

Abstract
Per-and polyfluoroalkyl substances (PFAS) occurrence in drinking water and treatment methods for their removal are reviewed. PFAS are fluorinated substances whose unique properties make them effective surface-active agents with uses ranging from stain repellants to fire-fighting foams. In response to concerns about drinking water contamination and health risks from PFAS exposure, the United States Environmental Protection Agency published Health Advisories (HAs) for perfluorooctanoic acid and perfluorooctane sulfonic acid. The occurrence of six PFAS in drinking water has been reported in the Third Unregulated Contaminant Monitoring Rule (UCMR3), and subsequent analysis of the dataset suggested that four percent of water systems reported at least one detectable PFAS compound and 1.3 percent of water systems reported results above the HAs. Many treatment technologies have been evaluated in the literature, with the most promising and readily applied treatment technologies being activated carbon, anion exchange resins, and high-pressure membrane systems. From these data and literature reports, research and data gaps were identified and suggestions for future research are provided.
Keywords: Per- and Polyfluoroalkyl Substances, PFAS, Activated Carbon Adsorption, Anion Exchange Resin, Reverse Osmosis, Drinking Water Treatment, UCMR3
1.0. INTRODUCTION
1.1. History, Properties, and Exposure Pathways of Per-and polyfluoroalkyl Substances
Per-and polyfluoroalkyl substances (PFAS) are fluorinated aliphatic substances with unique properties, such as being hydrophobic, lipophobic, and extremely stable due to the strength of the C-F bond (Buck et al. 2011). These properties led to their extensive use as surface active agents in products like stain repellants and fire-fighting foams (Xiao et al. 2017, Zhi & Liu 2015, Buck et al. 2011). Perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are two of the most well-known and broadly researched PFAS compounds. They first came into production in the 1940s, and related health concerns were raised as early as the 1960s (Grandjean & Clapp 2015).
Exposure to PFOA has been associated with high cholesterol, increased liver enzymes, decreased vaccination response, thyroid disorders, pregnancy-induced hypertension and preeclampsia, and testicular and kidney cancer (USEPA 2016b). Similarly, exposure to PFOS is associated with high cholesterol and thyroid disease as well as immune suppression and reduced fertility and fecundity (USEPA 2016a).
PFOA and PFOS contaminated food intake has been found to be the primary exposure pathway for adults while dust and dietary ingestion are for children (USEPA 2016b, 2016a). However, drinking water is the major exposure pathway in communities with contaminated water (Vestergren & Cousins 2009). PFAS are bioaccumulative, and PFOS and PFOA have serum elimination half-lives of 5.4 and 3.8 years, respectively (Olsen et al. 2007) and were detected in the blood serum of 99% of the United States (US) general population that was tested between 1999 and 2012 (USEPA 2016b, 2016a, Olsen et al. 2007).
Concerns about PFOS and PFOA health effects led to the voluntary phase out of PFOS production by the 3M Company in 2000. In 2006, the United States Environmental Protection Agency (USEPA) began the PFOA Stewardship Program which worked with eight leading PFAS manufacturers to achieve a 95% reduction from a year 2000 baseline of PFOA, PFOA precursors, and higher homologues from emissions and media by 2010 and to completely eliminate PFOA from emissions and products by 2015 (USEPA 2006). In 2009, the first provisional health advisories (HAs) for PFOS and PFOA were issued by the USEPA (Grandjean & Clapp 2015) and finalized drinking water HAs of 70 ng/L for PFOS and PFOA, both individually and combined, were issued in 2016 (USEPA 2016b, 2016a). It should be noted, however, that HA levels have only been established for PFOA and PFOS, and the risks to human health for other PFAS have not been determined by the USEPA, although many have been detected in drinking water (USEPA 2017). USEPA has released draft toxicity assessments for perfluorobutane sulfonic acid (PFBS) and perfluoro-2-propoxypropanoic acid (PFPrOPrA or “GenX”). All names, classifications, and abbreviations of PFAS referenced in this paper can be found in Table S1 in the Supplementary Information (SI).
The purpose of this review is to provide a comprehensive survey and analysis of the occurrence of PFAS in drinking water sources in the US and their subsequent treatment in drinking water. PFAS review articles published to date have typically assessed PFAS occurrence in a broad suite of aqueous matrices and often lump together source water, treated drinking water, and surface and ground waters with point source contamination, or other environmental samples & matrices from around the globe (Ross et al. 2018, Kucharzyk et al. 2017, Dickenson & Higgins 2016, Cummings et al. 2015, Appleman et al. 2014, Du et al. 2014, Rahman et al. 2014, Buck et al. 2011, McLaughlin et al. 2011). Additionally, evaluation of treatment methods is often broad enough in scope to include both drinking water treatment and remediation technologies conducted with a small suite of PFAS at concentrations several orders of magnitude higher than those observed in drinking water sources and treated drinking waters. To provide a more detailed and focused assessment of PFAS issues related to drinking water in the US, this literature review focuses on the occurrence of a broad suite of PFAS compounds in drinking water sources, in treated drinking water, and on technologies shown to be effective in removing PFAS from drinking water at relevant concentrations. From these data, a summary of gaps in the literature, and recommendations for future research was compiled.
1.2. PFAS Occurrence in Drinking, Surface, and Groundwater in the United States
Occurrence data for six PFAS (perfluorobutanesulfonic acid (PFBS), perfluorohexane sulfonic acid (PFHxS), perfluoroheptanoic acid (PFHpA), PFOA, PFOS, and perfluorononanoic acid (PFNA)) were reported in the USEPA’s third Unregulated Contaminant Monitoring Rule (UCMR3) report released in January 2017 (USEPA 2017). PFBS, PFHxS, and PFOS are perfluoroalkyl sulfonic acids (PFSAs) and are considered long chain when they contain 6 or more carbons (e.g., PFHxS and PFOS). PFHpA, PFOA, and PFNA are perfluoroalkyl carboxylic acids (PFCAs) and considered long chain when they contain 8 or more carbons (e.g. PFOA and PFNA) (Buck et al. 2011). The UCMR was instituted in response to the 1996 amendments to the Safe Drinking Water Act to collect data to support the USEPA Administrator’s determination of whether to regulate particular contaminants that do not have health based standards (USEPA 2016c). The UCMR3 dataset is of considerable significance to PFAS research because there are few national-level sources in the literature reporting on PFAS occurrence in the environment, and of the available sources, many have focused on sampling areas outside the US or on matrices other than drinking water. UCMR3, however, provided nearly 37,000 PFAS sample results, originating from 4,920 US drinking water utilities between 2013 and 2015 (USEPA 2017). All large water systems (i.e., those serving >10,000 people) conducted PFAS monitoring under UCMR3. The selection process for small systems (i.e., those serving ≥10,000) sought to account for differences in system size, source water types, and geographic location to achieve a representative set of small system results. The public water systems (PWS) selected for inclusion in UCMR3 supplied approximately 79% of the US population (USEPA 2017). Therefore, the UCMR3 data represents the most directly-relevant information on PFAS occurrence in US drinking water.
Table 1 summarizes the detected PFAS concentrations during UCMR3 sampling, broken out both by system size and source (i.e., surface or groundwater) to highlight variations in matrices based on these parameters. Note that all references to means from this dataset pertain only to reported detections above the minimum reporting level (MRL). PFBS was found only in large systems with low detection frequency (0.05%) but with notably high mean concentrations of 212 (surface water) and 136 ng/L (groundwater). PFBS also had the highest MRL of all monitored PFAS at 90 ng/L; therefore, reported PFBS detection may have been underreported relative to other PFAS. PFOA and PFOS were detected most frequently across all system sizes and sources at 1.03% and 0.79%, respectively, and PFOS also had the highest maximum concentration of 7,000 ng/L. The mean PFHxS concentration was highest in small groundwater systems (409 ng/L), but a higher detection frequency (0.86%) and maximum concentration (1,600 ng/L) were found in large groundwater systems. Conversely, PFHpA exhibited the lowest mean concentration in large surface (19 ng/L) and groundwater (28 ng/L) systems. Mean PFNA concentrations were highest in large surface (54 ng/L) and groundwater (35 ng/L) systems.
Table 1 –
| Small Systems – Surface Water (n = 1,199) | Small Systems – Groundwater (n = 2,075) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PFAS | # Detects | Det. Freq.c (%) | Concentration (ng/L) |
PFAS | # Detects | Det. Freq. (%) | Concentration (ng/L) |
||||||||||
| MRLd | Mean | SDe | Median | P90f | Max.g | MRL | Mean | SD | Median | P90 | Max. | ||||||
| PFBS | 0 | 0 | 90 | -- | -- | -- | -- | -- | PFBS | 0 | 0 | 90 | -- | -- | -- | -- | -- |
| PFHxS | 0 | 0 | 30 | -- | -- | -- | -- | -- | PFHxS | 4 | 0.19 | 30 | 409 | 348 | 404 | 718 | 730 |
| PFHpA | 0 | 0 | 10 | -- | -- | -- | -- | -- | PFHpA | 4 | 0.19 | 10 | 41 | 35 | 33 | 76 | 87 |
| PFOA | 0 | 0 | 20 | -- | -- | -- | -- | -- | PFOA | 4 | 0.19 | 20 | 100 | 85 | 81 | 183 | 206 |
| PFOS | 2 | 0.17 | 40 | 54 | 6 | 54 | 58 | 59 | PFOS | 4 | 0.19 | 40 | 158 | 127 | 142 | 279 | 300 |
| PFNA | 0 | 0 | 20 | -- | -- | -- | -- | -- | PFNA | 1 | 0.05 | 20 | 26 | -- | 26 | 26 | 26 |
| Large Systems – Surface Water (n = 13,279) | Large Systems – Groundwater (n = 20,419) | ||||||||||||||||
| PFAS | # Detects | Det. Freq. (%) |
Concentration (ng/L) |
PFAS | # Detects | Det. Freq. (%) |
Concentration (ng/L) |
||||||||||
| MRL | Mean | SD | Median | P90 | Max. | MRL | Mean | SD | Median | P90 | Max. | ||||||
| PFBS | 12 | 0.09 | 90 | 212 | 94 | 185 | 357 | 370 | PFBS | 7 | 0.03 | 90 | 136 | 49 | 110 | 196 | 220 |
| PFHxS | 28 | 0.21 | 30 | 69 | 35 | 62 | 79 | 190 | PFHxS | 175 | 0.86 | 30 | 144 | 174 | 79 | 330 | 1,600 |
| PFHpA | 92 | 0.69 | 10 | 19 | 11 | 15 | 39 | 60 | PFHpA | 140 | 0.69 | 10 | 28 | 43 | 20 | 53 | 410 |
| PFOA | 101 | 0.76 | 20 | 31 | 13 | 29 | 41 | 100 | PFOA | 274 | 1.34 | 20 | 45 | 46 | 30 | 74 | 349 |
| PFOS | 66 | 0.50 | 40 | 77 | 56 | 57 | 140 | 400 | PFOS | 220 | 1.08 | 40 | 200 | 603 | 64 | 383 | 7,000 |
| PFNA | 1 | 0.01 | 20 | 54 | -- | 54 | 54 | 54 | PFNA | 17 | 0.08 | 20 | 35 | 11 | 32 | 52 | 56 |
Systems ≤10,000 population served = “Small” and >10,000 = “Large”;
“Surface water” also includes groundwater under the influence of surface water and mixed sources;
Detection frequency, only detections ≥MRL reported;
Minimum reporting level;
Standard deviation;
90th Percentile;
Maximum; “--” = not applicable
Guelfo and Adamson (2018) performed an extensive analysis of the UCMR3 data and revealed an increasing trend in overall PFAS detection rates over time (2013–2015 quarterly). This was observed for PFOS and PFOA independently as well, despite their phase-outs in 2002 and 2006, respectively. Sun et al. (2016) reported stable PFOA and PFOS concentrations in source water in the Cape Fear watershed, NC from 2006 to 2013. These observations suggest the persistence of PFAS in the environment, which may arise from ongoing generation, historic releases, precursor transformation, or lack of attenuation processes.
Knowledge of PFAS sources can help predict both where contamination may occur and which class of PFAS may be present. Guelfo and Adamson (2018) showed PFAS mixtures in groundwater were likely to be dominated by PFSAs based on the ratio of PFSAs and PFCAs concentrations to total PFAS concentrations. This is not surprising as PFSAs are majority constituents in some aqueous film-forming foams (AFFFs) and have been shown to migrate into groundwater from the application site (Guelfo & Adamson 2018, Xiao et al. 2017). PFCAs, conversely, occurred at higher percentages in surface waters as compared to PFSAs (Guelfo & Adamson 2018). PFCAs enter surface waters from primary or secondary manufacturing facilities through runoff and effluent discharge, atmospheric deposition, from landfills through leachate sent to wastewater treatment plants (WWTPs), and by WWTPs themselves which are ineffective at PFAS removal in general (Guelfo & Adamson 2018, Hu et al. 2016, Taniyasu et al. 2013). Notably, fluoropolymer manufacturing is the source of the highest PFAS concentrations but even relatively minor contributions from WWTPs are significant because they are numerous and typically continuously discharged (Guelfo & Adamson 2018).
