

Abstract
The profound energetic demand of prolonged exercise imposed upon skeletal muscle and the heart is met by oxidation of substrate within mitochondria. As such, several coordinated events are initiated in order to maintain mitochondria, collectively known as mitochondrial quality control. In this review, we discuss how mitochondrial quality control functions to maintain the integrity of the reticulum and energy production in response to prolonged exercise, as well as the relevant signaling events that dictate these responses. Based upon the prevailing data in the field, we propose a model where exercise-mediated quality control may be chiefly regulated through local mechanisms, thus allowing for the remarkable precision in mitochondrial quality control events.
Keywords: exercise, muscle, myofiber, mitochondria, mitophagy, quality control
1. Myofiber mitochondria: function follows form
Skeletal muscle and heart consist primarily of specialized cells called myofibers, which are capable of generating mechanical force through contraction. Force produced by skeletal muscles facilitates movement, ranging from activities of daily living to feats of athleticism while increased force production in the heart assists other peripheral tissue function, including skeletal muscles, by increasing blood flow, thus providing more oxygen and metabolic substrates for oxidation. Sufficient ATP production is tantamount to increased force productions in both skeletal and cardiac myofibers. Energy production can increase as much as 100 fold in skeletal muscle1 and approximately 3 fold in the heart2 during prolonged periods of increased contraction, as can occur during exercise. Such a profound increase in energetic demand during prolonged exercise is met by oxidation of metabolic substrates within mitochondria.
Mitochondria are double membrane organelles, consisting of an outer membrane (OMM), an inner membrane (IMM), and an interior region known as the matrix. OMM contains various transport proteins for import and export of protons, metabolic intermediates and substrates, whereas IMM forms an elaborate, folded membrane structure called cristae, which contains macromolecules of the electron transport chain proteins where oxidative phosphorylation of metabolic intermediates takes place. Mitochondrial matrix contains all the proteins and enzymes required for generation of ATP via the Kreb’s Cycle as well as proteins and enzymes for other metabolic pathways. Myofiber mitochondria are particularly enriched in proteins involved in the TCA cycle, electron transport chain, and oxidative phosphorylation3–7, illustrating a physiologic safeguard for meeting energetic demands in these tissues.
In their native state, myofiber mitochondria form an intricate reticulum that extends along the length of the cell8–11, which in humans can be in the tens of centimeters in length. Myofiber mitochondria function as a syncytium with potential energy distributed across the reticulum9,10,12. Recent evidence suggests that morphology of the reticulum is directly related to the metabolic properties of a given myofiber. For example, glycolytic myofibers (e.g. plantaris muscle in the hindlimb), which rely primarily on glycolysis for ATP production, have a less robust mitochondrial reticulum among myofibrils and it interacts more with the sarcoplasmic reticulum, presumably owing to the high demands of Ca2+ cycling during repeated contractions11. In contrast, mitochondrial reticulum in oxidative myofibers (e.g. soleus muscle in the hindlimb and the heart) take up approximately a third of cell volume and is geometrically associated with lipid droplets and the contractile proteins11. Such phenotypic variation between myofiber reticula is thought to be optimal for tissue function, as contractile power is of greater importance in glycolytic muscle compared to preferential maintenance of energetic homeostasis in oxidative myofibers, such as the heart9,11.
2. Mitochondrial quality control maintains both structure and function
The high degree of structure to function specialization in myofiber mitochondria implies that it is closely monitored and regulated to maintain energy production capacity. These processes that regulate the mitochondrial reticulum are collectively referred to as mitochondrial quality control. Mitochondrial quality control largely consists of biogenesis, dynamics (i.e. fission and fusion), and selective degradation via proteolysis and mitophagy. In a non-pathological state, these processes are temporally upregulated in response to prolonged energetic stress, such as exercise13–16. This upregulation during prolonged energetic stress accomplishes at least two goals: 1) ensures energetic production and homeostasis by the mitochondrial reticulum during high demand, and 2) orchestrates adaptation of the reticulum to better meet similar energetic demands in the future. As we have discussed in depth elsewhere the various signaling factors involved in mitochondrial quality control pertaining to exercise15,16, we will provide only an abridged discussion of the main processes here, though framed within the current context of mitochondrial reticulum structure and function.
