

Abstract
In human lung cancer progression, the EMT process is characterized by the transformation of cancer cells into invasive forms that migrate to other organs. Targeting to EMT-related molecules is emerging as a novel therapeutic approach for the prevention of lung cancer cell migration and invasion. Traf2- and Nck-interacting kinase (TNIK) has recently been considered as an anti-proliferative target molecule to regulate the Wnt signaling pathway in several types of cancer cells. In the present study, we evaluated the inhibitory effect of a tyrosine kinase inhibitor sunitinib and the integrin-αⅤβ3 targeted cyclic peptide (cRGDfK) on EMT in human lung cancer cells. Sunitinib strongly inhibited the TGF-β1-activated EMT through suppression of Wnt signaling, Smad and non-Smad signaling pathways. In addition, the cRGDfK also inhibited the expression of TGFβ1-induced mesenchymal marker genes and proteins. The anti-EMT effect of sunitinib was enhanced when cRGDfK was treated together. When sunitinib was treated with cRGDfK, the mRNA and protein expression levels of mesenchymal markers were decreased compared to the treatment with sunitinib alone. Co-treatment of cRGDfK has shown the potential to improve the efficacy of anticancer agents in combination with therapeutic agents that may be toxic at high concentrations. These results provide new and improved therapies for treating and preventing EMT-related disorders, such as lung fibrosis and cancer metastasis, and relapse.
Introduction
Epithelial-to-mesenchymal transition (EMT) is a process in which closely packed epithelial cells with polarity become more motile and invasive and turn into spindle-shaped mesenchymal cells. In general, EMT can be seen in the complex process of transformation that epithelial cells must undergo to acquire mesenchymal cell characteristics during embryogenesis, development, wound healing, and organ fibrosis [1,2]. Notably, EMT-induced mobility and invasion potential play an important role in cancer metastasis to other organs. Because the metastatic process is a major cause of death and poor prognosis in cancer patients, suppression of signaling pathways involved in the EMT process is emerging as a new therapeutic strategy in cancer.
Non-small cell lung cancer (NSCLC) accounts for approximately 80–85% of total lung cancer and has high mortality due to invasion and metastatic ability [3]. Despite the development of chemotherapy, radiotherapy, and surgery, the 5-year survival rate for lung cancer has not increased significantly. In lung cancer progression, transforming growth factor (TGF)-β is a major cytokine that induces invasion and metastasis through an EMT process [4–7]. In the induction of EMT by TGF-β, TGF-β phosphorylates TβR-I by binding to transmembrane Ser/Thr receptors TGF-β type I (TβR-I) and type II (TβR-II), subsequently phosphorylating the downstream mediators Smad2 and Smad3 [8,9]. The phosphorylated Smad2/3 complex recruits Smad4, which translocates to the nucleus and binds to transcription factors, such as Snail/Slug and Twist, to activate the TGF-β-reactive genes [10–12]. TGF-β-mediated non-Smad signaling pathways, including the Wnt/β-catenin signaling pathway, are also involved in the EMT process in several types of cancer cells [13–15].
In the last decade, Traf2- and Nck-interacting kinase (TNIK) has been reported as a first-in-class cancer target molecule [16]. TNIK kinase activity and expression have been shown to be involved in the maintenance of cancer growth in colorectal cancer, blood cancer, and lung adenocarcinoma [17–20]. Notably, we previously reported the potential of TNIK as an anti-cancer target molecule on the TGF-β induced EMT process, migration, and invasion of human lung adenocarcinoma cells [18]. In that study, TNIK inhibitor inhibited the TGF-β-induced EMT, migration, and invasion process through the suppression of Smad and non-Smad signaling pathways, including the TCF4/β-catenin involved Wnt signaling pathway. Although the association of TNIK with EMT in non-small cell lung adenocarcinoma has not been fully studied in many reports, inhibition of metastasis and invasion through TNIK inhibition is expected to play a role in increasing the therapeutic effect.
In this study, we evaluated the effect of sunitinib, a multi-targeted receptor tyrosine kinase inhibitor with high affinity for TNIK [21]. Sunitinib is a clinically approved small molecule drug that exerts anti-angiogenic and anti-proliferative activity against NSCLC through the inhibition of certain receptor tyrosine kinases, such as the platelet-derived growth factor receptors (PDGFRs), the vascular endothelial growth factor receptors (VEGFRs), and the stem-cell factor receptor KIT [22–25]. In addition, we evaluated the anti-proliferative and anti-EMT effect in human lung adenocarcinoma cells of the combination of sunitinib with an αⅤβ3 integrin receptor-targeted cyclic-pentapeptide RGDfK (cRGDfK) [26].
The cRGDfK peptide has been identified as an integrin αⅤβ3 inhibitor that acts as a targeting peptide for enhancing the permeability of tumor cells to drugs in anti-angiogenic cancer therapy [27]. The peptide ligand containing the arginin-glycin-aspartic acid (RGD) shows a strong binding affinity and selectivity to integrin αⅤβ3 which sequence was discovered in fibronectin [28,29]. The RGD sequence is the cell attachment site with integrins that serves a recognition site for cell adhesion. The RGD-recognition sites were found in adhesive extra cellular matrix (ECM), blood, and cell surface proteins. Particularly, integrins recognize this RGD-containing sequence in their adhesion protein ligands [30]. Among the integrins, integrin αⅤβ3 is consist of two subunits, αⅤ subunit and β3 subunit, which binds to RGD-containing ECM proteins such as vitronectin, fibronection and thrombopondin [31,32]. Based on these findings, linear or cyclic RGD-containing peptides have been developed as integrin αⅤβ3 targeted ligands. Especially, cyclic peptide cRGDfK and cRGDyK showed high binding affinity for integrin αⅤβ3 that act as vectors for delivery of chemotherapeutics [33]. These cyclic peptides have been used to ameliorate the cytotoxicity of drugs and reduce the side effects caused by toxicity without killing healthy cells. For instance, cyclic peptide conjugated antitumor small molecules, doxorubicin and paclitaxel, showed improved inhibition of growth and metastasis in cancer models [27,34,35].
Our results provide new information that the combination of sunitinib and integrin-targeted cyclic peptides exerts a synergistic anti-cancer effect for the treatment of EMT-mediated diseases, including pulmonary fibrosis and lung cancer metastasis.
