposomes containing 10 ng of recombinant SOCS3, since this approximates the amount secreted by 1×106 primary AMs that we previously determined to be capable of inhibiting STAT3 activation in normal alveolar ECs (13). For both allergic mouse models, mice were treated o.p. with either synthetic empty (i.e., PBS) or SOCS3 liposomes in a volume of 30 μl PBS 2 h prior to challenge with PBS or HDM (o.p.) or OVA (nebulization) on both days 7 and 8 (see Supplemental Fig 3). The dose of liposomes administered in vivo contained 20 ng of recombinant SOCS3, as this approximates the amount secreted by primary AMs that we previously determined capable of inhibiting STAT3 activation in cytokine-treated mouse lungs (13).
SOCS3 and cytokine analysis in lavage fluids
SOCS3 was measured in cell-free human or mouse lavage fluid samples and in primary AM CM that both had been concentrated using 100 kD Amicon Ultra exclusion filters (MilliporeSigma). After sonication of the samples (Branson Sonifier 250: 40% duty cycle, output 3) to disrupt EVs, SOCS3 levels were determined using a SOCS3 ELISA kit according to the instructions of the manufacturer (Cloud-Clone). Cell-free mouse lavage fluids (not sonicated) were also used to measure levels of IL-4, IL-5, IL-6, KC, MCP-1, IP10 and eotaxin-1 by multiplex ELISA (Milliplex, MilliporeSigma) and to measure IL-33 levels by ELISA (R&D Systems).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). To determine normality of the data a D’Agostino & Pearson omnibus normality test was used. Data were log transformed to fit a normal distribution when not distributed normally. Differences between groups were tested using a one-way ANOVA followed by Sidak’s test for multiple comparisons, or by a Student t test, as appropriate. A two way ANOVA was performed to analyze the effect of both treatment and sex (and their interaction) using Prism 8.0 (GraphPad Software). P-values below 0.05 were considered to be significant.
Results
Reduced levels of AM-derived SOCS3 in BALF of subjects with asthma and mice in two models of allergic inflammation
We have previously shown that constitutive AM secretion of vesicular SOCS3 can be bidirectionally modulated in various acute and chronic inflammatory environments (14, 33). Since asthma likewise represents an inflammatory environment in which AMs are known to be dysregulated (9, 34), we hypothesized that AM secretion of this natural brake on JAK-STAT signaling within the lung might be impaired. BALF was collected from the lungs of both asthmatic (n=6; Table 1) and non-asthmatic control (n=10; Table 2) subjects and subjected to sequential centrifugation and sonication steps before analysis via ELISA (Fig.1A). Asthmatic subject BALF demonstrated significantly lower levels of secreted SOCS3 than that of normal controls (Fig. 1B).
Table 1.
Demographic and clinical characteristics of asthmatic subjects
| Sex | Age | Smoking* history | ICS† | % predicted FEV1 | ACQ‡ score |
|---|---|---|---|---|---|
| Female | 47 | N | No | 96% | 0.57 |
| Female | 30 | N | Yes | 53% | 2.71 |
| Female | 50 | F | No | 93% | 0.71 |
| Male | 50 | N | Yes | 97% | 0.42 |
| Female | 27 | N | No | 117% | 0.57 |
| Female | 49 | N | Yes | 123% | 0 |
N= never; F= former
ICS= inhaled corticosteroid
ACQ= asthma control questionnaire
Table 2.
Control subject demographics
| Sample size (n): | 10 |
| Median age: | 52 years |
| Age range: | 23–71 years |
| % Male: | 40 |
| % Smoker: | 40 |
| Former (n): | 4 |
| Current (n): | 0 |
Figure 1. SOCS3 secretion by AMs is reduced in the lungs of asthmatic subjects and allergen-challenged mice.
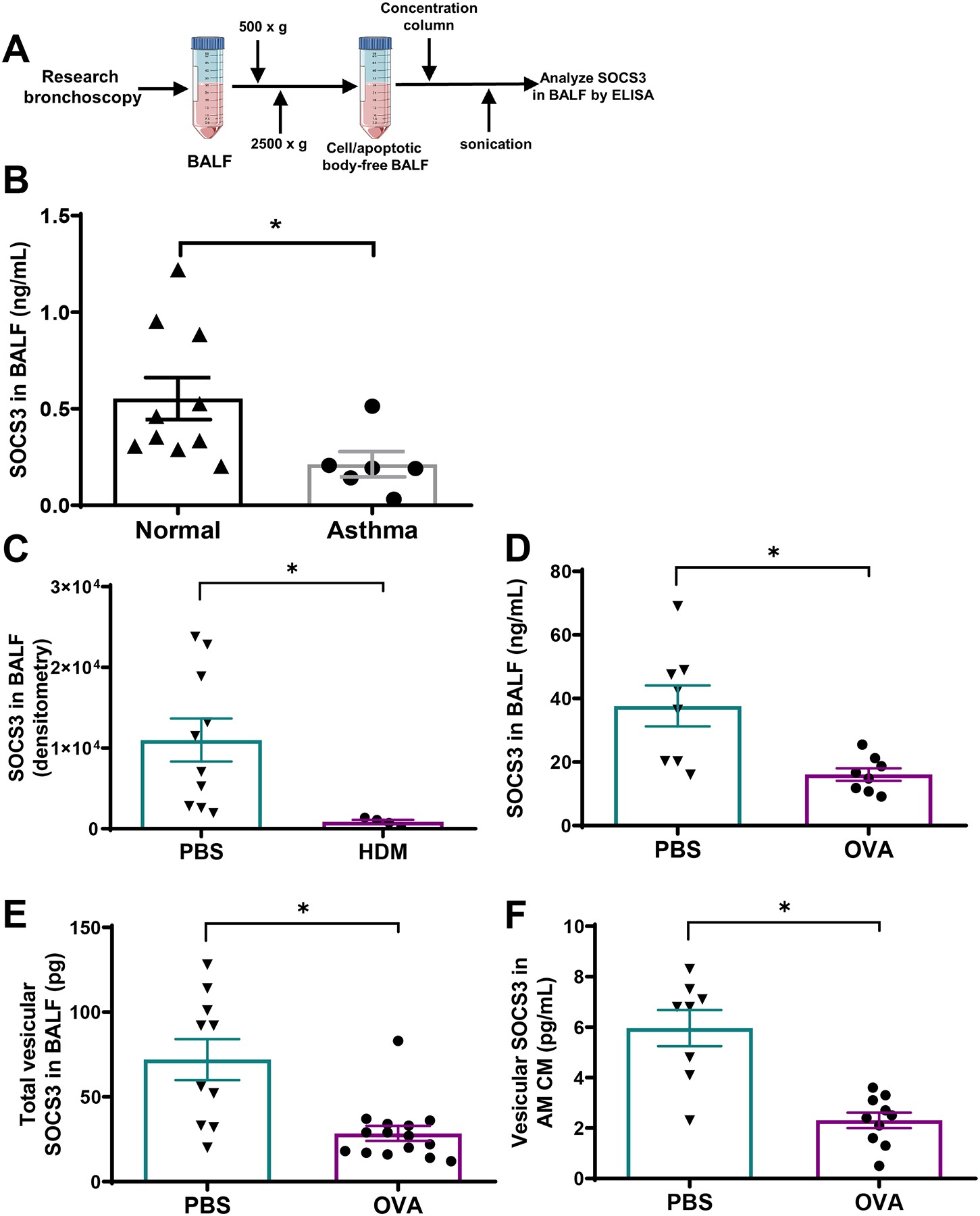
(A) Schematic depiction of human subject BALF processing for SOCS3 ELISA. SOCS3 protein levels in sonicated lavage BALF of both normal and asthmatic subjects (B), HDM-challenged mice (C), and OVA-challenged mice (D). SOCS3 protein levels in vesicular fraction of BALF of OVA-challenged mice (E) and in vesicular fraction of CM isolated from AMs of OVA-challenged mice. For (C), (D), and (E), SOCS3 was quantitated in samples obtained 24 h after the second of two consecutive daily intrapulmonary allergen challenges. SOCS3 was determined by western blot in (C) and by ELISA in all other panels. Each point represents an individual subject or mouse, and the mean ± SEM from each group is shown. *p<0.05, using a Student t test.
