

1. Introduction
Randomized clinical trials (RCTs) of treatments for pain have a long and distinguished history. The earliest clinical trials not only identified analgesic medications and their efficacious dosages but also contributed to the development of clinical trial research designs and methods that came to be used throughout medicine. The ground-breaking investigators who designed and conducted these early studies recognized that various sources of bias must be addressed,68,69,78,105 and appreciation of the fundamental roles of study design and statistical principles became widespread as experience conducting RCTs grew.
In this article, we first present analyses of a sample of chronic pain trials that show a decline in treatment effect estimates over the past few decades and discuss the implications of these results for determining sample sizes for future chronic pain trials. We then review explanations for the failure of RCTs to demonstrate the efficacy of truly efficacious treatments and address the role of excessive placebo group improvement. Finally, we consider various approaches that have the potential to improve the informativeness of clinical trials and their assay sensitivity, that is, their ability to distinguish an effective treatment from a less effective or ineffective treatment.
2. “The greatest teacher, failure is”: falsely negative and inconclusive clinical trial results
It has been recognized for at least 2 decades that clinical trials of psychiatric medications often fail to show a statistically significant difference between an active medication and placebo.29,53,63,74,82 Although some of these RCTs might have investigated treatments that truly lack efficacy, many were for medications that had demonstrated efficacy in multiple previous RCTs and had been approved by regulatory agencies around the world (eg, selective serotonin reuptake inhibitors for depression). Similarly, many RCTs of treatments for chronic pain have failed to demonstrate efficacy.19,22,31 Some of these results also might reflect a true lack of efficacy—either in general or for the specific dosage studied—but some RCTs have failed to show efficacy of medications at dosages that had demonstrated efficacy in previous trials, had been approved by multiple regulatory agencies, and are generally considered first-line treatments.19,22,31
It is common to refer to clinical trial results that fail to show the efficacy of truly efficacious treatments as “false negatives.” However, the failure of a clinical trial to reject the null hypothesis of no difference between an active treatment and placebo at a prespecified level of statistical significance does not necessarily indicate that the active treatment lacks efficacy.86 Such nonsignificant study results can be accompanied by confidence intervals that are consistent with the possibility of a clinically meaningful treatment effect. When there is such an outcome, the results of the trial should be considered “inconclusive” rather than “negative.”39 A failure to reject the null hypothesis can also be a result of chance, reflected in the type II error probability of failing to reject the null hypothesis of no difference between treatment groups when one truly exists.
Table 1 presents a list of potential explanations for the failure of clinical trials of truly efficacious treatments to show their efficacy (see also Ref. 86). We focus on the roles of statistical power, excessive improvement in placebo groups, and various study methods and patient characteristics in contributing to falsely negative and inconclusive clinical trial outcomes. An additional explanation for such clinical trial results is the possibility that existing outcome measures have limited responsiveness to detect treatment effects. Most chronic pain RCTs have used numerical or visual analogue scales of pain intensity as primary outcome measures,101 but other measures that could serve as primary outcomes—for example, ratings of pain relief, global improvement, or disease-specific pain-related symptoms—might have greater responsiveness.18,44,97,98,102,109 Furthermore, chronic pain RCTs have typically not been designed to study patients selected on the basis of genotypes or phenotypes targeted by “precision” or “personalized” pain treatments. Although we believe that the development of improved clinical outcome assessments and of mechanism-based treatments16,25,100 may make important contributions to the identification of pain treatments with greater efficacy or safety, further discussion of these issues is beyond the scope of this article.
Table 1.
Why can clinical trials of truly efficacious treatments fail to show their efficacy?

| 1. Chance |
| 2. Placebo group patients improved “too much” |
| 3. The optimal patients and phenotypes were not studied |
| 4. Existing outcome measures have limited responsiveness to treatment effects |
| 5. Temporal changes in characteristics of patients enrolling in trials |
| 6. Temporal changes in types of clinical sites conducting trials |
| 7. Research subject misbehaviour |
| 8. Research site unintentional bias and misconduct |
| 9. Inadequate sample sizes |
3. Treatment effects and sample size determination
Twenty years ago, Moore et al.79 concluded on the basis of a series of simulations that “size is everything” if the samples of patients enrolled in RCTs are to have adequate statistical power to provide credible estimates of the efficacy of acute pain treatments. The results of recent meta-analyses of assay sensitivity and placebo group changes in RCTs of chronic neuropathic pain have found that treatment effects have decreased and placebo group changes have increased over the past several decades, perhaps especially in the United States. Tuttle et al.110 concluded that from 1990 to 2013, placebo group changes increased while active treatment group changes remained relatively stable; as a consequence, “treatment advantage” vs placebo decreased substantially. Figure 1 presents the results of a second recent meta-analysis, on the basis of which Finnerup et al.32 concluded that, from 1982 to 2017, there was an increase in mean numbers-needed-to-treat (NNTs) that was associated with increases in placebo group change, study duration, and sample size (note that we refer to active and placebo group “changes” rather than “responses” because the term “responses” fails to encompass regression to the mean, spontaneous improvement, and other nonspecific sources of improvement or worsening that are not actual responses to active or placebo treatments).
Figure 1.
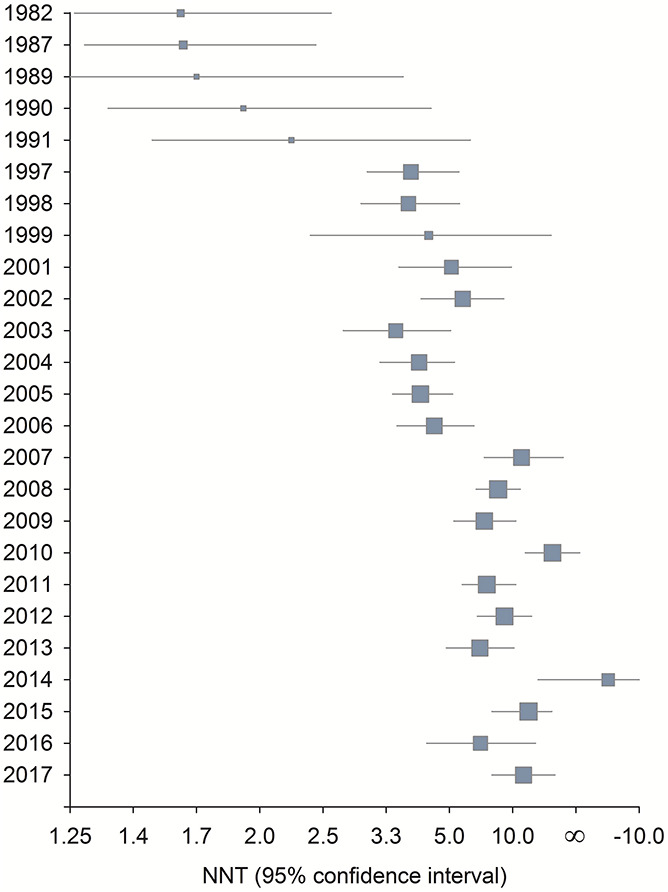
Combined number-needed-to-treat (NNT) per year from a meta-analysis of randomized clinical trials of pharmacologic treatments for chronic neuropathic pain.32
Although the results of these meta-analyses are generally consistent with what has been observed for RCTs in major depression13,53,106,119 and other therapeutic areas,5,52 only treatments for chronic neuropathic pain were examined and few such analyses have examined other chronic pain conditions.24 Nevertheless, the results suggest that factors such as increasing placebo group change and changes in study methods may be limiting or reducing estimates of the effects of chronic pain treatments, which would necessitate larger sample sizes for adequate statistical power to detect minimally clinically important effects.
When planning a clinical trial, appropriate sample size determination is necessary to avoid exposing more patients than necessary to a potentially nonefficacious or harmful treatment, while also including a sufficient number of participants to demonstrate a true treatment effect, if one exists.26,77 Tuttle et al.110 presented differences between medications and placebo in the percentage decrease in pain intensity from baseline, and Finnerup et al.32 presented NNTs. Such data, however, are of limited value for determining sample sizes for analyses of continuous pain outcomes, for example, analysis of covariance adjusting for baseline pain, which is a common primary efficacy analysis used in confirmatory RCTs of chronic pain treatments.21 In addition to type I and type II error probabilities—typically prespecified as 5% and 10% to 20%, respectively—sample size calculations for continuous variables require specification of the magnitude of the treatment effect and the variability of the outcome measure. A well-accepted approach to sample size determination for such a primary efficacy analysis involves the standardized effect size (SES),26 which for a parallel group RCT is the mean change from baseline in the active group minus that in the placebo group divided by the pooled SD.