Materials and methods
The study was approved by the International Review Board of Eulji University (EU 18–14). All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Materials
Sunitinib was obtained from Selleck Chemicals (USA), and cRGDfK peptide was synthesized by Dr. Park (CHA Meditech Co., Ltd, Korea). Recombinant human TGF-β1 was purchased from R&D Systems, Inc. (USA). Fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) were purchased from Corning, Inc. (USA). Antibodies against p-ERK1/2, ERK1/2, p-Smad2/3, and Smad2/3 were purchased from Cell Signaling Technology, Inc. (USA). Antibodies against E-cadherin, N-cadherin, vimentin, TNIK, β-catenin, lamin B1, horseradish peroxidase (HRP)-conjugated secondary antibodies, and HRP-conjugated actin were purchased from Santa Cruz Biotechnology, Inc. (USA), and α-smooth muscle actin (α-SMA) was purchased from Abcam (UK).
Cell culture and tissue sample preparation
Human NSCLC A549 cells were purchased from ATCC (USA) and H358 (KCLB No. 25807), H1299 (KCLB No. 25803), and human lung fibroblast IMR90 cells from Korean Cell Line Bank (KCLB No. 10186). A549, H358, and H1299 cells were maintained in DMEM and IMR90 cells in Minimum Essential Medium (MEM; Corning, USA) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). All cells were maintained at 37˚C in a humidified atmosphere of 5% CO2. The use of human NSCLC patient’s tissue samples for this study was approved by the International Review Board of Eulji University (EU 18–14). The NSCLC tissues were originated from human lung adenocarcinoma (10), bronchoalveolar carcinoma (3), squamous cell carcinoma (2), and large cell carcinoma (2). The cancer tissues of lung cancer patients used were obtained in pairs with normal lung tissues, of which eight normal lung tissues were pooled and used as a normal group in the qRT-PCR experiment.
Cell viability assay
To assess cell viability, cells (5 × 103 cells/well) were seeded in a 96-well plate for 24 h. Cells were treated with or without TGF-β1 (5 ng/mL) and sunitinib or cRGDfK for 24–72 h in culture media containing 10% FBS. Cell viability was assessed using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Japan) according to the manufacturer’s instructions. The absorbance was measured with a MultiscanTM FC microplate photometer (Thermo Fisher Scientific, USA). Cell viability is presented as the percentage of control (untreated cells). Experiments were performed in triplicate.
Molecular docking
Molecular docking was carried out using Discovery Studio 2017 R2 and the crystal structure of TNIK (PDB ID 5AX9) (http://www.rcsb.org). The protein was prepared using the Prepare Protein module of Discovery Studio 2017 R2 under CHARMm force field [36] and default conditions. The TNIK binding site sphere was defined as a volume using the applicable module in Discovery Studio 2017 R2. The backbone carbonyl group of Glu106, the backbone nitrogen, and the carbonyl groups of Cys108 were selected as hydrogen bond constraints.
RNA interference
A549 cells were transfected with TNIK siRNA or non-targeting control siRNA using the siRNA Reagent System (Santa Cruz Biotechnology, Inc., USA). After 48 h of incubation, the mRNA expression of Wnt target genes and mesenchymal marker genes was measured by qRT-PCR analysis (see the “qRT-PCR” section below).
TCF/LEF reporter assay
A549 cells were seeded in a 96-well plate for 24 h. Cells were treated with or without TGF-β1 (5 ng/mL) and sunitinib or cRGDfK. The relative TCF/LEF reporter activity was measured by the CignalTM TCF/LEF Reporter Assay Kit (Qiagen, Germany) according to the manufacturer’s instructions. After 48 h, cells were washed with PBS, and then lysed using passive lysis buffer (Promega, USA). Luciferase activity was evaluated using the Dual Luciferase Reporter Assay kit (Promega, USA) and normalized by the total lysate of negative control-transfected cells. All experiments were performed in triplicate.
Western blot analysis
Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) and sunitinib or cRGDfK for 72 h. Cytoplasmic and nuclear fractions were prepared from lysates using Nuclear and Cytoplasmic Extraction Reagent (Thermo Fisher Scientific, USA). Cytoplasmic or nuclear fractions were loaded on 8–15% polyacrylamide gels and transferred to nitrocellulose membranes. Specific primary antibodies were used to detect the expression of proteins. After incubation with HRP-conjugated secondary antibodies, the signals were visualized using LuminataTM Forte Western HRP Substrate (Merck Millipore, Germany). The band intensities were measured to determine the relative protein expression using X-ray films and development solution (Fujifilm, Japan). Actin or lamin B1 were used as loading controls. The detected bands were quantified based on the ImageJ software, and the relative ratio between each sample and loading controls was presented in the figures. The average band intensities of the independent three western blot results are shown in S1 Fig.
Quantitative Real-Time PCR (qRT-PCR) analysis
Cells were incubated in 60 mm2 dishes for 24 h. Total RNA was isolated using the AccuPrep® RNA Extraction Kit (Bioneer Corp., Korea) and cDNA synthesized from 1 μg of total RNA using oligo (dT) primers (Bioneer Corp., Korea) and the RocketScriptTM Reverse Transcriptase Kit (Bioneer Corp., Korea). qRT-PCR was performed with the ExcelTaq 2X Q-PCR Master Mix (SMOBiO, Taiwan) and CFX96TM Real-Time System (Bio-Rad, USA). The cycling conditions were as follows: 95°C for 3 min followed by 40 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. The primer sequences used in this study are provided in S1 Table. All reactions were run in triplicate, and data were analyzed using the 2−ΔΔCT method [37]. The internal standard was GAPDH. Significance was determined by the Student’s t-test with GAPDH-normalized 2−ΔΔCT values [37].
Invasion assay
A549 cells were incubated in a 24-well plate for 24 h. After serum starvation for 24 h, cells were treated with TGF-β1 and sunitinib/cRGDfK or combination for 48 h. Collected cells in culture medium containing 5% FBS was added to the bottom of a Boyden chamber. After placing over the gelatin-coated membrane filter, the silicone gasket, and the top chamber, the cell suspension was added to the top chamber, followed by incubation at 37°C in 5% CO2 for 6 h. The membrane filter was stained using the Diff-Quick staining kit (Dade Behring) and filter was dried and stabilized on a glass slide using 30% glycerol solution, the migrated cells were counted in three randomly selected fields at 400× magnification. This experimental procedure is referred to our previous study [18].
Analysis of combined drug effects
We analyzed the effects of the drug combination using the CalcuSyn software program (Biosoft, UK). To determine whether the result of treatment with the two compounds was additive or synergistic, we applied combination index (CI) methods derived from the median effect principle of Chou and Talalay [38]. The CI was calculated by the formula published by Zhao et al. [39]. A CI of 1 indicated an additive effect between the two compounds, a CI > 1 indicated antagonism, and a CI < 1 indicated synergism.