We next evaluated SOCS3 secretion in two mouse models of acute allergic inflammation. Mice were sensitized (day 0) and challenged (day 7 and 8) with either PBS, HDM, or OVA, and lavage fluid was collected on day 9. We have previously reported that both of these protocols cause leukocyte influx and cytokine generation in the lungs (5, 35). As compared to control challenge with PBS, both allergen challenge models exhibited decreases in SOCS3 protein levels contained in either neat BALF subjected to sonication (Fig. 1C and 1D) or in EVs purified from BALF (Fig. 1E). AMs retrieved from allergen-challenged mice exhibited no difference in intracellular SOCS3 relative to cells from PBS-challenged control mice (Supplemental Fig. 1A). However, AMs from allergen-challenged mice secreted significantly less SOCS3 into CM in vitro (Fig. 1F) despite elaborating more EVs (Supplemental Fig. 1B) than AMs from PBS-challenged mice. Thus, the ratio of SOCS3 per EV elaborated was clearly reduced in CM from AMs of challenged mice. These data indicate that homeostatic secretion of SOCS3 by AMs is impaired in animal models of allergic asthma, as it is in human asthma.
Mediators implicated in asthma decrease AM SOCS3 secretion in vitro
The decreases in vesicular SOCS3 secretion observed in asthmatics and allergen-challenged mice could reflect modulation by inflammatory mediators present in the lungs. To explore this possibility, we studied the effects of exogenous addition of the type 2 cytokine IL-4 as well as the EC-derived type 2-promoting cytokines IL-33, TSLP, and IL-25 on in vitro AM SOCS3 expression and secretion. For comparison, we also examined the effect of the anti-inflammatory glucocorticoid dexamethasone. J2-immortalized primary mouse AMs incubated for 48 h with IL-4, IL-33, or TSLP exhibited significantly lower SOCS3 protein secretion (Fig. 2A) in association with a possible reduction in intracellular expression (Fig. 2B); qualitatively similar effects of IL-25 did not reach statistical significance. By contrast, incubation with dexamethasone significantly increased basal SOCS3 secretion without increasing intracellular levels (Fig. 2A and B). These data suggest that inflammatory cytokines are capable of inhibiting SOCS3 secretion by AMs, and this may contribute to the observed defects in asthmatics and allergen-challenged mice.
Figure 2. SOCS3 secretion and expression is reduced in AMs exposed to inflammatory cytokines.
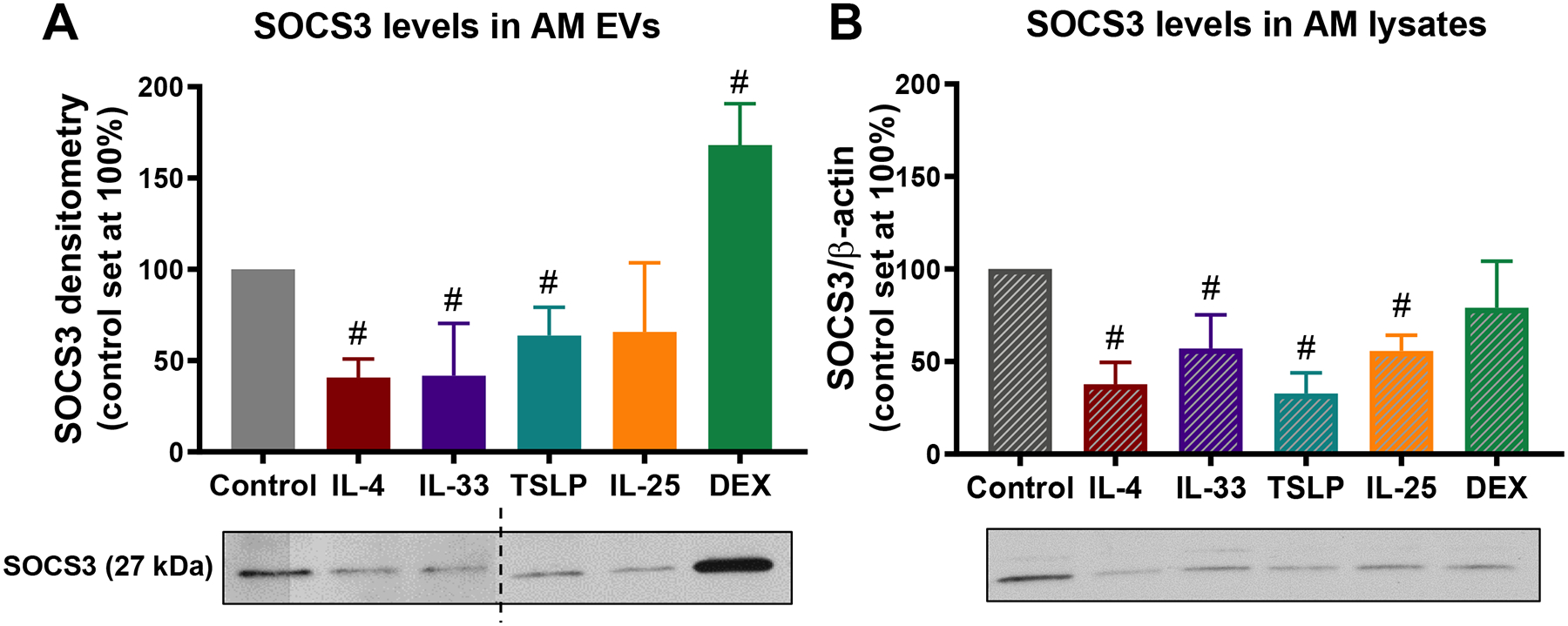
(Top) Relative SOCS3 protein levels determined by western blot in (A) vesicular fraction of CM, and in (B) lysates from AM cell line exposed for 48 h to IL-4 (10 ng/mL), IL-33 (20 ng/mL), TSLP (50 ng/mL), IL-25 (100 ng/mL) or dexamethasone (DEX, 1 μM). (Bottom) Representative SOCS3 WBs from vesicular fraction of CM (A) and cell lysates (B). Dashed line represents non-contiguous lanes from two separate blots. #p<0.05 as compared to unstimulated control, using a one-way ANOVA followed by Sidak’s multiple comparisons test.
IL4 and IL-33 treatment of AMs abrogates the ability of AM-derived CM and EVs to inhibit bronchial EC signaling and mediator gene expression
Previously we reported that SOCS3 protein secreted within AM-derived EVs has the ability to inhibit JAK STAT signaling and expression of its downstream inflammatory genes in alveolar ECs (13). We tested whether these findings extend to bronchial ECs by using the BEAS-2B human bronchial EC line, and to stimuli relevant to allergic airway inflammation, namely the type 2 cytolines IL-4/IL-13 and HDM. Both CM and EVs (added at a typically employed ratio of 5 EVs:1 EC) constitutively elaborated by AMs inhibited bronchial EC STAT3 activation in response to IL-4/IL-13 and HDM (Supplemental Fig. 2A and Fig. 3A), while only EVs significantly inhibited STAT6 activation in response to IL-4/IL-13 (Supplemental Fig. 2B and Fig. 3B). CM and EVs also inhibited BEAS-2B expression of eotaxin-1 (Supplemental Fig. 2C and Fig. 3C), IL-33 (Supplemental Fig. 2E and Fig. 3E), GM-CSF (Supplemental Fig. 2F and Fig. 3F), and IL-6 (Supplemental Fig. 2G and Fig. 3G) induced by IL-4/IL-13 and/or HDM. TSLP expression in BEAS-2B cells was only significantly induced by HDM stimulation, and pre-treatment with either CM or EVs prevented this induction (Supplemental Fig. 2D and Fig. 3D). These data demonstrate that EVs derived from resting AMs and known to contain SOCS3 are capable of inhibiting signaling and gene expression responses of bronchial ECs pertinent to allergic inflammation.