The UCMR3 data also showed that 72% of all PFAS detections occurred in groundwater and average total PFAS concentrations were higher in groundwater than in surface water across all system sizes (210 ng/L vs. 90 ng/L). Lower surface water concentrations have been attributed to dilution by the receiving water body and potential complexation with the sediment and natural matter, and this may explain the lower detection frequency in surface waters. One notable exception is PFBS which, likely due to its weaker sorption than the longer-chain PFAS, was the only UCMR3 PFAS compound to be detected more frequently in surface water than groundwater and was only found in large systems (Guelfo & Adamson 2018). Detections of one or more PFAS were 5.6 times more frequent in large than small PWS in UCMR3 when considering both surface and groundwater sources together but small PWS had greater total PFAS concentrations when detected (300 vs. 170 ng/L) (Guelfo & Adamson 2018).
Multiple PFAS were present in approximately 50% of samples with PFAS detections (Guelfo & Adamson 2018). This co-occurrence may result from multiple sources, multiple PFAS within individual products, PFAS substitutions, and/or changing use over time. Several PFAS pairs were found to correlate significantly, notably PFOS and PFHxS, which were both historically used in AFFF and are released from industry and WWTPs (Guelfo & Adamson 2018). Additionally, shifts in PFAS production towards shorter chain compounds and development of fluorinated alternatives to PFOA and PFOS, including perfluoro-2-methoxyacetic acid (PFMOAA) and perfluoro-2-propoxypropanoic acid (PFPrOPrA, or “GenX”), creates a situation where additional PFAS may be present in the future (Sun et al. 2016). Information on many PFAS is extremely limited and no peer-reviewed data exist regarding their fate and transport, while still other PFAS have yet to be identified due to proprietary restrictions or because they are formed as byproducts during manufacturing. Also, the health implications of novel PFAS are largely unknown (Xiao et al. 2017).
Some non-PFAS contaminants are known to co-occur with PFAS (e.g., PFAS from AFFFs and hydrocarbons from fuel fires). Correlations between PFAS and several other non-PFAS contaminants were observed during UCMR3, including 1,4-dioxane (strongest correlation), chlorodifluoromethane, hexavalent chromium, chlorate, and 1,1-dichloroethane (Guelfo & Adamson 2018). However, this does not necessarily mean that these compounds are used or released in conjunction with PFAS, and many environmental factors affect their occurrence and distribution (Guelfo & Adamson 2018).
To illustrate results of additional studies of PFAS occurrence evaluation in the US, data from 13 recent peer-reviewed publications, excluding UCMR3, reporting PFAS occurrence in drinking, surface, and ground water are summarized in Table 2 (Sun et al. 2016, Cummings et al. 2015, Appleman et al. 2014, Lindstrom 2009, Post et al. 2009, Quinones & Snyder 2009, Plumlee et al. 2008, Nakayama et al. 2007, Sinclair et al. 2006, Kannan et al. 2005, Simcik & Dorweiler 2005, Boulanger et al. 2004, Hansen et al. 2002). Note that not all the reported values were source waters for drinking water utilities but were included to illustrate occurrence of a broad range of emerging PFAS in the environment. Studies that characterized known contaminated sites for remediation were considered outside the scope of this review and were not included. In Table 2, the minimum, maximum, median, and mean concentrations are given for each PFAS by water source type where available. For each value, the number of contributing data points is given in parentheses (n). Because the original publications vary in how data are presented, note that (n) represents the number of reported values and not specifically the number of waters, sites, or samples.
Table 2 –
Reported PFAS Concentrations in the United States by Water Source in ng/La
| PFAS Compound | Finished Drinking Water | Groundwater | Surface Water | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Min. | Max. | Median | Mean | Min. | Max. | Median | Mean | Min. | Max. | Median | Mean | |
| PFBA | <5.0 (3) |
27 (2) |
-- | -- | <5.0 (2) |
1,260 (2) |
38.6 (1) |
201 (1) |
<5.0 (5) |
1750 (5) |
12–102 (4) |
12–257 (4) |
| PFBS | 0.43 (3) |
3.6 (3) |
-- | -- | <10 (2) |
76.6 (2) |
26.4 (1) |
36.0 (1) |
<0.25 (6) |
208 (6) |
<10–25.0 (5) |
<10–42.3 (5) |
| PFDA | <0.50 (3) |
1.6 (2) |
-- | <1.0–3.3 (7) |
nd (3) |
19 (8) |
<50 (1) |
<1.0–15 (7) |
<0.50 (8) |
838 (9) |
<25–127 (6) |
<1.0–236 (11) |
| PFHxA | 0.52 (3) |
62 (3) |
-- | <1.0–23 (7) |
<10 (2) |
3970 (7) |
360 (1) |
1–1,144 (7) |
<0.50 (7) |
6,710 (8) |
<10–153 (6) |
<1.0–772 (11) |
| PFHxS | <0.25 (3) |
5.6 (2) |
-- | <1.0–12 (7) |
nd (3) |
87.5 (8) |
46.4 (1) |
1.8–44.8 (7) |
nd (18) |
218 (19) |
<1–17.5 (16) |
<1.0–48.3 (12) |
| PFNA | <0.50 (3) |
55 (3) |
-- | <1.0–9.7 (7) |
<0.50 (2) |
47 (7) |
25.7 (1) |
<1.0–25.7 (7) |
<0.50 (8) |
286 (9) |
<10–52 (7) |
<1.0–70.2 (12) |
| PFOA | nd (12) |
4300 (12) |
nd-5.5 (3) |
<5.0–30 (7) |
nd (14) |
6410 (19) |
nd-1332 (5) |
10–2,305 (14) |
nd (25) |
11,000 (26) |
<10–155 (20) |
<5.0–821 (16) |
| PFOS | <0.25 (3) |
15 (3) |
-- | <1.0–57 (7) |
<10 (3) |
192 (8) |
14.1 (1) |
3.7–59.0 (7) |
<0.25 (21) |
1,090 (22) |
<25–756 (18) |
<1.0–69.2 (15) |
| PFPrOPrA | -- | -- | -- | -- | -- | -- | -- | -- | <10 (3) |
4,560 (3) |
<10–304 (3) |
<10–631 (3) |
| PFPeA | <2.0 (3) |
43 (3) |
-- | -- | <10 (2) |
2,330 (2) |
333 (1) |
782 (1) |
<2.0 (5) |
3,770 (5) |
19–180 (4) |
19–497 (4) |
| PFHpA | <0.50 (3) |
34 (3) |
-- | -- | nd (3) |
5,220 (3) |
1,360 (1) |
2,004 (1) |
nd (8) |
8,250 (8) |
<10–446 (6) |
<10–1,127 (6) |
| PFDS | <0.10 (3) |
-- | -- | -- | nd (2) |
15 (1) |
-- | -- | nd (2) |
44 (1) |
-- | -- |
| FOSA | <0.25 (3) |
1.7 (1) |
-- | -- | nd (2) |
4.3 (1) |
-- | -- | nd (3) |
3.5 (3) |
1.1 (1) |
1.0 (1) |
| EtFOSAA | <0.25 (3) |
2 (1) |
-- | -- | nd (2) |
26 (1) |
-- | -- | nd (3) |
31 (3) |
7.4 (1) |
7.0 (1) |
| MeFOSAA | <0.25 (3) |
0.9 (1) |
-- | -- | <0.25 (1) |
-- | -- | -- | <0.25 (1) |
1.2 (1) |
-- | -- |
| 4:2 FTUCA | <2.0 (3) |
-- | -- | <1.0–1.6 (7) |
<2.0 (1) |
-- | -- | <1.0 (6) |
<2.0 (1) |
-- | 5.67 (1) |
<1.0–10.4 (6) |
| 6:2 FTUCA | <2.0 (3) |
-- | -- | <1.0 (7) |
<2.0 (1) |
-- | -- | <1.0 (6) |
<2.0 (1) |
-- | 1.95 (1) |
<1.0–2.17 (6) |
| 8:2 FTUCA | <2.0 (3) |
-- | -- | -- | <2.0 (1) |
-- | -- | -- | <2.0 (1) |
-- | -- | -- |
| 10:2 FTUCA | <2.0 (3) |
-- | -- | -- | <2.0 (1) |
-- | -- | -- | <2.0 (1) |
-- | -- | -- |
| 4:2 FtS | <0.50 (3) |
-- | -- | -- | <0.50 (1) |
-- | -- | -- | <0.50 (1) |
-- | -- | -- |
| 6:2 FtS | <0.50 (3) |
2.2 (2) |
-- | -- | <0.50 (1) |
1.2 (1) |
-- | -- | <0.50 (1) |
3.7 (1) |
-- | -- |
| 8:2 FtS | <0.50 (3) |
-- | -- | -- | <0.50 (1) |
-- | -- | -- | <0.50 (1) |
-- | -- | -- |
| PFOSulfinate | -- | -- | -- | -- | -- | -- | -- | -- | 2.5 (1) |
18 (1) |
-- | -- |
| PFOSAA | -- | -- | -- | -- | -- | -- | -- | -- | 0.2 (1) |
6.5 (1) |
-- | -- |
| N-EtFOSE | -- | -- | -- | -- | -- | -- | -- | -- | nd (1) |
<2.2 (1) |
17 (1) |
12.5 (1) |
| N-EtFOSA | -- | -- | -- | -- | -- | -- | -- | -- | nd (1) |
<0.6 (1) |
0.3 (1) |
1.3 (1) |
| PFOSA | -- | -- | -- | -- | -- | -- | -- | -- | <10 (2) |
<10 (2) |
<2.2 (1) |
<2.2 (1) |
| PFUnDA | <0.50 (3) |
0.64 (2) |
-- | -- | <0.50 (1) |
0.63 (6) |
-- | -- | <0.50 (2) |
52.1 (2) |
<0.6 (1) |
<0.6 (1) |
| PFDoDA | <0.25 (3) |
-- | -- | -- | <0.25 (1) |
<1.0 (5) |
-- | -- | <0.25 (2) |
4.46 (2) |
<10 (2) |
<10 (2) |
(n) = number of reported values; nd = not detected
Concentrations of PFBS, PFHxS, and PFOS reported in the occurrence data were in the same range as reported in UCMR3. The largest variations from UCMR3 reported values were for PFHpA, PFOA, and PFNA; and the highest concentrations of these were reported in a study that evaluated PFAS transport from land applied sewage sludge known to have elevated PFAS concentrations into public drinking water, private wells, springs, ponds, and soils (Lindstrom 2009). Because UCMR3 did not target “vulnerable” PWSs, it is not surprising that a targeted study would find these higher levels. PFOA had the highest reported concentration in both surface and ground water at 11,000 and 6,410 ng/L, respectively (Lindstrom 2009). Although neither of those samples was taken from a drinking water source, a maximum PFOA concentration in drinking water sampled from a private well was in the same order of magnitude at 2,070 ng/L (Lindstrom 2009).
In addition to the six PFAS monitored in UCMR3, 23 other PFAS were reported in the literature. These PFAS can be divided into PFSAs, PFCAs, perfluoroalkyl ether carboxylic acids (PFECAs), fluorinated telomer sulfonates, perfluorooctane sulfonic acids, fluorinated telomer unsaturated carboxylic acids, and perfluorooctane sulfonamide ethanols, some of which are PFAS precursors and can be a significant source of PFAS in the environment (Buck et al. 2011). Sun et al. (2016) reported on PFAS drinking water contamination in the Cape Fear watershed, NC, in response to the discovery of PFECAs in a drinking water source downstream from a PFAS manufacturing plant. Sun et al. (2016) analyzed concentrations of the following PFAS at three community drinking water intakes: perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), PFHpA, PFOA, PFNA, perfluorodecanoic acid (PFDA), PFBS, PFHxS, PFOS, and PFPrOPrA (GenX). Mean concentrations ranged from below quantitation limits, which were 25 ng/L for PFOS and 10 ng/L for all others, up to 631 ng/L (GenX) at one of the community drinking water intakes. Other PFECAs were present in these samples but could not be quantified. It is important to note that Sun et al. (2016) was the sole reference available for GenX occurrence data. Analysis of UCMR3 data shows that 4.0percent of water systems reported at least one detectable PFAS compound and 1.3 percent of water systems reported drinking water containing concentrations of PFOA and PFOS above USEPA’s lifetime HAs. This, along with site specific PFAS contamination events that impact drinking water facilities, illustrate the need for drinking water treatment schemes that are reliable, robust, sustainable, produce minimal residuals, and avoid unintended consequences such as detrimentally affecting other treatment processes or distribution systems.
2.0. TREATMENT OVERVIEW
Many treatment technologies for removing PFAS from drinking water matrices have been evaluated and reported in the literature, including conventional treatment, aeration, biological treatment, oxidation, disinfection, sonochemical destruction, fungal degradation, ion exchange resin, high- and low-pressure membranes, and activated carbon adsorption (Kucharzyk et al. 2017, Dickenson & Higgins 2016). Additional literature sources and discussion can be found on USEPA’s Drinking Water Treatability Database (https://iaspub.epa.gov/tdb/pages/general/home.do).
As previously mentioned, the strength of the C–F bond makes PFAS hard to remove through many common or low-cost technologies such as coagulation/flocculation/sedimentation (conventional treatment), oxidation/disinfection, and biodegradation inefficient. For example, conventional drinking water treatment has been shown to be inefficient for PFAS removal (Appleman et al. 2014, Xiao et al. 2013, Thompson et al. 2011, Shivakoti et al. 2010, Tabe et al. 2010, Quinones & Snyder 2009, Takagi et al. 2008, Loos et al. 2007, Skutlarek et al. 2006). Also, both aerobic and anaerobic biological treatment are ineffective for PFAS removal, particularly when considering contact time constraints observed in drinking water facilities (Kwon et al. 2014, Thompson et al. 2011, Sáez et al. 2008).
