

Abstract
The fungal natural product Aspergillomarasmine A (AMA) has been identified as a non-competitive inhibitor of New Delhi Metallo-β-lactamase-1 (NDM-1) that inhibits via active site Zn(II) removal. The non-selective metal-chelating properties and the difficult synthesis and derivatization of AMA have hindered the development of this scaffold into a potent and selective inhibitor of NDM-1. Iminodiacetic acid (IDA) has been identified as the metal-binding pharmacophore (MBP) core of AMA that can be leveraged for inhibitor development. Herein, we report the utilization of IDA for the fragment-based drug discovery (FBDD) of NDM-1 inhibitors. IDA (IC50 = 122 μM) was developed into inhibitor 23f (IC50 = 8.6 μM, Ki, = 2.6 μM) and displayed the formation of a ternary complex with NDM-1, as evidenced by protein thermal shift and native state electrospray ionization mass spectrometry (ESI-MS) experiments. Combining mechanistic analysis in tandem with inhibitor derivatization, the utilization of IDA as an alternative AMA scaffold for NDM-1 inhibitor development is detailed.
Keywords: Metal-binding Pharmacophore (MBP), Aspergillomarasmine A (AMA), Iminodiacetic Acid (IDA), Metal Chelator, New Delhi Metallo-β-lactamase (NDM), antibiotic resistance
Graphical Abstract

Iminodiacetic acid (IDA) was identified as a novel lead fragment for New Delhi Metallo-β-lactamase (NDM) inhibitor development. Through a series of fragment-based drug design, synthesis, and mechanistic analysis, 23f was identified as a potent inhibitor. This inhibitor represents the potential to convert traditional metal chelators to one that displays the formation of a ternary complex. The IDA fragment and inhibitors reported provide a roadmap for future metallô-lactamase inhibitor development.
Introduction
A recent report published by the Centers for Disease Control and Prevention (CDC) estimated that antibiotic-resistant bacteria and fungi cause >2.8 million cases of infections in the United States each year, with >35,000 of those cases resulting in death.[1] Resistance mechanisms (including mutation of penicillinbinding proteins, production of efflux pumps, and expression of β-lactamases) evolved and employed by pathogens are a prime example of bacterial adaptability and pose an urgent threat to the public health.[2] The most valuable class of drugs for combating bacterial infections include β-lactam antibiotics. This class of antibiotics acts as a substrate analogue to obstruct peptidoglycan chain cross-linking in bacterial cell wall biosynthesis and accounts for ~65% of all injectable antibiotics prescribed in the United States.[3] However, the over-use of β-lactams has led to the evolution of β-lactamases, enzymes that hydrolyze the β-lactam ring to render the drug ineffective. Three classes of β-lactamases, Ambler class A, C, and (serin-β-lactamases, SBLs) utilize an active site serine residue for hydrolysis, while one class of β-lactamase, Ambler class B (metallo-β-lactamase, MBL) utilizes a metal center to initiate ring cleavage.[2a, 4] Merely two decades after the introduction of penicillin in the 1940s, the first observed MBL was reported.[5] Currently, there are >80 unique MBL families.[3] With their wide substrate profile (able to hydrolyze virtually all clinically used bicyclic p-lactam antibiotics), MBLs have risen to become one of the most problematic resistance mechanisms.[6] Detailed reviews on MBLs can be found elsewhere.[7]
Depending on protein sequence homology and number of Zn(II) ions in the catalytic site, MBLs are divided into three subclasses (B1, B2, and B3). The most prevalent members belong to subclass B1 and include IMiPenemase (IMP), Verona Integron-encoded Metallo-β-lactamase (VIM), and New Delhi Metallo-β-lactamase (NDM).[6, 8] NDM is the most recent member of the trio, with its genetic and biochemical characterization first reported in 2009 upon isolation from carbapenem-resistant Klebsiella pneumoniae.[9] The rapid spread of NDM is attributed to many factors, including the ability for blandm-gene bearing plasmids to undergo horizontal gene transfer between different species of microorganisms and to co-harbor genes that encode for other resistance factors.[9–10] In contrast to other MBLs (which are soluble periplasmic proteins), NDM is a lipoprotein that anchors to bacterial outer membrane and displays increased protein stability and secretion.[11] Additionally, NDM variants (>24 reported to date) have evolved to overcome metal scarcity and increased thermal stability.[11c, 12] The NDM active site bears two Zn(II) ions, with Zn1 ligated by H116, H118, H196, and a bridging hydroxide in a tetrahedral coordination geometry, and Zn2 ligated by D120, C221, H263, the bridging hydroxide, and an apical H2O in a trigonal bipyramidal coordination geometry (standard BBL numbering, Figure 1).[13] The binding pocket of NDM has a volume of 591 Å3, which is nearly 2-fold larger in comparison to that of IMP (303 Å3) and almost 4-fold larger compared to that of VIM (140 Å3).[14] This highly plastic and large cavity accommodates a wide range of antibiotic substrates and allows for the efficient hydrolysis of nearly all β-lactam antibiotics.[15]
Figure 1.