Figure 3. AM-derived EVs inhibit STAT3 and STAT6 activation as well as inflammatory gene expression in challenged bronchial ECs.
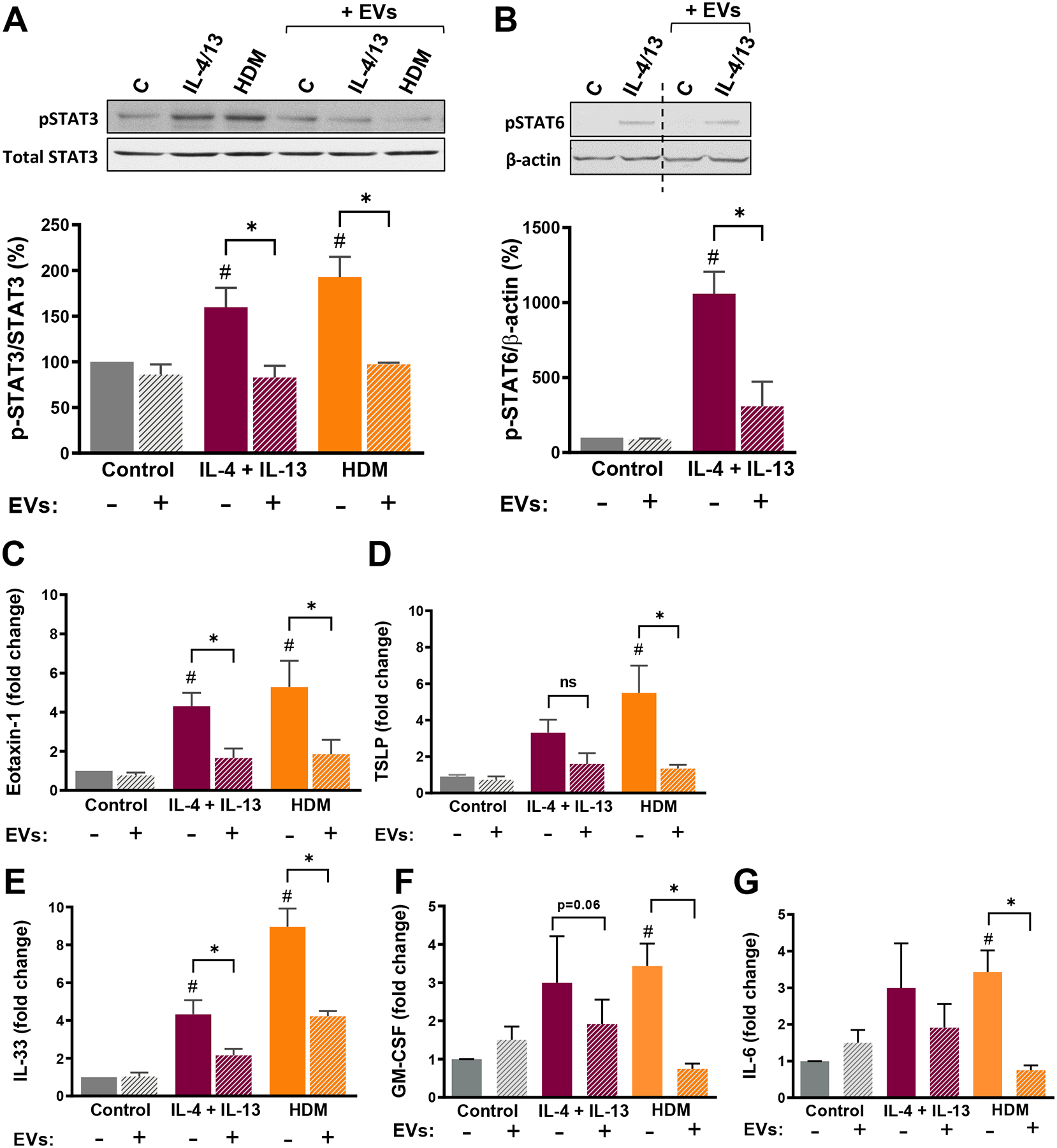
(A) (Top) Representative WBs; (Bottom) Activation of STAT3 (phosphorylated as percentage of total STAT3), and (B) (Top) Representative WBs; (Bottom) activation of STAT6 (phosphorylated as percentage of β-actin) – both determined by densitometry – in BEAS-2B ECs pretreated with or without AM EVs (EV:cell ratio 5:1) and thereafter stimulated with IL-4/IL-13 (10 ng/mL each) or HDM (10 μg/mL) for 1 h. Data represent mean ± SEM from 3–5 independent experiments. Dashed line represents non-contiguous lanes from the same blot. Relative expression of (C) eotaxin-1, (D) TSLP, (E) IL-33, (F) GM-CSF and (G) IL-6 mRNA in BEAS-2B ECs pretreated with or without AM EVs and thereafter stimulated with IL-4/IL-13 (10 ng/mL each) or HDM (10 μg/mL) for 6 h. All data represent fold change relative to untreated control, and are mean ± SEM from 5–6 independent experiments. #p<0.05 as compared to non-pretreated control ECs. *p<0.05 as compared to non-EV-treated cells, using a one-way ANOVA followed by Sidak’s multiple comparisons test.
Next, we asked whether pretreatment of AMs with IL-4, IL-33, or dexamethasone – which altered EV SOCS3 levels – influenced the ability of their secreted EVs to inhibit IL-4/IL-13-induced JAK-STAT signaling in BEAS-2B cells. EVs from dexamethasone-pretreated AMs, containing higher SOCS3 levels than basal AMs, retained the inhibitory effect on IL-4/IL-13-induced STAT3 signaling (Fig. 4A). By contrast, EVs from IL-4-pretreated AMs exhibited no inhibitory effect, while those from IL-33-pretreated AMs actually augmented STAT3 and STAT6 activation in response to IL-4/IL-13 (Fig. 4A and B). These data suggest that impaired secretion of AM SOCS3 in the context of allergic inflammation may result in a failure to restrain STAT-induced inflammation.
Figure 4. EVs from cytokine-pretreated AMs lose their capacity to inhibit STAT activation in response to inflammatory stimuli.