Statistical analysis
Data are presented as the mean ± SD of at least three independent experiments. Significant differences were evaluated by the One-way ANOVA with post-hoc Tukey HSD Test, with P < 0.05 considered significant. # p < 0.01 versus control, * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
Results
Sunitinib has high affinity for the ATP binding site of TNIK
We performed molecular docking studies to gain insight into the binding mode of sunitinib to TNIK at the atomic level. This molecular docking study proposed binding mode that E106 and C108 in the hinge region interact with three hydrogen bonds (Fig 1A). The E106 and C108 backbone hydrogen bonds interact with the pyrrolidone of sunitinib at distances of 2.81 Å and 2.03 Å, respectively. The pyrrole of sunitinib interacts with C108 in the backbone at a distance of 2.91 Å. Sunitinib binding to TNIK is further stabilized by the CH/π interaction with the hydrophobic properties of TNIK A52, V31, L160, and V170.
Fig 1. Binding mode and Ki of sunitinib for TNIK.
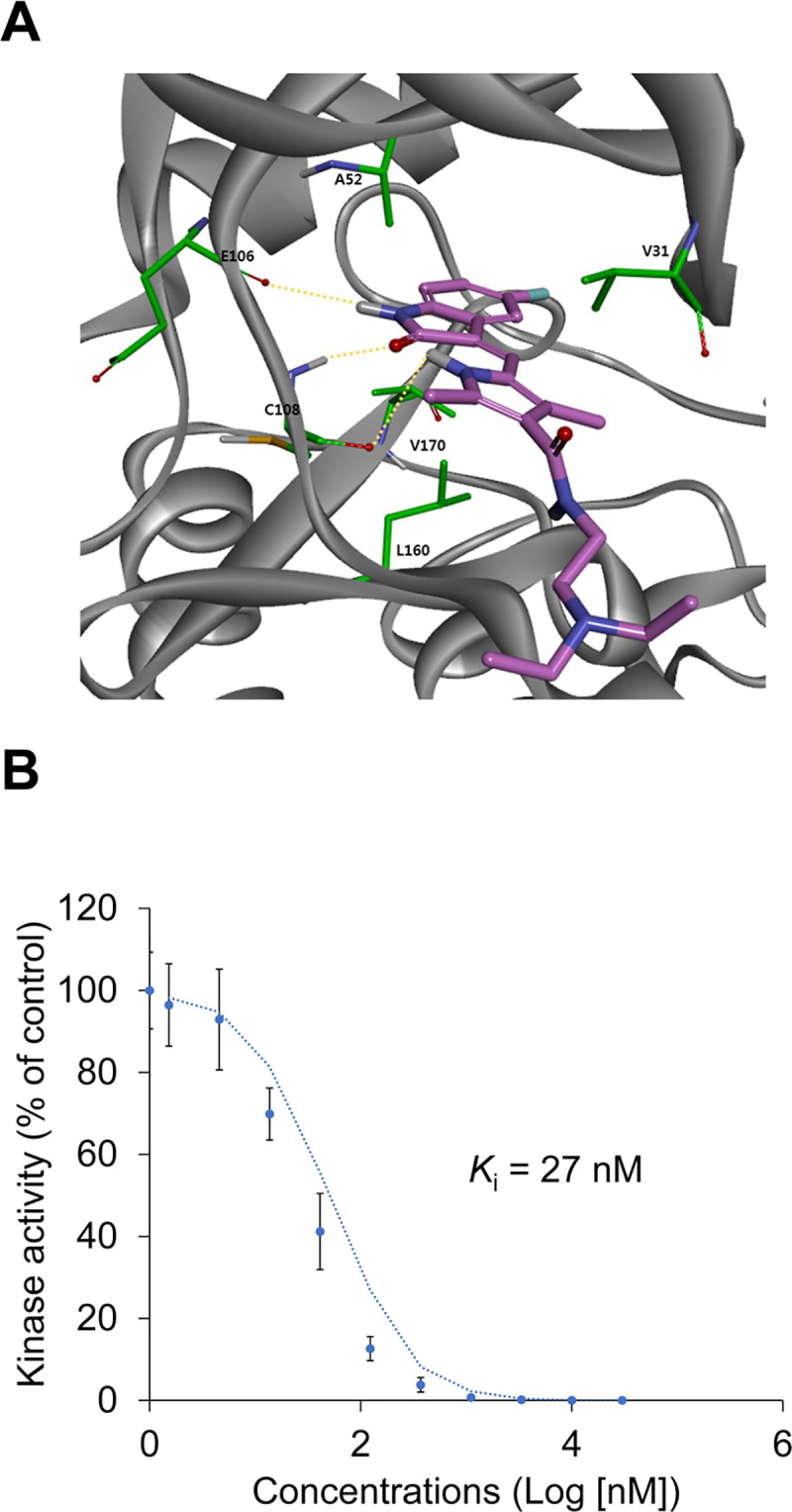
(A) The proposed binding mode of sunitinib (pink) in the ATP-binding pocket of TNIK. TNIK and its interacting residues are represented by ribbons and sticks, respectively. Hydrogen bonding interactions appear as dashed yellow lines. (B) The binding constant, Ki, of sunitinib for TNIK was determined using an ATP competition assay.
The inhibitory binding constant, Ki, of sunitinib for TNIK was also calculated from triplicate, 11-point dose-response curves (Fig 1B). Sunitinib inhibited the binding of ATP to TNIK in a dose-dependent manner (Ki = 27 nM). These results indicate that the high affinity of sunitinib for TNIK inhibits ATP binding and leads to a decrease in TNIK kinase activity.
Sunitinib inhibits TNIK-expressing NSCLC cell proliferation and TGF-β1-induced activation of Wnt signaling
To evaluate the inhibitory effect of sunitinib on TNIK-expressing NSCLC cell proliferation, we confirmed the mRNA expression of TNIK in NSCLC patient’s tissue cells and protein expression in normal lung fibroblast IMR90 and NSCLC A549, H1299, and H358 cells. TNIK mRNA expression was higher in NSCLC tissue cells than in normal lung tissues (Fig 2A). In addition, IMR90 cells did not express TNIK, but A549, H1299, and H358 cells did express the protein (Fig 2B). Sunitinib time- and dose-dependently inhibited the survival of A549, H1299, and H358 cells (Fig 2C–2E). However, IMR90 cells were not significantly affected by sunitinib compared to NSCLC cells (Fig 2F). This result suggests that sunitinib has stronger anti-proliferative effect in TNIK-expressing NSCLC cells than normal lung fibroblasts.
Fig 2. Sunitinib inhibits the proliferation of TNIK-expressing human NSCLC cells.