Scheme of the NDM active site and a proposed hydrolysis mechanism of the β-lactam antibiotic penicillin.
There are currently >500 distinct NDM inhibitors reported in literature (representative structures show in Figure 2).[16] An important class of compounds bear a sulfhydryl-motif (including d/l-captopril and bisthiazolidines) that act via a competitive inhibition mechanism by displacing the bridging hydroxide ion to form a μ-bridging species between the Zn(II) ions.[17] Another important class of inhibitors includes the cyclic boronates, which have been shown to successfully pan-inhibit SBLs and MBLs via a tetrahedral anionic transition state mimetic.[18] Notably, taniborbactam (VNRX-5133) is the only candidate to have advanced to the clinic (currently in phase III clinical trials).[18c]
Figure 2.

Representative inhibitors of NDM-1.
The last class includes compounds that bear metal-chelating motifs.[19] Of these, the fungal natural product Aspergillomarasmine A (AMA, IC50 = 4 – 7 μM, Figure 2), an aminopolycarboxylic acid, has gained attention due to its ability to restore meropenem activity in a mouse infected with NDM-1 expressing Klebsiella pneumoniae.[19e, 20] AMA inhibitor development has focused on modification of the carboxylic acid functional groups through removal or conversion to an ester motif, or exploration of related aminocarboxylic acid analogues.[20b, 21] AMA-1 and AMA-2 (Figure 2), where one of the carboxylate groups is replaced with a methyl substituent yielded weaker inhibition (IC50 = 22 and 94 μM, respectively) compared to that of AMA, validating the requirement of free carboxylic acids for enzyme inactivation and supporting the mechanism of action of AMA is via non-selective Zn(II) sequestration (similar to that of EDTA). The metal-chelating properties of AMA along with difficult synthesis and derivatization routes has hindered the development of this motif into NDM-1 inhibitors.[21a, 22] Structural comparison of AMA and EDTA reveals iminodiacetic acid (IDA) as a privileged scaffold that could be leveraged for NDM-1 inhibitor development (Figure 2, highlighted in bold). IDA is a strong tridentate metal chelator (via its O,N,O-donor atoms), as evidenced by its role in immobilized metal affinity chromatography (IMAC)[23] and its development for the sequestration of Zn(II) in IDA-modified human lysozyme (IDA-hLys) against Zn(II)-mediated Aβ-aggregation for the treatment of Alzheimer’s disease.[24] Utilization of IDA for inhibitor development allows for greater synthetic accessibility of derivatives to probe the NDM-1 active site and to develop inhibitors that form stable ternary complexes. In addition, IDA bears structural resemblance to the hydrolyzed antibiotic β-lactam ring, suggesting the potential for the development of transition-state analogue inhibitors. Notably, IDA is an aliphatic derivative of the previously investigated dipicolinic acid inhibitor,[19d] further justifying its use as a scaffold for novel NDM-1 inhibitor development (Figure 2, highlighted in bold).
Herein, we report the utilization of IDA as a novel metal-binding pharmacophore (MBP) for NDM-1 inhibitor development. Through fragment-based drug discovery (FBDD), the IDA MBP (IC50 = 122 μM) was developed into the lead inhibitor 23f (IC50 = 8.6 μM) against NDM-1. Protein thermal shift and native state electrospray ionization mass spectrometry (ESI-MS) experiments revealed 23f, and related derivatives, inhibits NDM-1 via the formation of a stable NDM-1:Zn(II):23f ternary complex. This work demonstrates the potential in the IDA scaffold for NDM-1 inhibitor development and provides a roadmap for future IDA derived inhibitors.
Results and Discussion
IDA MBP Design and Synthesis.