Because PFAS are often very stable compounds, standard oxidation/disinfection processes can neither change the structure of most PFAS, although precursors are a notable exception, nor can PFAS be oxidized into an insoluble form that can be removed by particulate control technologies, such as conventional treatment. Also, there is no data showing that PFAS can be removed by aeration or air stripping, although there are many PFAS for which relevant physical/chemical parameters are unknown. It may be that some PFAS precursors and some short chain PFAS may be amenable to being removed by air stripping. In conclusion, common drinking water approaches such as conventional treatment, aeration, disinfection (e.g., free chlorine, chloramines, ultraviolet (UV) light, and ozone), and other oxidants (e.g., potassium permanganate) are ineffective. Literature shows these technologies generally remove from 0 to 5 percent of the PFAS studied. (Appleman et al. 2014, Liu & Avendaño 2013, McLaughlin et al. 2011, Thompson et al. 2011, Shivakoti et al. 2010, Quinones & Snyder 2009, Schröder & Meesters 2005, Hori et al. 2004).
Advanced oxidation processes (AOPs) also have shown limited effectiveness for PFAS removal at doses and contact times that would be reasonable to be employed at drinking water treatment plants (Tellez 2014, Benotti et al. 2009, Schröder & Meesters 2005, Hori et al. 2004). This includes the formation of non-selective hydroxyl radicals by combining technologies such as hydrogen peroxide plus iron (Fenton’s reagent), ozone plus peroxide, UV plus titanium dioxide, UV plus ozone, and UV plus peroxide. Reported removals varied generally between 0 and 15 percent at conditions that would be observed in drinking water treatment plants. As with standard oxidation processes, it is possible that PFAS precursors can be oxidized by advanced oxidation technologies.
Given that conventional, aeration, oxidation, and biological treatment are ineffective, the focus of the rest of this review will be on technologies that have reported removal efficiencies of greater than 90% for certain PFAS at drinking water conditions: high pressure membranes, anion exchange (AEX) resins, and activated carbons (Dickenson & Higgins 2016). A visual comparison of the removal mechanisms of these technologies and important advantages and disadvantages of each can be found in Figure 1. Of these technologies, activated carbon has been studied the most extensively and has been used more frequently than the others in water treatment plants. The following sections will review relevant findings reported in the literature regarding treatment methods that have been shown to be effective for PFAS removal from drinking water and identify research gaps.
Figure 1 –
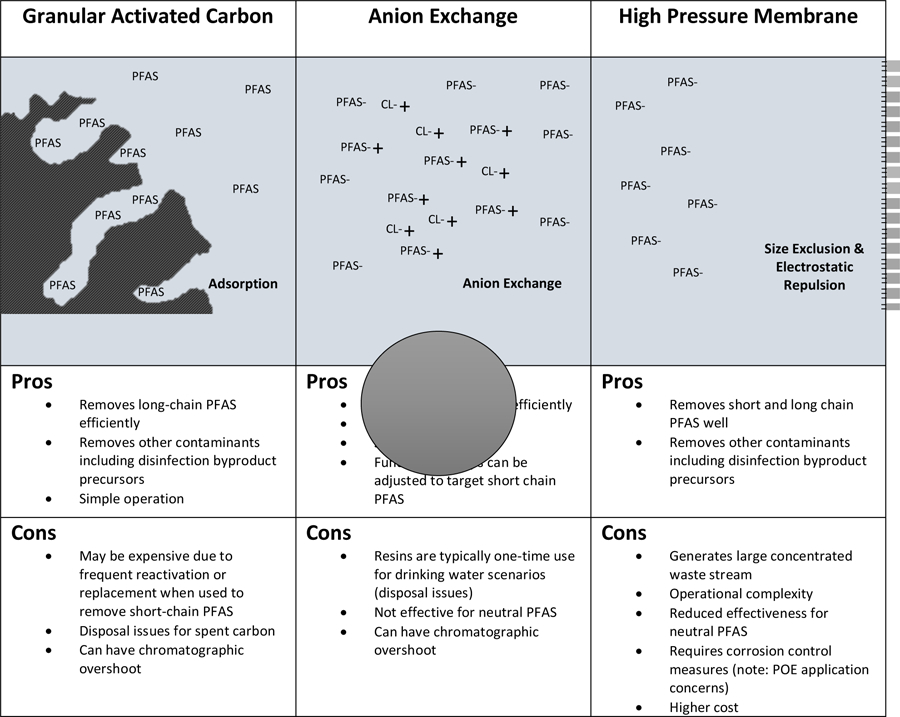
Comparison of Major Treatment Technologies (Not to Scale)
3.0. MEMBRANE TREATMENT
Membrane technologies can be grouped into low- or high-pressure categories. This grouping is helpful because these designations also indicate the general efficiencies of contaminant rejection. Low-pressure membranes remove particulates and microbials while passing dissolved constituents with the cutoff at the roughly colloidal/particulate size (Van der Bruggen et al. 2003). High-pressure membranes remove all particulate, colloidal, and dissolved constituents with the cutoff being based on molecular size and/or charge (Van der Bruggen et al. 2003).
Low-pressure membranes such as cartridge, microfiltration, or ultrafiltration membranes are designed for particulate control. Of these, ultrafiltration has the tightest pore structure and, in many instances, can remove colloids and viruses. However, even ultrafiltration membranes will not remove dissolved constituents such as PFAS (Rahman et al. 2014). Therefore, none of these types of low-pressure membranes will be able to control PFAS. Literature values for PFOA and PFOS removal by low-pressure membranes are typically between 0 and 23 percent, but these membranes may be more effective when used in conjunction with adsorbents, such as powdered activated carbon (PAC), which are retained by the membrane (Dickenson & Higgins 2016, McLaughlin et al. 2011, Takagi et al. 2008). Smaller chain PFAS will have even lower range of percent rejections due to their smaller size and reduced ability to adsorb to retained PAC.
High-pressure membranes such as nanofiltration or reverse osmosis can reject dissolved constituents such as PFAS. Reverse Osmosis can remove (>99%) PFAS, regardless of chain length via size exclusion and charge rejection, but is costly due to the high pressure required for operation (Appleman et al. 2014). Nanofiltration membranes have larger pores and therefore operate at lower pressures and rely on mechanisms such as electrostatic repulsion, hydrophobicity, and dipole moments for removing short-chain PFAS, which are too small to be rejected by size exclusion alone (Rahman et al. 2014, Appleman et al. 2013, Steinle-Darling et al. 2010, Steinle-Darling & Reinhard 2008). While lower than reverse osmosis, nanofiltration PFAS removal efficiencies are still typically greater than 90% even for short-chain PFAS (Dickenson & Higgins 2016). Neutral PFAS, such as perfluorooctane sulfonamide (FOSA) have limited electrostatic interactions and therefore low rejection rates in spite of their high molecular weight (Steinle-Darling et al. 2010, Steinle-Darling & Reinhard 2008). Studies with high-pressure membranes regularly report very high rejection of PFOA and PFOS, ranging from 93 to greater than 99 percent removal (Zeng et al. 2017, Yan et al. 2015, Appleman et al. 2014, Appleman et al. 2013, Flores et al. 2013, Thompson et al. 2011, Busch et al. 2010, Lipp et al. 2010, Steinle-Darling et al. 2010, Quinones & Snyder 2009, Steinle-Darling & Reinhard 2008, Tang et al. 2007, Tang et al. 2006). Other PFAS such as PFBA, PFPeA, PFHxA, PFNA, PFDA, PFBS, and PFHxS show rejections generally greater than 90% as shown in Table 3 (Zeng et al. 2017, Yan et al. 2015, Appleman et al. 2013, Thompson et al. 2011, Busch et al. 2010, Lipp et al. 2010, Quinones & Snyder 2009).
Table 3 –
Summary of High Pressure Membrane Studies
| Reference | Scale and Number | Water | pH | % Rejectionb |
|---|---|---|---|---|
| Tang et al., 2006 | Bench-scale Multiple membranes |
Diluted semiconductor manufacturing wastewater (25°C) |
4 | PFOS 93 to 99.91% * |
| Tang et al., 2007 | Bench-scale Multiple membranes |
Diluted semiconductor manufacturing wastewater (25°C) |
4 | PFOS: NF membranes 90 to 99% PFOS: RO membranes > 99% |
| Olsen & Paulson, 2008 | Multiple POE devices 12–47% recovery |
Spiked tap water (4.4–13.3 °C) | 7.3 7.9 |
PFOA >93 to >98% PFOS 93 to >98% |
| Steinle-Darling & Reinhard, 2008 | Bench-scale Multiple membranes |
Deionized waterd 15 PFAS reported |
2.8 5–6 |
pH 2.8: 60–90% MW<525 & >90% MW>525 pH 5–6: PFPnA 72% pH 5–6: >95% for >263 dalton FOSA: 40 to 93% FOSA: 70 (pH=2.8) to >99% (pH=10) |
| Quinones & Snyder, 2009 | Full scale | Secondary treated wastewaterd | NR | PFOA >67% PFOS >98% |
| Lipp et al., 2010 | Bench-scale Multiple membranes |
Diluted tap water (22 to 28°C) | 7.4 | PFBA 96 to 99.9% PFBS 95 to 99.8% PFOA 95.5 to 99.9% PFOS 99.8 to >99.9% |
| Thompson et al., 2011 | Full scale, single utility 85% recovery |
Reclaimed domestic wastewaterd | NR | PFHxA >99.4%a PFHpA >97.8%a PFOA 97.3%, >97.9%a PFNA >92.5%a PFDA >96.7%a PFBS >98.0%a PFHxS >99.2%a PFOS >99.7%a |
| Appleman et al., 2013 | Bench-scale One membrane type (virgin and fouled) |
Artificial groundwater (18°C) | 6.7 | PFBA virgin >97%, fouled >97% PFPeA virgin >97%, fouled >97% PFHxA virgin >95%, fouled >95% PFOA virgin >97%, fouled >97% PFNA virgin >98%, fouled >98%a PFDA virgin >97%, fouled >98% PFBS virgin 99, >99%, fouled 97, 98, >99%a PFHxS virgin >99%, fouled 98, >99%a PFOS virgin >99%, fouled >99% |
| Flores et al., 2013 | Full scale, one system | Conventional treated surface water blended with groundwaterd |
NR | PFOA >99%c PFOS >99%c |
| Appleman et al., 2014 | Full scale, multiple systems 80–85% recovery |
Artificial groundwaterd | NR | Utility A Utility B PFBA >82% >95% PFPeA >82% >99%a PFHxA >98% >99% PFHpA >86% >98%a PFOA >47% >98% PFNA >87% >98%a PFDA >67% >99% PFUnA NA >77%a PFDoA NA >87%a PFBS >98% >96%a PFHxS >94% >96%a PFOS >99% >96% N-MeFOSAA >36% >84%a N-EtFOSAA NA >58%a |
| Yan et al, 2015 | Full scale, multiple systems 80–85% recovery |
Landfill leachate 11 PFAS reportedd |
7.3–8.6 | Sum of 11 PFAS: 90- >99% |
| Zeng et al., 2017 | Bench-scale Multiple membranes |
Laboratory clean waterd | 7.0 | PFXxA 69–99.2% |
Extrapolated from figure;
Highest greater than percent rejection reported;
From lowest to highest molecular mass;
Impact of intermediate ultrafiltration difficult to quantify;
temperature not reported
Both size, charge, and other forms of rejection can be impacted by membrane fouling due to, but not limited to, pore blockage, charge exclusion, charge neutralization, adsorption, and resulting membrane degradation (Rahman et al. 2014, Appleman et al. 2013). While fouling typically results in reduced performance, instances of increased PFAS removal have been reported for fouled membranes (Appleman et al. 2013, Steinle-Darling & Reinhard 2008). Therefore, care should be taken when evaluating data from laboratory studies that used clean (e.g., distilled/deionized) water or were run for short time periods. Specifically, foulants from surface waters can be particularly impactful on membrane processes. Beyond rejection issues, fouling will impact the desired water production and thereby increase the operational cost and complexity.
Although it is recognized that high pressure membranes can be extremely effective for treating a wide range of PFAS, an additional factor to high-pressure membrane processes is water recovery. A high recovery (typically around 80 percent for non-desalination systems (Baruth 2005)) will produce a concentrated retentate stream (20 percent of feed volume), which must be treated prior to disposal. Dealing with the retentate represents a significant challenge and cost because of its higher concentrations, significant volume, and lack of technologies to treat it. Therefore, a significant research gap for high-pressure membrane systems treating PFAS remains how to manage the retentate stream. Considering the high operational cost of membrane treatment, the residual stream treatment scheme should be as cost efficient as possible. Corrosion control needs to be addressed for high-pressure membrane systems. Although typically controlled at the plant or irrelevant at the point-of-use (POU) applications, this could be problematic for point-of-entry (POE) systems for buildings or residences. Also, the impact of membrane fouling is a concern for water production and PFAS breakthrough and continues to be a research need, especially for surface water systems or ground waters with high organic or biofouling potential.
