

Abstract
Purpose
Brain tumors have become the leading cause of cancer-related mortality in young patients. Novel effective therapies on the basis of the unique biology of each tumor are urgently needed. The goal of this study was to evaluate the feasibility, utility, and clinical impact of integrative clinical sequencing and genetic counseling in children and young adults with high-risk brain tumors.
Patients and Methods
Fifty-two children and young adults with brain tumors designated by the treating neuro-oncologist to be high risk (> 25% chance for treatment failure; mean age, 10.2 years; range, 0 to 39 years) were enrolled in a prospective, observational, consecutive case series, in which participants underwent integrative clinical exome (tumor and germline DNA) and transcriptome (tumor RNA) sequencing and genetic counseling. Results were discussed in a multi-institutional brain tumor precision medicine teleconference.
Results
Sequencing revealed a potentially actionable germline or tumor alteration in 25 (63%) of 40 tumors with adequate tissue, of which 21 (53%) resulted in an impact on treatment or change of diagnosis. Platelet-derived growth factor receptor or fibroblast growth factor receptor pathway alterations were seen in nine of 20 (45%) glial tumors. Eight (20%) sequenced tumors harbored an oncogenic fusion isolated on RNA sequencing. Seventeen of 20 patients (85%) with glial tumors were found to have a potentially actionable result, which resulted in change of therapy in 14 (70%) patients. Patients with recurrent brain tumors receiving targeted therapy had a median progression-free survival (from time on therapy) of 4 months.
Conclusion
Selection of personalized agents for children and young adults with high-risk brain tumors on the basis of integrative clinical sequencing is feasible and resulted in a change in therapy in more than two thirds of children and young adults with high-risk glial tumors.
INTRODUCTION
Outcomes for younger patients with brain tumors remain poor, and these tumors have become the leading cause of cancer-related mortality in the young.1 This is due, in large part, to barriers of using many of the effective oncology treatments to this patient population.1-3 The location of many pediatric brain tumors in the thalamus and brainstem constrains the surgeon from performing a safe surgical resection or even a biopsy in some cases.4,5 Finally, the blood-brain barrier (BBB) restricts access to the CNS to all but 5% of chemical compounds screened for drug development.6
At the molecular level, pediatric and adult brain tumors are dissimilar.2,4 Recurrent mutations and gene expression profiles of pediatric brain tumors are clearly distinct from their adult counterparts.2,7,8 Moreover, molecular characterization of pediatric brain tumors has disclosed key differences in tumors with the same pathology, among their subgroups defined by age and location.2,4,9 Younger patients with high-risk or refractory brain tumors are in urgent need of novel effective therapies that are based, ideally, on the unique biology of each tumor.
In contrast to adult tumors, recent surveys of pediatric tumors have shown that most are driven by relatively few mutational events, many of which may be targetable with personalized clinically available agents.2,10 Although precision medicine is an exciting potential therapeutic avenue for younger patients with brain tumors, this approach requires substantial optimization to successfully improve outcomes for children with these aggressive tumors. Our group has successfully shown the feasibility and potential clinical utility of integrative clinical sequencing in the practice of precision oncology in pediatric and adult patients with relapsed and refractory disease,11,12 and other pediatric groups have recently reported similar findings.13,14 Two recent studies have reported targeted DNA sequencing in pediatric patients with brain tumors.15,16 However, to our knowledge, no studies have been primarily designed to study the role of DNA and RNA sequencing of children and adolescents with high-risk brain tumors, with a focus on their treatment and follow-up.
We therefore launched a clinical sequencing study to better understand the role of personalized genomic characterization and precision oncology in the management of these patients. We performed clinically integrated tumor (DNA/RNA) and germline (DNA) sequencing and genetic counseling for a prospective cohort of children and young adults with high-risk brain tumors. As part of this study, we discussed these results in a multidisciplinary and multi-institutional brain tumor precision medicine conference, with a special emphasis on discussion of the CNS penetration of potential targeted agents.
PATIENTS AND METHODS
Patients
We performed a single-site prospective, observational consecutive case series of patients younger than 40 years with a diagnosis of a high-risk primary brain tumor believed to have at least a 25% chance of treatment failure by the treating oncologist (including patients with tumors with unclear diagnosis or grading—if one of the diagnostic possibilities met our criteria for high risk). The Pediatric MiOncoSeq study was approved by the Institutional Review Board of the University of Michigan Medical School, and all patients or their parents or legal guardians provided informed consent (written assent if > 10 years). All patients were seen by a physician investigator and a genetic counselor. Clinically integrated sequencing was performed according to previous published methodology (Data Supplement).11,12,17 Our study began using a whole-exome gene panel, as previously described. Partway through the study of our cohort, the MiOncoseq sequencing laboratory (Michigan Center for Translational Pathology) adjusted their panel from whole exome to a panel of 1,700 genes (OncoSeq1700) that covered all cancer consensus genes and fusion partners seen in their initially sequenced cohort (> 1,000 tumors). The Michigan Center for Translational Pathology prospectively performed paired whole exome and 1,700 gene panel on a prospective cohort of tumors and confirmed no notable difference in reporting of cancer consensus gene alterations to clinicians.
As previously described,11 potentially actionable findings were defined as any genomic finding that could lead to a change in patient management by providing a targetable tumor molecular aberration, a change in diagnosis or risk stratification (leading to an immediate potential therapeutic application), or a change in patient or family counseling by identifying cancer-related germline findings. For medulloblastoma, sonic hedgehog (SHH) was the only subgroup designation considered actionable by itself.