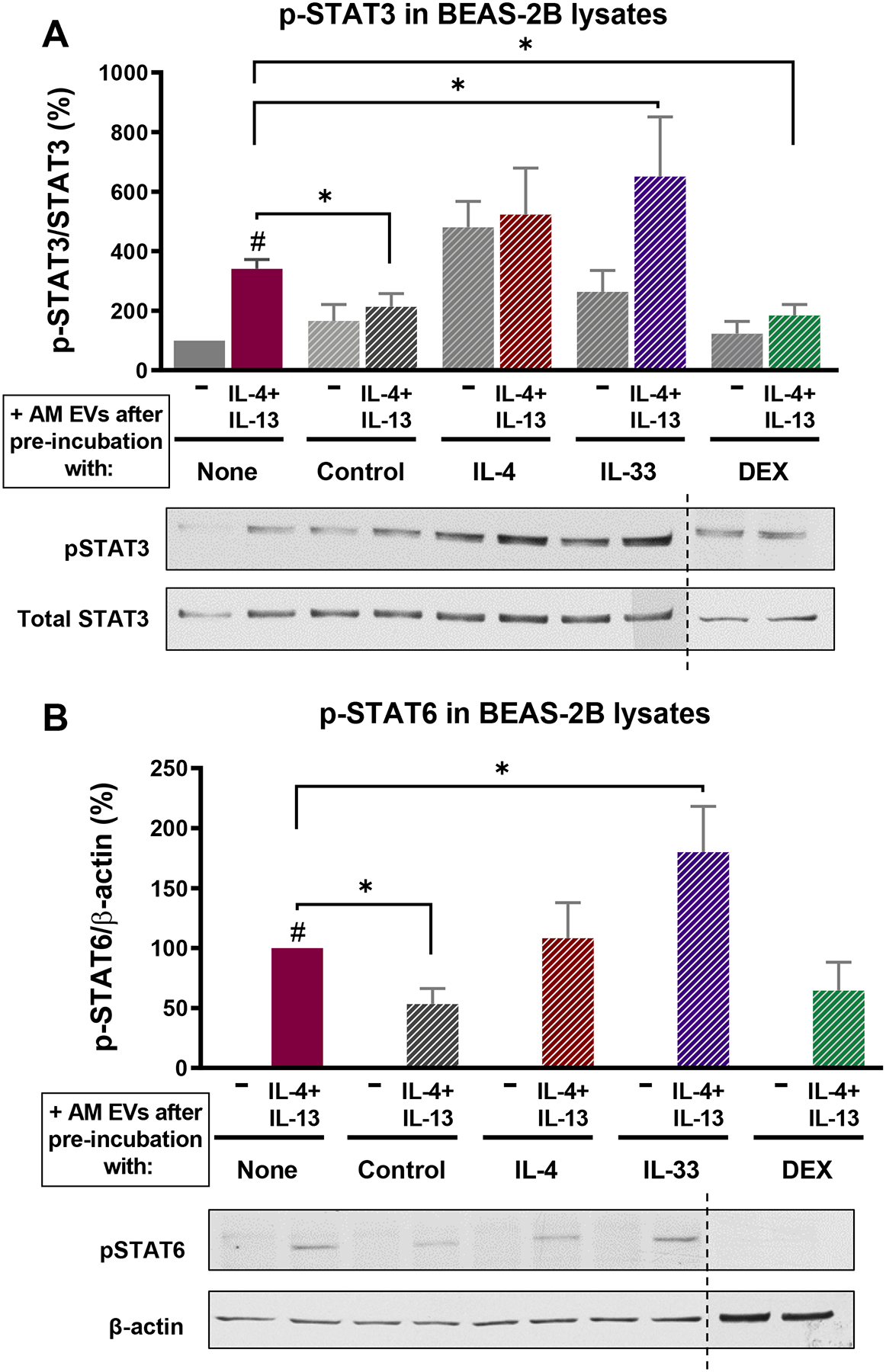
(A) (Top) Activation of STAT3 (as percentage of total STAT3) (Bottom) representative WBs, and (B) (Top) activation of STAT6 (as percentage of β-actin) (Bottom) representative WBs – both determined by densitometry – in BEAS-2B ECs pretreated with EVs from AM cell line exposed for 48 h to either IL-4, IL-33 or DEX, and thereafter stimulated with IL-4/IL-13 (10 ng/mL each) for 1 h. Dashed line represents non-contiguous lanes from two separate blots. All data are mean ± SEM from 5–6 independent experiments. # p<0.05 vs untreated cells; *p<0.05, using a one-way ANOVA followed by Sidak’s multiple comparisons test.
Synthetic SOCS3 liposomes inhibit bronchial EC signaling and mediator expression
Natural AM-derived EVs contain a myriad of other cargo molecules besides SOCS3. In order to determine if SOCS3 itself was sufficient to attenuate bronchial EC signaling, we generated synthetic phospholipid vesicles in which recombinant SOCS3 was the lone cargo molecule, and tested their effects on EC inflammatory responses in vitro. We previously determined that 10 ng of liposomal recombinant SOCS3 protein yielded a degree of STAT3 inhibition bioequivalent to that contained in natural AM EVs from 1 × 106 cells (33). As compared to empty liposomes loaded with PBS alone, SOCS3 liposomes inhibited EC activation of both STAT3 (Fig. 5A) and STAT6 (Fig. 5B) in response to IL-4/13. SOCS3 liposomes also inhibited gene expression of eotaxin-1 (Fig. 5C), TSLP (Fig. 5D) and IL-33 (Fig. 5E) elicited by stimulation with both IL-4/13 and HDM. Thus, these synthetic SOCS3 liposomes exert similar anti-inflammatory actions on bronchial ECs as do natural AM EVs.
Figure 5. Synthetic liposomes containing recombinant SOCS3 inhibit STAT activation and inflammatory cytokine production by BEAS-2B cells.
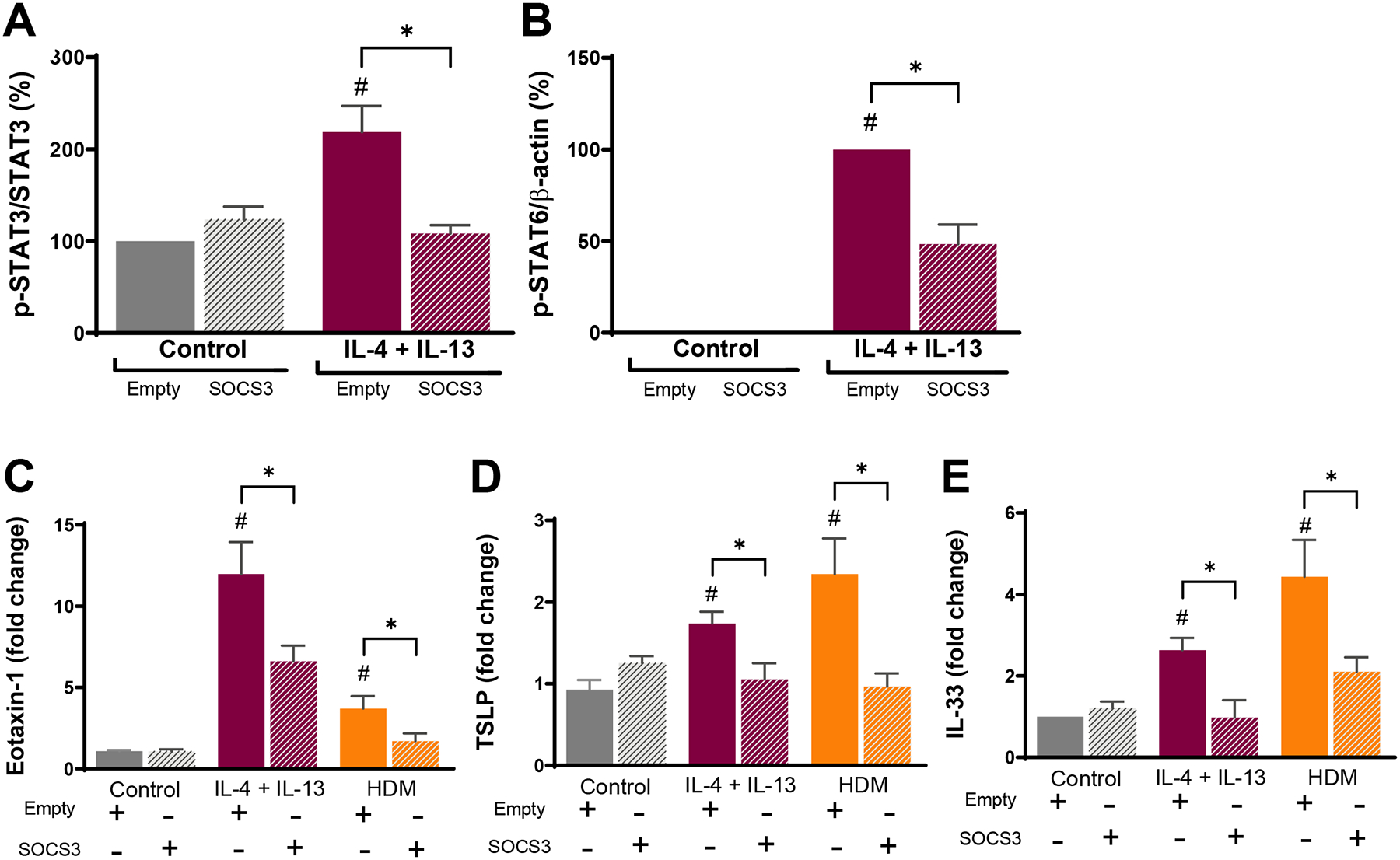
(A) Activation of STAT3 (phosphorylated as percentage of total STAT3) and (B) activation of STAT6 (phosphorylated as percentage of β-actin) – both determined by WB – in BEAS-2B ECs pretreated with empty or SOCS3 liposomes and thereafter stimulated with or without IL-4/IL-13 (10ng/mL each) for 1 h. Relative expression of (C) eotaxin-1, (D) TSLP, and (E) IL-33 mRNA in BEAS-2B ECs pretreated with empty or SOCS3 liposomes and thereafter stimulated with IL-4/IL-13 (10ng/mL each) or HDM (10 μg/mL) for 6 h; data are presented as fold change relative to untreated control. Data from 5 independent experiments. #p<0.05 as compared to control untreated ECs. *p<0.05, using a one-way ANOVA followed by Sidak’s multiple comparisons test.