(A) TNIK mRNA expression in NSCLC tissue cells from lung cancer patients. TNIK mRNA expression levels were measured by qRT-PCR analysis in pool normal lung tissue cells (n = 8) and in NSCLC cells from patients with lung cancer (n = 17). The normal group represents the average qRT-PCR results obtained from eight healthy lung tissue cells without cancer cells. Data represent the mean ± SD of raw results. Experiments were performed in triplicate. * p < 0.05 and ** p < 0.001 (vs. control). (B) The protein expression of TNIK in normal fibroblast IMR90 and NSCLC H1299, H358, and A549 cells was measured by Western blot. Actin was used as a loading control. A549 (C), H1299 (D), and H358 (E) cells and IMR90 (F) cells were treated with sunitinib for 24‒72 h. After incubation, cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent the mean ± SD of raw results.
Next, we confirmed the cytotoxicity and mRNA expression using qRT-PCR analysis whether silencing of TNIK affect the cell proliferation and transcriptional activity of TNIK and Wnt target genes in NSCLC cells. Silencing of TNIK enhanced the sensitivity to sunitinib and inhibited TGF-β1-induced mRNA expression of TNIK, CTNNB1, TCF4, and c-MYC (S2 and S3 Figs). Based on these results, we assessed the effect of sunitinib on the TGF-β1-induced activation of Wnt signaling pathways and TNIK expression. We previously demonstrated that TGF-β1 activates TCF4-mediated transcription and TNIK expression [18]. To confirm the effect of sunitinib on TCF4-mediated Wnt signaling pathways, we assessed the TGF-β1-induced β-catenin luciferase activity and transcriptional activity of Wnt target genes. TGF-β1-induced β-catenin luciferase activity was inhibited by sunitinib in a dose-dependent manner (Fig 3A). Activation of TNIK is mediated by the binding of β-catenin to TCF4 and subsequent phosphorylation of TCF4, which activates Wnt target gene expression [16,17]. We confirmed that TGF-β1 induced the transcription of TNIK and Wnt target genes. Increased mRNA expression of TNIK and Wnt target genes was strongly inhibited by sunitinib (Fig 3B). Our previous study also demonstrated that the expression of TNIK by TGF-β1 is related to the nuclear accumulation of β-catenin [18]. This study represented that KY-05009, a TNIK inhibitor, suppressed TNIK/β-catenin-mediated Wnt signaling in A549 cells. Sunitinib suppressed cytosolic β-catenin and nuclear TNIK and β-catenin protein expression (Fig 3C). These results indicate that sunitinib has an anti-proliferative effect and inhibits TGF-β1-induced activation of TNIK//β-catenin-mediated Wnt signaling in NSCLC A549 cells.
Fig 3. Sunitinib inhibits TGF-β1-induced Wnt signaling.

(A) Effect of sunitinib on TCF4 transcriptional activity. A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with sunitinib for 48 h before measuring TOPflash luciferase activity. FOPflash-normalized TOPflash luciferase activity is represented as the relative TCF/LEF luciferase activity. (B) A549 cells were pretreated with sunitinib for 3 h, and then incubated with TGF-β1 (5 ng/mL) for 1 h. After RNA extraction and cDNA synthesis, the mRNA expression of TNIK and Wnt target genes was measured by qRT-PCR. (C) The protein levels of TNIK and β-catenin in cytosolic and nuclear fractions were measured by Western blot. Actin and lamin B1 were used as loading controls for the cytosolic and nuclear fractions, respectively. The results are representative of triplicate experiments. # p < 0.01 versus control; * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
Sunitinib inhibits TGF-β1-induced EMT and Smad/non-Smad signaling
As in the above result, we also confirmed the effect of TNIK silencing on EMT phenotype by qRT-PCR analysis. Silencing of endogenous TNIK suppressed TGF-β1-induced mRNA expression of mesenchymal maker genes CDH2 and VIM encoding N-cadherin and vimentin, respectively (S4 Fig). However, transfection of TNIK siRNA did not significantly affect the mRNA expression of CDH1 encoding E-cadherin. Based on these results, to verify the inhibitory effect of sunitinib on TGF-β1-induced EMT, we evaluated the protein expression of epithelial and mesenchymal markers. TGF-β1 suppressed the expression of epithelial marker E-cadherin, but stimulated mesenchymal markers N-cadherin, vimentin, and α-smooth muscle actin (SMA) [1,2]. Sunitinib dose-dependently inhibited TGF-β1-induced protein expression of EMT markers in A549 cells (Fig 4A). We also confirmed these inhibitory effects of sunitinib on the TGF-β1-mediated transcription of mesenchymal marker genes CDH2 and VIM, encoding N-cadherin and vimentin, respectively (Fig 4B and 4C); the TGF-β1-induced mRNA expression of both CDH2 and VIM was suppressed.
Fig 4. Sunitinib inhibits TGF-β1-induced expression of EMT markers and Smad signaling.

(A) Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with sunitinib for 72 h. The protein expression of TGF-β1-mediatead EMT markers was evaluated by Western blot. Actin was used as a loading control. (B and C) Serum-deprived A549 cells were pretreated with sunitinib for 3 h, and then incubated with TGF-β1 (5 ng/mL) for 48 h. After RNA extraction and cDNA synthesis, we performed qRT-PCR to measure mRNA expression of CDH2 and VIM using GAPDH as an internal control. (D) Effects of sunitinib on TGF-β1-activated Smad signaling. A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with sunitinib for 24 h, and then the levels of p-Smad2/3 and endogenous Smad2/3 in cytosolic fraction were evaluated by Western blot. (E and F) The transcriptional activity of SNAI1 and TWIST1 was measured by qRT-PCR using GAPDH as an internal control. Serum-deprived A549 cells were pretreated with sunitinib for 3 h, and then incubated with TGF-β1 (5 ng/mL) for 1 h. (G) The effect of sunitinib on TGF-β1-induced phosphorylation of ERK1/2 and p38 was determined by Western blot. Actin was used as a loading control. # p < 0.01 versus control, * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
To elucidate the inhibitory effect of sunitinib on the TGF-β1-mediated EMT-related signaling pathway, we examined the level of TGF-β1-induced phosphorylation of Smad2/3 involved in the EMT process and the transcriptional activity of Smad signaling target genes SNAI1 (also referred to as Snail) and TWIST1 (also referred to as Twist), upregulating mesenchymal markers and downregulating epithelial markers. Sunitinib inhibited TGF-β1-induced phosphorylation of Smad2/3 (Fig 4D). We also confirmed that TGF-β1-induced mRNA expression of SNAI1 and TWIST1 was suppressed by sunitinib (Fig 4E and 4F). These results demonstrate that the TGF-β1-induced EMT phenotype was strongly attenuated by sunitinib through suppression of Smad-mediated signaling.