Brain Tumor Precision Medicine Conference
Tumor sequencing results were reviewed in a multi-institutional brain tumor precision medicine teleconference held at the University of Michigan and attended by Seattle Children’s Hospital, University of California, San Francisco, Baylor, Colorado Children’s Hospital, Lurie Children’s, Children’s National, and Children’s Hospital of Michigan. Clinical sequencing results and potential treatment options were discussed with a multidisciplinary team including clinicians from pediatric and adult neuro-oncology, pediatric neurosurgery, neuropathology, pathology/cytogenetics, pharmacology, bioinformaticians, genetic counselors, and study coordinators, to reach a consensus treatment opinion and to generate discussion of clinical trial availability.
For discussion of targeted agents to be considered for patients, special attention was paid to likelihood of their penetration of BBB. For all agents (available on or off trial), the University of Michigan CNS Targeted Agent Prediction (CNS-TAP) rating system was developed, with seven criteria to score utility of the agent, including targeted pathway/relevance to sequencing results, preclinical data, pediatric phase I data, clinical data in CNS tumors, active clinical trials for which patient is eligible, CNS/BBB penetration, and relevant formulation (pill v suspension; see Data Supplement for sample checkbox evaluation of everolimus for a PIK3CA mutation in a patient with diffuse intrinsic pontine glioma [DIPG]). Scores are given as total number of checks for each agent. For patients with multiple targetable lesions, preference was given to lesions with higher variant allele fraction in tumor, when known.
RESULTS
Feasibility of Clinically Integrated Sequencing for Children and Young Adults With Brain Tumors
We screened 52 patients and enrolled 50 patients with high-risk primary brain tumors (mean age, 10.2 years; median age, 9.0 years; range, 1 to 39 years) between January 2014 and March 2017 (Fig 1). Two families of patients with DIPG declined participation because of the need for biopsy (offered for clinical and research purposes). Patients were referred with a wide variety of brain tumor diagnoses, as long as their treating neuro-oncologist denoted high-risk status (> 25% risk of progression or treatment failure).
Fig 1.
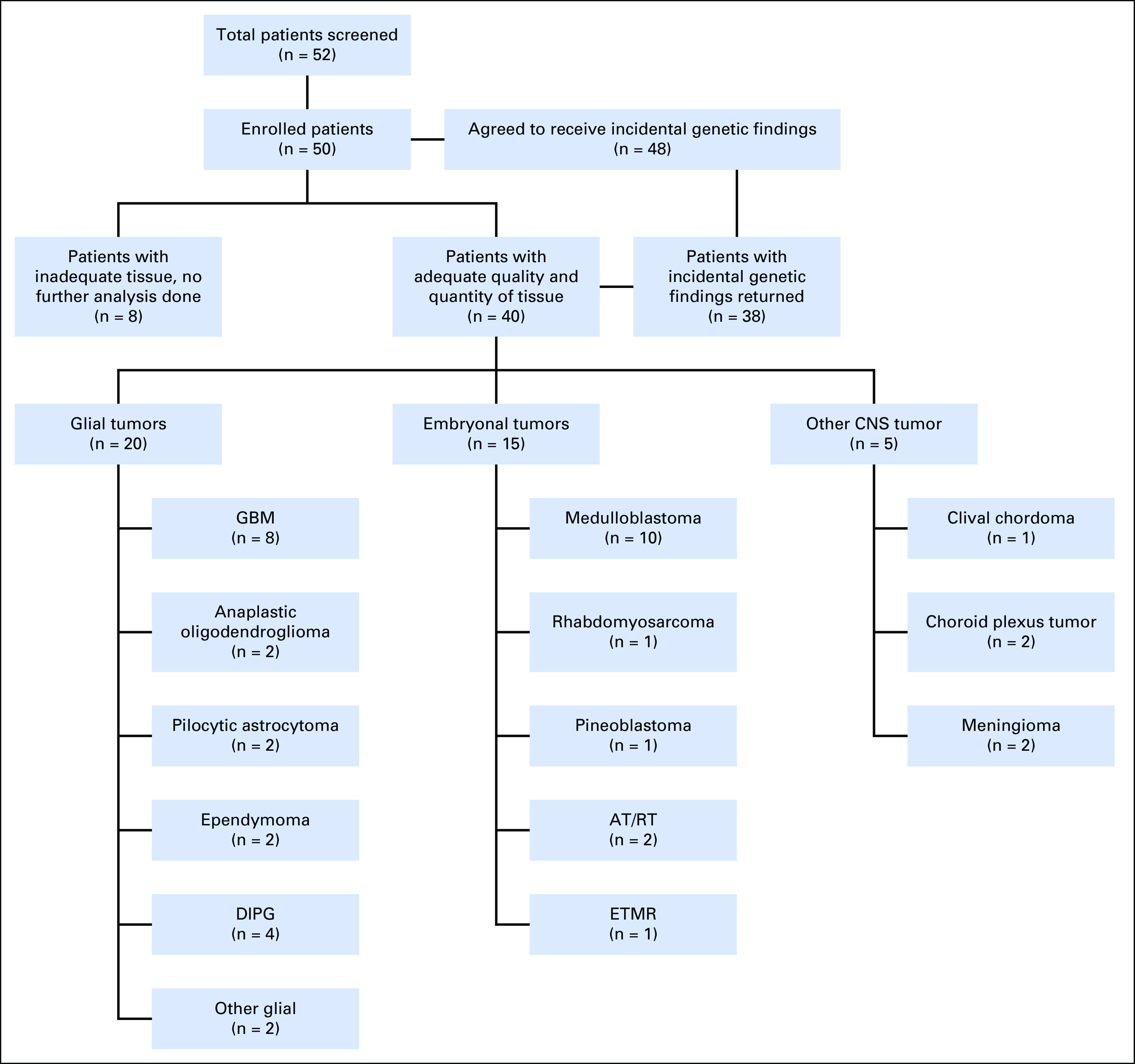
Study enrollment schematic. Children and their families were offered enrollment if they had a diagnosis of a primary brain tumor believed to have at least a 25% chance of treatment failure by their treating oncologist. Approximately half of tumors enrolled were glial by histology. AT/RT, atypical teratoid/rhabdoid tumor; DIPG, diffuse intrinsic pontine glioma; ETMR, embryonal tumor with multilayered rosettes; GBM, glioblastoma multiforme.