3.1. Methods and results
We examined whether SESs of published neuropathic and non-neuropathic chronic pain trials have decreased over the past several decades by performing a secondary analysis of data from a recent meta-analysis of RCTs of efficacious medications conducted from 1980 to 2016 for low back pain, fibromyalgia, osteoarthritis pain, painful diabetic peripheral neuropathy, and postherpetic neuralgia.102 The purpose of the initial meta-analysis was to compare the responsiveness of ratings of average pain intensity (API) and worst pain intensity (WPI), and in the current analysis, we explored the trajectories of API and WPI SESs over time. Twenty-three articles were identified for inclusion, with publication dates from 1999 to 2013. SESs were extracted or calculated using other reported data, and positive values indicate that the treatment reduced API or WPI more than placebo.102
Mixed-effects meta-regression was used to test the significance of the relationship between time and both API SES and WPI SES. Preliminary analysis suggested that the relationships between time and both API SES and WPI SES were not linear. We therefore fit quadratic models regressing API SES and WPI SES on time and the square of time, where time is the number of years from 1999. Four articles included 2 active treatments compared with the same placebo arm. A robust variance estimator was used to account for correlations among the dependent effect size estimates in these 4 articles. All analyses were conducted using R version 3.5.1 with the robust.se function for robust variance estimation.46,47
Table 2 presents the parameter estimates for time and the square of time for the API SES and WPI SES models. Figure 2 shows that API SES and WPI SES both increased slightly for a short time, but on average, the slopes decreased for every additional year after 1999. These results are consistent with the results of the meta-analyses of neuropathic pain trials32,110 and demonstrate that the average benefit of efficacious analgesic medications shown in recent RCTs is modest. It is unknown whether the SESs for API and WPI will level off at approximately 0.30 or whether there will be a continued downward slope that will result in even lower SESs.
Table 2.
Parameter estimates for the pain intensity models.

| Average pain intensity SES | Worst pain intensity SES | |||
|---|---|---|---|---|
| Estimate (95% CI) | P | Estimate (95% CI) | P | |
| Intercept | 0.379 (0.308 to 0.451) | <0.0001 | 0.401 (0.344 to 0.457) | <0.0001 |
| Time | 0.036 (−0.002 to 0.074) | 0.07 | 0.027 (−0.007 to 0.060) | 0.13 |
| Time2 | −0.003 (−0.007 to −0.0003) | 0.04 | −0.003 (−0.006 to −0.000) | 0.06 |
Time is the number of years since 1999.
CI, confidence interval; SES, standardized effect size.
Figure 2.
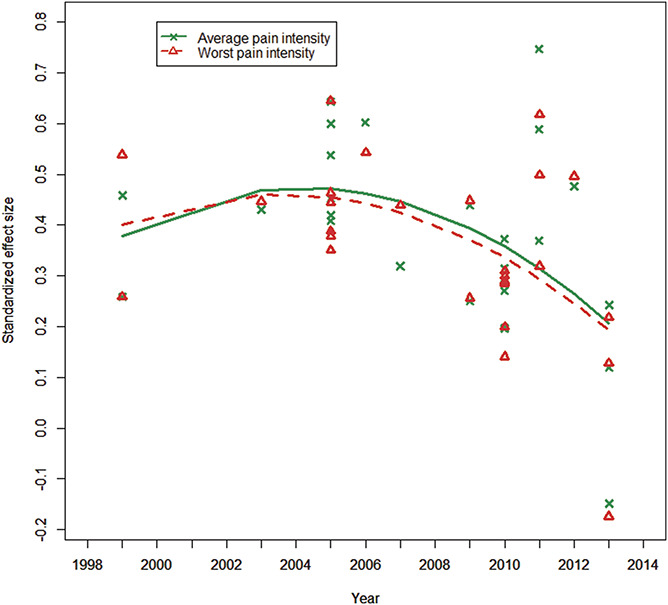
Standardized effect sizes for average and worst pain intensity in randomized clinical trials of chronic pain treatments from 1999 to 2013.
3.2. Implications
The results of our analysis do not address the causes of the decline in the SESs found in RCTs of efficacious medications for chronic pain. It is possible to speculate that this decline is due to efforts by the scientific community and government regulators to increase the rigor of clinical trial design, execution, and analysis through methods such as comprehensive prespecification of study methodology and analysis, limiting multiple hypothesis testing unless proper statistical adjustments are used, and principled methods to accommodate missing data.41,42,50,51,99,103 Declines in SESs may also result from greater availability of pain treatments over time, which could reduce the pool of eligible patients and increase the percentage of study participants who have refractory pain.22 Given the evidence that expectations are a major source of placebo effects, it is also possible that placebo group changes increase as evidence for a treatment's efficacy accumulates and becomes publicly available.2
One important limitation of the present analyses is that they are based on published trials of 5 chronic conditions that reported both API and WPI. Although our results provide some information about the temporal trajectories of SESs from chronic pain trials, analyses that examine SESs for different chronic pain conditions or that include a larger sample of RCTs might produce different results; indeed, because clinical trials with nonsignificant results are less likely to be published, meta-analyses that include unpublished studies might show even greater declines. In addition, because the clinical trials we examined were limited to studies of efficacious medications for chronic pain, analyses of clinical trials of devices (eg, spinal cord stimulators) or of other nonpharmacologic treatments (eg, cognitive-behavior therapy and physical therapy) might also produce different results. For example, it has been observed that treatment effect estimates from RCTs of psychosocial treatments for depression are generally greater than those from trials of antidepressant medications; this observation may be explained by attenuation of the antidepressant treatment effect in trials in which a medication is compared with placebo and both groups are receiving intensive clinical management, which can be “substantially more therapeutic for patients with depression than doing nothing.”90
The mean SES of approximately 0.30 for the most recent published chronic pain trials mirrors the mean SESs reported in meta-analyses of efficacious antidepressants for major depression.43,61,62 Antidepressant trials share with analgesic RCTs several methodologic characteristics that might contribute to decreased assay sensitivity, including subjective outcomes, considerable placebo group improvements, and appreciable missing data.41,61,110 Given the consistent meta-analysis results, it is crucial that analgesic and antidepressant RCTs be designed with realistic treatment effect estimates. To detect an SES of 0.30 with 80% power (α = 0.05, 2 tailed) in a parallel group trial, at least 175 patients per group would need to be randomized. An SES of 0.30 can be considered a modest treatment effect, and its clinical importance will depend on the risks and benefits of the treatment and its clinical context.15,20 Such SESs reflect not only the specific effects of the treatments (eg, the pharmacologic activity of a medication) but also any methodologic characteristics of the clinical trials that decrease their assay sensitivity.19,22
In designing chronic pain RCTs, an SES of 0.30 can serve as a benchmark that could be considered when performing sample size determinations. This approach addresses both the modest apparent efficacy of existing treatments and any limitations of the clinical trial methods that have been used to study them. It is important to acknowledge, however, that it is usually recommended that sample size determination be based on specifying an effect size that would be of minimal clinical importance to patients, clinicians, and other stakeholders. Given the often poor tolerability and risks of many existing treatments, doing so might be challenging because even a minimal treatment effect could be considered meaningful for a novel treatment that is well tolerated and safe.15,20
4. Three eras of analgesic clinical trials
The observation that clinical trials of medications with well-established efficacy are sometimes unable to demonstrate that efficacy provided the impetus for ongoing efforts to explain such results by examining associations between the research methods and patient characteristics of RCTs and their assay sensitivity. As can be seen from Figure 1, 3 eras of analgesic clinical trials can be identified from the NNTs associated with pharmacologic treatments for neuropathic pain.32 The first era—from the early 1980s through the early 1990s—has the lowest NNTs (ie, greatest treatment vs placebo differences) and consists primarily of relatively small cross-over trials conducted by investigators such as Mitchell Max, Michael Rowbotham and Howard Fields, Søren Sindrup, and Peter Watson. These studies were typically conducted at a single clinical site with patients who were either personally known by the researchers or carefully assessed by clinician investigators with substantial expertise. The second era—from the mid-1990s to the mid-2000s—reflects the involvement of pharmaceutical companies in developing drugs for chronic pain. The early clinical trials of gabapentin, duloxetine, and pregabalin were conducted at multiple sites but often included investigators at academic medical centers with experience treating or researching the specific pain condition being studied. The third era—from the late 2000s to the present—has the highest NNTs and includes multinational RCTs with large sample sizes using primarily for-profit clinical research centers that conduct clinical trials across a wide range of therapeutic areas.
The decrease in treatment effects reflected in these increasing NNTs could be a result of changes over time in research methods, study sites, and/or the patients enrolled in the trials.32 Meta-analyses of RCTs of chronic neuropathic23,32 and musculoskeletal pain24 have found that greater trial assay sensitivity was associated with shorter trial durations and also smaller sample sizes. It is possible, however, that smaller trials that are negative or inconclusive are less likely to be published, and such publication bias might contribute to the results of these meta-analyses. Nevertheless, on the basis of data such as these, it has been suggested that larger and longer trials are not necessarily better at demonstrating whether a treatment is truly efficacious.72,88 The decreased treatment effects observed over the past several decades could be a result of the pharmaceutical industry conducting an increasing number of appropriately powered RCTs intended to fulfill regulatory requirements for study durations that can examine durability of treatment effects.