Synthetic SOCS3 liposomes attenuate development of allergic airway inflammation
The impaired in vivo secretion of AM-derived SOCS3 available for delivery to ECs in the setting of allergic inflammation (Fig. 1) would be expected to disrupt homeostasis and thereby favor inflammatory responses. Since synthetic SOCS3 liposomes proved to be effective in vitro, we sought to test whether they could be used to rescue the endogenous SOCS3 secretory defect in an allergic model of airway inflammation in vivo. Therefore, both male (both PBS groups: n=5; both OVA groups: n=10) and female mice (PBS: n=3; OVA: n=5) were treated o.p. with either empty or SOCS3 liposomes 2 h prior to challenge with PBS or OVA (aerosolized) on both day 7 and 8 (Supplemental Fig. 3); we employed a dose of 20 ng of liposomal SOCS3 per mouse, a dose determined to be bioequivalent to that contained in natural AM EVs which was capable of inhibiting STAT activation in cytokine-treated lungs in vivo (13). PBS challenge elicited no inflammatory response, and neither empty nor SOCS3 liposomes had any effects in PBS-challenged mice (not shown). Increased total cells, eosinophils, and neutrophils were detected in lavage fluid of OVA-challenged mice treated with empty liposomes as compared to PBS challenged mice treated with empty liposomes (Supplemental Fig. 4A and Fig. 6A and B). As previously reported (36, 37), OVA-challenged female mice had significantly higher eosinophil numbers than their male counterparts (Fig. 6A). SOCS3 liposome treatment reduced the number of total cells, eosinophils, and neutrophils in BALF in OVA-challenged mice as compared to empty liposome treatment (Supplemental Fig. 4A and Fig. 6A and B). No differences were observed in total AM numbers in BALF (Supplemental Fig. 4B). SOCS3 liposome treatment affected numbers of total cells, eosinophils and neutrophils similarly in OVA-challenged males and females (Supplemental Fig. 4A and Fig. 6A and B).
Figure 6. Intrapulmonary administration of synthetic liposomes containing recombinant SOCS3 inhibits eosinophil and neutrophil recruitment and inflammatory cytokine generation in the allergen-challenged lung in vivo.
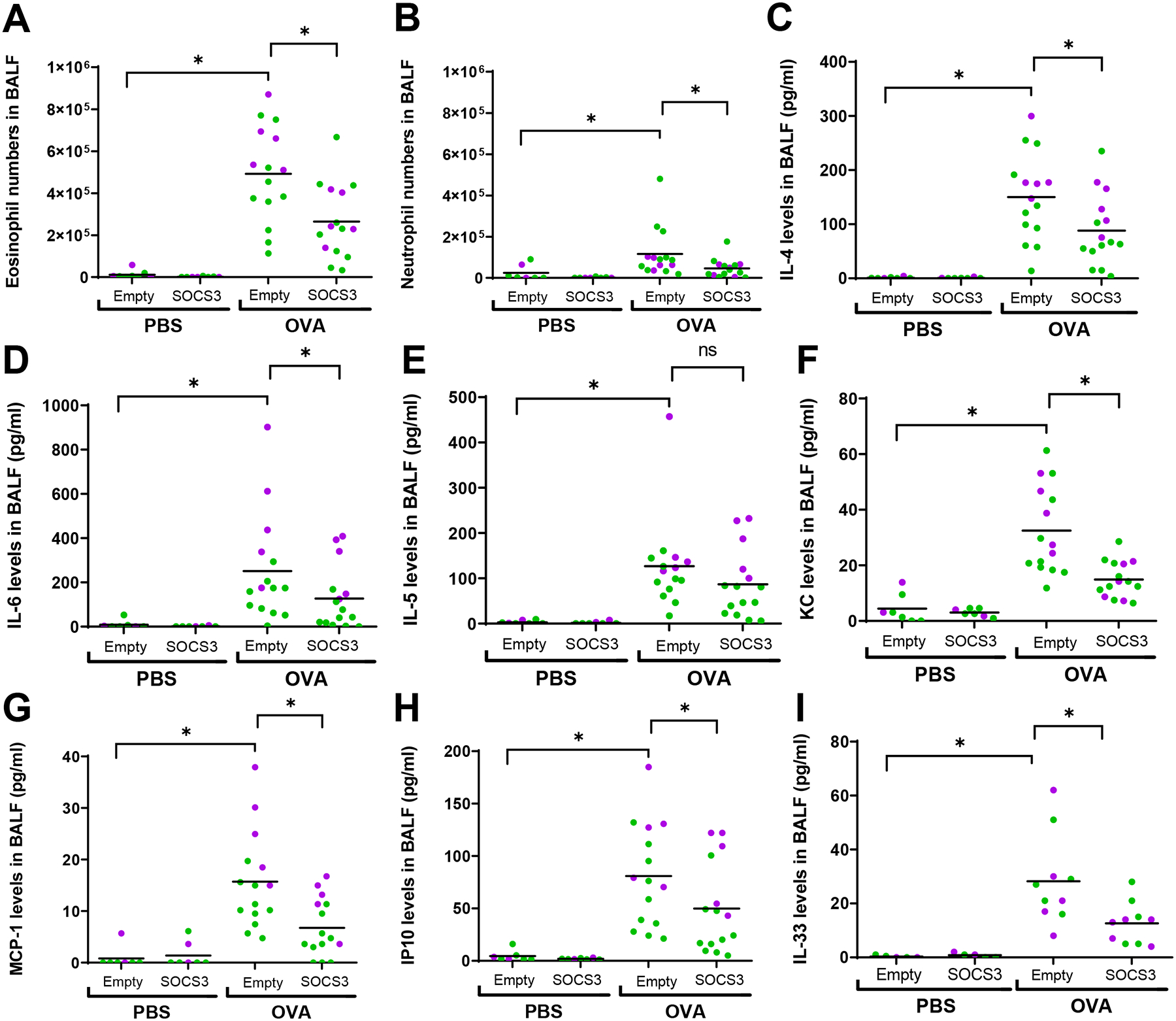
(A) Eosinophil numbers and (B) neutrophil numbers in lavage fluid of OVA-challenged male (green) and female (purple) mice determined 24 h after the second of two consecutive daily intrapulmonary PBS or OVA challenges. Protein levels of (C) IL-4, (D) IL-6, (E) IL-5, (F) KC, (G) MCP-1, (H) IP10 and (I) IL-33 in lavage fluid of PBS- and OVA-challenged male (green) and female (purple) mice determined 24 h after the second of two consecutive daily intrapulmonary PBS or OVA challenges. Empty (PBS) or SOCS3 liposomes were administered o.p. 2 h before challenges on both days. *p<0.05, using a two-way ANOVA followed by Sidak’s multiple comparisons test.
Lavage fluid of empty liposome-treated and OVA-challenged mice exhibited elevated levels of inflammatory cytokines (IL-4, IL-5, IL-6, and IL-33) and chemokines (KC, MCP-1 and IP10) (Fig. 6C–I). As compared to treatment with empty liposomes, SOCS3 liposome treatment in OVA-challenged mice reduced the levels of IL-4, IL-6, KC, MCP-1, IP10 (Fig. 6C, E, F, G, H), and IL-33 (Fig. 6I). As was also true for eosinophil numbers, we observed higher levels of IL-6, IL-5, MCP-1 and IP10 in OVA-challenged females as compared to males (Fig. 6D, E, G and H). For most of these cytokines/chemokines, the magnitude of the inhibitory effect of SOCS3 liposome treatment was similar in both sexes; however, SOCS3 liposomes lowered MCP-1 levels more in OVA-challenged females than in OVA-challenged males (Fig. 6G). These data demonstrate that intrapulmonary treatment with SOCS3 liposomes abrogates both the cellular and molecular components of allergic inflammation.