4.0. ANION EXCHANGE TREATMENT
4.1. AEX Resin Treatment Overview
Several resin types for PFAS removal have been reported in the literature, including those with and without anionic exchange properties, both of which can be single use and regenerable (Ross et al. 2018). Non ion exchange resins, such as those evaluated by Senevirathna et al. (2010b), rely on hydrophobic interactions and van der Waals interactions to adsorb PFAS. They are typically comprised of synthetic polymeric materials and with their lower bonding energy, they are more amenable to regeneration than resins with ion exchange properties (Ross et al. 2018). Resins with ion exchange properties are charged polystyrene or polyacrylic beads with polystyrene having slightly higher adsorption capacity and polyacrylic having significantly faster kinetics (Dudley 2012, Deng et al. 2010). Polystyrene can either be in a gel or microporous solid form, and there is no measurable difference reported between the two forms (Dudley 2012). The beads contain charged functional groups saturated with a counter ion, typically chloride, that can be exchanged with PFAS, which primarily exist as anions in solution at drinking water relevant pH values (e.g., pH 6.5 to 8.5 for raw water (APHA 1989)). Resin properties and their principal applications from literature reported studies are summarized in Table S2 in the SI. Researchers suggested that essential resin characteristics that affect exchange capacity include functional groups, polymer matrix, and porosity (Deng et al. 2010).
While the primary removal mechanism in AEX resins is ionic interaction, important secondary mechanisms include: adsorption, agglomeration, hydrophobic interactions, PFAS functional group interactions, and resin functional group interactions (Ross et al. 2018, Zaggia et al. 2016). These secondary removal mechanisms, and especially hydrophobicity, are responsible for reported variability in removal efficiencies among PFAS anions of varying chain lengths (Zaggia et al. 2016). PFAS hydrophobicity and removal during anion exchange treatment increases with fluorinated chain length. Hydrophobicity of the resin can also be adjusted by increasing the chain length of the alkyl substituents when quaternary ammonium functional groups are employed. Zaggia et al. (2016) demonstrated this through increased removal of more hydrophilic PFBA and PFBS when using a resin with triethyl alkyl groups, which had increased hydrophobicity compared to a resin with trimethyl alkyl groups. The strength of the PFAS functional group also impacts removal as illustrated by the increased removal of PFSAs compared to PFCAs (McCleaf et al. 2017, Zaggia et al. 2016, Dudley 2012). One notable exception being a five-fold increase in adsorption capacity of PFOA over PFOS reported by Yu et al. (2009).
AEX resins also have the advantage of having higher adsorption capacity than granular activated carbon (GAC), but adsorption tends to be hysteretic and non-reversible, often limiting resins to single use (Carter & Farrell 2010, Senevirathna et al. 2010b). Deng et al. (2010) and Carter and Farrell (2010) found that conventional resin regeneration methods were not effective, but Liu (2017) found improved resin regeneration in short-chain compared to long-chain PFAS because of their reduced hydrophobicity (Du et al. 2014). While these studies report varying success for regeneration via methanol, brine, or liquid rinses, single-use AEX resins remain the most feasible option for low concentration drinking water applications (Ross et al. 2018, Liu 2017, Carter & Farrell 2010, Deng et al. 2010). Also, one major issue with regeneration is the production of a concentrated waste stream that must be disposed of or further treated, like membrane treatment.
Although resins are used in fixed-bed adsorbers in general drinking water treatment practice, most studies focused on batch isotherm and kinetic tests for removing PFOS, though several studies report values for PFOA and PFBS as well (Liu 2017, Chularueangaksorn et al. 2014, Senevirathna et al. 2010a, Senevirathna et al. 2010b, Yu et al. 2009). Only a limited number of studies were conducted as continuous-flow column experiments and most of them focused on comparing the performance of different resin types to each other and to GAC (McCleaf et al. 2017, Woodard et al. 2017, Zaggia et al. 2016, Chularueangaksorn et al. 2014, Dudley 2012, Senevirathna et al. 2010a).
Time-to-equilibrium reported in the literature varied significantly by resin type and ranged from 4– to 168 hours (Senevirathna et al. 2010b, Yu et al. 2009). The shorter-chain PFBS was reported as having faster kinetics and higher affinity than long-chain PFOS, which was attributed to size exclusion factors (Deng et al. 2010). AEX removal of PFAS is reported to follow pseudo second order kinetics (Dudley 2012, Yu et al. 2009). As previously mentioned, properties of the adsorbate compounds and solution chemistry will play important roles. In ion exchange, resins do not prefer all ions equally and background ions such as sulfate, nitrate, and bicarbonate will occur at concentrations orders of magnitude greater than PFAS and compete for binding sites (Tripp et al. 2003). Ion preference for a resin can be mathematically represented by selectivity coefficients or separation factors which assume a linear isotherm (Ghurye et al. 1999). In a similar manner to Freundlich parameters for GAC isotherms, knowledge of the separation factors for each PFAS on each resin demonstrates the effectiveness and feasibility of a given resin/PFAS pair. There is a lack of information in the literature on PFAS selectivity coefficients which is important for assessing competition between PFAS chemicals and common inorganic anions (e.g., nitrate and sulfate). In addition, competition may lead to chromatographic effects where effluent PFAS concentrations may exceed influent concentrations. Specifically, in water containing mixtures of PFAS, PFAS with lower selectivities would be prone to the chromatographic effect as they are replaced with PFAS, or other anions, with greater selectivities.
Reported natural organic matter (NOM) impacts show varying results with Liu (2017) reporting reduced uptake due to competition with NOM and inorganic anions, but Dudley (2012) reported increased PFAS uptake with NOM present. In pre-equilibrium studies, Kothawala et al. (2017) found NOM presence to have a slight negative impact on PFAS adsorption over a dissolved organic carbon (DOC) ranging from 2 to 8 mg C/L. Further work is needed to elucidate the complex interactions between NOM, PFAS, important anions, and AEX resins.
4.2. Summary of Pilot and Full-Scale AEX Resin Evaluations for PFAS Removal
Several studies have evaluated AEX at the pilot- or full-scale level (Table S3 in the SI). Many of the studies have evaluated multiple medias or have evaluated the relative time to breakthrough for various PFAS. For example, Senevirathna et al. (2010b) compared the effectiveness of three non-ionic resins, including DOW V493, Amb XAD 4, and Dow L493 with Calgon F400 GAC for PFOS removal with parallel pilot scale column experiments. Dow V493 reached 10% breakthrough first at 13,000 bed volumes (BVs), followed by F400 at 25,000 BVs. The two other resins outperformed the GAC by reaching 10% breakthrough at 35,000 BVs (Dow L493) and 55,000 BVs (Amb XAD 4). Conte et al. (2015) compared AEX and non-ionic resins and found AEX to perform better, and similarly to Zaggia et al. (2016), decreasing removal efficiency was seen with decreasing PFAS chain length. Chularueangaksorn et al. (2014) compared five types of AEX resins (PFA300, Dow Marathon A, IRA400, PFA400, and PFA 444) with F400 GAC using parallel column tests similar to Senevirathna et al. (2010b). GAC reached 10% breakthrough first at 40 days, followed by the AEX resins in the order of FPA 444, PFA 400, IRA 400, Dow Marathon A and PFA 300, with PFA 300 exhibiting less than 1% breakthrough until day 52. Woodard et al. (2017) also found AEX to outperform GAC, except for PFBA, which behaved similarly in each system.
McCleaf et al. (2017) compared the removal of 14 PFAS using a pilot-scale AEX column and a pilot scale GAC column. AEX resin had better removal efficiencies for longer chained PFAS compared to shorter chained ones and for PFSAs compared to PFCAs. The authors concluded that AEX had higher removal for PFNA, PFDA, perfluoroundecanoic acid (PFUnDA), perfluorododecanoic acid (PFDoDA), PFBS, PFHxS, PFOS and FOSA, but GAC had higher removal efficiencies for PFBA, PFPeA, PFHxA, PFHpA, and perfluorotetradecanoic acid (PFTeDA).
Appleman et al. (2014) investigated PFAS removal at a full-scale water treatment plant in New Jersey using a strong base, porous AEX resin (Purolie FerrIX A33E) typically used for arsenic removal. Two sampling events occurred at 5 and 9 months of operation and similar to the observations from the pilot scale study by McCleaf et al. (2017), PFSAs and longer chain PFCAs had appreciable removal. Specifically, the resin demonstrated PFAS removals of more than 83% (PFBS), 97% (PFHxs), 90% (PFOS), 54% (PFHpA), and 76% (PFOA). However, the resin lacked effectiveness in removing for shorter chain PFCAs, including PFBA, PFPeA and PFHxA. Appleman et al. (2014) also reported on a full-scale water treatment plant using magnetic AEX resins in an up-flow fluidized-bed contactor for DOC removal. This AEX system showed negligible removal (<10%) for all 14 PFAS monitored, and the authors suggested the continuous regeneration method (as opposed to a complete resin replacement) or improper operation as possible reasons for the poor performance.
While AEX resins typically have higher capacities than GAC, several important limitations and knowledge gaps remain. Improvements in the removal of short-chain PFAS, most likely through the modification of the AEX resin functional group is needed. Assessment of PFAS competition with background ions such as sulfate, nitrate, and bicarbonate, determination of selectivity coefficients, and further elucidation of NOM interactions will increase fundamental understanding of process mechanics and improve modeling predictions. Improvements in resin regeneration and handling of related waste streams could allow resins to be used multiple times and reduce environmental impact and cost.
5.0. ACTIVATED CARBON TREATMENT
5.1. Activated Carbon Treatment Overview
Activated carbon adsorption is used to remove natural organic compounds, taste and odor compounds, and synthetic organic compounds from drinking water (USEPA 2018). Typically, either PAC is dosed early in the treatment train and removed through sedimentation, or GAC is used in fixed beds either in combination with filtration or as a post-filtration step (USEPA 2018, Chowdhury 2013). PAC generally has a diameter of less than 0.1 mm and GAC typically ranges from 1.2–1.6 mm in diameter (USEPA 2018). Many types of activated carbon have been evaluated for PFAS removal, including GAC, PAC, biochar, ash, carbon nanotubes, and carbon fibers (Chen et al. 2017, Inyang & Dickenson 2017, Dudley et al. 2015, Chen et al. 2011). Several activated carbon source materials have been evaluated as well, including bamboo, wood, coconut, and coal (Xiao et al. 2017, Deng et al. 2015, Zhi & Liu 2015, Qiu et al. 2007).
Several PFAS adsorption mechanisms have been identified, including multi-layer adsorption, micelle and hemi-micelle formation (Chen et al. 2017), acid base interactions, and the electrostatic and hydrophobic effects (Zhi & Liu 2015, Du et al. 2014). Adsorption is primarily controlled by surface chemistry, and according to Zhi and Liu (2015), the best predictor of PFAS adsorption is surface basicity. This was further illustrated by Deng et al. (2015) when a potassium hydroxide activated bamboo was evaluated and found to outperform other GACs reported in the literature. Zhi and Liu (2016) reported an increase in capacity through high temperature and ammonia activation. The size used has been shown to significantly impact the PFAS adsorption capacity with PAC (<0.1 mm) having nearly double the capacity of GAC (0.9–1 mm) with the same specific surface area. While these results may be impacted by pore blockage and steric hinderance, they have significant implications for scaling since capacity is usually assumed to be independent of particle size (Du et al. 2014, Crittenden et al. 1986).
It is important to note that the dominant mechanism may be different for different PFAS. For instance, short chain PFAS are more hydrophilic than their long chain counterparts and may be repelled by the hydrophobic effect, which has been identified as a dominant mechanism of adsorption for longer-chain PFAS (Rahman et al. 2014). Perfluorinated sulfonates are considered “hard bases” and more hydrophobic than their carboxylic counterparts, making them easier to adsorb under conditions in drinking water treatment (Du et al. 2014). This was clearly demonstrated by Ochoa-Herrera and Sierra-Alvarez (2008) who reported a capacity of 57 mg PFOA/g GAC compared to 182 mg PFOS/g GAC.
As previously mentioned and at drinking water relevant pH conditions, PFAS primarily exist as anions with a net negative charge and will be repelled by GAC when the pH is above the GAC’s point of zero charge (PZC), which is another weak but important predictor in PFAS adsorption (Deng et al. 2015, Zhi & Liu 2015). Several studies have shown increased adsorption capacity at lower pH and at positive zeta potential values (Chen et al. 2017, Deng et al. 2015). More work is needed to differentiate the effects of lower pH on sorbent properties from changes in sorbates at lower pH values. The impact of pH becomes increasingly important in the presence of even small amounts of NOM which in one case was shown to reduce the pH of a GAC’s PZC from 7 to 2.2 because of the NOM’s negative charge (Inyang & Dickenson 2017, Newcombe et al. 1997).
Additionally, NOM and PFAS can have overlapping molecular structure, allowing them to directly interact both electrostatically (e.g., cation bridge effect) and through hydrophobic or hydrophilic forces (Inyang & Dickenson 2017). For example, in pre-equilibrium conditions, increasing NOM (measured as DOC) concentration has been shown to improve PFAS retention on GAC through what was postulated to be co-adsorption of NOM and PFAS (Kothawala et al. 2017). Highest retention was observed when a more hydrophobic NOM was used. A more hydrophilic NOM led to reduced long chain retention, highlighting the importance of electrostatic and hydrophobic forces and the variation that is possible based on overall net charge and PFAS hydrophobicity/hydrophilicity (Kothawala et al. 2017). Co-adsorption could also explain the increase in PFAS adsorption when simultaneously adsorbed with NOM compared to PFAS adsorption onto GAC preloaded with NOM (Hansen et al. 2010). These results coupled with other reports in the literature of NOM significantly reducing PFAS adsorption capacity indicate that NOM-PFAS interactions are complex and may shift with time (Kothawala et al. 2017). Further work is needed to understand NOM-PFAS-GAC interactions throughout the adsorption process for optimization and application to different matrices and sorbents.
