

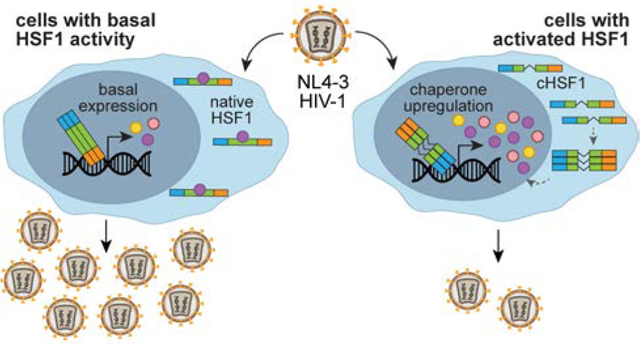
Abstract
Host protein folding stress responses can play important roles in RNA virus replication and evolution. Prior work suggested a complicated interplay between the cytosolic proteostasis stress response, controlled by the transcriptional master regulator heat shock factor 1 (HSF1), and human immunodeficiency virus-1 (HIV-1). We sought to uncouple HSF1 transcription factor activity from cytotoxic proteostasis stress and thereby better elucidate the proposed role(s) of HSF1 in the HIV-1 lifecycle. To achieve this objective, we used chemical genetic, stress-independent control of HSF1 activity to establish whether and how HSF1 influences HIV-1 replication. Stress-independent HSF1 induction decreased both the total quantity and infectivity of HIV-1 virions. Moreover, HIV-1 was unable to escape HSF1-mediated restriction over the course of several serial passages. These results clarify the interplay between the host’s heat shock response and HIV-1 infection and motivate continued investigation of chaperones as potential antiviral therapeutic targets.
Keywords: human immunodeficiency virus-1 (HIV-1), heat shock factor 1 (HSF1), heat shock response (HSR), cytosolic proteostasis, protein folding and assembly
Graphical Abstract
Human immunodeficiency virus-1 (HIV-1) remains a serious global health threat, with approximately 37 million people currently living with HIV/AIDS.1 While the number of HIV-related deaths continues to decline, owing to advances in treatment and prevention strategies in the past decades,2 the epidemic still claims nearly one million lives annually. The problems of latent infection and drug resistance remain, as does the continued failure to develop an effective HIV vaccine.
With respect to the development of new therapeutic modalities impervious to antiviral resistance mechanisms, not just for HIV but also for other RNA viruses, the alternative strategy of targeting host systems instead of the rapidly mutating virus itself has gained increasing traction.3–4 As a minimalistic pathogen, HIV-1 requires complex interactions with host systems for replication.5–6 A clear understanding of the intimate interplay between the host and the virus is essential to provide an effective roadmap for viable, host-targeted antiviral therapeutics.4, 7
Stress responses evolved to defend cells against damaging internal and external stimuli. In some cases, stress responses can provide defenses against invading pathogens. However, numerous viral pathogens have also developed strategies to take advantage of these same host stress signaling pathways. A prominent example of the latter is the cellular heat shock response (HSR), which is responsible for maintaining proteostasis in the cytosol and nucleus.8 The HSR is controlled by its master regulator, the heat shock factor 1 (HSF1) transcription factor. High levels of HSF1 activity can be triggered by a variety of stressors, including protein misfolding in the cytosol. HSF1-mediated upregulation of numerous heat shock protein (HSP) chaperones and quality control proteins serves to restore proteostasis, after which HSF1 activity is reduced to basal levels.9 Many host chaperones, including HSP70 and HSP90, are hijacked by diverse viruses to assist viral protein folding and thereby enable virion production.10–13 Inhibition of these same chaperones can suppress viral replication.14–18 Moreover, chaperones can potentiate the evolution of viral proteins. Changes in cellular proteostasis capacity can modulate viral evolutionary trajectories,19–21 and even define the accessibility of destabilized viral protein variants that can enable innate immune system escape.22
Hence, host HSF1 activity and the functions of HSF1-regulated host chaperones are often beneficial for viruses.10–13, 19–21 However, this conclusion derives largely from studies on just a few viruses, including influenza, Dengue, Zika, and polio – with limited studies on retroviruses. Similar phenomena might be expected for retroviruses, which also have high mutation rates and a need to fold their proteins. On the other hand, the requirement for host genome integration in particular adds an additional step that could be differentially influenced by HSF1 and other HSPs.
Prior work has suggested an intimate role for the host cell’s HSR in multiple steps of the HIV-1 lifecycle. The complexity of HSF1 engagement during HIV-1 replication is perhaps best illustrated by HSF1’s apparent ability to either assist12, 23–26 or restrict27–28 HIV-1 propagation depending on the method used to perturb HSF1 activity. For example, heat stress stimulates HIV-1 gene transcription23 and viral replication.25–26 In other work, transient overexpression of wild-type HSF1 assisted HIV-1 generation24 and reactivation from latency,12 while HSF1 knockdown proved deleterious for HIV-1 production. Alternatively, transient overexpression of a constitutively active variant of HSF1 suppressed long terminal repeat (LTR)-driven viral transcription27 and downregulated HIV-induced inflammation.28 Similarly, the reported roles of individual HSF1-controlled chaperones in HIV-1 replication extensively vary between different experimental systems.29–36 In sum, although the details are still unclear, there is clearly a complicated interplay between the host’s HSR and the HIV-1 lifecycle.
Our objective was to isolate the direct effects of HSF1 activation from the indirect effects of the cellular stressors that are traditionally used to activate HSF1, thereby gaining a clear understanding of the consequences of HSF1 activity for HIV-1 replication. The achievement of this goal requires a tool for stress-independent HSF1 activation. Heat induction of HSF1 activity is unsuitable because heat is a pleiotropic stress that causes acute and severe protein misfolding throughout the proteome. Genetic methods are preferred as they avoid HSR activation, however the extent of HSF1 activation is limited by cellular compensation mechanisms. For example, overexpression of wild-type HSF1 increases the protein levels of the transcription factor, but the excess HSF1 protein is subject to chaperone-mediated regulation and is thus kept in an inactive state.37 Genetic HSF1 knockdown is also inefficient, owing to compensating proteostasis mechanisms.38 Finally, unregulated overexpression of constitutively active HSF1 variants must be employed with great caution to avoid nonphysiologic levels of HSF1 induction and consequent pleiotropic remodeling of the transcriptome.39 Chemical methods for directly regulating HSF1 activity are preferred.40–42
We first sought to generate a system in which stress-independent, small molecule-mediated induction of HSF1 activity was possible. We engineered a stable, single-colony human T lymphocyte (CEM) cell line in which the expression of a constitutively active variant of HSF1 (cHSF1)39, 43 was placed under control of the doxycycline (dox)-dependent tetracycline (tet) repressor.39 Treatment of the resulting CEMcHSF1 cell line (Figure 1A) with dox resulted in the expression of HSF1 target genes, as demonstrated by the increased transcript levels of HSP90, HSP70, and HSP40 (Figure 1B). The single colony cell line was carefully chosen to ensure that the upregulation of these downstream chaperones was similar in magnitude to that caused by HSF1 activation by the prototypical chemical stressor As(III)44 (Figure 1B), ensuring that HSF1 activity was not induced beyond physiologically accessible levels.39 We also generated a fluorescent control cell line (CEMCtrl) in which cyan fluorescent protein (CFP) expression was similarly placed under control of the tet repressor.
Figure 1.