2.1. Exercise-induced Mitochondrial Dynamics
One of the first events of mitochondrial quality control in skeletal muscle during prolonged exercise appears to be mitochondrial fission. This is evidenced in that phosphorylation of the fission protein, dynamin-related protein 1 (Drp1) at its activating S616 site is increased following acute exercise in both mice and humans13,14,17,18 and is likely activated early during a prolonged exercise bout14. Ex vivo evidence suggests that the mitochondrial reticulum can physical separate regions of itself within minutes9, though whether mitochondrial fission is activated with similar urgency under physiological stimuli is unknown, but not unreasonable to speculate. Fission of selected regions of mitochondrial reticulum serves both to acutely preserve the reticulum and to pave the way for degradation of selected mitochondria through mitophagy (discussed in subsequent section)15,16. It is perhaps not surprising then that impaired fission in skeletal muscle, through muscle-specific deletion of Drp1, reduces exercise capacity14. Likewise, conditional deletion of Drp1 in the heart impairs not only mitochondrial respiratory function but also cardiac function19, as well as exacerbates the development of cardiac pathologies20.
Inversely to fission, an adaptive response to periods of increased oxidative phosphorylation in myofibers is increased fusion of mitochondria to support exchange of matrix components21–23, thus aiding energetic efficiency of the reticulum. Loss of fusion protein optic atrophy 1 (OPA1) impairs mitochondrial function in vitro14, and skeletal muscle-specific loss of the primary fusion proteins (Mitofusin 1 and 2) impairs exercise training-induced improvements in exercise capacity and impairs mitochondrial respiratory function in mice25. Exercise training in humans, increases the fusion to fission protein ratio in skeletal muscle, suggesting a condition favoring a more fused mitochondria reticulum26. Taken together, the capacity to precisely reorient the architecture of mitochondrial reticulum is essential for myofiber energetic homeostasis, with fission potentially more important during acute periods of stress and fusion more involved in the adaptive responses to exercise training.
2.2. Exercise-induced Mitophagy
A possible outcome for regions of mitochondrial reticulum to be separated from the whole via fission is degradation through mitophagy. Evidence from our lab in mouse skeletal muscle suggests that exercise-induced mitophagy is likely initiated during exercise but does not resolve (i.e. fusion of autophagosome containing mitochondria with lysosome) until well into the recovery period13. Upregulation of mitophagy in response to prolonged exercise is mediated through the energetic nucleotide sensor, 5’ AMP-activated Protein Kinase (AMPK). While AMPK is involved in various metabolic processes related to energetic stress, in the context of mitophagy, activated AMPK phosphorylates Unc51-like autophagy activating kinase 1 (Ulk1) at Ser55513,27,28. Phosphorylation at Ser555 activates Ulk1, initiating the formation of the autophagosome through various downstream effectors15,29–31. Loss of Ulk1 in skeletal muscle not only blocks mitophagy induced by acute exercise but also impairs metabolic adaptation to exercise training13. While exercise-induced mitophagy through Ulk1 in cardiac myofibers has not been described in the literature, Ulk1 has been recently proposed to be the predominant mechanism by which mitophagy protects cardiac myofibers from ischemia and starvation32.