Ionic strength can also have a significant impact on PFAS adsorption onto GAC, reducing solubility and ability to adsorb with increasing ionic strength. Carter and Farrell (2010) reported a 17-fold reduction in PFOS solubility when comparing ultrapure water with 100 mM sodium perchlorate electrolyte solution. Although the ionic strength value far exceeds expected ionic strengths in drinking water (low mM), impacts from a range of relevant concentrations on adsorption requires evaluation.
One significant advantage of GAC over other treatment technologies is that it shows promise for reactivation with respect to PFAS. A study evaluating the reactivation of GAC containing PFOS, PFOA, and PFHxA at the bench scale reported destruction of PFAS adsorbed onto GAC at 700°C and complete destruction of the PFAS not adsorbed onto GAC at 1,000°C (Watanabe et al. 2016). It was also noted that when regenerating between 800–900°C, some short-chain PFAS escaped into the off-gas by volatile release before they could be destroyed, leading to the recommendation to heat the off-gas to 1,000°C to ensure complete mineralization of adsorbed and volatilized PFAS. Further work is needed to verify this result for a broader range of PFAS and in a full-scale reactivation facility.
5.2. Activated Carbon Batch Studies
Isotherms are used to evaluate the equilibrium adsorption capacity of specific sorbate and sorbent combinations to optimize adsorbent selection and predict CURs (Tchobanoglous et al. 2003). Reported isotherms evaluating PFAS adsorption onto GAC have primarily focused on PFOS and PFOA at μg/L to mg/L concentration ranges (Chen et al. 2017, Inyang & Dickenson 2017, Kothawala et al. 2017, Xiao et al. 2017, Zhang et al. 2016, Deng et al. 2015, Chen et al. 2011, Carter & Farrell 2010, Yu et al. 2009, Ochoa-Herrera & Sierra-Alvarez 2008, Qiu et al. 2007), but several studies have isotherm and kinetic data of additional PFAS at concentrations similar to those reported in UCMR3 (i.e., ng/L range) (Zhi & Liu 2016, 2015, Chularueangaksorn et al. 2014, Hansen et al. 2010, Senevirathna et al. 2010a).
Freundlich isotherms have been consistently found to provide a better representation of the experimental data, especially at lower concentrations, than Langmuir isotherms (Zhi & Liu 2016, 2015, Chularueangaksorn et al. 2014, Hansen et al. 2010, Senevirathna et al. 2010a). Reported Freundlich isotherm parameters for single PFAS in deionized water (DI) water can be found in Table 4. These data were converted from their originally reported units to those listed in Table 4 according to the method described by Bowman (1982). Reported values of K, which is the capacity factor, range over 8 orders of magnitude for PFOA and large variations are seen for other PFAS as well. This large variation may be due to differences in carbon size, source, and treatment as well as differences in experimental conditions such as PFAS concentrations, ionic strength, duration, presence or absence of NOM, and potential errors in experimental and analytical procedures making them not directly comparable. It should also be noted that variation in reported K values for a single compound with different surface treatments evaluated by the same research group under consistent experimental conditions resulted in up to 5 orders of magnitude variation (Zhi & Liu 2016). This variation highlights the need for reproducible isotherm data in which equilibrium is confirmed for carbons tested at ambient water PFAS concentrations, like those reported in UCMR3, for a broader range of PFAS.
Table 4 –
Freundlich Parameters for PFAS adsorption onto GAC reported in the literature a
| Reference | PFAS | Carbon Type |
Carbon | Source Material |
Carbon Mesh |
K (ng1−1/nmg−1L1/n) |
1/n | Duration (days) |
Initial Concentration (µg/L) |
|---|---|---|---|---|---|---|---|---|---|
| Zhi and Liu (2015) | PFOS | GAC | F400 | Bituminous Coal | 18×20 | 25.44 | 0.61 | 7–14 | 5–5,000 |
| PFOS | GAC | WVB | Wood | 18×20 | 30.47 | 0.54 | 7–14 | 5–5,000 | |
| PFOS | GAC | BioNC | Wood | 18×20 | 16.47 | 0.59 | 7–14 | 5–5,000 | |
| PFOS | GAC | 1240c | Coconut Shell |
18×20 | 10.12 | 0.68 | 7–14 | 5–5,000 | |
| Zhi and Liu (2016) | PFOS | GAC | F400 | Bituminous Coal | 18×20 | 20.20 | 0.58 | 7–14 | 5–5,000 |
| PFOS | GAC | WVB | Wood | 18×20 | 30.47 | 0.54 | 7–14 | 5–5,000 | |
| PFOS | GAC | BioNC | Wood | 18×20 | 16.47 | 0.59 | 7–14 | 5–5,000 | |
| PFOS | GAC | 1240c | Coconut Shell |
18×20 | 25.44 | 0.61 | 7–14 | 5–5,000 | |
| PFOS | GAC | F400 HT | Bituminous Coal | 18×20 | 3.71 | 0.75 | 7–14 | 5–5,000 | |
| PFOS | GAC | WVB HT | Wood | 18×20 | 10.03 | 0.81 | 7–14 | 5–5,000 | |
| PFOS | GAC | BioNC HT | Wood | 18×20 | 8.38 | 0.77 | 7–14 | 5–5,000 | |
| PFOS | GAC | 1240c HT | Coconut Shell |
18×20 | 0.054 | 1.84 | 7–14 | 5–5,000 | |
| PFOS | GAC | F400 AT | Bituminous Coal | 18×20 | 0.064 | 1.51 | 7–14 | 5–5,000 | |
| PFOS | GAC | WVB AT | Wood | 18×20 | 0.0087 | 2.33 | 7–14 | 5–5,000 | |
| PFOS | GAC | BioNC AT | Wood | 18×20 | 0.604 | 1.53 | 7–14 | 5–5,000 | |
| PFOS | GAC | 1240c AT | Coconut Shell |
18×20 | 0.50 | 1.51 | 7–14 | 5–5,000 | |
| Chularueangaksorn et al. (2014) | PFOS | GAC | F400 | Bituminous Coal | 18×20 | 1.97 | 0.82 | 4 | 10–1,000 |
| Ochoa-Herrera (2008) | PFOS | GAC | F300 | Bituminous Coal | 12×20 | 390.54 | 0.33 | 2 | 15,000–150,000 |
| PFOS | GAC | F400 | Bituminous Coal | 12×20 | 1,123.61 | 0.29 | 2 | 15,000–150,000 | |
| PFOS | GAC | URV-MOD1 | Bituminous Coal | 12×20 | 218.11 | 0.37 | 2 | 15,000–150,000 | |
| PFOS | GAC | F400 low Ce |
Bituminous Coal | 12×20 | 0.0047 | 1.12 | 2 | 50–500 | |
| Yu et al. (2009) | PFOS | GAC | Pengcheng | Bituminous Coal | ~18×20 | 5844 | 0.18 | 7 | 20,000–250,000 |
| PFOS | PAC | Pengcheng | Bituminous Coal | >150 | 17,260 | 0.18 | 7 | 20,000–250,000 | |
| Deng et al. (2015) | PFOS | GAC | Bamboo Derived |
Bamboo | 20×30 | 4499 | 0.29 | 2 | 20,000–250,000 |
| Zhang et al. (2016) | PFOS | GAC | Not Reported |
Not Reported |
20×40 | 0.019 | 0.89 | 6 | 500–10,000 |
| Chen et al. (2017) | PFOS | GAC | Not Reported |
Not Reported |
~18×20 | 8,558 | 0.20 | Equilibrium | 20,000–300,000 |
| PFOS | PAC | Not Reported |
Not Reported |
>150 | 8,112 | 0.22 | Equilibrium | 20,000–300,000 | |
| Dudley et al. (2015) | PFOS | PAC | Not Reported |
Wood | >400 | 181 | 0.46 | 14–21 | 0.500 |
| Zhi and Liu (2015) | PFOA | GAC | F400 | Bituminous Coal | 18×20 | 0.0043 | 1.96 | 7–14 | 5–5,000 |
| PFOA | GAC | WVB | Wood | 18×20 | 0.787 | 0.75 | 7–14 | 5–5,000 | |
| PFOA | GAC | BioNC | Wood | 18×20 | 1.33 | 0.71 | 7–14 | 5–5,000 | |
| PFOA | GAC | 1240c | Coconut Shell |
18×20 | 0.782 | 1.13 | 7–14 | 5–5,000 | |
| Zhi and Liu (2016) | PFOA | GAC | F400 | Bituminous Coal | 18×20 | 0.0035 | 1.98 | 7–14 | 5–5,000 |
| PFOA | GAC | WVB | Wood | 18×20 | 0.787 | 0.75 | 7–14 | 5–5,000 | |
| PFOA | GAC | BioNC | Wood | 18×20 | 1.334 | 0.71 | 7–14 | 5–5,000 | |
| PFOA | GAC | 1240c | Coconut Shell |
18×20 | 0.782 | 1.13 | 7–14 | 5–5,000 | |
| PFOA | GAC | F400 HT | Bituminous Coal | 18×20 | 1.104 | 1.12 | 7–14 | 5–5,000 | |
| PFOA | GAC | WVB HT | Wood | 18×20 | 9.87 | 0.69 | 7–14 | 5–5,000 | |
| PFOA | GAC | BioNC HT | Wood | 18×20 | 5.92 | 0.68 | 7–14 | 5–5,000 | |
| PFOA | GAC | 1240c HT | Coconut Shell |
18×20 | 5.29 | 0.73 | 7–14 | 5–5,000 | |
| PFOA | GAC | F400 AT | Bituminous Coal | 18×20 | 8.32 | 0.87 | 7–14 | 5–5,000 | |
| PFOA | GAC | WVB AT | Wood | 18×20 | 4.41 | 1.06 | 7–14 | 5–5,000 | |
| PFOA | GAC | BioNC AT | Wood | 18×20 | 7.55 | 0.98 | 7–14 | 5–5,000 | |
| PFOA | GAC | 1240c AT | Coconut Shell |
18×20 | 5.58 | 0.77 | 7–14 | 5–5,000 | |
| Ochoa-Herrera (2008) | PFOA | GAC | F400 | Bituminous Coal | 12×20 | 25.93 | 0.44 | 2 | 15,000–150,000 |
| Yu et al. (2009) | PFOA | GAC | Pengcheng | Bituminous Coal | ~18×20 | 752 | 0.28 | 7 | 20,000–250,000 |
| PFOA | PAC | Pengcheng | Bituminous Coal | >150 | 6,497 | 0.20 | 7 | 20,000–250,000 | |
| Deng et al. (2015) | PFOA | GAC | Bamboo Derived |
Bamboo | 20×30 | 12,946 | 0.19 | 2 | 20,000–250,000 |
| Zhang et al. (2016) | PFOA | GAC | Not Reported |
Not Reported |
20×40 | 0.010 | 0.89 | 6 | 500–10,000 |
| Chen et al. (2017) | PFOA | GAC | Not Reported |
Not Reported |
~18×20 | 7,963 | 0.16 | Equilibrium | 20,000–300,000 |
| PFOA | PAC | Not Reported |
Not Reported |
>150 | 24,744 | 0.11 | Equilibrium | 20,000–300,000 | |
| Dudley et al. (2015) | PFOA | PAC | Not Reported |
Wood | >400 | 248 | 0.27 | 14–21 | 0.500 |
| Ochoa-Herrera (2008) | PFBS | GAC | F400 | Bituminous Coal | 12×20 | 0.0093 | 0.46 | 2 | 15,000–150,000 |
| Dudley et al. (2015) | PFBS | PAC | Not Reported |
Wood | >400 | 19.10 | 0.38 | 14–21 | 0.500 |
| PFNA | PAC | Not Reported |
Wood | >400 | 234 | 0.38 | 14–21 | 0.500 | |
| PFHpA | PAC | Not Reported |
Wood | >400 | 71.80 | 0.44 | 14–21 | 0.500 | |
| Zhang et al. (2016) | PFHpA | GAC | Not Reported |
Not Reported |
20×40 | 0.0067 | 0.89 | 6 | 500–10,000 |
| Dudley et al. (2015) | PFHxA | PAC | Not Reported |
Wood | >400 | 19.60 | 0.48 | 14–21 | 0.500 |
| PFBA | PAC | Not Reported |
Wood | >400 | 0.0016 | 1.52 | 14–21 | 0.500 | |
| PFPeA | PAC | Not Reported |
Wood | >400 | 1.25 | 0.69 | 14–21 | 0.500 | |
| PFDA | PAC | Not Reported |
Wood | >400 | 261 | 0.45 | 14–21 | 0.500 | |
| PFHxS | PAC | Not Reported |
Wood | >400 | 208 | 0.27 | 14–21 | 0.500 | |
Notably lacking are data on short chain PFAS which are an emerging concern due to their increased production as an alternative to the more bioaccumulative, longer chain PFAS (Xiao et al. 2017). One study compared the adsorption of single PFAS and a PFAS mixture onto GAC, and did not find differences in the adsorption constants between the two solutions, indicating that competition among PFAS was minimal in an unsaturated state (Qiu et al. 2007). However, further work is needed to determine a broader suite of single solute adsorption constants to predict potential competition with other PFAS in mixtures using modeling.