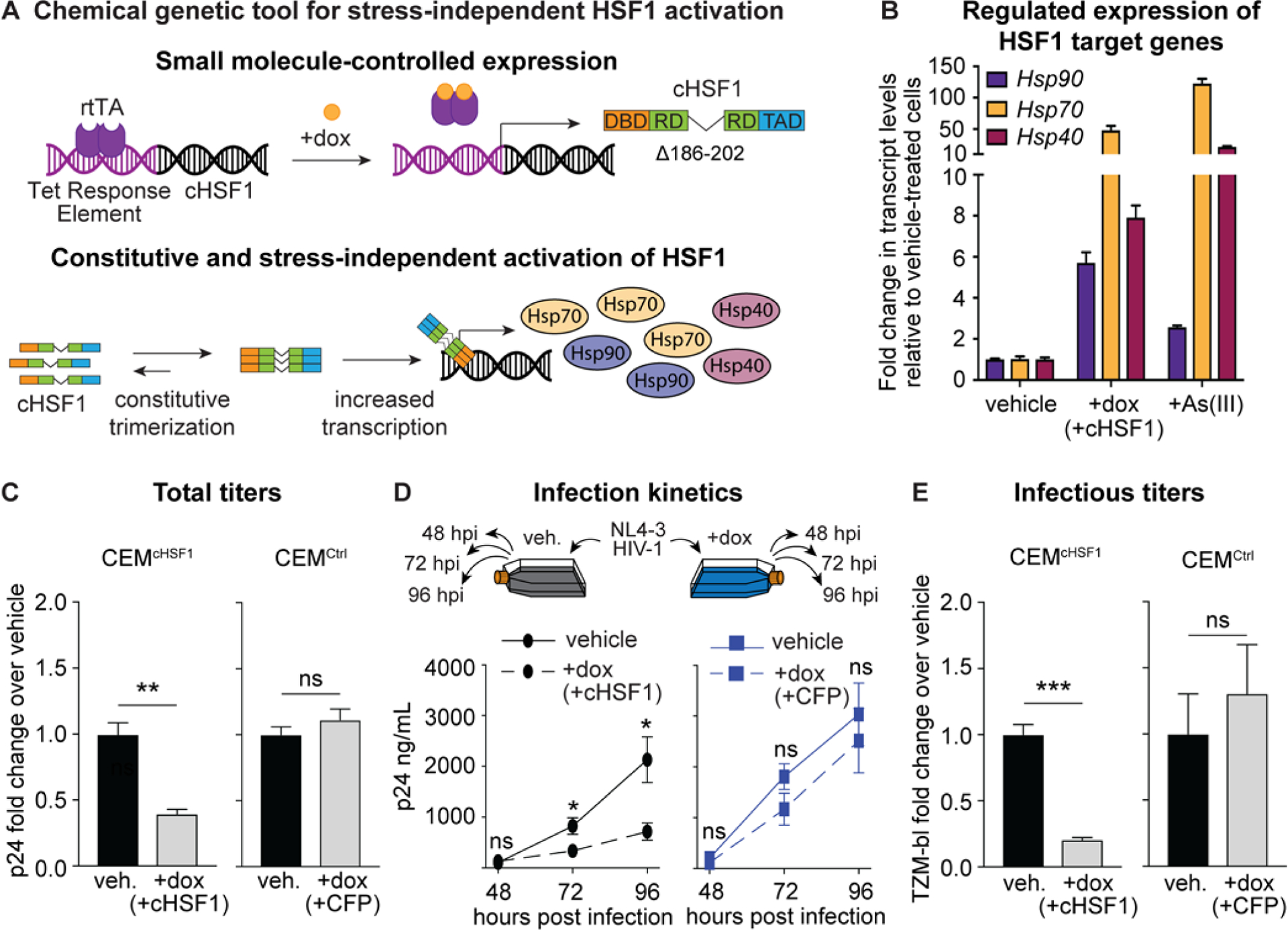
Stress-independent cHSF1 activation decreases HIV-1 replication and the infectivity of produced virions. (A) Chemical genetic tool for stress-independent, small molecule-regulated activation of HSF1. Treatment of CEMcHSF1 cells with dox induces expression of cHSF1, which constitutively trimerizes and upregulates the expression of HSF1 target genes in the absence of acute proteostatic stress. (B) qPCR analysis of Hsp90 (HSP90AA1), Hsp70 (HSPA1A), and Hsp40 (DNAJB1) expression in CEMcHSF1 after 18 h of treatment with 1 μg/mL dox or 2 h of treatment with 100 μM sodium arsenite as a positive heat shock control. (C) Fold-change in p24 titers after 96 h of HIV-1 infection at an MOI of 0.04 in CEMcHSF1 and CEMCtrl cells, treated with 1 μg/mL dox, relative to vehicle-treated cells. (D) Schematic of a timecourse infection and total p24 viral titers during different infection time points in CEMcHSF1 and CEMCtrl cells. (E) Fold-change in infectious TZM-bl titers after 96 h of HIV-1 propagation in CEMcHSF1 and CEMCtrl cells, treated with 1 μg/mL dox, relative to vehicle-treated cells. *, **, ***, and ns correspond to p-values <0.05, <0.001, <0.0001, and not significant, respectively.
With CEMcHSF1 and CEMCtrl cell lines in hand, we next sought to test whether stress-independent HSF1 activation impacted HIV-1 replication. We began by treating CEMcHSF1 and CEMCtrl cells with dox for 18 h to activate cHSF1 or CFP expression, respectively. Next, we infected these preactivated cells with NL4–3 HIV-1 at a multiplicity of infection (MOI) of 0.04 for 96 h, followed by harvesting the infectious supernatant and titering using a p24 enzyme-linked immunosorbent assay (ELISA).
We observed that cHSF1 activation significantly reduced total p24 viral titers relative to cells with basal HSF1 activity (Figure 1C). In contrast, dox-induced expression of CFP in the CEMCtrl cell line did not alter p24 titers, showing that the result was attributable to cHSF1 activity and not to dox treatment. We further assessed infection kinetics by harvesting the viral supernatant at successive time points and titering using the p24 assay. The relative difference in p24 titers between cHSF1-activated versus vehicle-treated CEMcHSF1 cells became more pronounced as the infection progressed, with no significant difference observed in dox- versus vehicle-treated CEMCtrl cells at any time point (Figure 1D). Finally, we used the TZM-bl assay45 to quantify the infectious titers of collected viral supernatants. Successful infection of reporter TZM-bl cells activates the expression of β-galactosidase in an HIV-1 Tat-dependent manner, turning reporter cells blue in the presence of a 5-bromo-4-chloro-3-indolyl-p-D-galactopyranoside (X-Gal) chromogenic substrate. The fraction of stained cells is then proportional to the number of infectious viral particles.45 We observed that, as also occurred with the p24 titers, infectious titers were indeed decreased by cHSF1 activation in CEMcHSF1 cells, whereas they did not change upon CFP activation in CEMCtrl cells (Figure 1E).
The high mutation rate of HIV-1 often promotes rapid escape from inhibitory pressure. Therefore, we next asked whether continuous propagation of HIV-1 under pressure from cHSF1 activity would result in rapid antiviral escape. We performed three serial passages in cHSF1-activated versus vehicle-treated CEMcHSF1 cells (Figure 2A). At each passage, the pre-activated cells were infected at an MOI of 0.04 for 96 h, followed by harvesting the viral supernatant and titering. The infectious (TZM-bl) titers were used to initiate the subsequent passage at the same MOI. Notably, both total and infectious viral titers were still decreased in +cHSF1 cells relative to vehicle-treated cells even after the third serial passage (Figures 2B and 2C), indicating that the virus cannot readily adapt to cHSF1-mediated replication restriction.