To verify IDA as a potential MBP lead for inhibitor design, a small library of MBP compounds bearing structural similarity to IDA was assembled and their inhibitory activity against NDM-1 was assessed (Table 1). This library included compounds that have at least one nitrogen atom and one carboxylate functional group for bidentate metal-binding (N,O-donor atoms). The N-donor atom in the MBPs were either a tertiary amine (1), secondary amine (IDA, 2 – 4), or aromatic amine (5 – 10). These MBPs were screened at a single concentration of 200 μM via an enzymatic assay which monitored the NDM-1 mediated hydrolysis of the substrate meropenem.[25] Evaluation of this library revealed IDA to be the only MBP with a secondary amine N-donor atom to yield significant inhibition activity (48%). MBPs 2 – 4, where the secondary amine is a part of the saturated ring, did not display any appreciable inhibition (≤8%). A methylated IDA derivative (1), was the most potent of this library, reaching 80% inhibition. Some MBPs bearing the aromatic amine motif showed some inhibitory activity, with 8 displaying the second highest inhibition value (57%). Data from this small set of MBPs suggest a preference for a tertiary or aromatic heterocycle N-donor atom. These findings verified the IDA MBP as a viable scaffold for NDM-1 inhibitor development. Based on these findings, MBP 1 was chosen for further investigation.
Table 1.
Percent Inhibition of IDA derived MBPs (at 200 μM) against NDM-1.[a]
 |
 |
Values are the average of triplicates experiments with standard deviations shown.
A second-generation of IDA derivatives (Table 2) were synthesized according to Scheme 1 – Scheme 3. Compounds in this sublibrary incorporated a benzyl substituent, as aromatic rings have previously been shown to form favorable hydrophobic interactions with the NDM β-hairpin loop.[16] Utilizing the concept of bioisosteric replacement, IDA derivatives where one carboxylic acid was modified (13a, 13b, 15, 17) were prepared to determine if both carboxylic acids were necessary for inhibition and if alternative MBPs could achieve increase potency. Compounds where the methyl- or benzyl-substituent (19a – 19d) was placed at the α-carbon were explored as well. The compounds were screened against at a single concentration of 250 μM against NDM-1. The methylated-IDA (1) remained the most potent of the series, exhibiting 90% inhibition at 250 μm. The benzylated-IDA (11) was the second most potent (65%). Interestingly, bioisosteres with a propionic acid motif (13a and 13b, which are most structurally similar to AMA) displayed a complete loss of activity. While the phosphate isostere (15) showed no inhibition against NDM-1, the less acidic tetrazole isostere (17) showed inhibition that was comparable to that of 11. Notably, there was no preference for R- or S- stereoisomers at the α-carbon position of IDA, as evident by similar inhibitory values (51 – 65%) displayed by derivatives 19a – 19d.
Table 2.
Percent Inhibition of IDA derivatives (at 250 μM) against NDM-1.[a]
 |
Values are the average of triplicates experiments with standard deviations shown.
Scheme 1.

Synthesis of 13a and 13b. Reagents and conditions: (a) t-butyl acrylate, TEA, EtOH, 65 °C, 16 h, 43 – 90%; (b) TFA:CH2Cl2, 25 °C, 16 h, ~99%.
Scheme 3.
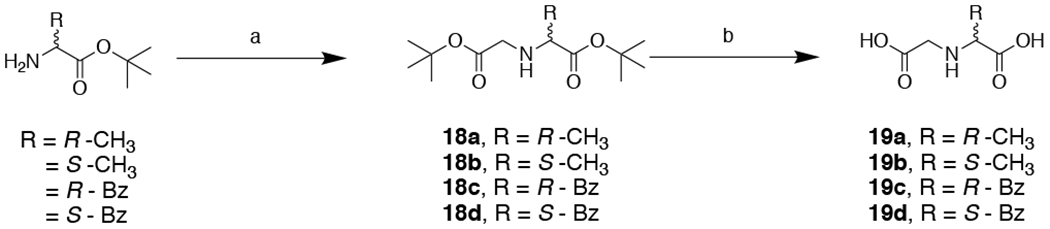
Synthesis of 19a – 19d. Reagents and conditions: (a) t-butyl 2-bromoacetate, TEA, DMF, 0 – 25 °C, 16 h, 40 – 54%; (b) TFA, CH2Cl2, 25 °C, 16 h, ~99%.
IDA Derivative Synthesis and Inhibitory Activity.