While it is clear that Ulk1 is integral to the mitophagy response following acute exercise, the downstream effectors of Ulk1 are still being elucidated. One downstream substrate of Ulk1 that has emerged recently as a critical factor for energetic stress-induced mitophagy in myofibers is FUN14-domain containing 1 (Fundc1). When activated, Ulk1 translocates to mitochondria where it phosphorylates Fundc1 at Ser17, enhancing its binding capacity with the autophagosome membrane via microtubule-associated protein light chain 3 (LC3) in vitro33. Skeletal muscle-specific loss of Fundc1 impairs mitochondrial energetics, exercise performance, and adipose metabolism34. In drosophila m. knock-down of other suggested Ulk1 substrates (e.g. Atg2, Atg9, and Atg18) dramatically disrupts mitochondrial reticulum in indirect flight muscle and heart, resulting in poor tissue function and reduced lifespan35. While much is still unknown how the downstream mitophagy process is mediated in response to acute exercise, it is clear that recognition and degradation of damaged and/or dysfunctional regions of mitochondrial reticulum is critical for tissue function, as well as systemic health, and this is a key component of how the beneficial effects of exercise are mediated.
2.3. Exercise-induced Mitochondrial Biogenesis
Finally, and arguably the most studied aspect of mitochondrial quality control in response to exercise in myofibers is mitochondrial biogenesis. Exercise is a potent inducer of the main transcriptional regulator of mitochondria, peroxisome proliferator-activated receptor gamma coactivated 1-alpha (PGC1α), resulting in increased expression of mitochondria-related mRNA36. Due to the energetic expense of prolonged exercise, mRNA translation is slowed to conserve ATP37, 38, in part due to increased AMPK activity, presumably through its antagonistic action on protein synthesis by inhibiting the mechanistic target of rapamycin complex I (mTORC1)39. However, long-term inhibition of mTORC1 through rapamycin feeding does not impair mitochondrial biogenesis in either skeletal muscle or heart40,41, suggesting an alternative translational mechanism for the bulk of mitochondria-related mRNA. Presumably, once exercise is stopped and energetics return to a new homeostasis, bulk synthesis of mRNA resumes, and incorporation of newly translated proteins into mitochondrial reticulum is aided through fusion, leading to an expansion of the reticulum in myofibers15.
In summary, mitochondria are able to respond to energetic stress caused by acute exercise through a rapid change of their confirmation to maintain energetic output. Simultaneously, this remodeling of the reticulum aids in the removal and degradation of areas that would otherwise impede the capacity for mitochondria to match the energetic demands. Likewise, these events lead to adaptive changes in the structure of the reticulum to better meet future energetic challenges. Therefore, exercise-regulated mitochondrial quality control illustrates the interdependency between the structure and the function in myofiber mitochondria.
3. Spatial specificity of mitochondrial quality control
Mitochondria in myofibers functions as a syncytium, with energy distributed across the reticulum9,10,12. During exercise, mitochondrial function dramatically increases42. In agreement with an optimal distribution of potential energy across the reticulum, acute energetic stress as a result of different stimuli (including exercise) also appears to be uniform across mitochondrial reticulum13,43–15. Interestingly though, advances in molecular imaging technology with the development of mitochondrial reporter genes (mt-Kiema46, MitoTimer43,44, and mitoQC47) for visualizing mitochondrial quality control processes has revealed a high degree of spatial specificity in these processes. For example, acute exercise results in only a small percentage of the overall mitochondrial reticulum being targeted for degradation through mitophagy in skeletal muscle, evidenced by distinct mitochondria-originating puncta encased in autolysosomes13,43 (Fig. 1A and B). Similarly, acute ischemia or starvation also results in spatially distinct mitophagy events in the heart and skeletal muscle32,44,45. Spatial specificity in mitophagy is in agreement with the spatial variability that is observed in fission events, a prerequisite for mitophagy, within myofibers9,21. Furthermore, synthesis rates of mitochondrial proteins in the heart and skeletal muscle displays a great deal of heterogeneity48–50, and turnover of the fluorescent mitochondria reporter MitoTimer in the heart is also spatially variable51. In that light, it is reasonable to speculate that biogenesis and incorporation of new mitochondria proteins in response to exercise occurs in a spatially heterogeneous manner as well.
Figure 1.