Synthetic SOCS3 liposomes attenuate macrophage polarization during allergic airway inflammation
BALF cells from empty and SOCS3 liposome-treated PBS and OVA-challenged mice were pooled and plated, and those remaining adherent following 18-h culture were predominantly macrophages. By qPCR analysis, cells from OVA-challenged mice that had been treated with empty liposomes showed substantial upregulation of mRNA for the M1 marker inducible nitric oxide synthase (iNOS) (Fig. 7A) and the M2 markers FIZZ1, YM1 and IRF4 relative to that observed following PBS challenge (Fig. 7D–F). As compared to empty liposome treatment, SOCS3 liposome administration to OVA-challenged mice attenuated ex vivo macrophage expression of M1 markers iNOS, TLR4 and IRF5 (Fig. 7A–C) as well as M2 markers FIZZ1, YM1 and IRF5 (Fig. 7D–F). These data indicate that while M2 polarization predominates in OVA-sensitized and challenged mice, treatment with SOCS3 liposomes is able to inhibit macrophage polarization to both M1 and M2 phenotypes, as reflected by reduced expression of cell surface markers (iNOS, TLR4, FIZZ1, and YM1) as well as of key responsible transcription factors (IRF4 and IRF5).
Figure 7. Intrapulmonary administration of synthetic liposomes containing recombinant SOCS3 inhibits AM polarization.
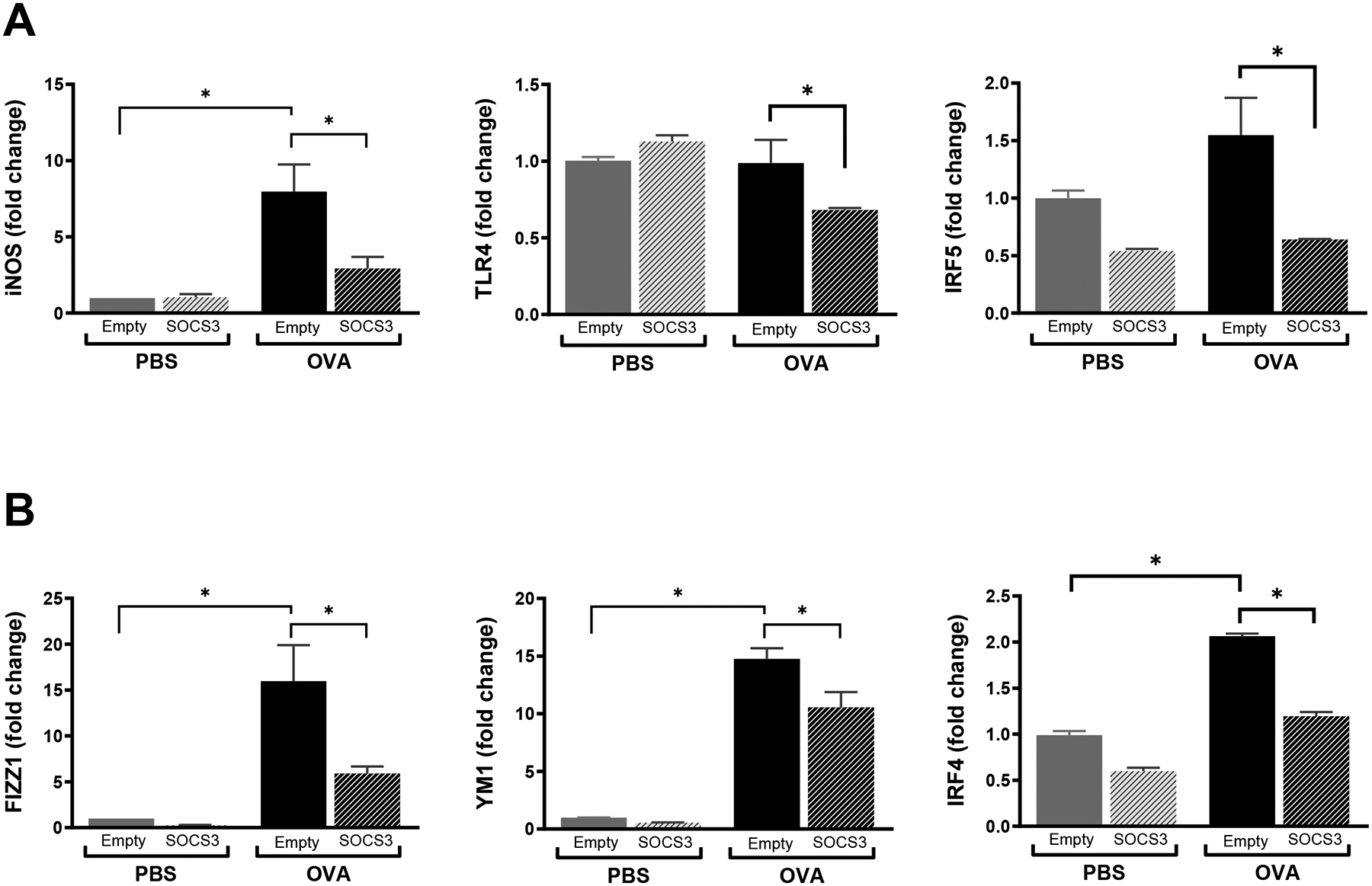
mRNA expression of M1 markers (A) iNOS, (B), TLR4, (C) IRF5 and M2 markers (D) FIZZ1, (E) YM1 and (F) IRF4 in AMs isolated from lavage fluid of PBS- and OVA-challenged mice pretreated with empty liposomes or SOCS3 liposomes. *p<0.05, using a one-way ANOVA followed by Sidak’s multiple comparisons test.
Discussion
Here we interrogated the role of anti-inflammatory SOCS3, secreted within EVs by AMs, in allergic inflammatory responses in the airways. Our key findings were that: 1) basal SOCS3 secretion in the lung is impaired in both asthmatics and in mouse models of allergic asthma; 2) AM-derived SOCS3 dampens bronchial EC inflammatory responses in vitro; and 3) intrapulmonary administration of liposomal recombinant SOCS3 can rescue the secretory defect in mice and exert broad suppression of diverse inflammatory pathways and events.
The complex interplay between multiple cell types during airway inflammation in response to allergens remains incompletely defined. Clearly, however, due to their location in the airway and close proximity to each other, bronchial ECs and AMs are the first lines of contact for inhaled allergens. Their cooperation is essential for proper inflammatory responses and maintenance of homeostasis, and as the resident immune cell of the pulmonary mucosal surface, AMs are in the perfect position to orchestrate these responses. We previously demonstrated that constitutive secretion of vesicular SOCS3 by AMs could be further potentiated in response to a “request signal” received from ECs in order to dampen their inflammatory response during infection (14). Data presented herein show that during acute allergic airway inflammation, AMs are unable to answer such a request, and their impaired secretion of endogenous SOCS3 would be expected to facilitate airway inflammation.
We acknowledge two limitations of this study. First, we have not directly studied the inflammation-modulatory actions of EVs derived from human AMs, but instead draw inferences from the actions of EVs elaborated by mouse AM cell lines. This reflects the logistical realities of ready access to large numbers of mouse AMs from which EVs can be isolated, as opposed to the limited access to AMs from asthmatic and control humans. However, we have previously confirmed that AMs from human subjects do indeed secrete SOCS3 ex vivo (13), and would therefore anticipate similar anti-inflammatory potential. Second, we have not definitively determined that SOCS3 secreted onto the mucosal surface in vivo and recovered in BALF from normal humans or mice derives exclusively from resident AMs. However, we have previously reported (13, 14) that in contrast to the abundant capacity of AMs to secrete SOCS3, that exhibited by a variety of other cell types (alveolar ECs, neutrophils, monocytes, lung fibroblasts, and certain other macrophage populations) is minimal. While we anticipate that resident AMs are the predominant source of secreted SOCS3 in vivo, we cannot exclude contributions from other cell types.