In addition, analyses of RCTs of depression72 and Parkinson disease45 have suggested that effect sizes might be smaller for patients who are enrolled later in the trial than for those enrolled earlier, perhaps due to the enrollment of patients who do not fulfill eligibility criteria because of pressure on sites to complete enrollment requirements. Also, with longer trials—for example, durations of 12 weeks or more rather than 5 to 8 weeks—there may be greater placebo vs active group improvement resulting from, as discussed in the next section, a greater number of study visits90 and an increased opportunity for patients to develop supportive relationships with study staff.87,91
It is also possible that over the course of these 3 eras of analgesic trials, the quality of RCT procedures and data, including patient clinical evaluations and outcome assessments, became more variable as greater numbers of study sites participated.74 In addition, there has been increasing recognition of the potential roles of unintentional and intentional investigator bias64,67,81 and frank research misconduct27 in contributing to negative, inconclusive, and invalid study results. It has also become apparent that surprisingly large percentages of the participants enrolled in clinical trials are either professional subjects who are fabricating a clinical condition—and may be participating in more than one clinical trial at different sites, so-called “duplicate patients”—or are patients who intentionally falsify key eligibility criteria to be randomized.10,11,76,96 Information provided on social media71 and clinical trial websites can facilitate enrollment of such unqualified participants, and methods to identify professional subjects and mitigate patient misbehavior are now being developed, including the creation of research subject registries.76,96
5. Placebo group changes and their interpretation
The results of meta-analyses of RCTs have found meaningful relationships between placebo group changes and study methods and patient characteristics. Paralleling the results discussed above for treatment effects, greater placebo group changes in neuropathic pain trials were associated with longer trial durations and larger sample sizes.19,32,110 In a larger number of meta-analyses of major depression trials, greater placebo group changes were associated with larger numbers of study sites, larger samples, greater frequency of study visits, longer trials, lower probability of receiving placebo, and higher patient expectations for improvement.29,35,84,87,92,111,118
A robust finding that has emerged from multiple analyses of both pain and psychiatric treatments are associations between greater magnitudes of placebo group change and negative or inconclusive clinical trial outcomes, as evaluated, for example, by statistical significance, risk ratios, and NNTs.32,52,53,59,110 In considering such relationships, it is important to recognize that random variation in the magnitudes of placebo group change across a set of RCTs will cause an association between placebo group changes and treatment effect estimates that reflect the difference between that placebo group and an active treatment. As Senn93 observed many years ago, a “negative correlation between odds ratios and placebo rates in clinical trials does not of itself indicate the presence of a phenomenon of interest. Such an effect is to be expected on statistical grounds alone and there is thus no need to search for medical explanations.”
Despite the statistical basis of associations between placebo group changes and treatment effect estimates, these associations can also reflect characteristics of the clinical trials that potentially reduce assay sensitivity. For example, it is uncommon for the mean pain intensity to fall below a mean of 3 or 4 on a 0 to 10 numerical rating scale. Such a “floor” of symptom reduction may represent an unresponsive core of refractory pain that if reached by patients in the placebo group would make it difficult to show any further pain reduction from an efficacious treatment. If this floor effect occurs, it could account, at least in part, for the associations between greater magnitudes of placebo group change and decreased treatment effects that have been reported. Assuming that there is such a floor effect, the separation between an efficacious treatment and placebo in an RCT might be greater if nonspecific sources of improvement in both treatment groups—such as placebo effects and regression to the mean—could be reduced, which could make it less likely that the placebo group would reach the floor.
Another explanation for associations between placebo group changes and treatment effect estimates involves the presumption of additivity in placebo-controlled clinical trials. It is generally assumed that the specific effects of an active treatment provide an additive benefit to the nonspecific effects associated with treatment in the placebo group, which include placebo effects and regression to the mean. As noted by Kaptchuk,58 this premise takes “for granted that the active drug response results partly from a placebo effect and that the placebo effect buried in the active arm is identical to the placebo effect of the dummy treatment.” But it is possible that response to the active treatment supplants at least part of the placebo group response, in which case the specific effects of the active treatment and the non-specific effects of trial participation, including placebo treatment, would be subadditive, that is, some of the nonspecific effects that occur in the placebo group would not occur in the active treatment group.6,66,73
An example of such subadditivity is provided by Roose et al.,90 who noted that therapeutic contact with study staff—who have been reported to vary greatly in what they consider appropriate interactions with study participants14—may be “a potent contributor to symptomatic improvement in patients with depression, particularly patients in the placebo arm” of antidepressant RCTs. In several trials, number of study visits was more strongly associated with improvement in the placebo groups than in the antidepressant groups. It was concluded that “increasing the number of study visits significantly increases placebo response while leaving medication response generally unaffected,” for example, having only 6 rather than 10 visits over the course of a 12-week trial was associated with a difference in response rates between an antidepressant and placebo of 12.2% vs 0.4%.90 Such differential effects on active and placebo group changes, if indeed causal, could reduce the apparent benefit of an efficacious treatment when compared with placebo. Although this subadditivity would decrease the assay sensitivity of any trials in which it occurs, it does provide a basis for hypothesizing that assay sensitivity can be increased if study procedures such as excluding certain patients28 or training study participants98,108 have differential effects on active and placebo group changes.
6. “Always in motion is the future”: emerging evidence-based approaches to the design of pain clinical trials
Size does matter when determining the number of participants needed for an RCT to provide adequate statistical power to identify minimally clinically important effects26 and to estimate their magnitude.79 Nevertheless, it is important to recognize that various strategies for increasing the assay sensitivity of RCTs and decreasing the probability of inconclusive results should also be considered.22
6.1. General methodologic considerations
As recently emphasized in the International Council on Harmonisation E9 (R1) addendum on estimands in clinical trials, the preeminent consideration in designing clinical trials is to identify the scientific question of interest and the estimand, “a precise description of the treatment effect reflecting the clinical question posed by the trial objective.”49 The choice of estimand determines the clinical trial design and the statistical analysis plan, including methods for accommodating inter-current events and missing data and the selection and interpretation of sensitivity analyses.4,49,85 Discussion of the complex conceptual and statistical issues involved in determining estimands and prespecifying principled statistical analyses for their estimation is beyond the scope of this article; however, we believe it is important to emphasize that biostatisticians with expertise in clinical trials should be involved from the earliest consideration of conducting a clinical trial and continuing through its design, execution, analysis, interpretation, and reporting.
The evidence that knowledge of clinical trial eligibility criteria can lead to intentional and unintentional biases among study staff and potential participants has provided a basis for recommending that key aspects of the protocol that do not involve safety should be concealed from all study staff and patients.21,48,89 Blinding staff and patients to eligibility criteria could reduce the numbers of patients who are randomized but who do not actually fulfill these criteria because of inflated or falsified baseline assessments; use of electronic diaries and case report forms has made implementation of such blinding relatively straightforward. In addition, blinding study staff and patients to allocation ratios when patients are more likely to be randomized to active vs placebo treatment (eg, dose finding and active comparator trials) could also prevent the increases in placebo group improvements that have been found in trials in which patients know that their chance of receiving placebo is less than their chance of receiving an active treatment and, presumably as a result, have greater expectations for improvement.83 Blinding patients and staff to the allocation ratio requires considerable attention to the language used in consent forms and patient materials and also involves explaining to ethics committees the anticipated benefits on assay sensitivity that might result.
An important feature of clinical trials that is receiving increased attention as a source of poor data quality and of failures to demonstrate the efficacy of truly efficacious treatments is poor treatment adherence. Poor medication adherence can decrease estimates of efficacy and confound assessments of safety,3,9 but it has typically been assessed using pill counts, which are known to be inaccurate. Although there are now a variety of more sophisticated methods for assessing medication adherence that have greater validity,3,96 they have rarely been used in chronic pain RCTs.
Minimizing placebo group changes also has the potential to enhance assay sensitivity. For example, in neuropathic pain RCTs, the time to onset of pain reduction in placebo groups has been shown to be longer than that associated with analgesic medications.110 This is consistent with the observation that longer trials tend to have a progressive increase in placebo group changes19,88 and suggests that shorter treatment durations may be preferable for proof-of-concept trials; of course, RCTs with longer durations would still be necessary to evaluate the durability of any benefits. In addition, when recruiting potential participants for a clinical trial evaluating a new treatment, placebo effects should be minimized by neutrally describing the treatment rather than enhancing participant expectations about its efficacy.92,115,120 Placebo group changes might also be reduced by limiting the number of study visits and standardizing interactions between study staff and participants.14 Importantly, whether such techniques reduce retention and thereby increase the amount of missing data should also be considered. Developing methods to mitigate unrealistic patient expectations is consistent with the obligation to ensure that patients understand the difference between participating in a clinical trial and receiving clinical care; any such standardized protocols intended to diminish placebo group improvement would ideally be evaluated in RCTs designed to examine their effectiveness and any unintended negative consequences.