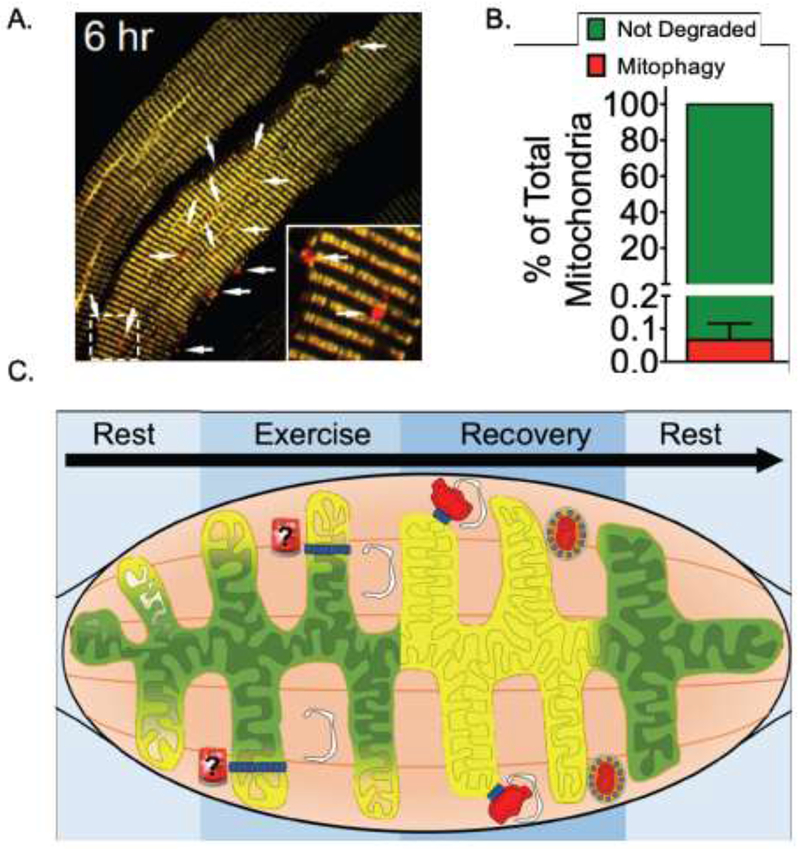
Model for domain-specific regulation of mitochondria quality control. A) Representative image of exercise-induced mitophagy in skeletal muscle (Laker and Drake, et al. Nat Commun. 2017, 8: 548). B) Quantification of mitochondria engulfed in autolysosomes (red puncta) relative to total mitochondrial area. Based on data published in Laker and Drake, et al. Nat Commun. 2017, 8: 548. While semi-quantitative, this calculation illustrates that the areas of the reticulum targeted for mitophagy in response to acute exercise is a small fraction of the network. C) Illustration of local-mediation of mitochondria quality control to maintain energetic homeostasis and adaptation to energetic stress.
Collectively, these observations suggest a model where, in a non-pathologic state, mitochondrial quality control is regulated by local mechanisms in distinct domains across the reticulum in myofibers (Fig. 1C). Given how closely mitochondrial quality control is related to mitochondrial energetics9,21, it is possible that fluctuations in local energetics may dictate spatial mitochondrial quality control. For example, in the heart, Ca2+ influx and efflux are spatially controlled across the mitochondrial reticulum to maintain its overall function52. Thus, local failures in energy production, possibly resulting in separation of that domain(s) from the whole9, and setting off a cascade of local events (i.e. mitophagy and biogenesis/fusion) to restore homeostasis at the affected domain. However, potential signaling regulator(s) for such a local mitochondrial quality control domain response are unknown.