Evaluation at relevant (i.e., ng/L) concentrations is important due to a wide range of sorbent-sorbate bonding energies resulting in nonlinear isotherms, such as PFOA (Zhi & Liu 2015), significant differences in isotherm constants extrapolated to relevant concentration ranges (Hansen et al. 2010), and changes in adsorption mechanism, such as micelle and hemi-micelle formation, at higher concentrations (Yu et al. 2009).
The time to reach equilibrium concentration of PFAS onto GAC reported in the literature ranges from 24–168 hours (Deng et al. 2015, Yu et al. 2009). Adsorption kinetics are strongly related to GAC surface area, particle and pore size, and to a lesser extent pH. In general, rates increase for smaller particles (e.g., PAC) and smaller pores which have shorter diffusional distances, and at lower pH (Chen et al. 2017, Zhi & Liu 2015, Du et al. 2014, Hansen et al. 2010, Yu et al. 2009). PFOS has been shown to have a faster initial adsorption rate than PFOA (Zhang et al. 2016), and kinetics also tend to be faster for shorter chain PFAS even though they have lower adsorption affinities (Inyang & Dickenson 2017, Du et al. 2014). Many authors have listed the pseudo second order rate model as the best for modeling PFOS, PFOA, and PFHpA adsorption rates (Chen et al. 2017, Zhang et al. 2016, Deng et al. 2015, Zhi & Liu 2015, Yu et al. 2009). Adsorption rates are typically divided into three steps: external diffusion, intraparticle diffusion, and adsorption onto active sites (Yu et al. 2009). Several authors have listed intraparticle diffusion as the rate limiting step but surface diffusion is more impactful for smaller size particles and may be controlling, depending on the GAC size used (Chen et al. 2017, Zhang et al. 2016, Du et al. 2014, Yu et al. 2009).
5.3. Powdered Activated Carbon Treatment
While several studies have evaluated PAC in isotherm and kinetics studies (Chen et al. 2017, Sun et al. 2016, Dudley et al. 2015, Yu et al. 2009), only one has used jar tests to simulate coagulation conditions typical of drinking water treatment plants (Dudley et al. 2015). The authors evaluated two different size PACs at different doses with different pH conditions for ten PFAS in surface water with a 55 mg/L alum concentration. The results from the jar tests were similar to results from batch kinetic testing, indicating alum addition did not impact performance. The pH did not significantly affect PFAS removal. A wood-based PAC dose of 50 mg/L with a 46-minute contact time was found to remove approximately 75% of PFOS and PFDA, 30% of PFBS and PFHxA, and no measurable removal for PFBA and PFPeA, indicating a decreasing removal efficiency when going from long to short chain PFAS. A superfine version (0.07–1.58 µm diameter) of the same PAC at the same dose also failed to remove PFBA and PFPeA, but showed a 10% increase in PFBS and PFHxA removal efficiency, and removal efficiencies of 78–95% for PFHS, PFOS, PFHpA, PFOA, PFNA, and PFDA. The authors concluded that to remove 90% of PFAS, the required PAC dose would be prohibitively high. However, for scenarios in which 50% removal is acceptable, PAC treatment for longer chain PFAS may be feasible. Unlike spent GAC, which can be regenerated, spent PAC exits the drinking water treatment train with the settled floc as sludge and will need to be disposed of according to State and Federal laws.
5.4. Activated Carbon Column Studies for PFAS Removal
Because GAC treatment in columns is a non-steady state process, efficiency will deteriorate over time as the contaminant chromatographically moves through the GAC bed. Pilot-scale adsorption tests are commonly used to investigate the continuous removal efficiency over time. Breakthrough data provided by pilot plant evaluation provides a clear picture of the GAC column performance at a specific time under a specific operational condition. An alternative to pilot-scale adsorption testing is the rapid small-scale column test (RSSCT). The small column used in RSSCTs is a scaled down physical model of a large GAC contactor (Sontheimer et al. 1988, Crittenden et al. 1987, Crittenden et al. 1986). RSSCTs require much less time for evaluation than pilot-scale tests. RSSCTs, pilot-scale tests, and full-scale systems have all been used to study the removal of PFAS by GAC adsorption (Table S4 in the SI).
Several authors compared different types of GAC in column tests and found bituminous coal-based GAC to outperform coconut-based GAC (McNamara et al. 2018, Ashani 2017, Inyang & Dickenson 2017, Appleman et al. 2013). Inyang and Dickenson (2017) also showed bituminous coal outperforms hardwood biochar and anthracite coal-based GAC. Appleman et al. (2013) proposed that the lower removal efficacy by coconut-based GAC might be due to its more microporous (i.e., tighter) structure which may have resulted in kinetic limitations. McNamara et al. (2018) similarly concluded that the well-developed transport pore structure of bituminous coal-based GACs resulted in their superior performance. McNamara et al. (2018) also found reactivated bituminous coal-based GAC outperformed virgin bituminous coal-based GAC.
As discussed earlier, the dominant adsorption mechanism may change depending on the properties of the PFAS being removed and can impact breakthrough patterns. Appleman et al. (2013) found that, in general, PFAS with longer chain lengths break through later than short chain PFAS, and PFSAs break through later than PFCAs. McCleaf et al. (2017) observed similar results to Appleman et al. (2013) and additionally found negative removal efficiencies for short-chain PFBA and PFPeA towards the end of the experiment. Negative removal efficiencies, or overshoot, was an indication of chromatographic displacement of the short-chain PFAS in favor of longer chain PFAS, other stronger adsorbing contaminants, and/or background NOM. The authors also suggested that during the later stages of adsorption, the dominant removal mechanism shifted to micelle formation.
NOM poses the main challenge for practical application of GAC adsorption in drinking water treatment processes due to its competition with other trace organic compounds for adsorption sites. NOM usually exists at much higher concentration and has a much longer mass transfer zone compared to trace organic compounds (Schideman et al. 2006, Kilduff et al. 1998); therefore, NOM preloads onto the lower portions of a GAC bed. The preloading causes a decrease in GAC adsorption capacity and kinetics for the target trace compounds because NOM blocks access to sorption sites. For example, Appleman et al. (2013) found that only short chain PFCAs reached notable breakthrough (>10%) at 125,000 BVs when using DI water. In contrast, when filtered surface water (1.7 mg/L DOC) was used, all PFAS evaluated had greater than 20% breakthrough by 11,000 BVs with the two shortest-chain PFCAs exceeding full (100%) breakthrough. Earlier PFAS breakthrough likely resulted from NOM preloading and competitive adsorption (Eschauzier et al. 2012). Because there was chromatographic displacement (overshoot) observed in the experiment conducted with NOM but not in DI water, the competition was attributed to NOM rather than PFAS. Pramanik et al. (2015) found PFOA and PFOS removal efficiency decreased with a background NOM concentration increase and that PFOS removal was affected more than PFOA.
5.5. PFAS Removal in Full-scale Activated Carbon Systems
Consistent with data reported for smaller scale systems, full-scale systems reported in the literature primarily focused on PFOS and PFOA and were generally effective in reaching target treatment concentrations. However, significant variation in carbon replacement frequency was reported and ranged from 3–24 months (Cummings et al. 2015, Rumsby et al. 2009) and one study reported no removal of shorter chained, more hydrophilic PFAS such as PFBA and PFBS. This is expected to be due to variations in previously discussed parameters such as NOM concentrations, differences in GAC used, and variation in other water quality parameters (McNamara et al. 2018, Cummings et al. 2015, Rumsby et al. 2009).
6.0. RESEARCH GAPS
While much excellent research has been reported in the literature, PFAS contamination is complex and evolving with shifts in production and usage and most of the work to date has focused primarily on PFOS and PFOA. Future research, especially for AEX and GAC treatment, needs to cover a much broader range of PFAS and source waters. More work is needed to understand co-adsorption of multiple PFAS and competition/interactions with NOM or other co-occurring contaminants. Improvements in removal of the more hydrophilic short-chain PFAS compounds are needed for both AEX and GAC systems. Work is also needed to characterize PFAS occurrence in small PWSs and private wells.
A significant research gap for high-pressure membrane systems treating PFAS is treatment of the concentrated retentate stream. Given the already high cost of membrane treatment itself, the residual stream treatment scheme should be as low cost as possible. Also, the impact of membrane fouling is a concern for water production, especially for surface water systems or ground waters with high organic loadings that have high organic or biofouling potential.
Additional AEX research needs include resolving conflicting reports of adsorption capacities among PFAS, competition between PFAS, PFAS-NOM interactions, evaluation of the fate and transport of PFAS upon incineration of spent AEX resin, and developing regeneration techniques that are economical, sustainable, and do not create problematic waste streams.
Future GAC research should include single solute isotherm and kinetic experiments using commonly employed GACs at relevant concentration ranges to develop data for modeling and column design. These data need to be evaluated in RSSCTs, pilot-scale, and full-scale treatment systems. Work is needed to elucidate the effect of influent concentrations, GAC particle size, removal of shorter chained PFAS, competition between different PFAS, and PFAS and NOM interactions. Reactivation of GAC needs to be evaluated in terms of the ultimate fate of the adsorbed PFAS. Finally, the combination of multiple technologies such as AEX and GAC for optimal treatment of specific contaminant and matrices while minimizing costs should also be evaluated.
Unintended consequences of PFAS treatment also need to be evaluated. Any treatment that modifies pH, bicarbonate/carbonate ions, sulfate, chloride, and NOM can influence distribution system corrosion. Corrosive water can arise from removing these competitive ions which can lead to changes in distribution system scale stability or solubility, potentially releasing scale embedded contaminants such as arsenic, lead, vanadium, and radionuclides (Speth & Schock 2007). Although the treatment technologies herein described have already been implemented into drinking water systems, corrosion issues need to be studied given the differences in water qualities and distribution system scales that have formed over multiple decades in cities across the US. These data will assist drinking water treatment utilities in decision making for efficient and cost-effective removal of a broad range of PFAS from varying source water matrices while protecting public health.
7.0. SUMMARY
PFAS are fluorinated compounds with unique properties that make them very effective as surface active agents leading to their utilization in a broad range of commercial products. Extensive usage has led to environmental release into drinking water sources from both consumer products and PFAS manufacturing. However, due to PFAS stability, which is derived primarily from the C-F bond’s strength, they are recalcitrant and not readily removed with conventional treatment methods, leading to potential human exposure to PFAS contaminated drinking water. Human exposure to PFOA has been linked with high cholesterol, increased liver enzymes, decreased vaccination response, thyroid disorders, pregnancy-induced hypertension and preeclampsia, and testicular and kidney cancer. PFOS is similarly associated with high cholesterol and thyroid disease as well as immune suppression and reduced fertility and fecundity. PFAS has become nearly ubiquitous as indicated by PFOS and PFOA being found in 99% of blood samples taken from the U.S. general population between 1999 and 2012. Known health effects of PFAS exposure led to the development of HAs for PFOS and PFOA in drinking water by the USEPA. However, the health implications of novel PFAS are largely unknown and require a concerted toxicological effort to better inform potential future HA decisions.
The occurrence of PFOS, PFOA, PFBS, PFHxS, PFHpA, and PFNA in drinking water was reported in UCMR3. Data were collected for UCMR3 from 2013–2015 and small drinking water systems were chosen to be representative of the US drinking water supply. PFOA and PFOS were detected most frequently across all systems at 1.03% and 0.79%, respectively. While the detection frequency was low, concentrations as high as 7,000 ng/L were reported and analysis of UCMR3 data shows that 4% of water systems reported at least one detectable PFAS compound and 1.3% of water systems reported results above the HA. Sources of these compounds include fluoropolymer manufacturers, wastewater treatment plants, and firefighting training sites. Shifts in production to shorter chain PFAS and replacement chemicals (e.g., GenX) have led to a broader range of reported PFAS compound occurrence and concentrations beyond what has been reported in UCMR3. While the toxicity of some shorter chain PFAS compounds are largely unknown, they are often harder to remove than long chain analogues and more work is needed to understand their human health and environmental impacts.
Reports on drinking water PFAS occurrence in UCMR3 and the broader literature coupled with the known health effects of PFOS and PFOA and questions about the toxicity of emerging PFAS compounds highlight the importance of evaluating and developing efficient and cost-effective drinking treatment technologies. High pressure membrane treatment, AEX resin, and activated carbon have all been evaluated and shown to have efficacy at a variety of scales.
High pressure membrane treatment has been shown to be highly effective for removing PFAS from drinking water, often achieving greater than 90% removal, but faces limitations due to being cost and energy intensive. They also produce concentrated retentate streams whose disposal are costly and difficult.
AEX resins can be effective at removing PFAS from drinking water due to high selectivity, rapid kinetics, increased affinity for short chain PFAS, and increased capacity when compared to GAC. Additionally, AEX can pair well with other technologies, such as GAC, for efficiently and cost effectively removing a broad range of PFAS that may not be readily removed with a single treatment technology. However, AEX resins face complications with regeneration or disposal and competition with NOM and other drinking water constituents that may bind to the resin more readily than PFAS.
The use of activated carbon for removing PFAS from drinking water is the most widely reported and evaluated treatment technology in the literature. GAC is of interest for treating PFAS contaminated drinking water because it is so commonly employed in drinking water treatment processes and can easily be reactivated. GAC has been reported to have varying levels of PFAS treatment efficacy that will be strongly dependent upon the PFAS present, the solution matrix, and the carbons physical characteristics and surface chemistry. This is exemplified by the broad range of reported isotherm parameters reported in the literature. Its widespread use and extensive evaluation in the literature suggest that it is the technology most suited to be a stop-gap treatment for PFAS, at least until other alternatives are evaluated and proven to be better.