Eighteen patients (36%) had relapsed or refractory tumors (of which we sequenced the original tumor in 12 cases), and 13 patients (26%) had metastatic tumors at the time of enrollment (Table 1). Thirty-two patients were enrolled at the time of their original diagnoses, when they presented with either a high-risk neoplasm or a possible diagnosis that met criteria for high-risk neoplasm after pathology review. For all patients, tissue was obtained by standard-of-care diagnostic or therapeutic surgical procedures done either at the time of enrollment or at the time of an earlier procedure. Of the 50 enrolled patients, 40 (80%) had adequate tumor specimen for DNA/RNA sequencing, including 20 (50%) glial tumors, 15 (38%) embryonal tumors, and five (13%) other tumors. Frozen samples were more often adequate than formalin-fixed paraffin-embedded (94% v 74%). In one case, paraffin-embedded tissue on two slides was sufficient to provide tissue for DNA, but not RNA, isolation. The 40 tumors with adequate specimen were used as a denominator for all subsequent analyses. For these tumors, results were provided to the clinician/family at a median of 78 days. A cause of delay for some cases was obtaining formalin-fixed paraffin-embedded tissue from patients at outside institutions. Patients with frozen tumor samples had a median time to results of 49 days.
Table 1.
Feasibility and Actionability of Clinically Integrated Sequencing, by Brain Tumor Subtype

Twenty-six tumors (65%) underwent DNA sequencing with a panel of 1,700 cancer-related genes (OncoSeq1700), rather than whole-exome sequencing. Rates of actionable findings were similar between both methodologies.
Clinical Utility of Integrated Sequencing for Children and Young Adults With Brain Tumors
Sequencing revealed a potentially actionable germline or tumor alteration in 25 (63%) tumors, including five germline and 22 somatic tumor alterations (two patients with both a distinct germline and tumor alteration; Table 1). For these patients, the results led to a change in clinical management/treatment in 21 patients (53%). This included 18 patients (45%) who underwent a change in therapy on the basis of sequencing results and three patients (8%) who received new genetic counseling for germline finding and future cancer risk. During the course of this study, none of these patients were found to have additional malignancies, although three of five of these patients (and the sibling of one of these patients) remain on annual surveillance programs for additional associated malignancies (Data Supplement).
We graded the actionable potential of the somatic mutations into category I to IV (Table A1) as described previously.13 Category I included somatic mutations of established clinical utility; category II: mutations of potential utility; category III: other consensus cancer gene mutations; and category IV: other genes. On the basis of this grading system, these brain tumors harbored an average of 2.3 grade I to III potentially actionable mutations per tumor (range, 0 to 8; median, 2). Glial tumors showed more grade I to III mutations per tumor (average, 3.0; range, 0 to 8; median, 3) than embryonal tumors (average, 1.6; range, 0 to 5; median, 1; Data Supplement).
Most targeted therapeutic interventions included use of tyrosine kinase inhibitors, histone deactylase inhibitors, or mechanistic target of rapamycin inhibitors. Fifteen patients (38%) underwent precision medicine–based therapy with agents selected through the CNS-TAP program, including 10 patients who also underwent radiation and one patient who underwent surgery and radiation (Data Supplement). Our group was heterogeneous in terms of tumor type, time of enrollment (diagnosis or relapse), and whether targeted therapy was given in isolation or was followed by radiation therapy, thus limiting our ability to analyze whether targeted treatments improved treatment outcomes. With the limits of our heterogeneous group, patients with recurrent/refractory brain tumors on targeted therapy had a median progression-free survival (from time on therapy) of 4 months (95% CI, 0.25 to 4 months; Data Supplement).
Despite persistent efforts and referrals to multiple centers with ongoing pediatric brain tumor clinical trials, no patient was able to enroll in a clinical trial on the basis of their sequencing results, and all targeted therapies were used on an off-trial basis. The most frequent reasons cited by clinicians or families of the five patients (of 22) for not pursuing treatment on the basis of potentially actionable findings included selecting a nontargeted salvage chemotherapy regimen (n = 3) or pursuing no therapy in the absence of clear clinical or radiologic progression (n = 1). For one patient with a recurrent medulloblastoma and SHH pathway upregulation, consideration of therapy with a targeted SHH inhibitor, such as vismodegib, was considered, but no clinical trial was available, and the provider and family decided not to pursue off-trial use of the agent.