TGF-β can also regulate EMT and invasion through non-Smad signaling pathways, including MAP kinase, Ras-ERK, and JNK [40–42]. In a previous study, we investigated the induction of ERK1/2 phosphorylation by TGF-β1 treatment [18]. We evaluated the inhibitory effects of sunitinib on TGF-β1-induced activation of ERK. As shown in Fig 4G, sunitinib inhibited the TGF-β1-induced phosphorylation of ERK1/2, suggesting that sunitinib can suppress the TGF-β-mediated EMT process through the inhibition of non-Smad signaling, as well as the Smad-signaling pathway.
cRGDfK inhibits TGF-β1-induced expression of mesenchymal markers in A549 cells
To improve the effect of sunitinib, A549 cells were co-treated with sunitinib and cyclic-RGDfK peptide (cRGDfK) (Fig 5A and S5 Fig). First, to confirm the therapeutic potency of cRGDfK as an integrin αⅤβ3 antagonist in NSCLC cells, we checked the mRNA expression of ITGAV and ITGB3 encoding integrin αⅤ and integrin β3, respectively. As shown in Fig 5B, A549 cells showed more ITGAV and ITGB3 mRNA expression than those of IMR90, H1299 and H358 cells. Next, to assess the cytotoxicity of cRGDfK, we conducted cell viability assays in both NSCLC cell lines and normal fibroblast IMR90 cells. cRGDfK suppressed proliferation of NSCLC cells, but IMR90 cells were not significantly affected (Fig 5C–5F). In addition, cRGDfK had an inhibitory effect on TGF-β1-induced expression of TNIK and β-catenin in A549 cells (S6 Fig).
Fig 5. Treatment with cRGDfK inhibits proliferation of human NSCLC cells.

(A) Chemical structure of cRGDfK. (B) The relative mRNA expression of integrin αⅤ and β3 in human NSCLC cells. (C‒F) NSCLC A549 (C), H1299 (D), and H358 (E) cells and normal fibroblast IMR90 cells (F) were treated with cRGDfK for 24‒72 h. After incubation, cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent the mean ± SD of raw results.
We assessed whether cRGDfK inhibits the expression of TGF-β1-induced EMT markers in A549 cells. As shown in Fig 6A, the TGF-β1-induced reduction of E-cadherin expression was not restored by cRGDfK, but the increase in N-cadherin, vimentin and α-SMA was inhibited. This result was confirmed by the mRNA expression of CDH2 and VIM encoding N-cadherin and vimentin, respectively. However, there were no concentration-dependent decreases in mRNA expression with cRGDfK treatment, and cRGDfK inhibited the TGF-β1-induced transcriptional activity of CDH2 and VIM (Fig 6B and 6C). The protein expression of phosphorylated Smad2/3 in the cytosol was also inhibited by cRGDfK (S7 Fig). Treatment with cRGDfK strongly suppressed TGF-β1-induced transcriptional activity of SNAI1 and TWIST1 (Fig 6D and 6E). These results demonstrate that cRGDfK has an inhibitory effect on the TGF-β1-mediated EMT process in NSCLC A549 cells.
Fig 6. Treatment with cRGDfK inhibits TGF-β1-induced expression of EMT markers.

(A) Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with cRGDfK for 72 h. The protein expression of TGF-β1-mediatead EMT markers was detected by Western blot. Actin was used as a loading control. (B‒E) Serum-deprived A549 cells were pretreated with cRGDfK for 3 h, and then treated with TGF-β1 (5 ng/mL) for 48 h (B and C) or 1 h (D and E). The mRNA expression of CDH2 and Vim (B and C) or Snail and Twist (D and E) was measured by qRT-PCR using GAPDH as an internal control. Experiments were performed in triplicate. Data represent the mean ± SD of raw results. # p < 0.01 versus control, * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
Combined treatment with cRGDfK improves the inhibitory effect of sunitinib on cell viability, TGF-β1-induced EMT process and invasion in A549 cells
To evaluate the effect of cRGDfK on the TGF-β1-induced EMT process in NSCLC cells with sunitinib, we evaluated cell viability. Compared to the single treatment with sunitinib, combined treatment with cRGDfK increased the inhibitory effect on A549 cell proliferation (Fig 7A) and other NSCLC cell lines (H358 and H1299) (S8 Fig and S2 Table). This result was confirmed by calculation of the combination index (CI) using the raw data (Fig 7A), which evaluated a synergistic effect at concentrations > 0.3 μM (Table 1). In addition, we confirmed the combination effect on the TGF-β1-induced protein expression of EMT markers and mRNA expression of target genes. Reduced expression of E-cadherin and CDH1 induced by TGF-β1 was recovered by combined treatment with sunitinib and cRGDfK (Fig 7B and 7C). The protein expression of N-cadherin and vimentin and transcriptional activity of CDH2 were synergistically inhibited by combined treatment (Fig 7B, 7D and 7E). The inhibitory effect of sunitinib on the mRNA expression of Smad signaling target genes SNAI1 and Twist was also enhanced by cRGDfK (Fig 7F and 7G). In addition, the effect of combined treatment on TGF-β1-induced invasion were investigated in A549 cells. As shown in Fig 8, the TGF-β1-induced invasion of A549 cells was significantly inhibited by combined treatment of sunitinib with cRGDfK.
Fig 7. Combination of sunitinib with cRGDfK shows enhanced inhibitory effects on cell viability and TGF-β1-induced EMT expression.

(A) A549 cells were treated with sunitinib or a combination of sunitinib plus cRGDfK for 24 h before measuring cell viability. Data represent mean ± SD. *p < 0.01 vs. sunitinib-free controls. (B) Serum-deprived A549 cells were pretreated for 3 h with sunitinib (3 μM) and cRGDfK (10 μM) individually or in combination, and then incubated with TGF-β1 (5 ng/mL) for 72 h. The expression of EMT markers was measured by Western blot. Actin was used as a loading control. (C‒G) A549 cells were pretreated with sunitinib or a combination of sunitinib plus cRGDfK for 3 h and incubated with TGF-β1 (5 ng/mL) for 48 h (C‒E) or 1 h (F and G). The mRNA expression of the indicated genes was measured by qRT-PCR using GAPDH as an internal control. Experiments were performed in triplicate. Data represent the mean ± SD of raw results. # p < 0.01 versus control, * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
Table 1. Combination Index (CI) values for the two-drug combination against A549 cell viability.
| Sunitinib (μM) | cRGDfK (μM) | CI value |
|---|---|---|
| 0.3 | 0.3 | 0.20468 |
| 1 | 1 | 0.37368 |
| 3 | 3 | 0.91230 |
| 10 | 10 | 0.66814 |
Fig 8. Combination of sunitinib with cRGDfK exerts enhanced inhibitory effect on TGF-β1-induced invasion of A549 cells.