Each of these technologies has benefits and limitations that need to be understood if they are to be used effectively when treating PFAS contaminated water. Further research is needed to improve each process and provide a fundamental understanding of the many variables involved in removing PFAS from drinking water. Cost effective and efficient treatment is needed immediately to reduce exposure and protect human health and the environment.
Supplementary Material
Footnotes
Publisher's Disclaimer: DISCLAIMER
The United States Environmental Protection Agency through its Office of Research and Development funded and managed the research described here. It has been subjected to the Agency’s administrative review and approved for publication. The views expressed in this article are those of the author(s) and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
REFERENCES
- APHA, 1989. Standard Methods for the Examination of Water and Wastewater.
- Appleman TD, Dickenson ER, Bellona C & Higgins CP, 2013. Nanofiltration and Granular Activated Carbon Treatment of Perfluoroalkyl Acids. Journal of hazardous materials, 260:740. [DOI] [PubMed] [Google Scholar]
- Appleman TD, Higgins CP, Quiñones O, Vanderford BJ, Kolstad C, Zeigler-Holady JC & Dickenson ERV, 2014. Treatment of Poly- and Perfluoroalkyl Substances in U.S. Full-Scale Water Treatment Systems. WATER RES, 51:246. [DOI] [PubMed] [Google Scholar]
- Ashani HS, 2017. Use of Granular Activated Carbon and Carbon Block Filters at Municipal and Point of Use Drinking Water Treatment for Removal of Organics. Arizona State University. [Google Scholar]
- Baruth EE, 2005. Water Treatment Plant Design (; American Water Works Association, and American Society of Civil Engineers. [Google Scholar]
- Benotti MJ, Stanford BD, Wert EC & Snyder SA, 2009. Evaluation of a Photocatalytic Reactor Membrane Pilot System for the Removal of Pharmaceuticals and Endocrine Disrupting Compounds from Water. Water research, 43:6:1513. [DOI] [PubMed] [Google Scholar]
- Boulanger B, Vargo J, Schnoor JL & Hornbuckle KC, 2004. Detection of Perfluorooctane Surfactants in Great Lakes Water. Environmental science & technology, 38:15:4064. [DOI] [PubMed] [Google Scholar]
- Bowman B, 1982. Conversion of Freundlich Adsorption K Values to the Mole Fraction Format and the Use of Sy Values to Express Relative Adsorption of Pesticides 1. Soil Science Society of America Journal, 46:4:740. [Google Scholar]
- Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, De Voogt P, Jensen AA, Kannan K, Mabury SA & van Leeuwen SP, 2011. Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins. Integrated environmental assessment and management, 7:4:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch J, Ahrens L, Sturm R & Ebinghaus R, 2010. Polyfluoroalkyl Compounds in Landfill Leachates. Environmental Pollution, 158:5:1467. [DOI] [PubMed] [Google Scholar]
- Carter KE & Farrell J, 2010. Removal of Perfluorooctane and Perfluorobutane Sulfonate from Water Via Carbon Adsorption and Ion Exchange. Separation Science and Technology, 45:6:762. [Google Scholar]
- Chen W, Zhang X, Mamadiev M & Wang Z, 2017. Sorption of Perfluorooctane Sulfonate and Perfluorooctanoate on Polyacrylonitrile Fiber-Derived Activated Carbon Fibers: In Comparison with Activated Carbon. RSC Advances, 7:2:927. [Google Scholar]
- Chen X, Xia X, Wang X, Qiao J & Chen H, 2011. A Comparative Study on Sorption of Perfluorooctane Sulfonate (Pfos) by Chars, Ash and Carbon Nanotubes. Chemosphere, 83:10:1313. [DOI] [PubMed] [Google Scholar]
- Chowdhury ZK, 2013. Activated Carbon: Solutions for Improving Water Quality. American Water Works Association. [Google Scholar]
- Chularueangaksorn P, Tanaka S, Fujii S & Kunacheva C, 2014. Batch and Column Adsorption of Perfluorooctane Sulfonate on Anion Exchange Resins and Granular Activated Carbon. Journal of Applied Polymer Science, 131:3:n/a. [Google Scholar]
- Conte L, Falletti L, Zaggia A & Milan M, 2015. Polyfluorinated Organic Micropollutants Removal from Water by Ion Exchange and Adsorption. CHEM ENG TRANS, 43:2257. [Google Scholar]
- Crittenden JC, Berrigan JK & Hand DW, 1986. Design of Rapid Small-Scale Adsorption Tests for a Constant Diffusivity. Journal (Water Pollution Control Federation):312. [Google Scholar]
- Crittenden JC, Hand DW, Arora H & Lykins BW Jr, 1987. Design Considerations for Gac Treatment of Organic Chemicals. Journal / American Water Works Association, 79:1:74. [Google Scholar]
- Cummings L, Matarazzo A, Nelson N, Sickels F & Storms C, 2015. Recommendation on Perfluorinated Compound Treatment Options for Drinking Water. New Jersey Drinking Water Quality Institute Treatment Subcommittee Report.
- Deng S, Nie Y, Du Z, Huang Q, Meng P, Wang B, Huang J & Yu G, 2015. Enhanced Adsorption of Perfluorooctane Sulfonate and Perfluorooctanoate by Bamboo-Derived Granular Activated Carbon. Journal of hazardous materials, 282:150. [DOI] [PubMed] [Google Scholar]
- Deng S, Yu Q, Huang J & Yu G, 2010. Removal of Perfluorooctane Sulfonate from Wastewater by Anion Exchange Resins: Effects of Resin Properties and Solution Chemistry. Water Research, 44:18:5188. [DOI] [PubMed] [Google Scholar]
- Dickenson E & Higgins C, 2016. Treatment Mitigation Strategies for Poly-and Perfluoroalkyl Substances. Water Research Foundation Web Report, 4322. [Google Scholar]
- Du Z, Deng S, Bei Y, Huang Q, Wang B, Huang J & Yu G, 2014. Adsorption Behavior and Mechanism of Perfluorinated Compounds on Various Adsorbents—a Review. Journal of hazardous materials, 274:443. [DOI] [PubMed] [Google Scholar]
- Dudley L-A, 2012. Removal of Perfluorinated Compounds by Powdered Activated Carbon, Superfine Powder Activated Carbon, and Anion Exchange Resin.
- Dudley L, Arevalo E & Knappe D, 2015. Removal of Perfluoroalkyl Substances by Pac Adsorption and Anion Exchange. Water Research Foundation: Denver. [Google Scholar]
- Eschauzier C, Beerendonk E, Scholte-Veenendaal P & De Voogt P, 2012. Impact of Treatment Processes on the Removal of Perfluoroalkyl Acids from the Drinking Water Production Chain. Environmental science & technology, 46:3:1708. [DOI] [PubMed] [Google Scholar]
- Flores C, Ventura F, Martin-Alonso J & Caixach J, 2013. Occurrence of Perfluorooctane Sulfonate (Pfos) and Perfluorooctanoate (Pfoa) in Ne Spanish Surface Waters and Their Removal in a Drinking Water Treatment Plant That Combines Conventional and Advanced Treatments in Parallel Lines. Science of the Total environment, 461:618. [DOI] [PubMed] [Google Scholar]
- Ghurye GL, Clifford DA & Tripp AR, 1999. Combined Arsenic and Nitrate Removal by Ion Exchange. Journal-American Water Works Association, 91:10:85. [Google Scholar]
- Grandjean P & Clapp R, 2015. Perfluorinated Alkyl Substances: Emerging Insights into Health Risks. New solutions: a journal of environmental and occupational health policy, 25:2:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelfo JL & Adamson DT, 2018. Evaluation of a National Data Set for Insights into Sources, Composition, and Concentrations of Per-and Polyfluoroalkyl Substances (Pfass) in Us Drinking Water. Environmental Pollution, 236:505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KJ, Johnson H, Eldridge J, Butenhoff J & Dick L, 2002. Quantitative Characterization of Trace Levels of Pfos and Pfoa in the Tennessee River. Environmental Science & Technology, 36:8:1681. [DOI] [PubMed] [Google Scholar]
- Hansen MC, Børresen MH, Schlabach M & Cornelissen G, 2010. Sorption of Perfluorinated Compounds from Contaminated Water to Activated Carbon. Journal of Soils and Sediments, 10:2:179. [Google Scholar]
- Hori H, Hayakawa E, Einaga H, Kutsuna S, Koike K, Ibusuki T, Kiatagawa H & Arakawa R, 2004. Decomposition of Environmentally Persistent Perfluorooctanoic Acid in Water by Photochemical Approaches. Environmental science & technology, 38:22:6118. [DOI] [PubMed] [Google Scholar]
- Hu XC, Andrews DQ, Lindstrom AB, Bruton TA, Schaider LA, Grandjean P, Lohmann R, Carignan CC, Blum A & Balan SA, 2016. Detection of Poly-and Perfluoroalkyl Substances (Pfass) in Us Drinking Water Linked to Industrial Sites, Military Fire Training Areas, and Wastewater Treatment Plants. Environmental science & technology letters, 3:10:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyang M & Dickenson ERV, 2017. The Use of Carbon Adsorbents for the Removal of Perfluoroalkyl Acids from Potable Reuse Systems. Chemosphere, 184:Supplement C:168. [DOI] [PubMed] [Google Scholar]
- Kannan K, Tao L, Sinclair E, Pastva SD, Jude DJ & Giesy JP, 2005. Perfluorinated Compounds in Aquatic Organisms at Various Trophic Levels in a Great Lakes Food Chain. Archives of environmental contamination and toxicology, 48:4:559. [DOI] [PubMed] [Google Scholar]
- Kilduff JE, Karanfil T & Weber WJ Jr, 1998. Competitive Effects of Nondisplaceable Organic Compounds on Trichloroethylene Uptake by Activated Carbon. I. Thermodynamic Predictions and Model Sensitivity Analyses. Journal of colloid and interface science, 205:2:271. [DOI] [PubMed] [Google Scholar]
- Kothawala DN, Köhler SJ, Östlund A, Wiberg K & Ahrens L, 2017. Influence of Dissolved Organic Matter Concentration and Composition on the Removal Efficiency of Perfluoroalkyl Substances (Pfass) During Drinking Water Treatment. Water Research, 121:Supplement C:320. [DOI] [PubMed] [Google Scholar]
- Kucharzyk KH, Darlington R, Benotti M, Deeb R & Hawley E, 2017. Novel Treatment Technologies for Pfas Compounds: A Critical Review. Journal of environmental management, 204:757. [DOI] [PubMed] [Google Scholar]
- Kwon BG, Lim H-J, Na S-H, Choi B-I, Shin D-S & Chung S-Y, 2014. Biodegradation of Perfluorooctanesulfonate (Pfos) as an Emerging Contaminant. Chemosphere, 109:221. [DOI] [PubMed] [Google Scholar]
- Lindstrom AB, Strynar MJ, Delinsky AD, and McMillan L, 2009. Results of the Analyses of Screening Surface and Well Water Samples from Decatur, Alabama for Selected Perfluorinated Compounds. Research Triangle Park, NC. [Google Scholar]
- Lipp P, Sacher F & Baldauf G, 2010. Removal of Organic Micro-Pollutants During Drinking Water Treatment by Nanofiltration and Reverse Osmosis. Desalination and Water Treatment, 13:1–3:226. [Google Scholar]
- Liu C, 2017. Removal of Perfluorinated Compounds in Drinking Water Treatment: A Study of Ion Exchange Resins and Magnetic Nanoparticles.