Figure 2.
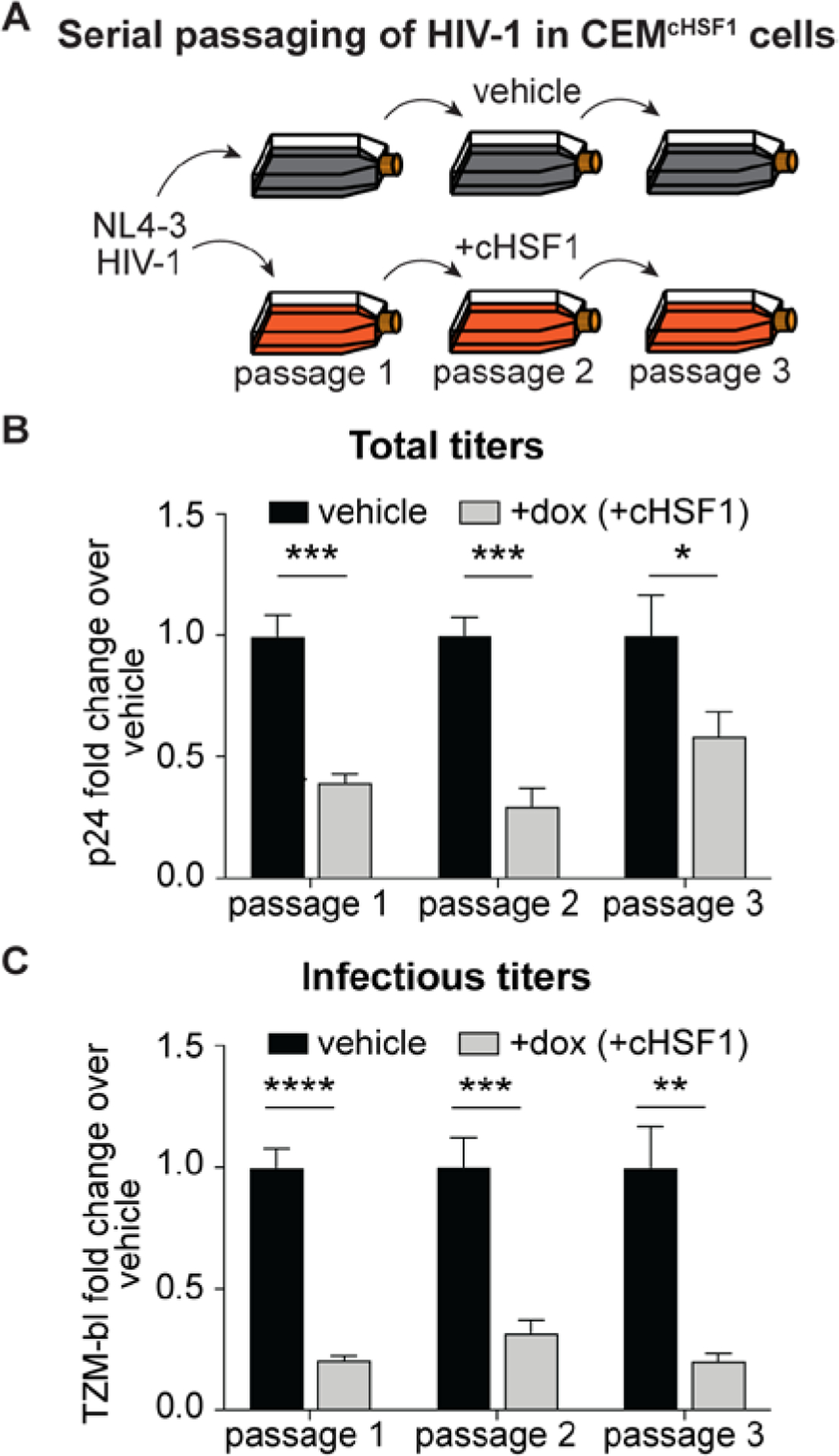
HIV-1 does not adapt to escape HSF1 activation over the course of three serial passages. (A) Schematic of NL4–3 HIV-1 serial passaging. CEMcHSF1 cells were pretreated with 1 μg/mL dox for 18 h prior to infection with NL4–3 HIV-1 for 96 h at an MOI of 0.04. Infectious titers of the viral supernatant were determined using the TZM-bl assay, and then, the supernatant was used to infect the subsequent passage at the same MOI. (B) Fold-change in total p24 and (C) infectious TZM-bl titers at each passage. *, **, ***, and **** correspond to p-values < 0.05, < 0.01, < 0.001, and < 0.0001, respectively.
One potential trivial explanation for HSF1’s effect on HIV-1 replication could be cytotoxicity. We assessed cell health in the conditions of our viral propagation experiments by using a CellTiter-Glo assay. We observed that cellular ATP levels were not significantly altered by stress-independent cHSF1 activation (Figure 3A).
Figure 3.
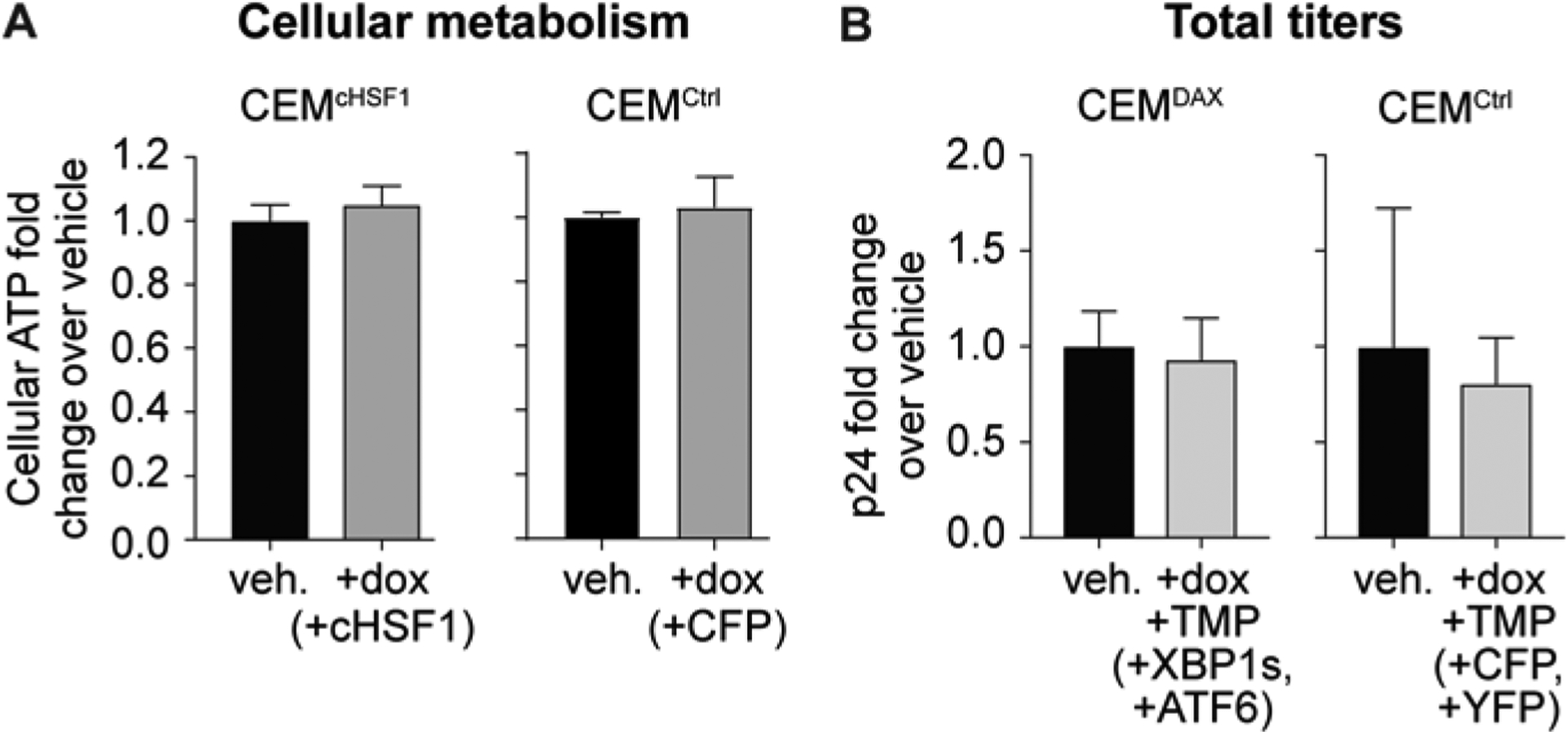
Decreased HIV-1 replication is not attributable to HSF1-induced cytotoxicity. (A) Cellular ATP levels upon 1 μg/mL dox treatment of CEMcHSF1 and CEMCtrl cells for 96 h, as assessed using a CellTiter-Glo assay. (B) Total p24 viral titers upon 1 μg/mL dox and 10 μM TMP treatment of CEMDAX and CEMCtrl cell lines.