The effect of combination on TGF-β1-induced invasion of A549 cells was evaluated using Boyden chambers. Numbers of invaded cells were represented by an average number of cells per randomly selected three high-power field (HPF). Experiments were performed in triplicate. Data represent the mean ± SD of raw results. # p < 0.01 versus control, * p < 0.05, ** p < 0.01 versus treatment with TGF-β1 only.
These results suggest that cRGDfK exerts an enhancing effect on A549 cell viability and TGF-β1-induced EMT as a targeting peptide that increases the drug efficacy of sunitinib in lung cancer cell death, the EMT process and invasion.
Discussion
Loss of epithelial characteristics and acquisition of mesenchymal features in the EMT is a key mechanism for metastatic and invasive changes in cancer cells [1,2]. EMT also stimulates anti-apoptotic signals that confer resistance to chemotherapy in NSCLC cells [42]. Thus, inhibition of EMT during tumor progression is a very important strategy for the treatment and prevention of NSCLC, together with the induction of cancer cell death. Here, we showed that TGF-β1 strongly induces the EMT and Smad/non-Smad signaling pathways of NSCLC A549 cells, and as a receptor tyrosine kinase (RTK) inhibitor sunitinib significantly inhibited these inductions. We previously demonstrated that TNIK is involved in TGF-β1-induced EMT, migration, and invasion of NSCLC A549 cells [18]. Inhibition of TNIK by siRNA or a specific TNIK inhibitor, KY-05009, suppressed cell proliferation and the TGF-β1-induced EMT process. Based on previous findings, we confirmed the effect of TNIK silencing on the proliferation inhibitory effect of sunitinib in NSCLC cells. Cells whose TNIK expression was suppressed by siRNA were found to have higher susceptibility to sunitinib than those not. These results suggest that expression of TNIK is related to NSCLC cell survival, and inhibition of TNIK expression may increase susceptibility to tyrosine kinase inhibitors such as sunitinib. As sunitinib also has been shown to inhibit TNIK kinase activity [21], and to address the relevance between TNIK inhibition and TGF-β1-induced EMT, we evaluated the effect of sunitinib as an anti-cancer agent inhibiting TNIK kinase activity and the EMT in NSCLC cells. As expected, sunitinib was found to have three hydrogen bonds with the hinge region of TNIK, which allows inhibition of kinase activity by strong interactions with TNIK. Therefore, we hypothesized that inhibition of TNIK by sunitinib can regulate TGF-β1-induced EMT and invasion in NSCLC cells. The results show that sunitinib has the potential to inhibit NSCLC cell proliferation and TGF-β1-induced EMT through Smad/non-Smad signaling and TNIK-mediated Wnt signaling.
Although several types of RTK inhibitors, such as erlotinib and gefitinib, are used to treat NSCLC, the EMT process in NSCLC is a major cause of resistance to long-term chemotherapy [42]. Therefore, to overcome the limitation of single drug chemotherapy, recent trends in NSCLC therapy have sought combinations with other NSCLC treatments that may increase drug efficacy, including immunotherapy, radiotherapy, and targeted therapy [43]. The use of peptides in these combination therapies is a good way to reduce side effects that can occur with multiple drug use. In particular, cyclic peptides have been actively studied recently as biochemical tools and therapeutic agents because of their excellent stability, high resistance to exogenous peptidases, and high affinity and selectivity for binding to target biomolecules [44].
Cyclic pentapeptides containing the RGD motif (cRGDfK) are used to improve the drug delivery and target selectivity of anti-cancer drugs [34,35,40,45,46]. Many studies have demonstrated the potential of RGD-containing peptides as vectors for delivering anticancer agents. In particular, cyclic peptides such as cRGDfK and cRGDyK showed high binding affinity and selectivity for integrin αⅤβ3. Thus, the use of cyclic peptides has been as one of the methods of delivery of therapeutic small molecules such as doxorubicin and paclitaxel in cancers [27]. However, the combined effect of RTK inhibitors and cRGDfK in the EMT process in the tumor microenvironment has not been fully investigated. Based on previous studies, we investigated whether cRGDfK enhances the inhibitory effect of sunitinib on cell proliferation and TGF-β1-induced EMT in NSCLC cells. We demonstrated that sunitinib itself exerts inhibitory effects on TNIK kinase activity and the TGF-β1-induced EMT in A549 cells. However, the inhibitory effects of sunitinib were increased by co-treatment with cRGDfK. A549 cell proliferation and the mRNA and protein expression of EMT markers were synergistically inhibited by sunitinib and cRGDfK co-treatment. These results demonstrate that the effect of cRGDfK, as a targeting peptide, can enhance the efficacy of sunitinib to NSCLC cells in anti-EMT.
Recent reports suggest new methods to improve the therapeutic efficacy of tyrosine kinase inhibitors (TKIs) for NSCLC using combination strategies [43,47]. Two or more TKIs or TKIs and other target therapies (e.g., immunotherapeutics) have been used in the combination regimens, but there is not much known to be used in combination with peptides. In addition, the use of TNIK inhibitors and cyclic peptides for the EMT process in NSCLC progression has not yet been studied. In this regard, the present study suggests that a combination of cRGDfK with TKIs may be a new applicable method for the treatment of NSCLC. In particular, the development of new drugs in the modern biopharmaceutical industry is increasingly challenged by the time and expense of R&D expenditures. To reduce the R&D costs, companies are diversifying how they develop new drugs. Drug repositioning is a new method to search existing drugs for the application of new indications [48,49]. The advantage of drug repositioning is that it reduces the time and cost of developing new drugs, in that safety and toxicity assessments have already been completed. Thus, the use of TKIs and cRGDfK in NSCLC could increase the therapeutic efficacy without the development of new therapies by using existing therapeutic agents and the synergistic effect of drug efficacy.
Although studies on the improvement of anticancer drug efficacy using cyclic peptides have not been actively conducted, our results suggest that cyclic peptides may help prevent the progression of NSCLC using TKIs as therapeutic agents. We plan to conduct studies on the anticancer effect using cyclic peptides in many cancers other than NSCLC.
Supporting information
The detected bands were quantified based on the ImageJ software, and the relative ratio between each sample and loading controls was presented in the figures.
(DOCX)
A549 (A) and H358 (B) cells were transfected with non-targeting control siRNA (siCont) or TNIK siRNA (siTNIK). Transfected cells were treated with sunitinib for 24 h. Cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent the mean ± SD of raw results. * p < 0.05 and ** p < 0.001 (vs. control).