6.2. Patient characteristics
Various inclusion and exclusion criteria seem to be associated with increased assay sensitivity; for example, greater baseline pain intensity and prohibition of concomitant analgesic medications were found to be associated with greater assay sensitivity in clinical trials of chronic neuropathic23,32 and musculoskeletal pain.24 In addition, analyses of individual patient data showed that the subgroup of patients with excessive variability of pain ratings at baseline had reduced separation between the active treatment and placebo.28,107
One approach to preventing the randomization of patients who do not fulfill eligibility criteria is to implement a central adjudication process, in which trial eligibility criteria are reviewed for each potential study patient.33,75 This approach has the potential to increase the response to efficacious treatments by eliminating individuals who are unlikely to respond because they do not have the condition for which the treatment is indicated. Independent adjudication of eligibility criteria may also decrease placebo group changes by eliminating professional subjects and others who might be more likely report improvement.33,76,96
6.3. Research designs
There are several clinical trial designs that have the potential to increase assay sensitivity and the efficiency of identifying efficacious pain treatments (Table 3). One relatively straightforward approach is to conduct an interim blinded sample size re-estimation to ensure that the variability of the primary outcome measure was not underestimated in the initial sample size determination.26 Interim futility analyses can also increase the efficiency of identifying efficacious treatments by determining whether a treatment is very unlikely to be statistically significantly different from the control treatment at the scheduled end of the trial.56,104 Although use of such interim analyses in chronic pain RCTs has rarely been reported, they are routinely implemented in other therapeutic areas, and it has been recommended that they be considered in the design of clinical trials of pain treatments.21,22,37
Table 3.
Clinical trial designs that can improve the efficiency and informativeness of clinical trials of pain treatments.

| 1. Interim blinded sample size re-estimation |
| 2. Interim futility analyses |
| 3. Cross-over and multiple N-of-1 designs |
| 4. Designs that might have greater assay sensitivity (eg, EERW, SPCD, and TED) |
| 5. Adaptive designs |
| 6. Master protocols, including, basket, umbrella, and platform designs |
EERW, enriched enrollment randomized withdrawal; SPCD, sequential parallel comparison design; TED, two-way enriched design.
Cross-over designs can be used to reduce sample size requirements when studying pain conditions that are expected to remain stable throughout the trial duration and treatments that have relatively fast onset and offset of their pharmacodynamic effects.26,40,94 However, cross-over trials also have several potential limitations, including carry-over effects, in which the effect of an active treatment in the first period may carry over to a placebo condition in the next period and reduce the second period treatment-placebo difference. Various methods for addressing these effects have been proposed, but the best approach is to design the trial to minimize potential carry-over effects and any other causes of treatment-by-period interaction.26,94
When a cross-over trial randomizes patients to at least 2 periods with an active treatment and 2 periods with placebo—also referred to as an N-of-1 design when used in clinical practice65—it becomes possible to examine whether there is evidence of treatment-by-patient interaction.17,38 Significant treatment-by-patient interaction indicates that there is heterogeneity of treatment effects among patients, that is, different patients truly respond differently to the treatment. Multiperiod cross-over trials, therefore, have the potential to identify those pain conditions and treatments for which efforts to determine genotypic and phenotypic predictors of treatment response could be worthwhile.95
Enrichment designs may increase clinical trial assay sensitivity by randomizing those patients who are expected to be more likely to respond to treatment and not withdraw because of adverse events.114 The most common type of enrichment design used in studying chronic pain treatments has been termed “enriched enrollment randomized withdrawal.”60,80 In this design, an initial enrichment phase in which patients receive the active treatment is followed by a double-blind phase in which patients who have tolerated the treatment and reported an improvement in pain intensity are randomized to continued active treatment or to placebo. The results of published trials suggest that the assay sensitivity of these trials may be greater than the assay sensitivity of standard parallel group trials, but the evidence is not conclusive.34,60,80
The sequential parallel-comparison design (SPCD) was developed to reduce placebo group improvements and thereby increase assay sensitivity in RCTs of antidepressant medications.29,30 In the most common version, patients are first randomized to active treatment and placebo groups, typically with more participants allocated to placebo. Patients in the placebo group who do not improve in this phase are then rerandomized to either the active treatment or placebo. The efficacy analysis typically includes all first-phase data and second-phase data only from the placebo group patients who did not improve in the first phase. Because some patients contribute outcome data from both phases and there is typically a reduced magnitude of change in the placebo group in the second phase, SPCD trials can reduce required sample sizes.12,29,54 The potential of this design for increasing the assay sensitivity of RCTs of chronic pain treatments has been discussed.37
A two-way enriched design that is an extension of SPCD has also been described.55 In this design, after randomization to either active or placebo treatment, patients in the active treatment group who improved and patients in the placebo group who did not improve are rerandomized to active or placebo treatment. The data from this second phase make it possible to test whether a treatment “that is significantly superior to placebo in achieving short-term efficacy will also be superior to placebo in the maintenance of efficacy.”55
Adaptive clinical trial designs can be used for exploratory studies as well as for confirmatory trials, and their objectives have included (1) dose finding; (2) bridging phases 1 and 2 or phases 2 and 3 with seamless designs (eg, using dose-finding data to transition to a confirmatory trial); (3) response adaptive randomization to increase the percentage of patients randomized to treatments with promising interim data; and (4) interim sample-size re-estimation and futility analyses, as discussed above.7,26,112 The benefits of adaptive designs can include smaller sample sizes, shorter durations, and an increased likelihood of achieving trial objectives. However, operational challenges include extensive simulation studies often required for study planning, medication supply, and monitoring of sites, data, and analyses.36 Although it has been suggested that adaptive dose-finding designs can play an important role in early analgesic drug development,57 there have been very few published RCTs of pain treatments that have used adaptive designs.
There has recently been considerable attention to the potential of master protocols to increase the efficiency of drug development by using “a single infrastructure, trial design, and protocol to simultaneously evaluate multiple drugs and/or disease populations in multiple substudies.”113 There are 3 different types of master protocols: (1) umbrella trials, in which multiple treatments are studied for a single disease; (2) basket trials, in which a single treatment is studied in multiple diseases or multiple subtypes of a single disease; and (3) platform trials, in which multiple treatments are studied for a single disease, as in umbrella trials, but in a perpetually continuing manner, and often with sharing of common control patients, treatments entering and exiting the platform on the basis prespecified decision algorithms, and early stopping for success or failure.116 The most frequent use of master protocols has been in oncology, in which different designs have been used to study novel drugs and drug combinations, often in biomarker-defined subgroups of patients. Master protocols could have particular value for novel treatments that potentially have efficacy in one or more different pain conditions given the prevailing expectation that predictive biomarkers will be developed that can identify subgroups of patients who respond more robustly to treatment,1,117
7. Discussion
Sample size does matter for ensuring that clinical trials of pain treatments have adequate assay sensitivity, but it is not everything. In designing, conducting, and analyzing RCTs, a large number of additional methodologic issues and advances should also be considered. Unfortunately, very few studies have formally examined whether modifying study methods increases assay sensitivity or decreases placebo group changes in RCTs of efficacious pain treatments. Providing preliminary support for the value of patient training, the results of recent studies in which patients were randomized to training or no training showed that training can improve the accuracy of pain ratings98,108; in addition, placebo group changes were reduced and there were numerically greater effect sizes in trained vs untrained patients in one of these studies, a clinical trial in painful diabetic peripheral neuropathy.108
The ultimate objective of the research discussed in this article is to develop an evidence-based approach to the design of clinical trials,19 and prospective RCTs must be conducted to test methods that are hypothesized to increase assay sensitivity. Nevertheless, on the basis of available evidence as well as general considerations involving study execution and data quality, recommendations have been presented for improving the design of acute8 and chronic21,37 pain trials and for increasing their assay sensitivity.22 Adopting such recommendations and giving careful consideration to optimizing study design has the potential to increase the assay sensitivity and informativeness of RCTs of pain treatments. The results of the clinical trials conducted over the next decade will hopefully demonstrate whether these approaches give rise to a fourth era of analgesic clinical trials, one in which meaningful increases in treatment effects will occur.
8. Summary
There is no better summary of our perspective on the current state of pain treatment than one provided by Paul Leber70 for psychiatric medications. Based on his wide-ranging experiences as director of the U.S. Food and Drug Administration's Division of Neuropharmacologic Drug Products, Leber maintained that “given how little we actually understand about the behaviors and affects we seek to manage through pharmacological interventions…we are exceedingly fortunate to possess the number of modestly effective drugs that we do.”