In our studies, we observed that AMs secreted less SOCS3 during acute allergic airway inflammation, and we sought to gain some insight into the responsible mechanism(s) using both our in vitro and in vivo models. In vitro exposure of the AM cell line to IL-4, IL-33 or TSLP – mediators shown to be increased in the milieu of the inflamed lung – reduced intracellular SOCS3 protein levels as compared to those observed in untreated AMs. This approach, albeit reductionist in nature, suggests that the decrease in SOCS3 secretion by AMs could reflect less intracellular SOCS3 protein available for packaging into EVs, perhaps because of interference with transcription/translation of the protein during inflammation. By contrast, in the in vivo HDM model, SOCS3 secretion by AMs was diminished despite these cells exhibiting similar intracellular SOCS3 protein levels and elaborating more total EVs as compared to AMs from PBS-challenged mice. These data suggest that reduced SOCS3 secretion was attributable instead to lower amounts of SOCS3 protein incorporated per secreted EV. Taken together, our data suggest that while inflammatory cytokines have the potential to reduce SOCS3 secretion by suppressing intracellular expression of the protein, the predominant operative mechanism in vivo may instead involve defective packaging of intracellular SOCS3 within EVs. Indeed, there is precedent for the amount of SOCS3 protein packaged per EV to be directly modulated independent of bulk intracellular levels (13). Further work is needed to definitively elucidate the molecular mechanisms responsible for diminished SOCS3 secretion within AM EVs in the setting of allergic inflammation.
We previously reported that various pro- and anti-inflammatory signaling molecules could bidirectionally regulate AM secretion of SOCS3 (13). Additionally, we demonstrated that innate immune activation and/or type I inflammation (i.e. acute bacterial and viral infections) results in a significant increase in SOCS3 secretion in the lungs of naïve mice (14). These current data extend the phenomenon of dynamic regulation of AM SOCS3 secretion to substances important in modulating type 2 inflammatory responses. Although the precise quantitative relationship between SOCS3 content and anti-inflammatory actions of EVs remains to be clarified, our data do point to a qualitative relationship in that EVs from AMs treated with type 2-associated cytokines such as IL-4, IL-13 and TSLP – exhibiting reduced SOCS3 – exhibited a parallel loss of their anti-inflammatory actions on ECs in vitro. We would anticipate that a similar loss of anti-inflammatory activity would accompany the reduced SOCS3 content of EVs elaborated by AMs from allergen-challenged mice in vivo. Unfortunately, this possibility could not be feasibly tested directly because the numbers of EVs that can be obtained from AMs isolated from these mice is limiting. However, the fact that supplementation of SOCS3 protein via synthetic liposomes resulted in reduced inflammatory cytokine production and cell recruitment in allergic mice indirectly suggests that loss of AM SOCS3 secretion facilitates enhanced allergic inflammation.
A variety of signaling pathways and transcription factors (such as STAT1, STAT3, and STAT6) up-regulated or activated in the airway epithelium have been shown to be important in mediating airway responses to allergens in both mouse models and in asthmatics (38–40). Our current studies demonstrate that endogenously released AM-derived EVs containing SOCS3 are capable of inhibiting the activation of both STAT3 and STAT6, indicating a broad anti-inflammatory repertoire. Importantly, we found that this homeostatic control mechanism is disrupted during allergic inflammation, prompting an opportunity for therapeutic rescue. Attempting to accomplish this with natural AM-derived EVs is potentially problematic. First, the presence of myriad cargo molecules other than SOCS3 in natural EVs could confound therapeutic studies. Second, the prospect of treatment with natural EVs poses a variety of immunologic, logistical, and regulatory challenges. To circumvent these limitations, we engineered synthetic phospholipid-based liposomes whose only cargo is recombinant SOCS3. Our findings indicate that these liposomes possess anti-inflammatory actions when delivered to either bronchial ECs in vitro or by intrapulmonary administration in mouse models of allergic inflammation. They inhibited activation of STAT3 and STAT6 in bronchial ECs. In vivo, they inhibited inflammatory cell influx, the expression of a wide range of cytokines and chemokines, as well as inhibition of AM polarization to both M1 and M2 phenotypes; the latter is important because polarized macrophages are key drivers of inflammatory responses (41, 42). Homogeneous liposomes of uniform phospholipid and SOCS3 cargo content and size could be readily synthesized to large scale. Moreover, synthetic SOCS3 liposomes appear to target a broader range of inflammatory signaling pathways than do pharmacologic inhibitors of JAK. These features endow SOCS3 liposomes with the potential to comprise an innovative anti-inflammatory approach for the treatment of asthma.
In conclusion, we have identified the constitutive secretion of SOCS3 within EVs as one potential mechanism by which resident AMs restrain allergic inflammatory responses in the airways. Vesicular SOCS3 exerts broad inhibitory effects on various aspects of inflammatory signaling and responses within airway ECs. AM incorporation of SOCS3 protein within EVs is known to be a regulatable phenomenon, and we demonstrate here that this process can be reduced by type 2 cytokines in vitro and during allergic inflammation in vivo. We speculate that the impairment of this homeostatic anti-inflammatory brake promotes the development and persistence of airway inflammation. Its restoration by intrapulmonary administration of SOCS3 within synthetic vesicles can attenuate inflammation. Future studies will be required to evaluate the importance of this defect in SOCS3 secretion and the therapeutic potential of synthetic SOCS3 liposomes in more chronic models of allergic airway inflammation.
Supplementary Material
Acknowledgments
We thank Dr. Jeanne Stuckney and Krishnapriya Chinnaswamy from the University of Michigan Center for Structural Biology for their help with recombinant SOCS3 purification. We also thank Molly Cook from the Huang lab for her help collecting and processing the asthma BALF specimens. Finally, we thank the members of the Peters-Golden lab for their input and support during this project.
Research funding:
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL 12555 and R35 H144979 (to M.P.G.), NIH Training Grant T32 HL 7749-23 (to C.D. and J.M.S.), an American Cancer Society Fellowship (to J.M.S.), and an AHA Fellowship Award (to L.R.P.).