We next asked whether the observed inhibition of HIV-1 was specific to the HSR or could be replicated by stress-independent activation of other protein misfolding stress responses. We engineered a stable cell line, termed CEMDAX, in which the IRE1-XBP1s and ATF6 arms of the unfolded protein response (UPR), which is responsible for maintaining proteostasis in the secretory pathway,46 could be activated by small molecules in a stress-independent manner. Our approach was to render XBP1s expression dox-inducible by placing the XBP1s gene under control of the tetracycline promoter.47 To control the ATF6 arm of the UPR, we fused the transcriptionally active form of ATF6 to an Escherichia coli dihydrofolate reductase (DHFR) destabilized domain.48 Treatment of CEMDAX cells with trimethoprim (TMP) stabilizes the DHFR domain, resulting in ATF6 transcriptional activity. This strategy is well-established for stress-independent control of the IRE1-XBP1s and ATF6 arms of the unfolded protein response.21, 41, 49–54 We verified the selective, dox-dependent induction of XBP1s target genes and the selective, TMP-dependent induction of ATF6 target genes in CEMDAX cells using qPCR (Figure S1).49 We also employed a fluorescent control CEMCtrl cell line stably engineered to express dox-inducible CFP and E. coli DHFR-fused yellow fluorescent protein (YFP). We then pretreated CEMDAX and CEMCtrl cells with dox and TMP for 18 h to activate the corresponding constructs, infected the cells with HIV-1 at an MOI of 0.04, and measured the resulting p24 titers after 96 h. No significant change in p24 titers was observed upon dox and TMP treatment in either the CEMDAX or the CEMCtrl cells (Figure 3B). Thus, HSF1-mediated abrogation of HIV-1 replication is a specific feature of HSR activation, not a general consequence of inducing protein misfolding stress responses.
Next, we used RNA-Seq to globally assess transcriptome remodeling owing to cHSF1 activation in CEMcHSF1 cells. In particular, we were interested in whether or not stress-independent cHSF1 induction might elicit an antiviral response in CEMcHSF1 cells. As expected, given the specificity of our stress-independent, chemical induction of cHSF1, the most prominent upregulated genes were all well-known components of the HSR (Figures 4 and S2, Table S1).39 Also, as expected, no significant induction of UPR target genes was observed (Figure 4 and Table S1).
Figure 4.
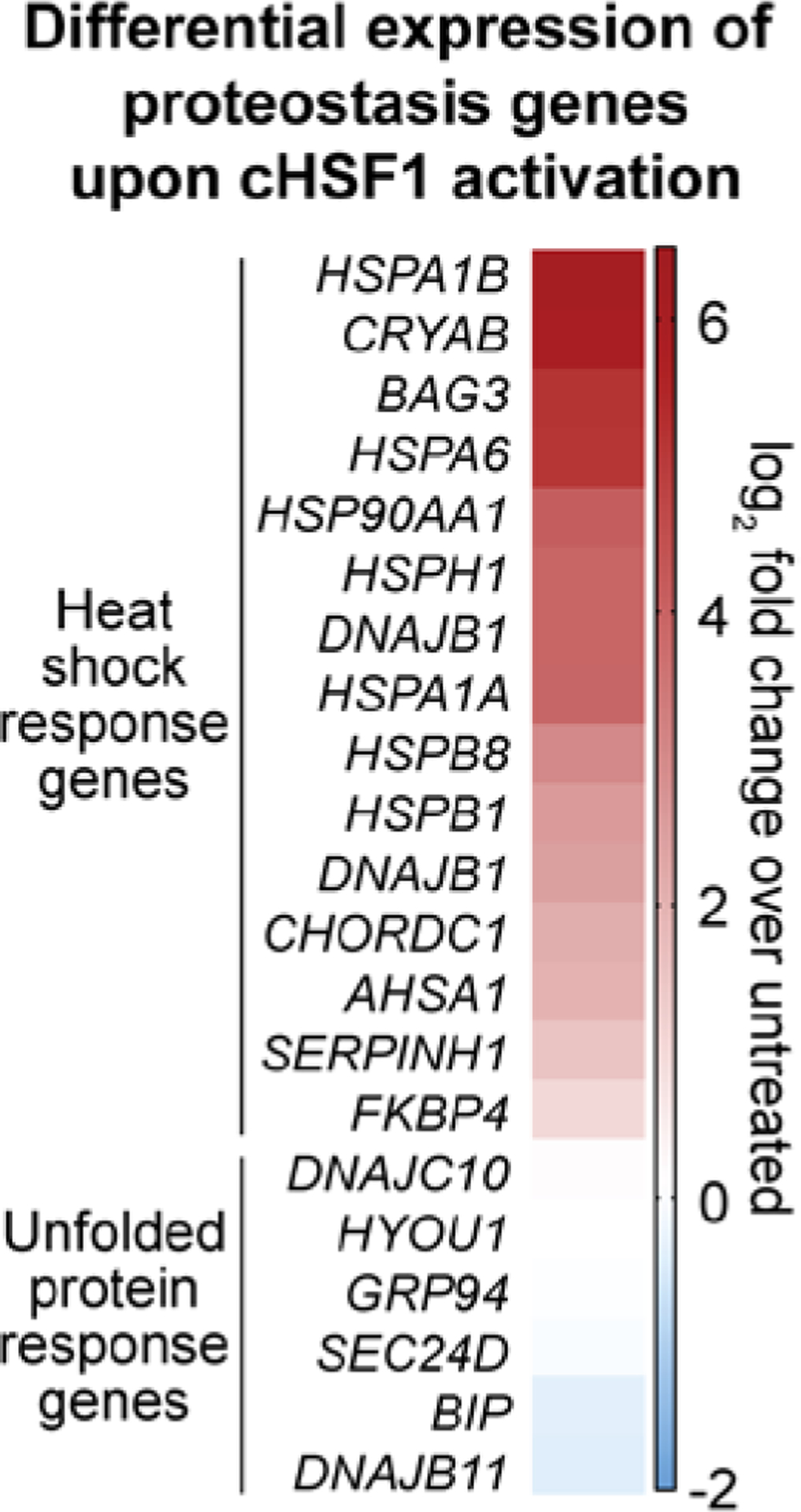
Heat map showing the differential expression of select proteostasis genes upon stress-independent cHSF1 activation with 1 μg/mL dox for 18 h in CEMcHSF1 cells relative to vehicle treatment.