Conflict of interest statement
S.M. Smith has received in the past 36 months a research grant from the Richard W. and Mae Stone Goode Foundation. For a complete list of lifetime disclosures for M. Fava, please see: https://mghcme.org/faculty/faculty-detail/maurizio_fava. M.P. Jensen has received in the past 36 months research grants from the U.S. National Institutes of Health, the U.S. Department of Education, the Administration of Community Living, the Patient-Centered Outcomes Institute, and National Multiple Sclerosis Society, the International Association for the Study of Pain, and the Washington State Spinal Injury Consortium, and compensation for consulting from Goalistics. O. Mbowe has no disclosures for the past 36 months. M.P. McDermott has been supported in the past 36 months by research grants from the U.S. National Institutes of Health, U.S. Food and Drug Administration, NYSTEM, SMA Foundation, Cure SMA, and PTC Therapeutics, has received compensation for consulting from Neuropore Therapies and Voyager Therapeutics, and has served on Data and Safety Monitoring Boards for U.S. National Institutes of Health, Novartis Pharmaceuticals Corporation, AstraZeneca, Eli Lilly, aTyr Pharma, Catabasis Pharmaceuticals, Vaccinex, Cynapsus Therapeutics, and Voyager Therapeutics. In the past 36 months, D.C. Turk has received research grants and contracts from U.S. Food and Drug Administration and U.S. National Institutes of Health, and compensation for consulting on clinical trial and patient preferences from AccelRx, Eli Lilly, GlaxoSmithKline, Nektar, Novartis, and Pfizer. R.H. Dworkin has received in the past 36 months research grants and contracts from U.S. Food and Drug Administration and U.S. National Institutes of Health, and compensation for serving on advisory boards or consulting on clinical trial methods from Abide, Acadia, Adynxx, Analgesic Solutions, Aptinyx, Aquinox, Asahi Kasei, Astellas, AstraZeneca, Biogen, Biohaven, Boston Scientific, Braeburn, Celgene, Centrexion, Chromocell, Clexio, Concert, Decibel, Dong-A, Editas, Eli Lilly, Eupraxia, Glenmark, Grace, Hope, Immune, Lotus Clinical Research, Mainstay, Merck, Neumentum, Neurana, NeuroBo, Novaremed, Novartis, Olatec, Pfizer, Phosphagenics, Quark, Reckitt Benckiser, Regenacy (also equity), Relmada, Sanifit, Scilex, Semnur, Sollis, Teva, Theranexus, Trevena, Vertex, and Vizuri.
Acknowledgments
The authors appreciate Yoda's contributions to the headings of sections 2 and 6. Financial support was provided by the Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks (ACTTION) public–private partnership with the U.S. Food and Drug Administration (FDA), which has received research contracts, grants, or other revenue from the FDA, multiple pharmaceutical and device companies, philanthropy, and other sources. The views expressed in this article are those of the authors and no official endorsement by the FDA or the pharmaceutical and device companies that provided unrestricted grants to support the activities of the ACTTION public–private partnership should be inferred.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
With apologies for the title to Godzilla (1998 movie) and Moore et al.79
References
- [1].Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, Finnerup NB, Haanpää M, Hansson P, Hüllemann P, Jensen TS, Freynhagen R, Kennedy JD, Magerl W, Mainka T, Reimer M, Rice ASC, Segerdahl M, Serra J, Sindrup S, Sommer C, Tölle T, Vollert J, Treede RD. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. PAIN 2017;158:361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bennett GJ. Does the word “placebo” evoke a placebo response? PAIN 2018;159:1928–31. [DOI] [PubMed] [Google Scholar]
- [3].Breckenridge A, Aronson JK, Blaschke TF, Hartman D, Peck CC, Vrijens B. Poor medication adherence in clinical trials: consequences and solutions. Nat Rev Drug Discov 2017;16:149–50. [DOI] [PubMed] [Google Scholar]
- [4].Callegari F, Akacha M, Quarg P, Pandhi S, von Raison F, Zuber E. Estimands in a chronic pain trial: challenges and opportunities. Stat Biopharm Res 2020;12:39–44. [Google Scholar]
- [5].Chuang-Stein C, Kirby S. The shrinking or disappearing observed treatment effect. Pharmaceut Statist 2014;13:277–80. [DOI] [PubMed] [Google Scholar]
- [6].Coleshill MJ, Sharpe L, Colloca L, Zachariae R, Colagiuri B. Placebo and active treatment additivity in placebo analgesia: research to date and future directions. Int Rev Neurobiol 2018;139:407–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Coffey CS, Levin B, Clark C, Timmerman C, Wittes J, Gilbert P, Harris S. Overview, hurdles, and future work in adaptive designs: perspectives from a National Institutes of Health-funded workshop. Clin Trials 2012;9:671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cooper SA, Desjardins PJ, Turk DC, Dworkin RH, Katz NP, Kehlet H, Ballantyne JC, Burke LB, Carragee E, Cowan P, Croll S, Dionne RA, Farrar JT, Gilron I, Gordon DB, Iyengar S, Jay GW, Kalso EA, Kerns RD, McDermott MP, Raja SN, Rappaport BA, Rauschkolb C, Royal MA, Segerdahl M, Stauffer JW, Todd KH, Vanhove GF, Wallace MS, West C, White RE, Wu C. Research design considerations for single-dose analgesic clinical trials in acute pain: IMMPACT recommendations. PAIN 2016;157:288–301. [DOI] [PubMed] [Google Scholar]
- [9].Czobor P, Skolnick P. The secrets of a successful clinical trial: compliance, compliance, and compliance. Mol Interv 2011;11:107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Devine EG, Peebles KR, Martini V. Strategies to exclude subjects who conceal and fabricate information when enrolling in clinical trials. Contemp Clin Trials Commun 2017;5:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Devine EG, Waters ME, Putnam M, Surprise C, O'Malley K, Richambault C, Fishman RL, Knapp CM, Patterson EH, Sarid-Segal O, Streeter C, Colanari L, Ciraulo DA. Concealment and fabrication by experienced research subjects. Clin Trials 2013;10:935–48. [DOI] [PubMed] [Google Scholar]
- [12].Doros G, Pencina M, Rybin D, Meisner A, Fava M. A repeated measures model for analysis of continuous outcomes in sequential parallel comparison design stdies. Stat Med 2013;32:2767–89. [DOI] [PubMed] [Google Scholar]
- [13].Dunlop BW, Rapaport MH. Antidepressant signal detection in the clinical trials vortex. J Clin Psychiatry 2015;76:e657–9. [DOI] [PubMed] [Google Scholar]
- [14].Dunlop BW, Vaughan CL. Survey of investigators' opinions on the acceptability of interactions with patients participating in clinical trials. J Clin Psychopharmacol 2010;30:323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dworkin RH. Two very different types of clinical importance. Contemp Clin Trials 2016;46:11. [DOI] [PubMed] [Google Scholar]
- [16].Dworkin RH, Edwards RR. Phenotypes and treatment response: it's difficult to make predictions, especially about the future. PAIN 2017;158:187–9. [DOI] [PubMed] [Google Scholar]
- [17].Dworkin RH, McDermott MP, Farrar JT, O'Connor AB, Senn S. Interpreting patient treatment response in analgesic clinical trials: implications for genotyping, phenotyping, and personalized pain treatment. PAIN 2014;155:457–60. [DOI] [PubMed] [Google Scholar]
- [18].Dworkin RH, Peirce-Sandner S, Turk DC, McDermott MP, Gibofsky A, Simon LS, Farrar JT, Katz NP. Outcome measures in placebo-controlled trials of osteoarthritis: responsiveness to treatment effects in the REPORT database. Osteoarthritis Cartilage 2011;19:483–92 (Corrigendum: Osteoarthritis Cartilage, 2011;19:919). [DOI] [PubMed] [Google Scholar]
- [19].Dworkin RH, Turk DC, Katz NP, Rowbotham MC, Peirce-Sandner S, Cerny I, Clingman CS, Eloff BC, Farrar JT, Kamp C, McDermott MP, Rappaport BA, Sanhai WR. Evidence-based clinical trial design for chronic pain pharmacotherapy: a blueprint for ACTION. PAIN 2011;152(suppl):S107–15. [DOI] [PubMed] [Google Scholar]
- [20].Dworkin RH, Turk DC, McDermott MP, Peirce-Sandner S, Burke LB, Cowan P, Farrar JT, Hertz S, Raja SN, Rappaport BA, Rauschkolb C, Sampaio C. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. PAIN 2009;146:238–44. [DOI] [PubMed] [Google Scholar]