4. Conclusion: Importance of targeted remodeling for disease intervention.
The molecular response in mitochondrial quality control to acute exercise in a healthy or nonpathological state represents the physiological ideal stress signaling response. That is to say exercise elicits the appropriate changes to mitochondrial reticulum that maximize energetic output and, overtime, results in adaptations to the system to better meet future energetic challenges (i.e. training adaptations). Under disease conditions, however, poor mitochondria quality in myofibers is a common characteristic, as are impairments in aspects of quality control mechanisms (i.e. aging, sarcopenia, neuromuscular and cardiovascular diseases)29,53–55. Therefore, understanding how mitochondrial quality control is spatially mediated by energetic stress could lead to novel interventions to treat disease or aid in recovery from injury (e.g. myocardial infarction). Elucidating the mechanisms that dictate how specific regions are distinguished from the vast, complex mitochondrial reticulum is an important step in developing efficacious therapies to improve mitochondrial quality in chronic disease.
Acknowledgements
This work was supported by NIH (R01-AR050429) to Z.Y. and NIH (K99-AG057825) to J.C.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
The authors declare no competing financial interests
References
* Of special interest
** Of outstanding interest
- 1.Weibel ER. & Hoppeler H. Exercise-induced maximal metabolic rate scales with muscle aerobic capacity. J. Exp. Biol. 208, 1635–1644 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Bittls JA & Ingwall J Reaction Rates of Creatine Kinase and ATP Synthesis in the Isolated Rat Heart. J Biol Chem 260, 3512–3517 (1985). [PubMed] [Google Scholar]
- 3.Johnson DT, Harris RA, Blair PV & Balaban RS Functional consequences of mitochondrial proteome heterogeneity. Am J Physiol Cell Physiol 292, C698–C707 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Benard G et al. Physiological diversity of mitochondrial oxidative phosphorylation. Am J Physiol Cell Physiol 291, C1172–C1182 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Mootha VK et al. Integrated Analysis of Protein Composition , Tissue Diversity , and Gene Regulation in Mouse Mitochondria. Cell 115, 629–640 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Bak S, Leon IR, Jensen ON & Højlund K Tissue Specific Phosphorylation of Mitochondrial Proteins Isolated from Rat Liver, Heart Muscle, and Skeletal Muscle. J Proteome Res 12, 4327–4339 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Forner F, Foster LJ, Campanaro S, Valle G & Mann M Quantitative Proteomic Comparison of Rat Mitochondria from Muscle , Heart , and Liver. Mol Cell Proteomics 608–619 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Kirkwood SP, Munn EA & Brooks GA Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 251, C395–402 (1986). [DOI] [PubMed] [Google Scholar]
- 9.**.Glancy B et al. Power Grid Protection of the Muscle Mitochondrial Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 19, 487–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors expanded upon previous work in skeletal muscle, and illustrated the level of complexity in the mitochondrial reticulum interconnectivity in cardiac myofibers via focused ion beam scanning electron microscopy. They also illustrated that the organization of the mitochondrial reticlum in myofibers is directly related to the energetic requirments of the tissue. Futhermore, the authors demonstrated that select areas of the reticulum under the condition of dysfunction, were physically separated from the whole reticulum, limiting the spread of dysfunction and preserving the functional capacity of the reticulum.
- 10.Glancy B et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617–620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.*.Bleck CKE, Kim Y, Willingham TB & Glancy B Subcellular connectomic analyses of energy networks in striated muscle. Nat. Commun. 9, 5111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors developed a connectomics approach to analyze mitochondria reticulum in relation to myofiber energy utilization and storage. Here, the authors demonstrated that mitochondria structure and electrical connectivity is related to metabolic and contractile properties of a given skeletal muscle fiber type, thus, optimizing the energetic output towards either contractile force (i.e. glycolytic fibers) versus homeostasis (i.e. oxidative fibers).
- 12.Ghosh S et al. Insights on the impact of mitochondrial organisation on bioenergetics in high-resolution computational models of cardiac cell architecture. PLoS Comput. Biol. 14, 1–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.**.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z Ampk phosphorylation of Ulk1 is required for lysosome targeting of mitochondria in in exercise-induced mitophagy. Nat. Commun. 8, 548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that acute exercise induces mitophagy in skeletal muscle through an AMPK-Ulk1 dependent mechanism. Interestingly, though mitochondrial oxidative stress was uniform across the fiber, mitophagy events were spatially variable. These data illustrate the high degree of specificity in the mitophagy machinery to recognize precise locations within the reticulum for degradation. Furthermore, loss of the the mitophagy regulator Ulk1 in skeletal muscle impaired metabolic adaptation to exercise training, suggesting mitochondrial quality control in myofibers is important for systemic metabolism.