We applied gene set enrichment analysis (GSEA)55 (Table S2) to better understand key features of the transcriptome remodeling caused by cHSF1 activation. We observed that known HSR-related gene sets were massively enriched (MSigDB c5 collection; Figure 5A and Tables S2A and S2B). Furthermore, the HSF1-binding motif itself was strongly enriched upstream of genes that were found to be responsive to stress-independent cHSF1 activation (MSigDB c3 collection, Figure S3 and Table S2C). However, we did not observe any significant enrichment of antiviral restriction factors using the MSigDB c5 collection (see Figure 5B for example enrichment plots and Tables S2A and S2B). Similarly, when other functional databases regrouped in the MSigDB c2 collections were interrogated, viral- and interferon-response pathways tended either not to display any bias or even to be enriched among downregulated gene sets (Tables S2D and S2E).
Figure 5.
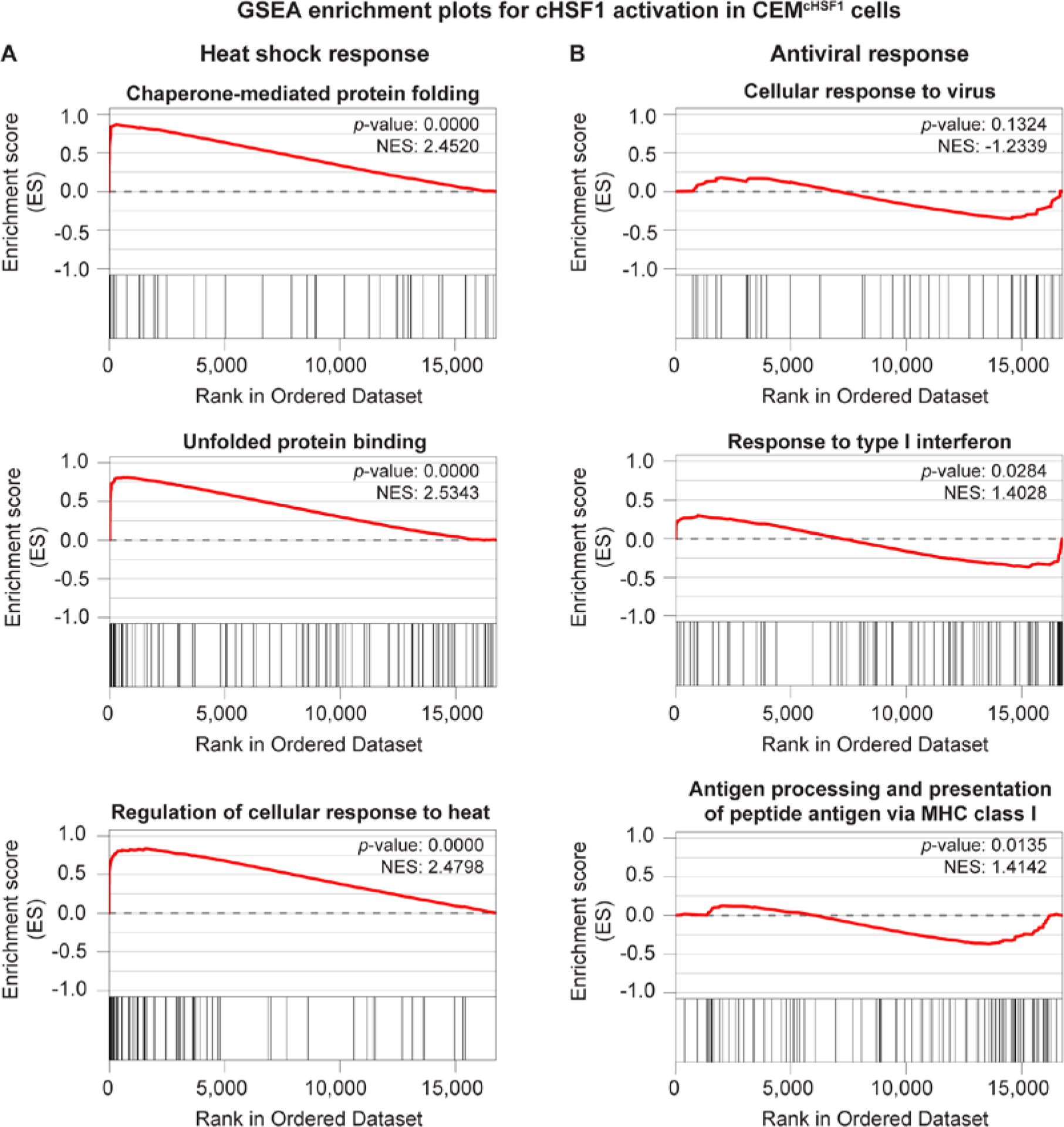
Stress-independent HSF1 induction activates heat shock response genes and does not trigger a broad-scale antiviral response. (A) Selected gene set enrichment plots for heat shock response-related and (B) antiviral response-related gene sets in CEMcHSF1 cells treated with 1 µg/mL doxycycline for 18 h to induce cHSF1. These enrichment plots are drawn from the MSigDB c5 collection. See Table S2 for the complete gene set enrichment analysis.
These observations suggest that stress-independent HSF1 activation in CEMcHSF1 cells does not inhibit HIV-1 replication by inducing a general antiviral response. We next examined individual genes within the broad gene ontology group “Defense Response to Virus” (Table S3). While typical components of the general antiviral defense response, including many interferon-related genes, were not enriched or even downregulated, we were intrigued to note that the most upregulated gene in the entire gene set was ZC3HAV1. ZC3HAV1 encodes the zinc finger antiviral protein ZAP (also known as PARP13), and was upregulated 3.2-fold in our RNA-Seq experiment upon cHSF1 induction. ZAP is known to restrict the replication of multiple viruses, particularly including HIV-1,56–57 by targeting viral mRNA in the cytoplasm for degradation.58 ZAP can also bind HSF159 and assist HSF1 binding to DNA prior to heat shock.60 Indeed, the first intron of ZAP possesses an HSF1-binding motif, located in a putative chromatin regulatory region denoted by a peak of H3K27-acetylated histones, as reported by the Encyclopedia of DNA Elements (ENCODE) consortium in an immortalized B-cell line (chromatin immunoprecipitation (ChIP)-Seq ENCODE track on the UCSC Genome Browser).61 We used qPCR to confirm that the induction of cHSF1 in CEMcHSF1 cells indeed triggered upregulation of ZAP mRNA (Figure S4). On the basis of these observations, ZAP induction is likely to contribute to cHSF1-mediated inhibition of HIV-1 replication.