- [21].Dworkin RH, Turk DC, Peirce-Sandner S, Baron R, Bellamy N, Burke LB, Chappell A, Chartier K, Cleeland CS, Costello A, Cowan P, Dimitrova R, Ellenberg S, Farrar JT, French JA, Gilron I, Hertz S, Jadad AR, Jay GW, Kalliomaki J, Katz NP, Kerns RD, Manning DC, McDermott MP, McGrath PJ, Narayana A, Porter L, Quessy S, Rappaport BA, Rauschkolb C, Reeve BB, Rhodes T, Sampaio C, Simpson DM, Stauffer JW, Stucki G, Tobias J, White RE, Witter J. Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. PAIN 2010;149:177–93. [DOI] [PubMed] [Google Scholar]
- [22].Dworkin RH, Turk DC, Peirce-Sandner S, Burke LB, Farrar JT, Gilron I, Jensen MP, Katz NP, Raja SN, Rappaport BA, Rowbotham MC, Backonja MM, Baron R, Bellamy N, Bhagwagar Z, Costello A, Cowan P, Fang WC, Hertz S, Jay GW, Junor R, Kerns RD, Kerwin R, Kopecky EA, Lissin D, Malamut R, Markman JD, McDermott MP, Munera C, Porter L, Rauschkolb C, Rice AS, Sampaio C, Skljarevski V, Sommerville K, Stacey BR, Steigerwald I, Tobias J, Trentacosti AM, Wasan AD, Wells GA, Williams J, Witter J, Ziegler D. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. PAIN 2012;153:1148–58. [DOI] [PubMed] [Google Scholar]
- [23].Dworkin RH, Turk DC, Peirce-Sandner S, He H, McDermott MP, Farrar JT, Katz NP, Lin AH, Rappaport BA, Rowbotham MC. Assay sensitivity and study features in neuropathic pain trials: an ACTTION meta-analysis. Neurology 2013;81:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dworkin RH, Turk DC, Peirce-Sandner S, He H, McDermott MP, Hochberg MC, Jordan JM, Katz NP, Lin AH, Neogi T, Rappaport BA, Simon LS, Strand V. Meta-analysis of assay sensitivity and study features in clinical trials of pharmacologic treatments for osteoarthritis pain. Arthritis Rheumatol 2014;66:3327–36. [DOI] [PubMed] [Google Scholar]
- [25].Edwards RR, Dworkin RH, Turk DC, Angst MS, Dionne R, Freeman R, Hansson P, Haroutounian S, Arendt-Nielsen L, Attal N, Baron R, Brell J, Bujanover S, Burke LB, Carr D, Chappell AS, Cowan P, Etropolski M, Fillingim RB, Gewandter JS, Katz NP, Kopecky EA, Markman JD, Nomikos G, Porter L, Rappaport BA, Rice ASC, Scavone JM, Scholz J, Simon LS, Smith SM, Tobias J, Tockarshewsky T, Veasley C, Versavel M, Wasan AD, Wen W, Yarnitsky D. Patient phenotyping in clinical trials of chronic pain treatments: IMMPACT recommendations. PAIN 2016;157:1851–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Evans S, Ting N. Fundamental concepts for new clinical trialists. Boca Raton, FL: CRC Press, 2016. [Google Scholar]
- [27].Fanelli D. How many scientists fabricate and falsify research: a systematic review and meta-analysis of survey data. PLoS One 2009;4:e5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Farrar JT, Troxel AB, Haynes K, Gilron I, Kerns RD, Katz NP, Rappaport BA, Rowbotham MC, Tierney AM, Turk DC, Dworkin RH. Effect of variability in the 7-day baseline pain diary on the assay sensitivity of neuropathic pain randomized clinical trials: an ACTTION study. PAIN 2014;155:1622–31. [DOI] [PubMed] [Google Scholar]
- [29].Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom 2003;72:115–27. [DOI] [PubMed] [Google Scholar]
- [30].Fava M, Mischoulon D, Iosifescu D, Witte J, Pencina M, Flynn M, Harper L, Levy M, Rickels K, Pollack M. A double-blind, placebo-controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPT-A Study). Psychother Psychosom 2012;81:87–97. [DOI] [PubMed] [Google Scholar]
- [31].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice ASC, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Finnerup NB, Haroutounian S, Baron R, Dworkin RH, Gilron I, Haanpaa M, Jensen TS, Kamerman PR, McNicol E, Moore A, Raja SN, Andersen NT, Sena ES, Smith BH, Rice ASC, Attal N. Neuropathic pain clinical trials: factors associated with decreases in estimated drug efficacy. PAIN 2018;159:2339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Freeman MP, Pooley J, Flynn MJ, Baer L, Mischoulon D, Mou D, Fava M. Guarding the gate: remote structured assessments to enhance enrollment precision in depression trials. J Clin Psychopharmacol 2017;37:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Furlan A, Chaparro LE, Irvin E, Mailis-Gagnon A. A comparison between enriched and nonenriched enrollment randomized withdrawal trials of opioids for chronic noncancer pain. Pain Res Manage 2011;16:337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Furukawa TA, Cipriani A, Atkinson LZ, Leucht S, Ogawa Y, Takeshima N, Hayasaka Y, Chaimani A, Salanti G. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double-blind randomised controlled studies. Lancet Psychiatry 2016;3:1059–66. [DOI] [PubMed] [Google Scholar]
- [36].Gaydos B, Anderson KM, Berry D, Burnham N, Chuang-Stein C, Dudinak J, Fardipour P, Gallo P, Givens S, Lewis R, Maca J, Pinheiro J, Pritchett Y, Krams M. Good practices for adaptive clinical trials in pharmaceutical product development. Drug Inf J 2009;43:539–56. [Google Scholar]
- [37].Gewandter JS, Dworkin RH, Turk DC, McDermott MP, Baron R, Gastonguay MR, Gilron I, Katz NP, Mehta C, Raja SN, Senn S, Taylor C, Cowan P, Desjardins P, Dimitrova R, Dionne R, Farrar JT, Hewitt DJ, Iyengar S, Jay GW, Kalso E, Kerns RD, Leff R, Leong M, Petersen KL, Ravina BM, Rauschkolb C, Rice AS, Rowbotham MC, Sampaio C, Sindrup SH, Stauffer JW, Steigerwald I, Stewart J, Tobias J, Treede RD, Wallace M, White RE. Research designs for proof-of-concept chronic pain clinical trials: IMMPACT recommendations. PAIN 2014;155:1683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gewandter JS, McDermott MP, He H, Gao S, Cai X, Farrar JT, Katz NP, Markman JD, Senn S, Turk DC, Dworkin RH. Demonstrating heterogeneity of treatment effects among patients: an overlooked but important step toward precision medicine. Clin Pharmacol Ther 2019;106:204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gewandter JS, McDermott MP, Kitt RA, Chaudari J, Koch JG, Evans SR, Gross RA, Markman JD, Turk DC, Dworkin RH. Interpretation of CIs in clinical trials with non-significant results: systematic review and recommendations. BMJ Open 2017;7:e017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gewandter JS, McDermott MP, McKeown A, Hoang K, Iwan K, Kralovic S, Rothstein D, Gilron I, Katz NP, Raja SN, Senn S, Smith SM, Turk DC, Dworkin RH. Reporting of cross-over clinical trials of analgesic treatments for chronic pain: analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks systematic review and recommendations. PAIN 2016;157:2544–51. [DOI] [PubMed] [Google Scholar]
- [41].Gewandter JS, McDermott MP, McKeown A, Smith SM, Williams MR, Hunsinger M, Farrar J, Turk DC, Dworkin RH. Reporting of missing data and methods used to accommodate them in recent analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2014;155:1871–7. [DOI] [PubMed] [Google Scholar]
- [42].Gewandter JS, Smith SM, McKeown A, Burke LB, Hertz SH, Hunsinger M, Katz NP, Lin AH, McDermott MP, Rappaport BA, Williams MR, Turk DC, Dworkin RH. Reporting of primary analyses and multiplicity adjustment in recent analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2014;155:461–6. [DOI] [PubMed] [Google Scholar]
- [43].Gilbertini M, Nations K, Wihitaker J. Obtained effect size as a function of sample size in approved antidepressants: a real-world illustration in support of better trial design. Clin Psychopharmacol 2012;27:100–6. [DOI] [PubMed] [Google Scholar]
- [44].Gordh TE, Stubhaug A, Jensen TS, Arnèr S, Biber B, Boivie J, Mannheimer C, Kalliomäki J, Kalso E. Gabapentin in traumatic nerve injury pain: a randomized, double-blind, placebo-controlled, cross-over, multi-center study. PAIN 2008;138:255–66. [DOI] [PubMed] [Google Scholar]
- [45].Hauser RA, Stocchi F, Rascol O, Huyck SB, Capece R, Ho TW, Sklar P, Lines C, Michelson D, Hewitt D. Preladenant as an adjunctive therapy with levodopa in Parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol 2015;72:1491–500. [DOI] [PubMed] [Google Scholar]
- [46].Hedges LV, Tipton E, Johnson MC. Robust variance estimation in meta-regression with dependent effect size estimates. Res Synth Methods 2010;1:39–65. [DOI] [PubMed] [Google Scholar]
- [47].Hedges LV, Tipton E, Johnson MC. Erratum: robust variance estimation in meta-regression with dependent effect size estimates. Res Synth Methods 2010;1:164–5. [DOI] [PubMed] [Google Scholar]