- 14.*.Moore TM et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol. Metab. 21, 51–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors illustrated that Drp1 S616 phosphorylation, indicative of mitochondrial fission, was increased early during prolonged exercise. Loss of Drp1 in skeletal muscle impaired exercise capacity and mitigated gains in exercise capacity with exercise training. Additionally, the authors showed evidenc of reduced Dmn1L expression, which encodes for Drp1, in skeletal muscle biopsies from dysglycemic humans, indicative of the importance for mitochondrial quality control in myofibers for proper metabolic health.
- 15.Drake JC, Wilson RJ & Yan Z Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 30, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan Y, Drake JC & Yan Z Exercise-Induced Mitophagy in Skeletal Muscle and Cardiomyocyte. Exerc. Sport Sci. Rev. In press, DOI: 10.1249/JES.0000000000000192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruse R, Pedersen AJT, Kristensen JM, Petersson SJ & Jørgen FP Intact initiation of autophagy and mitochondrial fission by acute exercise in skeletal muscle of patients with Type 2 diabetes. Clin Sci 131, 37–47 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Schwalm C, Deldicque L & Francaux M Lack of Activation of Mitophagy during Endurance Exercise in Human ´. Med. Sci. Sports Exerc. 3, 1552–1561 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Ikeda Y et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Shirakabe A et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133, 1249–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra P et al. Mitochondrial Dynamics Is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Article Mitochondrial Dynamics Is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 22, 1033–1044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisner V, Lenaers G & Hajnoczky G Mitochondrial fusion is frequent in skeletal muscle and supports excitation–contraction coupling. J Cell Biol 205, 179–195 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisner V et al. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci U S A. 114, E859–E868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishra P, Carelli V, Manfredi G & Chan DC Article Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 19, 630–641 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell MB, Bush Z, Mcginnis GR & Rowe XGC Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J Appl Physiol 126, 341–353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Axelrod CL, Fealy CE, Mulya A & Kirwan JP Exercise training remodels human skeletal muscle mitochondrial fission and fusion machinery towards a pro ‐ elongation phenotype. Acta Physiol (Oxf). 225, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egan DF et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 331, 456–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bujak AL et al. AMPK Activation of Muscle Autophagy Prevents Fasting-Induced Hypoglycemia and Myopathy during Aging. Cell Metab. 21, 883–890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drake JC, Yan Z Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol. 595, 6391–6399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weerasekara VK et al. Metabolic-stress-induced rearrangement of the 14-3-3ζ interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3ζ interaction with phosphorylated Atg9. Mol Cell Biol. 34, 4379–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou C et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 27, 184–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.**.Saito T et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest 129, 802–819 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; By using the Mt-Kiema reporter gene, this study illustrated the spatial specificity of mitophagy within cardiac myofibers following starvation or ischemia. Interestingly, the authors demonstrated that energetic stress-induced mitophagy was exclusively dependent upon Ulk1 and not other mitophagy regulators Atg7 or Parkin. The authors also demonstrated that Ulk1 acts simultaneoulsy through Rab9 to promote formation of the autophagosome and through Rip1/Drp1 to promote fission, both aiding in degredation of selective regions of the reticlum through mitophagy.
- 33.Wu W et al. ULK 1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.*.Fu T et al. Mitophagy Directs Muscle-Adipose Crosstalk to Article Mitophagy Directs Muscle-Adipose Crosstalk to Alleviate Dietary Obesity. Cell Rep. 23, 1357–1372 (2018). [DOI] [PubMed] [Google Scholar]; Here, the authors demonstrated that loss of the Ulk1 substrate, Fundc1, in both skeletal muscle and heart as well as just skeletal muscle alone impairs exercise performance and mitochondrial function. Interestingly, loss of Fundc1 also impaired fat utilization in skeletal muscle but resulted in protection from a high-fat diet due to FGF21-mediated white adipose thermogenesis, indicating a high level of communication myofiber mitochondrial qualtiy control and systemic metabolism.