Actionable Findings
Sequencing was of highest utility in glial tumors. Seventeen of 20 patients (85%) with glial tumors were found to have a potentially actionable result. Fourteen of these patients (70%) underwent a change in therapy on the basis of sequencing results (Table A1). As an example, patient PO-3151 is a 5-year-old with a unilateral thalamic glioblastoma who originally underwent partial resection and focal radiation. He was treated with adjuvant temozolomide and lomustine (CCNU), but magnetic resonance imaging obtained before third cycle (including magnetic resonance spectroscopy) was consistent with tumor progression before third cycle. Sequencing of his tumor revealed H3F3A K27M mutation, TP53 mutation and copy loss, and CDK4 amplification and outlier expression (all category III; Figs 2A to 2C). After discussion in our precision medicine conference, he was treated with monotherapy on the histone deactylase inhibitor panobinostat three times per week every other week, on the basis of preclinical data supporting its use for DIPG with H3.3 mutations.18 After establishing that he tolerated this therapy, the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitor palbociclib was added (three times per week, 2 weeks on/2 weeks off), which previously showed efficacy in a pediatric CNS teratoma.19 He has had an ongoing partial response now for 18 months (25 months from diagnosis; Fig 2D). He has tolerated therapy well, with the exception of mild hyperbilirubinemia and count suppression.
Fig 2.
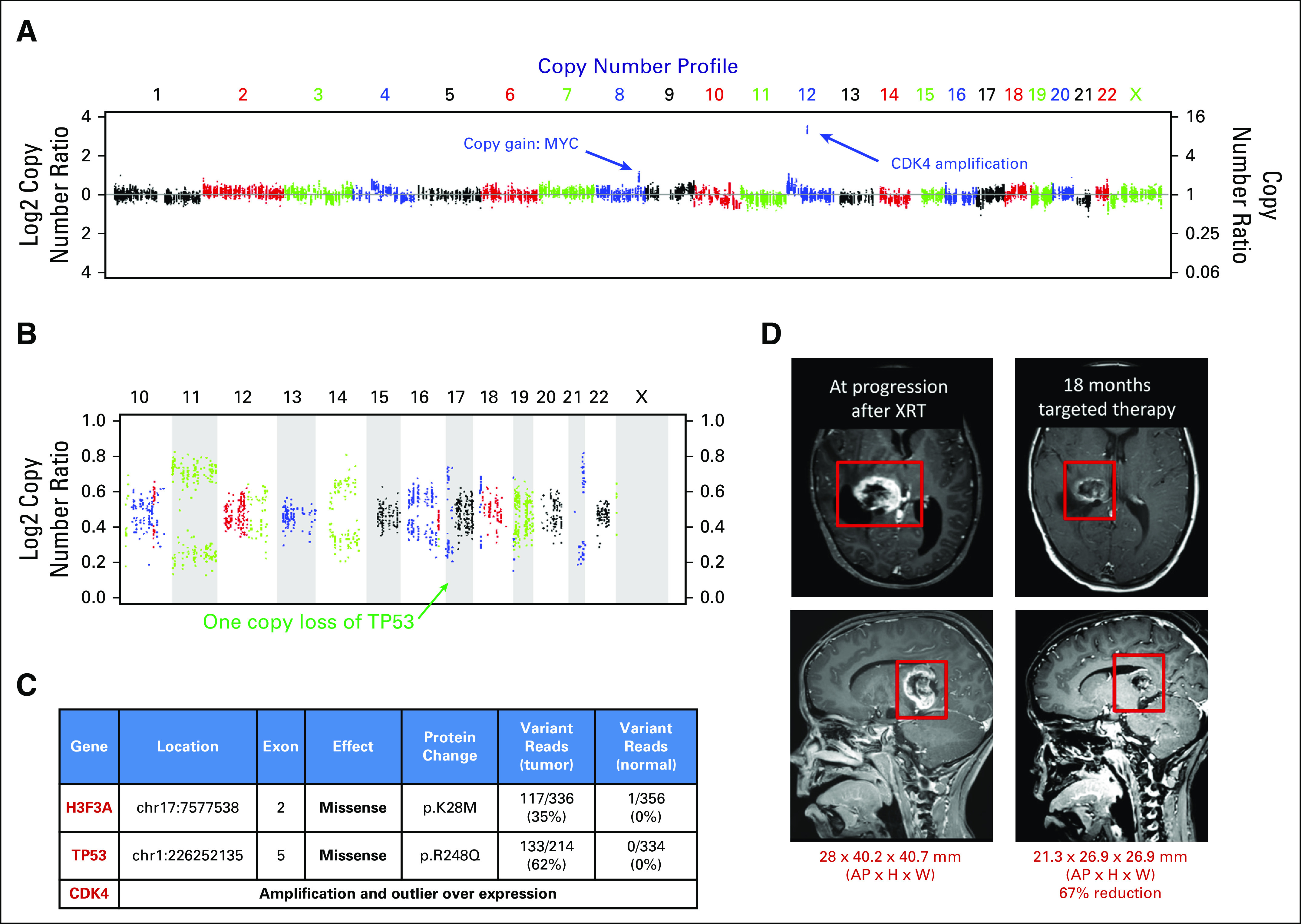
Clinical response to panobinostat and palbociclib in H3.3-K27M mutated, cyclin-dependent kinase 4 (CDK4)-amplified glioblastoma. (A) A 5-year-old patient (PO-3151) with a history of unilateral thalamic glioblastoma underwent partial resection and focal radiation. Sequencing of his tumor revealed CDK4 amplification. (B) Loss of heterozygosity plot shows one copy loss of TP53. (C) Somatic tumor mutations in H3F3A K27M mutation and TP53 mutation and outlier expression (RNA) of CDK4 were also seen. (D) Magnetic resonance imaging of the brain (T1 with contrast) showed increase in size in enhancing mass after three cycles of adjuvant lomustine (CCNU)/temozolomide. He was treated with the oral histone deacetylase (HDAC) inhibitor panobinostat, with later addition of the CDK 4/6 inhibitor palbociclib. After 18 months of targeted therapy, magnetic resonance imaging (T1 with contrast) showed 67% reduction in size of the lesion. TP53, tumor protein 53; XRT, radiation.