Although ZAP induction may play a role in the inhibition of HIV-1 replication, the key finding from our RNA-Seq analysis was that cHSF1 activation largely drives a transcriptional remodeling of the cellular chaperone network, with minimal impacts on immune responses and traditional viral restriction factors. A number of these chaperones have been implicated in the HIV lifecycle and play important roles in viral protein folding and assembly.62–64 Thus, it is possible that the remodeled cytosolic and nuclear proteostasis network, which did not evolve to support HIV-1 replication but rather to ensure cellular proteostasis, might disrupt these steps in the lifecycle by diverting viral proteins from function or the orchestrated virion assembly process. In this regard, it is noteworthy that comparing the total (p24) to the infectious (TZM-bl) viral titers, we observed that the fraction of produced virions that are infection-competent significantly decreased upon cHSF1 activation (Figure 6). This observation is consistent with cHSF1 activation disrupting steps in the viral lifecycle such as viral protein folding and/or virion assembly that could lead to the production of a larger fraction of defective viral particles. Because host chaperones not only directly modulate viral protein folding and assembly but also participate in earlier steps of the viral replication cycle, such as nuclear import,34 genome integration,65 and transcription,33, 36 we do not exclude the possibility of additional inhibitory roles of the cHSF1-remodeled proteostasis network in these processes.
Figure 6.
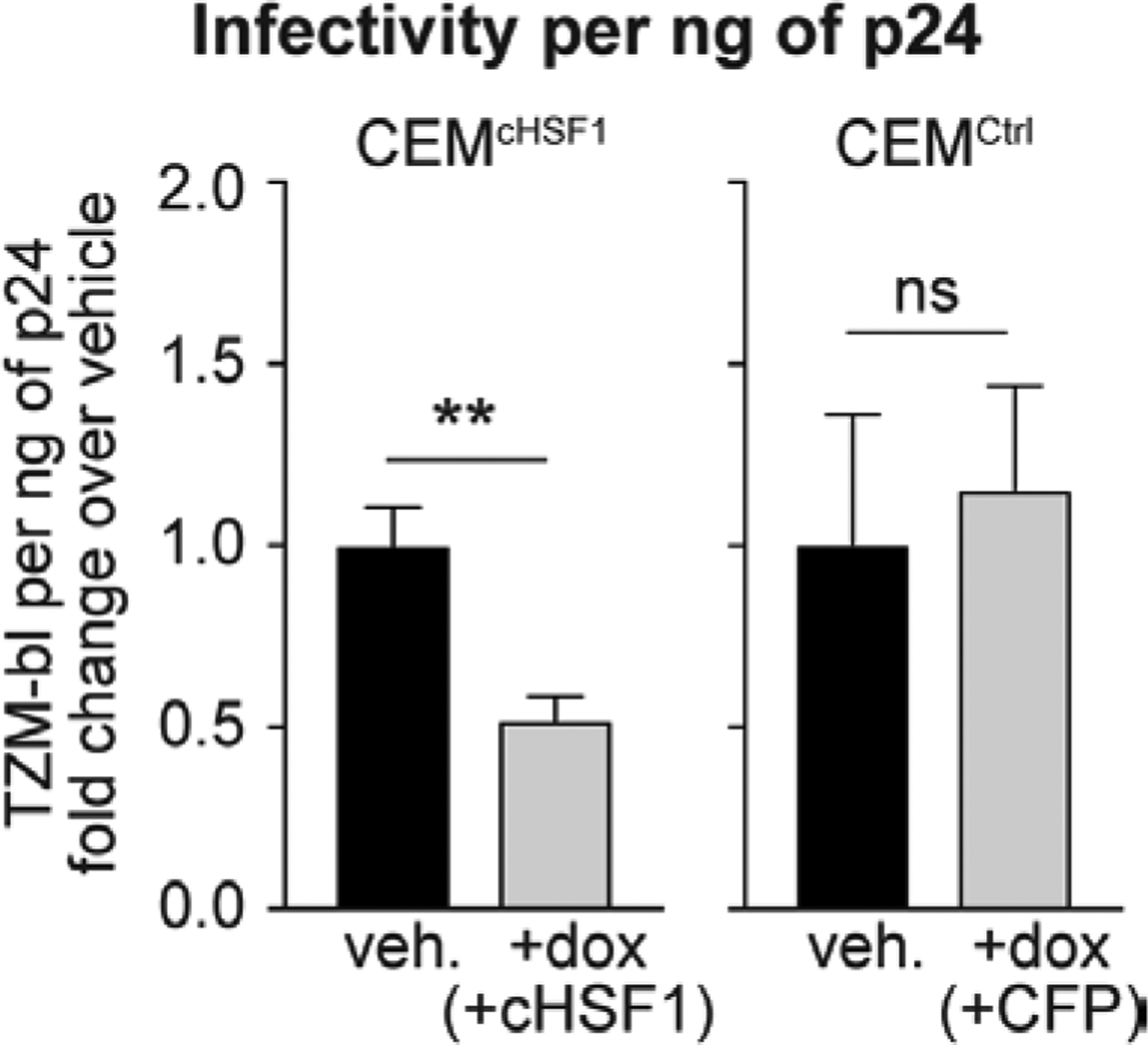
HSF1 reduces infectivity of newly produced virions. Fold-change in infectious TZM-bl titers per ng of p24 after 96 h of HIV-1 infection at an MOI of 0.04 in CEMcHSF1 and CEMCtrl cells treated with 1 μg/mL dox, relative to vehicle-treated cells. ** and ns correspond to p-values < 0.01 and not significant, respectively.
In summary, the use of a chemically controlled cHSF1 construct allowed us to investigate the direct consequences of HSF1 activation at physiologically relevant levels, eliminating the requirement for inducing global protein misfolding while also avoiding the off-target consequences of cHSF1 overexpression. We were also able to avoid the complications associated with transient overexpression of HSF1 or cHSF1,39, 42 including off-target gene induction, which convoluted prior studies. Using this approach, we demonstrated that stress-independent HSF1 activation restricts HIV-1 replication in CEM cells. When cHSF1 was activated, fewer total HIV-1 virions were produced and the proportion of infectious virions was also lowered. Moreover, cHSF1-mediated inhibition of HIV-1 replication persisted through three consecutive serial passages without detectable recovery of viral titers, suggesting that escape mechanisms are not readily available to the virus. The effects of cHSF1 activation were HSR-specific and not attributable to reductions in host cell health, off-target cHSF1 activity, or activation of protein misfolding stress responses in general.
The exact molecular mechanisms of HSF1-mediated restriction of HIV-1 replication remain an important subject for further study and are likely multifactorial. First, viral transcripts are known to be targeted to degradation by the HSF1-controlled host restriction factor ZAP, which has an HSF1-binding promoter and was transcriptionally upregulated in our system despite the absence of a general antiviral response induced by cHSF1. Second, cHSF1 activation reduces the infectivity of newly formed virions. This observation suggests that the remodeled host chaperone network promotes the formation of defective viral particles. While deciphering the origins of HSF1-mediated inhibition of HIV-1 replication and elucidating in vivo relevance of these findings requires future investigation, this work clearly implicates HSF1 as a host antiviral restriction factor for HIV-1 and motivates continued consideration of host HSR-targeted therapeutics to address retroviral infections.
Methods
Detailed protocols for the following procedures can be found in the Supporting Information: stable cell line construction, quantitative RT-PCR, HIV-1 infection, p24 assays, TZM-bl assays, CellTiter-Glo viability assays, RNA-Seq, GSEA, and statistical analyses.
Supplementary Material
Acknowledgements
This work was supported by the Smith Family Foundation Award for Excellence in Biomedical Research, the National Science Foundation (CAREER Award 1652390), the National Institutes of Health (Grants 1DP2GM119162 and 1R35GM136354) to M.D.S., the MIT Center for Environmental Health Sciences (P30-ES002109), and the Koch Institute (P30-CA14051). E.E.N. was supported by a UNCF-Merck Postdoctoral Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00166.