Abbreviations
- AM
alveolar macrophage
- DMEM
Dulbecco’s modified Eagle medium
- EC
epithelial cell
- EV
extracellular vesicle
- FIZZ1
found in inflammatory zone 1, also termed resistin like alpha
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HDM
house dust mite
- IRF
interferon regulatory factor
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IP10
interferon-γ-induced protein 10
- JAK
Janus kinase
- MCP-1
monocyte chemoattractant protein 1
- OVA
ovalbumin
- qPCR
quantitative real-time polymerase chain reaction
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- TLR
Toll-like receptor
- TSLP
thymic stromal lymphopoietin
- YM1
also termed “chitinase-like 3
References
- 1.Galli SJ, Tsai M, and Piliponsky AM (2008) The development of allergic inflammation. Nature 454, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thepen T, Kraal G, and Holt PG (1994) The role of alveolar macrophages in regulation of lung inflammation. Ann N Y Acad Sci 725, 200–206 [DOI] [PubMed] [Google Scholar]
- 3.Hussell T, and Bell TJ (2014) Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol 14, 81–93 [DOI] [PubMed] [Google Scholar]
- 4.Peters-Golden M (2004) The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol 31, 3–7 [DOI] [PubMed] [Google Scholar]
- 5.Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, and Peters-Golden M (2014) Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol 193, 4245–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Careau E, and Bissonnette EY (2004) Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. Am J Respir Cell Mol Biol 31, 22–27 [DOI] [PubMed] [Google Scholar]
- 7.Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G, and Thepen T (1993) Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med 177, 397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, Karpurapu M, Deng J, Qian F, Kelly EA, Jarjour NN, Ackerman SJ, Natarajan V, Christman JW, and Park GY (2015) Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol 52, 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draijer C, and Peters-Golden M (2017) Alveolar Macrophages in Allergic Asthma: the Forgotten Cell Awakes. Curr Allergy Asthma Rep 17, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, and Bhattacharya J (2014) Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506, 503–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyde DM, Tyler NK, Putney LF, Singh P, and Gundersen HJ (2004) Total number and mean size of alveoli in mammalian lung estimated using fractionator sampling and unbiased estimates of the Euler characteristic of alveolar openings. Anat Rec A Discov Mol Cell Evol Biol 277, 216–226 [DOI] [PubMed] [Google Scholar]
- 12.Colombo M, Raposo G, and Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30, 255–289 [DOI] [PubMed] [Google Scholar]
- 13.Bourdonnay E, Zaslona Z, Penke LR, Speth JM, Schneider DJ, Przybranowski S, Swanson JA, Mancuso P, Freeman CM, Curtis JL, and Peters-Golden M (2015) Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med 212, 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speth JM, Bourdonnay E, Penke LR, Mancuso P, Moore BB, Weinberg JB, and Peters-Golden M (2016) Alveolar Epithelial Cell-Derived Prostaglandin E2 Serves as a Request Signal for Macrophage Secretion of Suppressor of Cytokine Signaling 3 during Innate Inflammation. J Immunol 196, 5112–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimura A, Ito M, Chikuma S, Akanuma T, and Nakatsukasa H (2018) Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb Perspect Biol 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider DJ, Speth JM, Penke LR, Wettlaufer SH, Swanson JA, and Peters-Golden M (2017) Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J Biol Chem 292, 20897–20910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee S, Biehl A, Gadina M, Hasni S, and Schwartz DM (2017) JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 77, 521–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashino S, Takeda K, Li H, Taylor V, Joetham A, Pine PR, and Gelfand EW (2014) Janus kinase 1/3 signaling pathways are key initiators of TH2 differentiation and lung allergic responses. J Allergy Clin Immunol 133, 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsunaga Y, Inoue H, Fukuyama S, Yoshida H, Moriwaki A, Matsumoto T, Matsumoto K, Asai Y, Kubo M, Yoshimura A, and Nakanishi Y (2011) Effects of a Janus kinase inhibitor, pyridone 6, on airway responses in a murine model of asthma. Biochem Biophys Res Commun 404, 261–267 [DOI] [PubMed] [Google Scholar]
- 20.Vale K (2016) Targeting the JAK-STAT pathway in the treatment of ‘Th2-high’ severe asthma. Future Med Chem 8, 405–419 [DOI] [PubMed] [Google Scholar]
- 21.Calbet M, Ramis I, Calama E, Carreno C, Paris S, Maldonado M, Orellana A, Calaf E, Pauta M, De Alba J, Bach J, and Miralpeix M (2019) Novel Inhaled Pan-JAK Inhibitor, LAS194046, Reduces Allergen-Induced Airway Inflammation, Late Asthmatic Response, and pSTAT Activation in Brown Norway Rats. J Pharmacol Exp Ther 370, 137–147 [DOI] [PubMed] [Google Scholar]
- 22.Li RF, and Wang GF (2018) JAK/STAT5 signaling pathway inhibitor ruxolitinib reduces airway inflammation of neutrophilic asthma in mice model. Eur Rev Med Pharmacol Sci 22, 835–843 [DOI] [PubMed] [Google Scholar]
- 23.Kubo M, and Inoue H (2006) Suppressor of cytokine signaling 3 (SOCS3) in Th2 cells evokes Th2 cytokines, IgE, and eosinophilia. Curr Allergy Asthma Rep 6, 32–39 [DOI] [PubMed] [Google Scholar]
- 24.Zafra MP, Canas JA, Mazzeo C, Gamez C, Sanz V, Fernandez-Nieto M, Quirce S, Barranco P, Ruiz-Hornillos J, Sastre J, and del Pozo V (2015) SOCS3 silencing attenuates eosinophil functions in asthma patients. Int J Mol Sci 16, 5434–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Oliveira JR, Favarin DC, Tanaka SC, Balarin MA, Teixeira DN, Levy BD, and Rogerio Ade P (2015) AT-RvD1 modulates CCL-2 and CXCL-8 production and NF-kappaB, STAT-6, SOCS1, and SOCS3 expression on bronchial epithelial cells stimulated with IL-4. Biomed Res Int 2015, 178369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nasreen N, Khodayari N, Sukka-Ganesh B, Peruvemba S, and Mohammed KA (2012) Fluticasone propionate and Salmeterol combination induces SOCS-3 expression in airway epithelial cells. Int Immunopharmacol 12, 217–225 [DOI] [PubMed] [Google Scholar]
- 27.Lambrecht BN, and Hammad H (2012) The airway epithelium in asthma. Nat Med 18, 684–692 [DOI] [PubMed] [Google Scholar]
- 28.Holgate ST (2011) The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev 242, 205–219 [DOI] [PubMed] [Google Scholar]
- 29.Zaslona Z, Okunishi K, Bourdonnay E, Domingo-Gonzalez R, Moore BB, Lukacs NW, Aronoff DM, and Peters-Golden M (2014) Prostaglandin E(2) suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allergy Clin Immunol 133, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mbawuike IN, and Herscowitz HB (1989) MH-S, a murine alveolar macrophage cell line: morphological, cytochemical, and functional characteristics. J Leukoc Biol 46, 119–127 [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Imrich A, and Kobzik L (2008) Characterization of immortalized MARCO and SR AI/II-deficient murine alveolar macrophage cell lines. Part Fibre Toxicol 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spinelli C, Montermini L, Meehan B, Brisson AR, Tan S, Choi D, Nakano I, and Rak J (2018) Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J Extracell Vesicles 7, 1490144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speth JM, Penke LR, Bazzill JD, Park KS, de Rubio RG, Schneider DJ, Ouchi H, Moon JJ, Keshamouni VG, Zemans RL, Lama VN, Arenberg DA, and Peters-Golden M (2019) Alveolar macrophage secretion of vesicular SOCS3 represents a platform for lung cancer therapeutics. JCI Insight 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fricker M, and Gibson PG (2017) Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J 50 [DOI] [PubMed] [Google Scholar]
- 35.Salama N, and El-Sawy M (2013) Isolated low follicle stimulating hormone (FSH) in infertile males - a preliminary report. Arch Ital Urol Androl 85, 118–124 [DOI] [PubMed] [Google Scholar]
- 36.Draijer C, Robbe P, Boorsma CE, Hylkema MN, and Melgert BN (2013) Characterization of macrophage phenotypes in three murine models of house-dust-mite-induced asthma. Mediators Inflamm 2013, 632049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melgert BN, Postma DS, Kuipers I, Geerlings M, Luinge MA, van der Strate BW, Kerstjens HA, Timens W, and Hylkema MN (2005) Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin Exp Allergy 35, 1496–1503 [DOI] [PubMed] [Google Scholar]
- 38.Sampath D, Castro M, Look DC, and Holtzman MJ (1999) Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J Clin Invest 103, 1353–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simeone-Penney MC, Severgnini M, Tu P, Homer RJ, Mariani TJ, Cohn L, and Simon AR (2007) Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J Immunol 178, 6191–6199 [DOI] [PubMed] [Google Scholar]
- 40.Kaplan MH, Schindler U, Smiley ST, and Grusby MJ (1996) Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 4, 313–319 [DOI] [PubMed] [Google Scholar]
- 41.Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy B, Geerlings M, Hylkema MN, and Ray A (2010) Macrophages: regulators of sex differences in asthma? Am J Respir Cell Mol Biol 42, 595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung S, Lee TJ, Reader BF, Kim JY, Lee YG, Park GY, Karpurapu M, Ballinger MN, Qian F, Rusu L, Chung HY, Unterman TG, Croce CM, and Christman JW (2016) FoxO1 regulates allergic asthmatic inflammation through regulating polarization of the macrophage inflammatory phenotype. Oncotarget 7, 17532–17546 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.