As another example, a 1-year old girl presented with a recurrent posterior fossa glioblastoma multiforme. She originally underwent a gross total resection at 8 months of age, multiagent chemotherapy according to Children's Cancer Group protocol 99703,20 and then focal radiation therapy with concomitant temozolomide (after initial relapse). The patient developed additional progression 4 months after radiation therapy. Sequencing of her initial tumor revealed significant focal amplification of PDGFB (19 copies; Data Supplement), outlier expression of PDGFB and PDGFRA (Data Supplement), and somatic mutations in SETD2, with the loss-of-function mutation in SETD2 comprising 42% of the sequenced tumor fraction. She was treated with dasatinib for 6 months with no toxicity and resolution of her enhancing lesion on magnetic resonance imaging (Data Supplement). Unfortunately, she developed additional progression and changed to a phase I trial.
Embryonal tumors were found to have potentially actionable results in five of 15 patients (33%). Only two of these patients (13%) underwent a change in their therapy on the basis of sequencing results (Table A1) both cases involved clarification or revision of the original diagnosis.
Thirty-eight (95%) participating patients received germline sequencing results. These patients had both agreed to receive incidental genetic findings and had an adequate tumor sample to complete sequencing. Pathogenic germline findings identified in five patients (13%) are listed in Table A1. These included mutations associated with established syndromes (DICER1 syndrome, Li-Fraumeni syndrome, and familial SMARCB1/INI1 deficiency). All patients with a new actionable pathogenic germline variant were seen in a cancer genetics clinic for counseling and additional testing in family members.
Molecular Landscape of Brain Tumors in Cohort
Tumor DNA and RNA sequencing allowed us to explore the molecular landscape of a diverse group of high-risk brain tumors from children and young adults. Activating alterations (mutation, amplification, or fusion) in platelet-derived growth factor receptor (PDGFR) or fibroblast growth factor receptor (FGFR) pathways was seen in nine (45%) of 20 glial tumors (Fig 3), which is higher than previously published in any of these tumor subtypes.21-23 In addition, we saw potential tumor-driving RNA fusions in eight (20%) tumors, including novel fusions in three tumors (LRP6-ETV6 fusion in a pediatric glioblastoma [PO-3089], an FGFR3-PHGDH fusion in a thalamic anaplastic oligodendroglioma [PO-3124], and an ELF4-SMARCA fusion in a bithalamic pediatric glioblastoma [PO-3195]). All genes involved in these two fusions are expressed at approximately 99th percentile in comparison to 1,700 MI-ONCOSEQ samples. The FGFR3-PHGDH fusion results in the fusion of FGFR3 exon 17 to exon 9 of PHGDH. The location of the FGFR3 breakpoint in this fusion is similar to that of reported FGFR3-TACC3 fusions in adult glioblastoma multiforme.24 Of note, overexpression of phosphoglycerate dehydrogenase has been shown to be a negative prognostic funding in adult glioma, possibly related to increase in glioma invasion and proliferation.25
Fig 3.
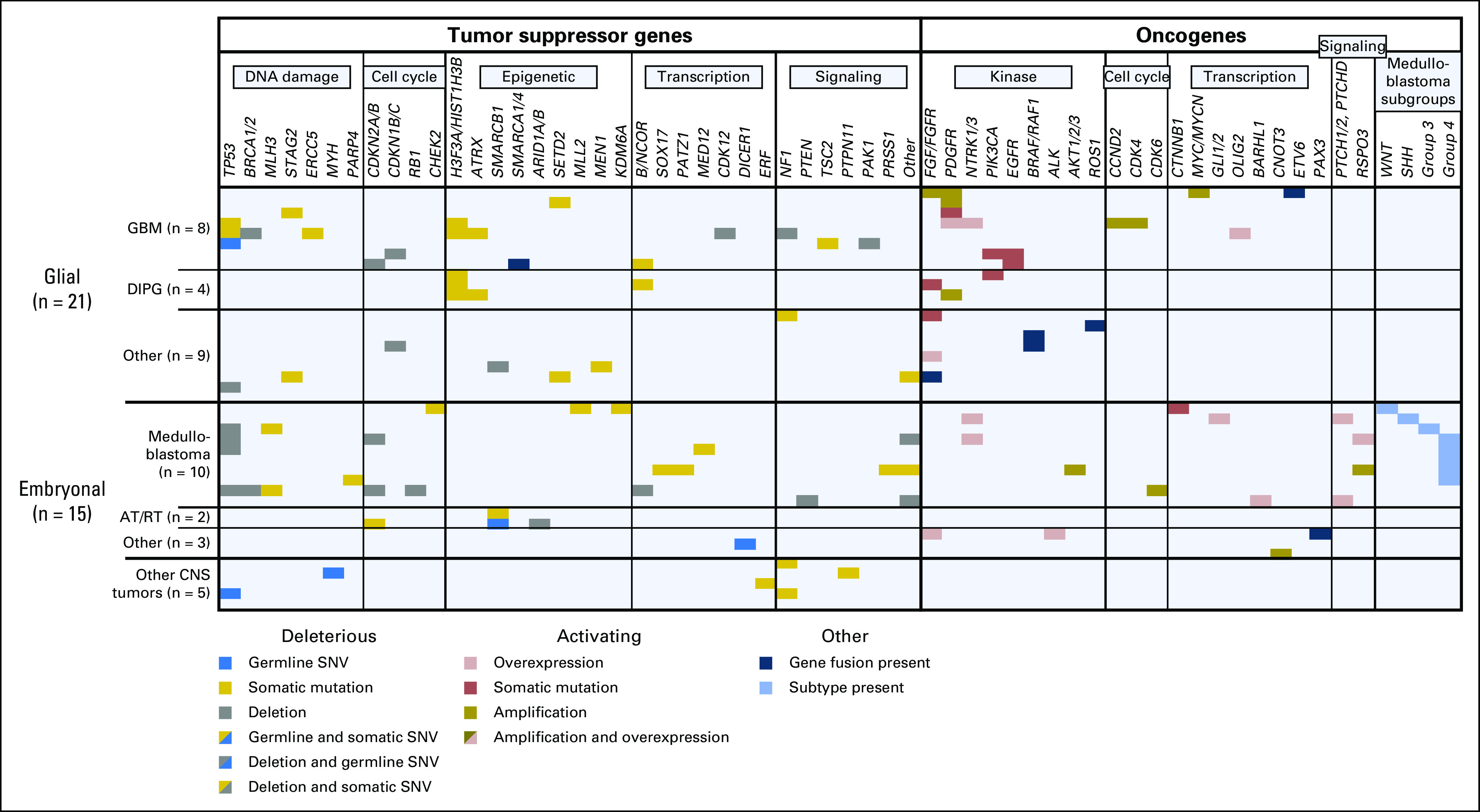
Genomic landscape of high-risk pediatric and young adult patients with brain tumors at diagnosis and recurrence. Activating DNA lesions (mutation, amplification, or fusion) were seen in 19 tumors (48%), most frequently in the mitogen-activated protein kinase signaling and mechanistic target of rapamycin/phosphoinositide 3-kinase pathways. Tumor suppressor mutations or deletions (germline and/or somatic) were seen in 32 tumors (80%), most frequently in DNA damage or apoptotic signaling pathways. AT/RT, atypical teratoid/rhabdoid tumor; DIPG, diffuse intrinsic pontine glioma; GBM, glioblastoma multiforme; SNV, somatic nucleotide variant.