- [48].Hewitt DJ, Ho TW, Galer B, Backonja M, Markovitz P, Gammaitoni A, Michelson D, Bolognese J, Alon A, Rosenberg E, Herman G, Wang H. Impact of responder definition on the enriched enrollment randomized withdrawal trial design for establishing proof of concept in neuropathic pain. PAIN 2011;152:514–21. [DOI] [PubMed] [Google Scholar]
- [49].International Council for Harmonisation of Technical Requirtements for Pharmaceuticals for Human Use. Addendum on estimands and sensitivity analysis in clinical trials to the guideline on statsistical principles for clinical trials: E9 (R1). 2019. Available at: https://database.ich.org/sites/default/files/E9-R1_Step4_Guideline_2019_1203.pdf. Accessed March 6, 2020. [Google Scholar]
- [50].Ioannidis JP. Why most published research findings are false. PLoS Med 2005;2:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ioannidis JPA. How to make more published research true. PLoS Med 2014;11:e1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Iovieno N, Nierenberg AA, Parkin SR, Kim H, Walker RS, Fava M, Papakostas GI. Relationship between placebo response rate and clinical trial outcome in bipolar depression. J Psychiatr Res 2016;74:38–44. [DOI] [PubMed] [Google Scholar]
- [53].Iovieno N, Papakostas GI. Correlation between different levels of placebo response rate and clinical trial outcome in major depressive disorder: a meta-analysis. J Clin Psychiatry 2012;73:1300–6. [DOI] [PubMed] [Google Scholar]
- [54].Ivanova A, Qaqish B, Schoenfeld D. Optimality, sample size, and power calculations for the sequential parallel comparison design. Stat Med 2011;30:2793–803. [DOI] [PubMed] [Google Scholar]
- [55].Ivanova A, Tamura RN. A two-way enriched clinical trial design: combininig advantages of placebo lead-in and randomized withdrawal. Stat Methods Med Res 2015;24:871–90. [DOI] [PubMed] [Google Scholar]
- [56].Jitlal M, Khan I, Lee SM, Hackshaw A. Stopping clinical trials early for futility: retrospective analysis of several randomised clinical studies. Br J Cancer 2012;107:910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kalliomäki J, Miller F, Kågedal M, Karlsten R. Early phase drug development for treatment of chronic pain: options for clinical trial and program design. Contemp Clin Trials 2012;33:689–99. [DOI] [PubMed] [Google Scholar]
- [58].Kaptchuk TJ. Powerful placebo: the dark side of the randomised controlled trial. Lancet 1998;351:1722–5. [DOI] [PubMed] [Google Scholar]
- [59].Katz J, Finnerup NB, Dworkin RH. Clinical trial outcome in neuropathic pain: relationship to study characteristics. Neurology 2008;70:263–72. [DOI] [PubMed] [Google Scholar]
- [60].Katz N. Enriched enrollment randomized withdrawal trial designs of analgesics: focus on methodology. Clin J Pain 2009;25:797–807. [DOI] [PubMed] [Google Scholar]
- [61].Khan A, Fahl Mar K, Faucett J, Khan Schilling S, Brown WA. Has the rising placebo response impacted antidepressant clinical trial outcome?: data from the US Food and Drug Administration 1987-2013. World Psychiatry 2017;16:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Khan A, Mar KF, Brown WA. The conundrum of depression clinical trials: one size does not fit all. Int Clin Psychopharmacol 2018;33:239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Khin NA, Chen YF, Yang Y, Yang P, Laughren TP. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry 2011;72:464–72. [DOI] [PubMed] [Google Scholar]
- [64].Kobak KA, Kane JM, Thase ME, Nierenberg AA. Why do clinical trials fail? The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharm 2007;27:1–5. [DOI] [PubMed] [Google Scholar]
- [65].Kronish IM, Hampsey M, Falzon L, Konrad B, Davidson KW. Personalized (N-of-1) trials for depression: a systematic review. J Clin Psychopharmacol 2018;38:218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kube T, Rief W. Are placebi and drug-specific effects additive?: questioning basic assumptions of double-blind randomized clinical trials and presenting novel study designs. Drug Discov Today 2017;22:729–35. [DOI] [PubMed] [Google Scholar]
- [67].Landin R, DeBrota DJ, DeVries TA, Potter WZ, Demitrack MA. The impact of restrictive entry criterion during the placebo lead-in period. Biometrics 2000;56:271–8. [DOI] [PubMed] [Google Scholar]
- [68].Lasagna L. The controlled clinical trial: theory and practice. J Chron Dis 1955;1:353–67. [DOI] [PubMed] [Google Scholar]
- [69].Lasagna L, Meieir P. Clinical evaluation of drugs. Ann Rev Med 1958;9:347–54. [DOI] [PubMed] [Google Scholar]
- [70].Leber P. Not in our methods, but in our ignorance. Arch Gen Psychiatry 2002;59:279–80. [DOI] [PubMed] [Google Scholar]
- [71].Lipset CH. Engage with research participants about social media. Nat Med 2014;20:231. [DOI] [PubMed] [Google Scholar]
- [72].Liu KS, Snavely DB, Ball WA, Lines CR, Reines SA, Potter WZ. Is bigger better for depression trials? J Psychiatr Res 2008;42:622–30. [DOI] [PubMed] [Google Scholar]
- [73].Lund K, Vase L, Petersen GL, Jensen TS, Finnerup NB. Randomized controlled trials may underestimate drug effects: blanced placebo trial design. PLoS One 2014;9:e84104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Marder SR, Laughren T, Romano SJ. Why are innovative drugs failing in phase III?. Am J Psychiatry 2017;174:829–31. [DOI] [PubMed] [Google Scholar]
- [75].Markman J, Resnick M, Greenberg S, Katz N, Yang R, Scavone J, Whalen E, Gregorian G, Parsons B, Knapp L. Efficacy of pregabalin in post-traumatic peripheral neuropathic pain: a randomized, double-blind, placebo-controlled phase 3 trial. J Neurol 2018;265:2815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].McCann DJ, Petry NM, Bresell A, Isacsson E, Wilson E, Alexander RC. Medication nonadherence, “professional subjects,” and apparent placebo responders: overlapping challenges for medications development. J Clin Psychopharmacol 2015;35:566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].McKeown A, Gewandter JS, McDermott MP, Pawlowski JR, Poli JJ, Rothstein D, Farrar JT, Gilron I, Katz NP, Lin AH, Rappaport BA, Rowbotham MC, Turk DC, Dworkin RH, Smith SM. Reporting of sample size calculations in analgesic clinical trials: ACTTION systematic review. J Pain 2017;16:199–206. [DOI] [PubMed] [Google Scholar]
- [78].Modell W, Houde RW. Factors influencing clinical evaluation of drugs: with special reference to the double-blind technique. JAMA 1958;167:2190–9. [DOI] [PubMed] [Google Scholar]
- [79].Moore RA, Gavaghan D, Tramer MR, Collins SL, McQuay HJ. Size is everything—large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. PAIN 1998;78:209–16. [DOI] [PubMed] [Google Scholar]
- [80].Moore RA, Wiffen PJ, Eccleston C, Derry S, Barin R, Bell RF, Furlan AD, Gilron I, Haroutounian S, Katz NP, Lipman AG, Morley S, Peloso PM, Quessy SN, Seers K, Strassels SA, Straube S. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. PAIN 2015;156:1382–95. [DOI] [PubMed] [Google Scholar]
- [81].Mundt JC, Greist JH, Jefferson JW, Katzelnick DJ, DeBrota DJ, Chappell PB, Modell JG. Is it easier to find what you are looking for if you think you know what it looks like? J Clin Psychopharmacol 2007;27:121–5. [DOI] [PubMed] [Google Scholar]
- [82].Otto MW, Nierenberg AA. Assay sensitiity, failed clinical trials, and the conduct of science. Psychother Psychosom 2002;71:241–3. [DOI] [PubMed] [Google Scholar]
- [83].Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome?: a meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol 2009;19:34–40. [DOI] [PubMed] [Google Scholar]
- [84].Papakostas GI, Østergaard SD, Iovieno N. The nature of placebo response in clinical studies of major depressive disorder. J Clin Psychiatry 2015;76:456–66. [DOI] [PubMed] [Google Scholar]
- [85].Permutt T. Sensitivity analysis for missing data in regulatory submisasions. Stat Med 2016;35:2876–9. [DOI] [PubMed] [Google Scholar]
- [86].Pocock SJ, Stone GW. The primary outcome fails—what next? N Engl J Med 2016;375:861–70. [DOI] [PubMed] [Google Scholar]
- [87].Posternak MA, Zimmerman M. Therapeutic effect of follow-up assessments on antidepressant and placebo response rates in antidepressant efficacy trials: meta-analysis. Br J Psychiatry 2007;190:287–92. [DOI] [PubMed] [Google Scholar]
- [88].Quessy SN, Rowbotham MC. Placebo response in neuropathic pain trials. PAIN 2008;138:479–83. [DOI] [PubMed] [Google Scholar]