- 35.*.Xu P et al. Atg2, Atg9 and Atg18 in mitochondrial integrity, cardiac function and healthspan in Drosophila. J Mol Cell Cardiol. 127, 116–124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; By pairing the MitoTimer reporter gene with Mef2-GAL4-RNAi-mediated knockdowns of selected Atg’s (e.g. Atg2, Atg18, and Atg9), this study demonstrated that impaired mitophagy in drosophila m. heart tube and intermediate flight muscle disrupts mitochondria quality and leads to poor mobility, or healthspan, and reduced lifespan.
- 36.Safdar A et al. Exercise increases mitochondrial PGC-1α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem. 286, 10605–10617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Pagano AF, Py G, Bernardi H, Candau RB & Sanchez AMJ Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med Sci Sports Exerc. 46, 1314–1325 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Williamson DL, Kubica N, Kimball SR & Jefferson LS Exercise-induced alterations in extracellular signal-regulated kinase 1/ 2 and mammalian target of rapamycin (mTOR) signalling to regulatory mechanisms of mRNA translation in mouse muscle. J. Physiol. 573, 497–510 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoki K, Zhu T & Guan KL TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 115, 577–590 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Drake JC et al. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J. Gerontol. A. Biol. Sci. Med. Sci. 68, 1493–501 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller BF, Drake JC, Naylor B, Price JC & Hamilton KL The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev. 18, 106–111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollozy J Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242, 2278–2282 (1967). [PubMed] [Google Scholar]
- 43.Laker RC et al. A novel mitotimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 289, 12005–12015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson RJ et al. Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion 44, 20–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson RJ et al. Mitochondrial protein S-nitrosation protects against ischemia reperfusion-induced denervation at neuromuscular junction in skeletal muscle. Free Radic Biol Med. 117, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun N et al. Measuring In Vivo Mitophagy. Mol. Cell 60, 685–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McWilliams TG et al. Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214, 333–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim T-Y et al. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics. 11, 1586–1594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaleel A et al. In vivo measurement of synthesis rate of individual skeletal muscle mitochondrial proteins. Am J Physiol Endocrinol Metab 295, E1255–E1268 (20008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kasumov T et al. Mitochondria in Cardiovascular Physiology and Disease Assessment of cardiac proteome dynamics with heavy water : slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Hear Circ Physiol 304, H1201–H1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stotland A & Gottlieb RA Journal of Molecular and Cellular Cardiology α -MHC MitoTimer mouse : In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J Mol Cell Cardiol. 90, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.*.De La Fuente S et al. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart Report Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling. Cell Rep. 24, 3099–3107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrated in cardiomyoctes that spatial separation of MCUC-mediated Ca2+ uptake sites within the mitochondria reticulum are spatially separated from NCLX-mediated Ca2+ extrusion sites. This spatial separation in Ca2+ influx and efflux allws for fine-tuning of mitochondrial energetics within heart myofibers.
- 53.Joseph AM et al. Dysregulation of Mitochondrial Quality Control Processes Contribute to Sarcopenia in a Mouse Model of Premature Aging. PLoS One 8, 1–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kowald A & Kirkwood T Accumulation of Defective Mitochondria through Delayed Degradation of Damaged Organelles and Its Possible Role in the Ageing of Post-mitotic and Dividing Cells. J Theor Biol 202, 145–160 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Beltran Valls MR, Wilkinson DJ, Narici MV, Smith K, Phillips BE, Caporossi D, A. P. Protein carbonylation and heat shock proteins in human skeletal muscle: relationships to age and sarcopenia. J Gerontol A Biol Sci Med Sci. 70, 174–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]