(DOCX)
A549 cells were transfected with non-targeting control siRNA or TNIK siRNA. Transfected cells were treated with TGF-β1 (5 ng/mL) for 48 h. The mRNA expression was measured by qRT-PCR analysis. Experiments were performed in triplicate. Data represent mean ± SD.
(DOCX)
A549 cells were transfected with non-targeting control siRNA or TNIK siRNA. Transfected cells were treated with TGF-β1 (5 ng/mL) for 48 h. The mRNA expression was measured by qRT-PCR analysis. Experiments were performed in triplicate. Data represent mean ± SD.
(DOCX)
The protected linear pentapeptide (1) bound to the resin was synthesized using the Fmoc solid phase peptide synthesis (SPPS) method. The linear peptide (2) was cleaved from the resin without affecting other protecting groups by using acetic acid/TFE/CH2Cl2 (1:1:3 ratio) solution. Finally, cyclic pentapeptide c(RGDfK) (4) was obtained by head-to tail cyclization under T3P, TEA, DAMP and elimination of the protecting group.
(DOCX)
Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with cRGDfK for 72 h. Actin was used as a loading control.
(DOCX)
Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with cRGDfK for 48 h (p-Smad2/3) or 72 h (p-ERK1/2 and p-p38). Actin was used as a loading control.
(DOCX)
H358 (A) and H1299 (B) cells were treated with sunitinib and cRGDfK for 24 h. After incubation, cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent mean ± SD. * p < 0.05 and ** p < 0.001 (vs. control).
(DOCX)
(PDF)
(PDF)
(PDF)
Acknowledgments
The biospecimens and data used for this study were provide by the Biobank of Chungnam National University Hospital, a member of the Korea Biobank Network.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and NCT; 2020R1A2C1006416 and Ministry of Education; 2017R1D1A1B03027968). This work has not received any financial investment by CHA Meditech, Co., Ltd. The funder provided support in the form of salaries for authors [KYP], but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘Author Contributions’ section.
References
- 1.Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ward E, Brawley O, Jemal A (2011) Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 61:212–236 10.3322/caac.20121 [DOI] [PubMed] [Google Scholar]
- 4.Behrens J, Mareel MM, Van Roy FM, Birchmeier W (1989) Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol 108:2435–2447 10.1083/jcb.108.6.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck MB, Knabbe C (2006) TGF-β signaling in breast cancer. Ann NY Acad Sci 1989:119–126 [DOI] [PubMed] [Google Scholar]
- 6.Barcellos-Hoff MH, Akhurst RJ (2009) TGF-β in breast cancer: too much, too late, Breast Cancer Res 11:202 10.1186/bcr2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drabsch Y, ten Dijke P (2012) TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 31:553–568 10.1007/s10555-012-9375-7 [DOI] [PubMed] [Google Scholar]
- 8.Heldin CH, Miyazono K, ten Dijke P (1997) TGF-beta signaling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471 10.1038/37284 [DOI] [PubMed] [Google Scholar]
- 9.Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19:2783–2810 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- 10.Massague J (1998) TGF-beta signal transduction. Annu Rev Biochem 67:753–791 10.1146/annurev.biochem.67.1.753 [DOI] [PubMed] [Google Scholar]
- 11.Peinado H, Portillo F, Cano A (2004) Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol 48:365–375 10.1387/ijdb.041794hp [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C et al. (2004) Twist a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927–939 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 13.Galliher AJ, Neil JR, Schiemann WP (2006) Role of TGF-β in cancer progression. Future Oncol 2:743–763 10.2217/14796694.2.6.743 [DOI] [PubMed] [Google Scholar]
- 14.Medici D, Hay ED, Olsen BR (2008) Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 19:4875–4887 10.1091/mbc.e08-05-0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen XF, Zhang HJ, Wang HB, Zhu J, Zhou WY, Zhang H et al. (2012) Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Mol Biol Rep 39:3549–3556 10.1007/s11033-011-1128-0 [DOI] [PubMed] [Google Scholar]
- 16.Mahmoudi T, Li VS, Ng SS, Taouatas N, Vries RG, Mohammed S et al. (2009) The kinase TNIK is an essential activator of Wnt target genes. EMBO J 28:3329–3340 10.1038/emboj.2009.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shitashige M, Satow R, Jigami T, Aoki K, Honda K, Shibata T et al. (2010) Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res 70:5024–5033 10.1158/0008-5472.CAN-10-0306 [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Moon SH, Kim BT, Chae CH, Lee JY, Kim SH (2014) A novel aminothiazole KY-05009 with potential to inhibit Traf2- and Nck-interacting kinase (TNIK) attenuates TGF-β1-mediated epithelial-to-mesenchymal transition in human lung adenocarcinoma A549 cells. PLoS One 9:e110180 10.1371/journal.pone.0110180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Chon, Bae KJ, Byun BJ, Kim SA, Kim J (2016) Traf2- and Nck-interacting kinase (TNIK) is involved in the anti-cancer mechanism of dovitinib in human multiple myeloma IM-9 cells. Amino Acids 48:1591–1599 10.1007/s00726-016-2214-3 [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Jung JI, Park KY, Kim SA, Kim J (2017) Synergistic inhibition effect of TNIK inhibitor KY-05009 and receptor tyrosine kinase inhibitor dovitinib on IL-6-induced proliferation and Wnt signaling pathway in human multiple myeloma cells. Oncotarget 8:41091–41101 10.18632/oncotarget.17056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G et al. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 29:1046–1051 10.1038/nbt.1990 [DOI] [PubMed] [Google Scholar]
- 22.Papaetis GS, Syrigos KN (2009) Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 23:377–389 10.2165/11318860-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 23.Novello S, Camps C, Grossi F, Mazieres J, Abrey L, Vernejoux JM et al. (2011) Phase II Study of Sunitinib in Patients With Non-Small Cell Lung Cancer and Irradiated Brain Metastases. J Thorac Oncol 6:1260–1266 10.1097/JTO.0b013e318219a973 [DOI] [PubMed] [Google Scholar]
- 24.Socinski MA, Wang XF, Baggstrom MQ, Gu L, Stinchcombe TE, Edelman MJ (2014) Sunitinib (S) switch maintenance in advanced non-small cell lung cancer (NSCLC): An ALLIANCE (CALGB 30607), randomized, placebo-controlled phase III trial. J Clin Oncol 32 (suppl):8040–8040 [Google Scholar]
- 25.Scagliotti GV, Krzakowski M, Szczesna A, Strausz J, Makhson A, Reck M et al. (2012) Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol 30:2070–2078 10.1200/JCO.2011.39.2993 [DOI] [PubMed] [Google Scholar]