Sequencing allowed for medulloblastoma molecular group analysis in nine of 10 medulloblastomas with tissue suitable for RNA library formation, by comparison with established groups of highly expressed genes in each group.26 Our population of medulloblastomas was from all four groups but was enriched for group 4 tumors (Table A1; Data Supplement). One tumor (PO-3063) had expression and copy number attributes of three subgroups (WNT, SHH, and group 3).
DISCUSSION
Younger patients with brain tumor diagnoses die as a result of their disease more commonly than those with any other tumor type.1 In an effort to tackle this problem, we sought to study a comprehensive integration of precision medicine into the management of this patient population. A few recent studies have assessed the utility of sequencing pediatric cancers,11,13,14 including targeted DNA sequencing at diagnosis in pediatric patients with brain tumors.15 To our knowledge, our study is the first to report on the feasibility of integrating DNA and RNA sequencing into the clinical management of children and young adults with high-risk brain tumors at both diagnosis and relapse, along with their treatment and response. We performed DNA and RNA sequencing and systematic selection of targeted therapies on the basis of discussions in a multi-institutional brain tumor precision medicine conference. Sequencing revealed a potentially actionable germline or somatic alteration in two thirds of brain tumors, of which one third resulted in an impact on treatment or change of diagnosis.
Our results demonstrate the utility of combining tumor exome DNA and RNA sequencing with germline DNA sequencing in pediatric brain tumors. We usually achieved more than 150× to 750× coverage of the tumor exome or 1,700 gene panel, which allowed us to detect subclonal populations < 5%. The brain tumors in our cohort harbored driving tumor mutations not previously known for their tumor type or seen only in a small number (< 5%) of tumors. This finding reinforces the need for broad (exome or > 1,000 gene panel) coverage for tumor profiling in younger patients with brain tumors. In addition, this series supports the use of RNA-seq, which identified fusions in 20% of tumors, allowed us study gene expression levels, and also helped us with identification of medulloblastoma subgroups. Germline sequencing revealed germline cancer predisposition alterations in 13% of tumors, which is consistent with previous studies of pediatric malignancies that have included germline sequencing.11,13 Our median turnaround time was suboptimal. Future efforts for molecular profiling will be clinically more relevant if available within 1 to 2 weeks, and novel platforms have been able to bring the time down to days.27
The brain tumors in our younger patient cohort were found to have relatively few somatic mutational events, as compared with mutational surveys of adult brain tumors, which are more heavily mutated.28,29 As seen in previous surveys of pediatric cancer,2,12 the somatic mutations that were present in our tumors, especially in our gliomas, were frequently targetable with personalized clinically available agents. Of note, 14 of 17 (83%) of our patients who underwent therapy changes on the basis of sequencing had a high-grade glial tumor, whereas only one of 23 (4%) of our patients who did not undergo therapy change had a high-grade glial tumor. The lack of efficacious treatments or available clinical trials for many of our patients with brain tumors influenced clinicians’ interest in pursuit of targeted therapies in this patient population. The multidisciplinary and multi-institutional conference also provided a consensus opinion that improved clinician and patient/family comfort with nonstandard therapy. Although no formal survey was performed on this topic, multiple families expressed comfort with pursuing a targeted therapy without a formal second opinion because of the attendance and input from clinicians from multiple other children’s hospitals around the country.