Complete experimental methods; selective induction of XBP1s and/or ATF6 target genes in CEMDAX cells; transcriptional profile of the HSF1-activated host environment; heat shock factor motif enrichment upon cHSF1 induction; induction of cHSF1 activating ZAP transcription; sequences of RT-PCR primers used (PDF)
RNA-Seq characterization of CEMcHSF1 cells: Differential expression analysis of the HSF1-activated environment (XLSX)
RNA-Seq characterization of CEMcHSF1 cells: Gene set enrichment analysis (XLSX)
Gene list for the “Defense Response to Virus” gene ontology group (XLSX)
The authors declare no competing financial interest.
References
- 1.World Health Organization (accessed June 3, 2019) Summary of the global HIV epidemic (2017), https://www.who.int/hiv/data/2017_summary-global-hiv-epidemic.png?ua=1.
- 2.Cihlar T; Fordyce M, Current status and prospects of HIV treatment. Curr. Opin. Virol 2016, 18, 50–56. [DOI] [PubMed] [Google Scholar]
- 3.Jean MJ; Fiches G; Hayashi T; Zhu J, Current strategies for elimination of HIV-1 latent reservoirs using chemical compounds targeting host and viral factors. AIDS Res. Hum. Retroviruses 2019, 35, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park RJ; Wang T; Koundakjian D; Hultquist JF; Lamothe-Molina P; Monel B; Schumann K; Yu H; Krupzcak KM; Garcia-Beltran W et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet 2017, 49, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brass AL; Dykxhoorn DM; Benita Y; Yan N; Engelman A; Xavier RJ; Lieberman J; Elledge SJ, Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [DOI] [PubMed] [Google Scholar]
- 6.Jäger S; Cimermancic P; Gulbahce N; Johnson JR; McGovern KE; Clarke SC; Shales M; Mercenne G; Pache L; Li K et al. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arhel N; Kirchhoff F, Host proteins involved in HIV infection: new therapeutic targets. Biochim. Biophys. Acta, Mol. Basis Dis 2010, 1802, 313–321. [DOI] [PubMed] [Google Scholar]
- 8.Aviner R; Frydman J, Proteostasis in viral infection: unfolding the complex virus–chaperone interplay. Cold Spring Harbor Perspect. Biol 2019, a034090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter K; Haslbeck M; Buchner J, The heat shock response: life on the verge of death. Mol. Cell 2010, 40, 253–266. [DOI] [PubMed] [Google Scholar]
- 10.Filone CM; Caballero IS; Dower K; Mendillo ML; Cowley GS; Santagata S; Rozelle DK; Yen J; Rubins KH; Hacohen N et al. The master regulator of the cellular stress response (HSF1) is critical for orthopoxvirus infection. PLoS Pathog 2014, 10, e1003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai T-T; Chen C-L; Tsai C-C; Lin C-F, Targeting heat shock factor 1 as an antiviral strategy against dengue virus replication in vitro and in vivo. Antiviral Res 2017, 145, 44–53. [DOI] [PubMed] [Google Scholar]
- 12.Pan X-Y; Zhao W; Zeng X-Y; Lin J; Li M-M; Shen X-T; Liu S-W, Heat shock factor 1 mediates latent HIV reactivation. Sci. Rep 2016, 6, 26294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F-W; Wu X-R; Liu W-J; Liao Y-J; Lin S; Zong Y-S; Zeng M-S; Zeng Y-X; Mai S-J; Xie D, Heat shock factor 1 upregulates transcription of Epstein–Barr Virus nuclear antigen 1 by binding to a heat shock element within the BamHI-Q promoter. Virology 2011, 421, 184–191. [DOI] [PubMed] [Google Scholar]
- 14.Geller R; Taguwa S; Frydman J, Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta, Mol. Cell Res 2012, 1823, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi P; Sloan B; Torbett BE; Stoddart CA, Heat shock protein 90AB1 and hyperthermia rescue infectivity of HIV with defective cores. Virology 2013, 436, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heaton NS; Moshkina N; Fenouil R; Gardner TJ; Aguirre S; Shah PS; Zhao N; Manganaro L; Hultquist JF; Noel J et al. Targeting viral proteostasis limits influenza virus, HIV, and dengue virus infection. Immunity 2016, 44, 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taguwa S; Maringer K; Li X; Bernal-Rubio D; Rauch JN; Gestwicki JE; Andino R; Fernandez-Sesma A; Frydman J, Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in flavivirus infection. Cell 2015, 163, 1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taguwa S; Yeh M-T; Rainbolt TK; Nayak A; Shao H; Gestwicki JE; Andino R; Frydman J, Zika virus dependence on host Hsp70 provides a protective strategy against infection and disease. Cell Rep 2019, 26, 906–920. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips AM; Gonzalez LO; Nekongo EE; Ponomarenko AI; McHugh SM; Butty VL; Levine SS; Lin Y-S; Mirny LA; Shoulders MD, Host proteostasis modulates influenza evolution. eLife 2017, 6, e28652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geller R; Pechmann S; Acevedo A; Andino R; Frydman J, Hsp90 shapes protein and RNA evolution to balance trade-offs between protein stability and aggregation. Nat. Commun 2018, 9, 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips AM; Doud MB; Gonzalez LO; Butty VL; Lin Y-S; Bloom JD; Shoulders MD, Enhanced ER proteostasis and temperature differentially impact the mutational tolerance of influenza hemagglutinin. eLife 2018, 7 e38795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips AM; Ponomarenko AI; Chen K; Ashenberg O; Miao J; McHugh SM; Butty VL; Whittaker CA; Moore CL; Bloom JD et al. Destabilized adaptive influenza variants critical for innate immune system escape are potentiated by host chaperones. PLoS Biol 2018, 16, e3000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kretz-Remy C; Munsch B; Arrigo A-P, NFκB-dependent transcriptional activation during heat shock recovery. Thermolability of the NFκB.IκB complex. J. Biol. Chem 2001, 276, 43723–43733. [DOI] [PubMed] [Google Scholar]
- 24.Rawat P; Mitra D, Cellular heat shock factor 1 positively regulates human immunodeficiency virus-1 gene expression and replication by two distinct pathways. Nucleic Acids Res 2011, 39, 5879–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roesch F; Meziane O; Kula A; Nisole S; Porrot F; Anderson I; Mammano F; Fassati A; Marcello A; Benkirane M et al. Hyperthermia stimulates HIV-1 replication. PLoS Pathog 2012, 8, e1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto K; Baba M; Gohnai K; Sato M; Shigeta S, Heat shock induces HIV-1 replication in chronically infected promyelocyte cell line OM10.1. Arch. Virol 1996, 141, 439–447. [DOI] [PubMed] [Google Scholar]
- 27.Ignatenko NA; Gerner EW, Regulation of the HIV1 long terminal repeat by mutant heat shock factor. Exp. Cell Res 2003, 288, 1–8. [DOI] [PubMed] [Google Scholar]
- 28.Pan X; Lin J; Zeng X; Li W; Wu W; Lu WZ; Liu J; Liu S, Heat shock factor 1 suppresses the HIV-induced inflammatory response by inhibiting nuclear factor-κB. Cell. Immunol 2018, 327, 26–35. [DOI] [PubMed] [Google Scholar]
- 29.Vozzolo L; Loh B; Gane PJ; Tribak M; Zhou L; Anderson I; Nyakatura E; Jenner RG; Selwood D; Fassati A Gyrase B inhibitor impairs HIV-1 replication by targeting HSP90 and the capsid protein. J. Biol. Chem 2010, 285, 39314–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson I; Low JS; Weston S; Weinberger M; Zhyvoloup A; Labokha AA; Corazza G; Kitson RA; Moody CJ; Marcello A et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E1528–E1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urano E; Morikawa Y; Komano J, Novel role of HSP40/DNAJ in the regulation of HIV-1 replication. JAIDS, J. Acquir. Immune Defic. Syndr 2013, 64, 154–162. [DOI] [PubMed] [Google Scholar]