- Liu J & Avendaño SM, 2013. Microbial Degradation of Polyfluoroalkyl Chemicals in the Environment: A Review. Environment international, 61:98. [DOI] [PubMed] [Google Scholar]
- Loos R, Wollgast J, Huber T & Hanke G, 2007. Polar Herbicides, Pharmaceutical Products, Perfluorooctanesulfonate (Pfos), Perfluorooctanoate (Pfoa), and Nonylphenol and Its Carboxylates and Ethoxylates in Surface and Tap Waters around Lake Maggiore in Northern Italy. Analytical and Bioanalytical Chemistry, 387:4:1469. [DOI] [PubMed] [Google Scholar]
- McCleaf P, Englund S, Ostlund A, Lindegren K, Wiberg K & Ahrens L, 2017. Removal Efficiency of Multiple Poly and Perluoroalkyl Substances (Pfass) in Drinking Water Using Granular Activated Carbon (Gac) and Anion Exchange (Ae) Column Tests. WATER RES, 120:77. [DOI] [PubMed] [Google Scholar]
- McLaughlin CL, Blake S, Hall T, Harman M, Kanda R, Foster J & Rumsby PC, 2011. Perfluorooctane Sulphonate in Raw and Drinking Water Sources in the United Kingdom. Water and Environment Journal, 25:1:13. [Google Scholar]
- McNamara JD, Franco R, Mimna R & Zappa L, 2018. Comparison of Activated Carbons for Removal of Perfluorinated Compounds from Drinking Water. Journal-American Water Works Association, 110:1:E2. [Google Scholar]
- Nakayama S, Strynar MJ, Helfant L, Egeghy P, Ye X & Lindstrom AB, 2007. Perfluorinated Compounds in the Cape Fear Drainage Basin in North Carolina. Environmental science & technology, 41:15:5271. [DOI] [PubMed] [Google Scholar]
- Newcombe G, Drikas M & Hayes R, 1997. Influence of Characterised Natural Organic Material on Activated Carbon Adsorption: Ii. Effect on Pore Volume Distribution and Adsorption of 2-Methylisoborneol. Water Research, 31:5:1065. [Google Scholar]
- Ochoa-Herrera V & Sierra-Alvarez R, 2008. Removal of Perfluorinated Surfactants by Sorption onto Granular Activated Carbon, Zeolite and Sludge. Chemosphere, 72:10:1588. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL & Zobel LR, 2007. Half-Life of Serum Elimination of Perfluorooctanesulfonate,Perfluorohexanesulfonate, and Perfluorooctanoate in Retired Fluorochemical Production Workers. Environmental health perspectives, 115:9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumlee MH, Larabee J & Reinhard M, 2008. Perfluorochemicals in Water Reuse. Chemosphere, 72:10:1541. [DOI] [PubMed] [Google Scholar]
- Post GB, Louis JB, Cooper KR, Boros-Russo BJ & Lippincott RL, 2009. Occurrence and Potential Significance of Perfluorooctanoic Acid (Pfoa) Detected in New Jersey Public Drinking Water Systems. Environmental science & technology, 43:12:4547. [DOI] [PubMed] [Google Scholar]
- Pramanik BK, Pramanik SK & Suja F, 2015. A Comparative Study of Coagulation, Granular-and Powdered-Activated Carbon for the Removal of Perfluorooctane Sulfonate and Perfluorooctanoate in Drinking Water Treatment. Environmental technology, 36:20:2610. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Fujii S & Tanaka S, 2007. Removal of Perfluorochemicals from Wastewater by Granular Activated Carbon Adsorption. Environemental Engineering Research, 44:185. [Google Scholar]
- Quinones O & Snyder SA, 2009. Occurrence of Perfluoroalkyl Carboxylates and Sulfonates in Drinking Water Utilities and Related Waters from the United States. Environmental science & technology, 43:24:9089. [DOI] [PubMed] [Google Scholar]
- Rahman MF, Peldszus S & Anderson WB, 2014. Behaviour and Fate of Perfluoroalkyl and Polyfluoroalkyl Substances (Pfass) in Drinking Water Treatment: A Review. Water Research, 50:318. [DOI] [PubMed] [Google Scholar]
- Ross I, McDonough J, Miles J, Storch P, Thelakkat Kochunarayanan P, Kalve E, Hurst J, Dasgupta S, S. & Burdick J, 2018. A Review of Emerging Technologies for Remediation of Pfass. Remediation Journal, 28:2:101. [Google Scholar]
- Rumsby PC, McLaughlin CL & Hall T, 2009. Perfluorooctane Sulphonate and Perfluorooctanoic Acid in Drinking and Environmental Waters. Philosophical Transactions of the Royal Society of London A: Mathematical, Physical and Engineering Sciences, 367:1904:4119. [DOI] [PubMed] [Google Scholar]
- Sáez M, de Voogt P & Parsons JR, 2008. Persistence of Perfluoroalkylated Substances in Closed Bottle Tests with Municipal Sewage Sludge. Environmental Science and Pollution Research, 15:6:472. [DOI] [PubMed] [Google Scholar]
- Schideman LC, Mariñas BJ, Snoeyink VL & Campos C, 2006. Three-Component Competitive Adsorption Model for Fixed-Bed and Moving-Bed Granular Activated Carbon Adsorbers. Part Ii. Model Parameterization and Verification. Environmental science & technology, 40:21:6812. [DOI] [PubMed] [Google Scholar]
- Schröder HF & Meesters RJ, 2005. Stability of Fluorinated Surfactants in Advanced Oxidation Processes—a Follow up of Degradation Products Using Flow Injection–Mass Spectrometry, Liquid Chromatography–Mass Spectrometry and Liquid Chromatography–Multiple Stage Mass Spectrometry. Journal of Chromatography A, 1082:1:110. [DOI] [PubMed] [Google Scholar]
- Senevirathna S, Tanaka S, Fujii S, Kunacheva C, Harada H, Ariyadasa B & Shivakoti B, 2010a. Adsorption of Perfluorooctane Sulfonate (N-Pfos) onto Non Ion-Exchange Polymers and Granular Activated Carbon: Batch and Column Test. Desalination, 260:1–3:29. [Google Scholar]
- Senevirathna STMLD, Tanaka S, Fujii S, Kunacheva C, Harada H, Shivakoti BR & Okamoto R, 2010b. A Comparative Study of Adsorption of Perfluorooctane Sulfonate (Pfos) onto Granular Activated Carbon, Ion-Exchange Polymers and Non-Ion-Exchange Polymers. Chemosphere, 80:6:647. [DOI] [PubMed] [Google Scholar]
- Shivakoti B, Fujii S, Nozoe M, Tanaka S & Kunacheva C, 2010. Perfluorinated Chemicals (Pfcs) in Water Purification Plants (Wpps) with Advanced Treatment Processes. Water Science and Technology: Water Supply, 10:1:87. [Google Scholar]
- Simcik MF & Dorweiler KJ, 2005. Ratio of Perfluorochemical Concentrations as a Tracer of Atmospheric Deposition to Surface Waters. Environmental science & technology, 39:22:8678. [DOI] [PubMed] [Google Scholar]
- Sinclair E, Mayack DT, Roblee K, Yamashita N & Kannan K, 2006. Occurrence of Perfluoroalkyl Surfactants in Water, Fish, and Birds from New York State. Archives of environmental contamination and toxicology, 50:3:398. [DOI] [PubMed] [Google Scholar]
- Skutlarek D, Exner M & Farber H, 2006. Perfluorinated Surfactants in Surface and Drinking Waters. Environmental science and pollution research international, 13:5:299. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Crittenden JC & Summers RS, 1988. Activated Carbon for Water Treatment. American Water Works Association. [Google Scholar]
- Speth TF & Schock MR, 2007. Removing Esoteric Contaminants from Drinking Waters: Impacts of Treatment Implementation.
- Steinle-Darling E, Litwiller E & Reinhard M, 2010. Effects of Sorption on the Rejection of Trace Organic Contaminants During Nanofiltration. Environmental science & technology, 44:7:2592. [DOI] [PubMed] [Google Scholar]
- Steinle-Darling E & Reinhard M, 2008. Nanofiltration for Trace Organic Contaminant Removal: Structure, Solution, and Membrane Fouling Effects on the Rejection of Perfluorochemicals. Environmental science & technology, 42:14:5292. [DOI] [PubMed] [Google Scholar]
- Sun M, Arevalo E, Strynar M, Lindstrom A, Richardson M, Kearns B, Pickett A, Smith C & Knappe DR, 2016. Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environmental science & technology letters, 3:12:415. [Google Scholar]
- Tabe S, Yang P, Zhao X, Hao C, Seth R, Schweitzer L & Jamal T, 2010. Occurrence and Removal of Ppcps and Edcs in the Detroit River Watershed. Water practice and technology, 5:1:wpt2010015. [Google Scholar]
- Takagi S, Adachi F, Miyano K, Koizumi Y, Tanaka H, Mimura M, Watanabe I, Tanabe S & Kannan K, 2008. Perfluorooctanesulfonate and Perfluorooctanoate in Raw and Treated Tap Water from Osaka, Japan. Chemosphere, 72:10:1409. [DOI] [PubMed] [Google Scholar]
- Tang CY, Fu QS, Criddle CS & Leckie JO, 2007. Effect of Flux (Transmembrane Pressure) and Membrane Properties on Fouling and Rejection of Reverse Osmosis and Nanofiltration Membranes Treating Perfluorooctane Sulfonate Containing Wastewater. Environmental science & technology, 41:6:2008. [DOI] [PubMed] [Google Scholar]
- Tang CY, Fu QS, Robertson A, Criddle CS & Leckie JO, 2006. Use of Reverse Osmosis Membranes to Remove Perfluorooctane Sulfonate (Pfos) from Semiconductor Wastewater. Environmental science & technology, 40:23:7343. [DOI] [PubMed] [Google Scholar]
- Taniyasu S, Yamashita N, Moon H-B, Kwok KY, Lam PKS, Horii Y, Petrick G & Kannan K, 2013. Does Wet Precipitation Represent Local and Regional Atmospheric Transportation by Perfluorinated Alkyl Substances? Environment International, 55:25. [DOI] [PubMed] [Google Scholar]
- Tchobanoglous G, Burton FL & Stensel HD, 2003. Metcalf & Eddy, Inc Wastewater Engineering: Treatment, Disposal, Reuse. [Google Scholar]
- Tellez MH, 2014. Treatment of Perfluorinated Compounds and Nitroaromatics by Photocatalysis in the Presence of Ultraviolet and Solar Light.
- Thompson J, Eaglesham G, Reungoat J, Poussade Y, Bartkow M, Lawrence M & Mueller JF, 2011. Removal of Pfos, Pfoa and Other Perfluoroalkyl Acids at Water Reclamation Plants in South East Queensland Australia. Chemosphere, 82:1:9. [DOI] [PubMed] [Google Scholar]
- Tripp A, Clifford D, Roberts D, Cang Y, Aldridge L, Gillogly T & Boulos L, 2003. Treatment of Perchlorate in Groundwater by Ion Exchange Technology AWWA and AwwaRF, Denver. [Google Scholar]
- USEPA, 2006. 2010/2015 Pfoa Stewardship Program.
- USEPA, 2016a. Drinking Water Health Advisory for Perfluorooctane Sulfonate (Pfos). Office of Water Health and Ecological Criteria Division.
- USEPA, 2016b. Drinking Water Health Advisory for Perfluorooctanoic Acid (Pfoa). Office of Water Health and Ecological Criteria Division.
- USEPA, 2016c. Third Unregulated Contaminant Monitoring Rule. United States Environmental Protection Agency.
- USEPA, 2017. The Third Unregulated Contaminant Monitoring Rule (Ucmr 3): Data Summary.
- USEPA, 2018. Powdered Activated Carbon Drinking Water Treatability Database
- Van der Bruggen B, Vandecasteele C, Van Gestel T, Doyen W & Leysen R, 2003. A Review of Pressure-Driven Membrane Processes in Wastewater Treatment and Drinking Water Production. Environmental progress, 22:1:46. [Google Scholar]
- Vestergren R & Cousins IT, 2009. Tracking the Pathways of Human Exposure to Perfluorocarboxylates. Environmental Science & Technology, 43:15:5565. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Takemine S, Yamamoto K, Haga Y & Takata M, 2016. Residual Organic Fluorinated Compounds from Thermal Treatment of Pfoa, Pfhxa and Pfos Adsorbed onto Granular Activated Carbon (Gac). Journal of Material Cycles and Waste Management, 18:4:625. [Google Scholar]
- Woodard S, Berry J & Newman B, 2017. Ion Exchange Resin for Pfas Removal and Pilot Test Comparison to Gac. Remediation Journal, 27:3:19. [Google Scholar]
- Xiao F, Simcik MF & Gulliver JS, 2013. Mechanisms for Removal of Perfluorooctane Sulfonate (Pfos) and Perfluorooctanoate (Pfoa) from Drinking Water by Conventional and Enhanced Coagulation. Water research, 47:1:49. [DOI] [PubMed] [Google Scholar]
- Xiao X, Ulrich BA, Chen B & Higgins CP, 2017. Sorption of Poly- and Perfluoroalkyl Substances (Pfass) Relevant to Aqueous Film-Forming Foam (Afff)-Impacted Groundwater by Biochars and Activated Carbon. Environmental Science and Technology, 51:11:6342. [DOI] [PubMed] [Google Scholar]
- Yan H, Cousins IT, Zhang C & Zhou Q, 2015. Perfluoroalkyl Acids in Municipal Landfill Leachates from China: Occurrence, Fate During Leachate Treatment and Potential Impact on Groundwater. Science of the Total Environment, 524:23. [DOI] [PubMed] [Google Scholar]
- Yu Q, Zhang R, Deng S, Huang J & Yu G, 2009. Sorption of Perfluorooctane Sulfonate and Perfluorooctanoate on Activated Carbons and Resin: Kinetic and Isotherm Study. Water research, 43:4:1150. [DOI] [PubMed] [Google Scholar]
- Zaggia A, Conte L, Falletti L, Fant M & A., C., 2016. Use of Strong Anion Exchange Resins for the Removal of Perfluoroalkylated Substances from Contaminated Drinking Water in Batch and Continuous Pilot Plants. WATER RES, 91:137. [DOI] [PubMed] [Google Scholar]
- Zeng C, Tanaka S, Suzuki Y, Yukioka S & Fujii S, 2017. Rejection of Trace Level Perfluorohexanoic Acid (Pfhxa) in Pure Water by Loose Nanofiltration Membrane. Journal of Water and Environment Technology, 15:3:120. [Google Scholar]
- Zhang D, Luo Q, Gao B, Chiang S-YD, Woodward D & Huang Q, 2016. Sorption of Perfluorooctanoic Acid, Perfluorooctane Sulfonate and Perfluoroheptanoic Acid on Granular Activated Carbon. Chemosphere, 144:2336. [DOI] [PubMed] [Google Scholar]
- Zhi Y & Liu J, 2015. Adsorption of Perfluoroalkyl Acids by Carbonaceous Adsorbents: Effect of Carbon Surface Chemistry. Environmental Pollution, 202:Supplement C:168. [DOI] [PubMed] [Google Scholar]
- Zhi Y & Liu J, 2016. Surface Modification of Activated Carbon for Enhanced Adsorption of Perfluoroalkyl Acids from Aqueous Solutions. Chemosphere, 144:1224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.