Pediatric and young adult patients with GBM and DIPG highlight the unique opportunities and drawbacks of precision medicine in this patient population. In our 12 patients with these diagnoses (and adequate tissue), all harbored potentially actionable mutations by our criteria, for which therapy was altered in 11. However, all of these tumors harbored multiple consensus cancer gene alterations (category I to III), with an average of 3.8 per tumor, and one with eight consensus cancer gene alterations. The selected targeted therapy may have led to the evolution of resistant subclonal tumor cell populations in tumors that progressed.30 Recent studies have shown a variable degree of heterogeneity within pediatric and adult high-grade gliomas, which could account for treatment failure with the use of a single agent.31,32 Current and future clinical trials will approach some of these issues by starting with multiple agents targeting nonredundant pathways.30
The timing of the biopsy is also likely important, because gliomas have also been shown to develop numerous mutations in response to cytotoxic chemotherapy and radiation.9 As a limitation of our study, a minority of our patients with progressive tumors underwent biopsy or resection at progression. Nevertheless, targeted therapies chosen for some of these patients in our study contributed to potential clinical benefit (although response attribution is uncertain because of previous irradiation in many cases). Therapies were generally administered orally and on an outpatient basis. Targeted agents were monitored for toxicity by standard-of-care guidelines (off study), with reduction or holding dose for grade 3 or 4 adverse events. None of the patients receiving targeted therapy had grade 3 or 4 adverse events, cytopenia requiring transfusion, or admission for fever and neutropenia.
The CNS-TAP tool provided a useful framework for selection of targeted agents on the basis of previously published data and the likelihood of BBB penetration. Despite this, the molecular tumor board was often conflicted after discussion of multiple promising agents, and the reproducibility of the board’s recommendations was not always fully consistent. Our group is currently optimizing the tool to provide a numerical score for all relevant agents that will be available to clinical researchers as a Web-based tool. We are also developing a prospective trial, which will compare CNS-TAP–selected therapy to clinician choice, as well as measuring outcomes for a more homogenous cohort prospectively compared with historical controls. These studies will be crucial to our understanding of the true value of targeted therapy for children with brain tumors.
In summary, selection of personalized agents on the basis of integrative clinical sequencing of high-risk brain tumors of children and young adults is feasible and frequently results in change of management. Our study highlights the utility of this approach, particularly in glial tumors. Going forward, continuing efforts to match therapies to the molecular profile of individual tumors should lead to improved outcomes for younger patients with brain tumors. Our results demonstrate the promise of this approach.
ACKNOWLEDGMENT
We thank the patients and families who participated in this study for their generosity and kindness. We also thank Steven Pipe, Valerie Opipari, Maria Castro, Pedro Lowenstein, Becky Sigler, Coral Grothe, Lilly Pritula, Justin Flees, Javed Siddiqui, Christine Brennan, Erica Rabban, Terrence Barrette, Christine Betts, Karen Giles, Pallavi Mohapatra, and Xiaoxuan Dong for their administrative or academic guidance; none of them received compensation beyond their salaries at the University of Michigan. We also thank participants and presenters in the University of Michigan Brain Tumor Precision Medicine Conference.
Appendix
Table A1.
Clinically Integrated Sequencing Results for Patients

Footnotes
Tempus was not involved with the conduct of this study. None of the sponsors played a role in the design; data collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Supported by National Institutes of Health (NIH) Clinical Sequencing Exploratory Research Grant No. NIH 1UM1HG006508, the Prostate Cancer Foundation, and Grant No. NIH 1UM1HG006508; National Institute of Neurological Disorders and Stroke Grant No. K08-NS099427-01 (C.K.); the University of Michigan (UM) Department of Pediatrics; and the UM Pediatric Brain Cancer Initiative. R.M. is a Hyundai Hope on Wheels Scholar. Funds from the UM Pediatric Brain Cancer Initiative and the UM Chad Tough Fund were used to support this work. A.M.C. is a Howard Hughes Medical Institute Investigator, an A. Alfred Taubman Scholar, and an American Cancer Society Professor.