- [89].Rice ASC, Dworkin RH, McCarthy TD, Anand P, Bountra C, McCloud PI, Hill J, Cutter G, Kitson G, Desem N, Raff M. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 2014;383:1637–47. [DOI] [PubMed] [Google Scholar]
- [90].Roose SP, Rutherford BR, Wall MM, Thase ME. Praticing evidence-based medicine in an era of high placebo response: number needed to treat reconsidered. Br J Psychiatry 2016;208:416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rutherford BR, Cooper TM, Persaud A, Brown PJ, Sneed JR, Roose SP. Less is more in antidepressant clinical trials: a meta-analysis of the effect of visit frequency on treatment response and dropout. J Clin Psychiatry 2013;74:703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry 2013;170:723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Senn S. Letters to the editor (Brand R, Kragt H. Importance of trends in the interpretation of an overall odds ratio in the meta-analysis of clinical trials. Stat Med 1992;11:2077-82). Stat Med 1994;13:293–6. [DOI] [PubMed] [Google Scholar]
- [94].Senn S. Cross-over trials in clinical research. Chichester, United Kingdom: John Wiley & Sons, 2002. [Google Scholar]
- [95].Senn S. Mastering variation: variance components and personalised medicine. Stat Med 2016;35:966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shiovitz TM, Bain EE, McCann DJ, Skolnick P, Laughren T, Hanina A, Burch D. Mitigating the effects of nonadherence in clinical trials. J Clin Pharmacol 2016;56:1151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Singla N, Hunsinger M, Chang PD, McDermott MP, Chowdhry AK, Desjardins PJ, Turk DC, Dworkin RH. Assay sensitivity of pain intensity versus pain relief in acute pain clinical trials: ACTTION systematic review and meta-analysis. J Pain 2015;16:683–91. [DOI] [PubMed] [Google Scholar]
- [98].Smith SM, Amtmann D, Askew RL, Gewandter JS, Hunsinger M, Jensen MP, McDermott MP, Patel KV, Williams M, Bacci ED, Burke LB, Chambers CT, Cooper SA, Cowan P, Desjardins P, Etropolski M, Farrar JT, Gilron I, Huang IZ, Katz M, Kerns RD, Kopecky EA, Rappaport BA, Resnick M, Strand V, Vanhove GF, Veasley C, Versavel M, Wasan AD, Turk DC, Dworkin RH. Pain intensity rating training: results from an exploratory study of the ACTTION PROTECCT system. PAIN 2016;157:1056–64. [DOI] [PubMed] [Google Scholar]
- [99].Smith SM, Dworkin RH. Prospective clinical trial registration: not sufficient, but always necessary. Anaesthesia 2018;73:538–41. [DOI] [PubMed] [Google Scholar]
- [100].Smith SM, Dworkin RH, Turk DC, Baron R, Polydefkis M, Tracey I, Borsook D, Edwards RR, Harris RE, Wager TD, Arendt-Nielsen L, Burke LB, Carr DB, Chappell A, Farrar JT, Freeman R, Gilron I, Goli V, Haeussler J, Jensen T, Katz NP, Kent J, Kopecky EA, Lee DA, Maixner W, Markman JD, McArthur JC, McDermott MP, Parvathenani L, Raja SN, Rappaport BA, Rice ASC, Rowbotham MC, Tobias JK, Wasan AD, Witter J. The potential role of sensory testing, skin biopsy, and functional brain imaging as biomarkers in chronic pain clinical trials: IMMPACT considerations. J Pain 2017;18:757–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Smith SM, Hunsinger M, McKeown A, Parkhurst M, Allen R, Kopko S, Lu Y, Wilson HD, Burke LB, Desjardins P, McDermott MP, Rappaport BA, Turk DC, Dworkin RH. Quality of pain intensity assessment reporting: ACTTION systematic review and recommendations. J Pain 2015;16:299–305. [DOI] [PubMed] [Google Scholar]
- [102].Smith SM, Jensen MP, He H, Kitt R, Koch J, Pan A, Burke LB, Farrar JT, McDermott MP, Turk DC, Dworkin RH. A comparison of the assay sensitivity of average and worst pain intensity in pharmacologic trials: an ACTTION systematic review and meta-analysis. J Pain 2018;19:953–60. [DOI] [PubMed] [Google Scholar]
- [103].Smith SM, Wang AT, Pereira A, Chang RD, McKeown A, Greene K, Rowbotham MC, Burke LB, Coplan P, Gilron I, Hertz SH, Katz NP, Lin AH, McDermott MP, Papadopoulos EJ, Rappaport BA, Sweeney M, Turk DC, Dworkin RH. Discrepancies between registered and published primary outcome specifications in analgesic trials: ACTTION systematic review and recommendations. PAIN 2013;154:2769–74. [DOI] [PubMed] [Google Scholar]
- [104].Snapinn S, Chen MG, Jiang Q, Koutsoukos T. Assessment of futility in clinical trials. Pharm Stat 2006;5:273–81. [DOI] [PubMed] [Google Scholar]
- [105].Sriwatankul K, Lasagna L, Cox C. Evaluation of current clinical trial methodology in analgesiometry based on experts' opinions and analysis of several analgesic studies. Clin Pharmacol Ther 1983;34:277–83. [DOI] [PubMed] [Google Scholar]
- [106].Thase ME. US Food and Drug Administration's review of the novel antidepressant vortioxeyine. J Clin Psychiatry 2015;76:e120–1. [DOI] [PubMed] [Google Scholar]
- [107].Treister R, Honigman L, Lawal OD, Lanier RK, Katz NP. A deeper look at pain variability and its relationship with the placebo response: results from a randomized, double-blind, placebo-controlled clinical trial of naproxen in osteoarthritis of the knee. PAIN 2019;160:1522–8. [DOI] [PubMed] [Google Scholar]
- [108].Treister R, Lawal OD, Shecter JD, Khurana N, Bothmer J, Field M, Harte SE, Kruger GH, Katz NP. Accurate pain reporting training diminishes the placebo response: results from a randomised, double-blind, crossover trial. PLoS One 2018;13:e0197844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Treister R, Suzan E, Lawal OD, Katz NP. Staircase-evoked pain may be more sensitive than traditional pain assessments in discriminating analgesic effects: a randomized, placebo-controlled trial of naproxen in patietns with osteoarthritis of the knee. Clin J Pain 2019;35:50–5. [DOI] [PubMed] [Google Scholar]
- [110].Tuttle AH, Tohyama S, Ramsay T, Kimmelman J, Schweinhardt P, Bennett GJ, Mogil JS. Increasing placebo responses over time in U.S. clinical trials of neuropathic pain. PAIN 2015;156:2616–26. [DOI] [PubMed] [Google Scholar]
- [111].Undurraga J. Baldessarini RJ Randomized, placebo-controlled trials of antidepressants for acute major depression: thirty-year meta-analytic review. Neuropsychopharmacology 2012;37:851–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].United States Department of Health and Human Services, Food and Drug Administration. Adaptive designs for clinical trials of drugs and biologics (draft guidance). Silver Spring, MD: United States Food and Drug Administration, 2018. [Google Scholar]
- [113].United States Department of Health and Human Services, Food and Drug Administration. Master protocols: efficient clinical trial design strategies to expedite development of oncology drugs and biologics (draft guidance). Silver Spring, MD: United States Food and Drug Administration, 2018. [Google Scholar]
- [114].United States Department of Health and Human Services, Food and Drug Administration. Enrichment strategies for clinical trials to support determination of effectiveness of human drugs and biological products. Silver Spring, MD: United States Food and Drug Administration, 2019. [Google Scholar]
- [115].Wise RA, Bartlett SJ, Brown ED, Castro M, Cohen R, Holbrook JT, Irvin CG, Rand CS, Sockrider MM, Sugar EA; American Lung Association Asthma Clinical Research C. Randomized trial of the effect of drug presentation on asthma outcomes: the American Lung Association Asthma Clinical Research Centers. J Allergy Clin Immunol 2009;124:436–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med 2017;377:62–70. [DOI] [PubMed] [Google Scholar]
- [117].Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 2017;16:545–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yildiz A, Vieta E, Tohen M, Baldessarini RJ. Factors modifying drug and placebo responses in randomized trials for bipolar mania. Int J Neuropsychopharmacol 2011;14:863–75. [DOI] [PubMed] [Google Scholar]
- [119].Zhang J, Mathis MV, Sellers JW, Kordzakhia G, Jackson AJ, Dow A, Yang P, Fossom L, Zhu H, Patel H, Unger EF, Temple RJ. The US Food and Drug Administration's perspective on the new antidepressant vortioxetine. J Clin Psychiatry 2015;76:8–14. [DOI] [PubMed] [Google Scholar]
- [120].Zimbroff DL. Patient and rater education of expectations in clinical trials (PREECT). J Clin Psychopharmacol 2001;21:251–2. [DOI] [PubMed] [Google Scholar]