- 26.Vansteenkiste J, Barlesi F, Waller CF, Bennouna J, Gridelli C, Goekkurt E et al. (2015) Cilengitide Combined With Cetuximab and Platinum-Based Chemotherapy as First-Line Treatment in Advanced Non-Small-Cell Lung Cancer (NSCLC) Patients: Results of an Open-Label, Randomized, Controlled Phase II Study (CERTO), Ann Oncol. 26:1734–1740 10.1093/annonc/mdv219 [DOI] [PubMed] [Google Scholar]
- 27.Chen K, Chen X (2011) Integrin targeted delivery of chemotherapeutics. Theranostics 1:189–200 10.7150/thno/v01p0189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierschbacher MD, Ruoslahti E (1984) Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 309:30–33 10.1038/309030a0 [DOI] [PubMed] [Google Scholar]
- 29.Ruoslahti E (2003) The RGD story: a personal account. Matrix Biol 22:459–465 10.1016/s0945-053x(03)00083-0 [DOI] [PubMed] [Google Scholar]
- 30.Ruoslahti E (1996) RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12:697–715 10.1146/annurev.cellbio.12.1.697 [DOI] [PubMed] [Google Scholar]
- 31.Ruoslahti E, Pierschbacher MD (1987) New perspectives in cell adhesion: RGD and integrins. Science 238:491–497 10.1126/science.2821619 [DOI] [PubMed] [Google Scholar]
- 32.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL et al. (2002) Crystal structure of the extracellular segment of integrin αⅤβ3 in complex with an Arg-Gly-Asp ligand. Science 296:151–155 10.1126/science.1069040 [DOI] [PubMed] [Google Scholar]
- 33.Temming K, Schiffelers RM, Molema G, Kok RJ (2005) RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist Updat 8:381–402 10.1016/j.drup.2005.10.002 [DOI] [PubMed] [Google Scholar]
- 34.Bianchini F, Portioli E, Ferlenghi F, Vacondio F, Andreucci E, Biagioni A et al. (2019) Cell-targeted c(AmpRGD)-sunitinib molecular conjugates impair tumor growth of melanoma. Cancer Lett 446:25–37 10.1016/j.canlet.2018.12.021 [DOI] [PubMed] [Google Scholar]
- 35.Sartori A, Corno C, De Cesare M, Scanziani E, Minoli L, Battistini L et al. (2019) Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models. Cancers 11:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem 4:187–217 [Google Scholar]
- 37.Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 38.Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
- 39.Zhao L, Wientjes MG, Au JL (2004) Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res 10:7994–8004 10.1158/1078-0432.CCR-04-1087 [DOI] [PubMed] [Google Scholar]
- 40.Kulhari H, Pooja D, Kota R, Reddy TS, Tabor RF, Shukla R et al. (2016) Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol Pharm 13:1491–1500 10.1021/acs.molpharmaceut.5b00935 [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Kuiatse I, Lee AV, Pan J, Giuliano A, Cui X (2010) Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol Cancer Res 8:266–277 10.1158/1541-7786.MCR-09-0221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakobsen KR, Demuth C, Sorensen BS, Nielsen AL (2016) The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl Lung Cancer Res 5:172–182 10.21037/tlcr.2016.04.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z, Tam KY (2018) Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int J Biol Sci 14:204–216 10.7150/ijbs.22955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ong YS, Gao L, Kalesh KA, Yu Z, Wang J, Liu C et al. (2017) Recent Advances in Synthesis and Identification of Cyclic Peptides for Bioapplications. Curr Top Med Chem 17:2302–2318 10.2174/1568026617666170224121658 [DOI] [PubMed] [Google Scholar]
- 45.Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y (2012) Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials 33:1627–1639 10.1016/j.biomaterials.2011.11.012 [DOI] [PubMed] [Google Scholar]
- 46.Morlieras J, Dufort S, Sancey L, Truillet C, Mignot A, Rossetti F et al. (2013) Functionalization of small rigid platforms with cyclic RGD peptides for targeting tumors overexpressing αvβ3-integrins. Bioconjug Chem 24:1584–1597 10.1021/bc4002097 [DOI] [PubMed] [Google Scholar]
- 47.Zhou C, Yao LD (2016) Strategies to Improve Outcomes of Patients with EGRF-Mutant Non-Small Cell Lung Cancer: Review of the Literature. J Thorac Oncol 11:174–186 10.1016/j.jtho.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 48.Chong CR, Sullivan DJ Jr (2007) New uses for old drugs. Nature 448:645–646 10.1038/448645a [DOI] [PubMed] [Google Scholar]
- 49.Hanusova V, Skalova L, Kralova V, Matouskova P (2015) Potential anti-cancer drugs commonly used for other indications. Curr Cancer Drug Targets 15:35–52 10.2174/1568009615666141229152812 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The detected bands were quantified based on the ImageJ software, and the relative ratio between each sample and loading controls was presented in the figures.
(DOCX)
A549 (A) and H358 (B) cells were transfected with non-targeting control siRNA (siCont) or TNIK siRNA (siTNIK). Transfected cells were treated with sunitinib for 24 h. Cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent the mean ± SD of raw results. * p < 0.05 and ** p < 0.001 (vs. control).
(DOCX)
A549 cells were transfected with non-targeting control siRNA or TNIK siRNA. Transfected cells were treated with TGF-β1 (5 ng/mL) for 48 h. The mRNA expression was measured by qRT-PCR analysis. Experiments were performed in triplicate. Data represent mean ± SD.
(DOCX)
A549 cells were transfected with non-targeting control siRNA or TNIK siRNA. Transfected cells were treated with TGF-β1 (5 ng/mL) for 48 h. The mRNA expression was measured by qRT-PCR analysis. Experiments were performed in triplicate. Data represent mean ± SD.
(DOCX)
The protected linear pentapeptide (1) bound to the resin was synthesized using the Fmoc solid phase peptide synthesis (SPPS) method. The linear peptide (2) was cleaved from the resin without affecting other protecting groups by using acetic acid/TFE/CH2Cl2 (1:1:3 ratio) solution. Finally, cyclic pentapeptide c(RGDfK) (4) was obtained by head-to tail cyclization under T3P, TEA, DAMP and elimination of the protecting group.
(DOCX)
Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with cRGDfK for 72 h. Actin was used as a loading control.
(DOCX)
Serum-deprived A549 cells were treated with TGF-β1 (5 ng/mL) or its combination with cRGDfK for 48 h (p-Smad2/3) or 72 h (p-ERK1/2 and p-p38). Actin was used as a loading control.
(DOCX)
H358 (A) and H1299 (B) cells were treated with sunitinib and cRGDfK for 24 h. After incubation, cell viability was measured by CCK-8 assay. Experiments were performed in triplicate. Data represent mean ± SD. * p < 0.05 and ** p < 0.001 (vs. control).
(DOCX)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.