AUTHOR CONTRIBUTIONS
Conception and design: Carl Koschmann, Bernard Marini, Hugh Garton, Dan Robinson, Arul M. Chinnaiyan, Rajen Mody
Financial support: Arul M. Chinnaiyan, Rajen Mody
Administrative support: Xuhong Cao, Karin Muraszko, Arul M. Chinnaiyan
Provision of study material or patients: Bailey Anderson, Sandra Camelo-Piragua, Sriram Venneti, Andrew P. Lieberman, Marcia Leonard, Cormac O. Maher, Karin Muraszko, Patricia Robertson
Collection and assembly of data: Carl Koschmann, Yi-Mi Wu, Marcin Cieslik, Xuhong Cao, Bailey Anderson, Kevin Frank, Andrew H. Zureick, Jessica Everett, Bailey Anderson, Brendan Mullan, Sandra Camelo-Piragua, Sriram Venneti, Paul McKeever, Marcia Leonard, Cormac O. Maher, Hugh Garton, Karin Muraszko, Patricia Robertson, Dan Robinson, Arul M. Chinnaiyan, Rajen Mody
Data analysis and interpretation: Carl Koschmann, Chandan Kumar-Sinha, Robert Lonigro, Pankaj Vats, Katayoon Kasaian, Bailey Anderson, Kevin Frank, Lili Zhao, John R. Prensner, Bernard Marini, Kathryn McFadden, Andrew P. Lieberman, Patricia Robertson, Dan Robinson, Arul M. Chinnaiyan, Rajen Mody
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Carl Koschmann
No relationship to disclose
Yi-Mi Wu
No relationship to disclose
Chandan Kumar-Sinha
No relationship to disclose
Robert Lonigro
No relationship to disclose
Pankaj Vats
No relationship to disclose
Katayoon Kasaian
No relationship to disclose
Marcin Cieslik
No relationship to disclose
Xuhong Cao
No relationship to disclose
Bailey Anderson
No relationship to disclose
Kevin Frank
No relationship to disclose
Lili Zhao
No relationship to disclose
John R. Prensner
Patents, Royalties, Other Intellectual Property: Patent and royalties for gene expression profiling of lncRNAs in prostate cancer to GenomeDx Biosciences
Andrew H. Zureick
No relationship to disclose
Jessica Everett
No relationship to disclose
Brendan Mullan
No relationship to disclose
Bernard Marini
Speakers' Bureau: Genentech/Abbvie, Shire
Sandra Camelo-Piragua
No relationship to disclose
Sriram Venneti
No relationship to disclose
Paul McKeever
No relationship to disclose
Kathryn McFadden
No relationship to disclose
Andrew P. Lieberman
No relationship to disclose
Marcia Leonard
No relationship to disclose
Cormac O. Maher
No relationship to disclose
Hugh Garton
No relationship to disclose
Karin Muraszko
No relationship to disclose
Patricia Robertson
No relationship to disclose
Dan Robinson
No relationship to disclose
Arul M. Chinnaiyan
Consulting or Advisory Role: Tempus
Rajen Mody
No relationship to disclose
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A: Cancer statistics, 2015. CA Cancer J Clin 65:5-29, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Gajjar A, Pfister SM, Taylor MD, et al. : Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res 20:5630-5640, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollack IF: Brain tumors in children. N Engl J Med 331:1500-1507, 1994 [DOI] [PubMed] [Google Scholar]
- 4.Jones DT, Jager N, Kool M, et al. : Dissecting the genomic complexity underlying medulloblastoma. Nature 488:100-105, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow LM, Baker SJ: Capturing the molecular and biological diversity of high-grade astrocytoma in genetically engineered mouse models. Oncotarget 3:67-77, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nau R, Sorgel F, Eiffert H: Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev 23:858-883, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartzentruber J, Korshunov A, Liu XY, et al. : Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226-231, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Paugh BS, Qu C, Jones C, et al. : Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061-3068, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson BE, Mazor T, Hong C, et al. : Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189-193, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janeway KA, Place AE, Kieran MW, et al. : Future of clinical genomics in pediatric oncology. J Clin Oncol 31:1893-1903, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Mody RJ, Wu YM, Lonigro RJ, et al. : Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314:913-925, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu YM, Su F, Kalyana-Sundaram S, et al. : Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 3:636-647, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons DW, Roy A, Yang Y, et al. : Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2:616-624, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris MH, DuBois SG, Glade Bender JL, et al. : Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: The Individualized Cancer Therapy (iCat) study. JAMA Oncol 10.1001/jamaoncol.2015.5689 [epub ahead of print on January 28, 2016] [DOI] [PubMed] [Google Scholar]
- 15.Ramkissoon SH, Bandopadhayay P, Hwang J, et al. : Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: Precision medicine analysis of 203 pediatric brain tumors. Neuro-oncol 19:986-996, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kline CN, Joseph NM, Grenert JP, et al. : Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-oncol 19:699-709, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson DR, Wu YM, Vats P, et al. : Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446-1451, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasso CS, Tang Y, Truffaux N, et al. : Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21:827, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Schultz KA, Petronio J, Bendel A, et al. : PD0332991 (palbociclib) for treatment of pediatric intracranial growing teratoma syndrome. Pediatr Blood Cancer 62:1072-1074, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Thorarinsdottir HK, Rood B, Kamani N, et al. : Outcome for children <4 years of age with malignant central nervous system tumors treated with high-dose chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer 48:278-284, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. : Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46:462-466, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu G, Diaz AK, Paugh BS, et al. : The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444-450, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Wu G, Miller CP, et al. : Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602-612, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh D, Chan JM, Zoppoli P, et al. : Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231-1235, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Guo S, Li Q, et al. : Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J Neurooncol 111:245-255, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northcott PA, Korshunov A, Witt H, et al. : Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408-1414, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Euskirchen P, Bielle F, Labreche K, et al. : Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 134:691-703, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennan CW, Verhaak RG, McKenna A, et al. : The somatic genomic landscape of glioblastoma. Cell 155:462-477, 2013 [Erratum: Cell 157:753, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciriello G, Miller ML, Aksoy BA, et al. : Emerging landscape of oncogenic signatures across human cancers. Nat Genet 45:1127-1133, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prados MD, Byron SA, Tran NL, et al. : Toward precision medicine in glioblastoma: The promise and the challenges. Neuro-oncol 17:1051-1063, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Lee IH, Cho HJ, et al. : Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28:318-328, 2015 [DOI] [PubMed] [Google Scholar]
- 32.Nikbakht H, Panditharatna E, Mikael LG, et al. : Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7:11185, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]