- 32.Kumar M; Mitra D, Heat shock protein 40 is necessary for human immunodeficiency virus-1 Nef-mediated enhancement of viral gene expression and replication. J. Biol. Chem 2005, 280, 40041–50. [DOI] [PubMed] [Google Scholar]
- 33.Kumar M; Rawat P; Khan SZ; Dhamija N; Chaudhary P; Ravi DS; Mitra D, Reciprocal regulation of human immunodeficiency virus-1 gene expression and replication by heat shock proteins 40 and 70. J. Mol. Biol 2011, 410, 944–958. [DOI] [PubMed] [Google Scholar]
- 34.Agostini I; Popov S; Li J; Dubrovsky L; Hao T; Bukrinsky M, Heat-shock protein 70 can replace viral protein R of HIV-1 during nuclear import of the viral preintegration complex. Exp. Cell Res 2000, 259, 398–403. [DOI] [PubMed] [Google Scholar]
- 35.Iordanskiy S; Zhao Y; DiMarzio P; Agostini I; Dubrovsky L; Bukrinsky M, Heat-shock protein 70 exerts opposing effects on Vpr-dependent and Vpr-independent HIV-1 replication in macrophages. Blood 2004, 104, 1867–1872. [DOI] [PubMed] [Google Scholar]
- 36.Chaudhary P; Khan SZ; Rawat P; Augustine T; Raynes DA; Guerriero V; Mitra D, Hsp70 binding protein 1 (HspBP1) suppresses HIV-1 replication by inhibiting NF-κB mediated activation of viral gene expression. Nucleic Acids Res 2016, 44, 1613–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuo J; Baler R; Dahl G; Voellmy R, Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol. Cell. Biol 1994, 14, 7557–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahat DB; Salamanca HH; Duarte FM; Danko CG; Lis JT, Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol. Cell 2016, 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryno LM; Genereux JC; Naito T; Morimoto RI; Powers ET; Shoulders MD; Wiseman RL, Characterizing the altered cellular proteome induced by the stress-independent activation of heat shock factor 1. ACS Chem. Biol 2014, 9, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore CL; Dewal MB; Nekongo EE; Santiago S; Lu NB; Levine SS; Shoulders MD, Transportable, chemical genetic methodology for the small molecule-mediated inhibition of heat shock factor 1. ACS Chem. Biol 2016, 11, 200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shoulders MD; Ryno LM; Cooley CB; Kelly JW; Wiseman RL, Broadly applicable methodology for the rapid and dosable small molecule-mediated regulation of transcription factors in human cells. J. Am. Chem. Soc 2013, 135, 8129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebastian RM; Shoulders MD, Chemical biology framework to illuminate proteostasis. Annu. Rev. Biochem 2020, 89, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voellmy R, Dominant-positive and dominant-negative heat shock factors. Methods 2005, 35, 199–207. [DOI] [PubMed] [Google Scholar]
- 44.Del Razo LM; Quintanilla-Vega B; Brambila-Colombres E; Calderón-Aranda ES; Manno M; Albores A, Stress proteins induced by arsenic. Toxicol. Appl. Pharmacol 2001, 177, 132–148. [DOI] [PubMed] [Google Scholar]
- 45.Kimpton J; Emerman M, Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol 1992, 66, 2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong MY; DiChiara AS; Suen PH; Chen K; Doan N-D; Shoulders MD, Adapting secretory proteostasis and function through the unfolded protein response. Curr. Top. Microbiol. Immunol 2017, 414, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee A-H; Iwakoshi NN; Glimcher LH, XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol 2003, 23, 7448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwamoto M; Björklund T; Lundberg C; Kirik D; Wandless TJ, A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol 2010, 17, 981–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shoulders MD; Ryno LM; Genereux JC; Moresco JJ; Tu PG; Wu C; Yates JR; Su AI; Kelly JW; Wiseman RL, Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 2013, 3, 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen JJ; Genereux JC; Qu S; Hulleman JD; Shoulders MD; Wiseman RL, ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem. Biol 2014, 21, 1564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dewal MB; DiChiara AS; Antonopoulos A; Taylor RJ; Harmon CJ; Haslam SM; Dell A; Shoulders MD, XBP1s links the unfolded protein response to the molecular architecture of mature N-glycans. Chem. Biol 2015, 22, 1301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong MY; Chen K; Antonopoulos A; Kasper BT; Dewal MB; Taylor RJ; Whittaker CA; Hein PP; Dell A; Genereux JC; Haslam SM; Mahal LK; Shoulders MD, XBP1s activation can globally remodel N-glycan structure distribution patterns. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E10089–E10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wires ES; Henderson MJ; Yan X; Bäck S; Trychta KA; Lutrey MH; Harvey BK, Longitudinal monitoring of Gaussia and Nano luciferase activities to concurrently assess ER calcium homeostasis and ER stress in vivo. PLoS One 2017, 12, e0175481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kroeger H; Grimsey N; Paxman R; Chiang W-C; Plate L; Jones Y; Shaw PX; Trejo J; Tsang SH; Powers E; Kelly JW; Wiseman RL; Lin JH, The unfolded protein response regulator ATF6 promotes mesodermal differentiation. Sci. Signaling 2018, 11, eaan5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Todorova T; Bock FJ; Chang P, Poly (ADP-ribose) polymerase-13 and RNA regulation in immunity and cancer. Trends Mol. Med 2015, 21, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghimire D; Rai M; Gaur R, Novel host restriction factors implicated in HIV-1 replication. J. Gen. Virol 2018, 99, 435–446. [DOI] [PubMed] [Google Scholar]
- 58.Zhu Y; Chen G; Lv F; Wang X; Ji X; Xu Y; Sun J; Wu L; Zheng Y-T; Gao G, Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 15834–15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujimoto M; Takii R; Takaki E; Katiyar A; Nakato R; Shirahige K; Nakai A, The HSF1–PARP13–PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nat. Commun 2017, 8, 1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujimoto M; Takii R; Katiyar A; Srivastava P; Nakai A, Poly (ADP-ribose) polymerase 1 promotes the human heat shock response by facilitating heat shock transcription factor 1 binding to DNA. Mol. Cell. Biol 2018, 38, e00051–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gurer C; Cimarelli A; Luban J, Specific incorporation of heat shock protein 70 family members into primate lentiviral virions. J. Virol 2002, 76, 4666–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franke EK; Yuan HEH; Luban J, Specific incorporation of cyclophilin A into HIV-1 virions. Nature 1994, 372, 359–362. [DOI] [PubMed] [Google Scholar]
- 64.Bartz SR; Pauza CD; Ivanyi J; Jindal S; Welch WJ; Malkovsky M, An Hsp60 related protein is associated with purified HIV and SIV. J. Med. Primatol 1994, 23, 151–154. [DOI] [PubMed] [Google Scholar]
- 65.Matysiak J; Lesbats P; Mauro E; Lapaillerie D; Dupuy J-W; Lopez AP; Benleulmi MS; Calmels C; Andreola M-L; Ruff M et al. Modulation of chromatin structure by the FACT histone chaperone complex regulates HIV-1 integration. Retrovirology 2017, 14, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.