Compounds 1 and 11 were selected as scaffolds for inhibitor development, and additional IDA derivatives with various substituents were prepared. This sublibrary was prepared using a double substitution reaction of various primary amines with t-butyl 2-bromoacetate to yield compounds 21a – 21m (Scheme 4, Table 3). The sublibrary was screened at a single inhibitor concentration of 250 μM. The majority of the compounds in this sublibrary inhibited NDM-1 at an appreciable level (~60%); however, no clear SAR could be elucidated. Compared to the percent inhibition of 11 (65%, Table 2), modification via an ethyl-linker (21a) or a bi-phenyl substituent (21b and 21c) did not result in substantial inhibition improvements (56 – 76%). Notably, compounds bearing a phenyl- or benzyl-sulfonamide motif (21j and 21k) displayed a complete loss of activity (most likely due to the reduced basicity of the central nitrogen); however, substitution with a thiophene substituent (21l and 21m) restored activity by ~20%. In addition, 21i stood out as the most potent inhibitor of this sublibrary with almost complete inhibition against NDM-1 (~99%). When the heterocyclic oxygen is swapped out for a sulfur (21h), the inhibition activity is reduced to 64%, showing a preference for the furan substituent.
Scheme 4.

Synthesis of IDA inhibitors 21a – 21m and 23a – 23h. Reagents and conditions: (a) t-butyl 2-bromoacetate, KHCO3, THF, 25 °C, 16 h, 25 – 98%; (b) TFA, CH2Cl2, 25 °C, 16 h or (c) MeOH:THF:1M NaOH, 100 °C, 16 h, 29 – 99%.
Table 3.
Inhibitory activity of 21a – 21m (at 250 μM) against NDM-1.[a]
 |
Values are the average of triplicates experiments with standard deviations shown.
To further develop inhibitors against NDM-1 and investigate the difference in inhibitory activity between the furan and thiophene substituents, a second library bearing analogues of 21h and 21i were synthesized and evaluated (23a – 23h, Table 4). In general, all derivatives bearing a furan motif exhibited a lower IC50 value compared to that of the corresponding thiophene derivative. These results validate a preference for an oxygen heteroatom. While the 1,2-furan (23b, IC50 = 22 μM) displayed a lowered IC50 value compared to that of the 1,3-furan (21i, IC50 = 32 μM), the introduction of a methyl substituent at the 5-positon (23d, IC50 = 47 μM) resulted in poorer activity. Extension from a methyl-linker (23b) to an ethyl-linker (23f) resulted in a 2.5-fold fold improvement in inhibitory activity and resulted in the most potent inhibitor of this sublibrary (IC50 = 8.6 μM). It is predicted that the ethyl linker allows for the furan substituent to make more favorable interactions with the base of the L3 β-hairpin loop of NDM-1, as observed in the crystal structure of hydrolyzed antibiotic cefuroxime complexed with NDM-1 (PDB 5O2E);[24] however, further experiments are required to confirm the specific ligand-protein interactions. The corresponding thiophene derivatives displayed the same trends, albeit with poorer inhibition values. Due to the strong affinity IDA has for Zn(II) ions (Kd = 3.2×10−5 M)[26] and the analysis of inhibition mechanism (vide infra), we propose that 23f binds via coordination to the Zn(II) ions at the NDM-1 protein active site via a competitive mechanism of action. The Cheng-Prusoff relationship27 for competitive inhibitors enables calculation of Ki of 2.6±0.3 μM for 23f. Two methods, thermal shift and native state ESI-MS, were utilized to interrogated the mode of inhibition of selected inhibitors. Compared to alternative methods (such as NMR, crystallography, equilibrium dialysis, and others), thermal shift assay and native state ESI-MS utilize relatively lower protein and inhibitor concentrations, and are more amenable to high-throughput analysis, making them a suitable approach for initial mechanistic studies.
Table 4.
Inhibitory activity of IDA derivatives 23a – 23h against NDM-1.[a]
 |
Values are the average of triplicate experiments with fitting errors shown.
Protein Thermal Shift Assay.
Protein thermal shift assay detects ligand-induced protein stabilization, and has emerged as a valuable tool for hit-identification and validation methodology in drug discovery.[28] Herein, we utilize this general method to validate IDA derivatives as inhibitors of NDM-1 and evaluate their propensity to remove Zn(II) from the active site of NDM-1. In this assay, a fluorescent dye is utilized to monitor the difference in the unfolding temperature of the native protein versus the inhibitor-bound protein. The inhibitor-bound protein generally has greater protein stability and increases the melting temperature (positive ∆Tm, as observed with L-captopril, Table 5).[29] In contrast, the removal of Zn(II) has been observed to destabilize the protein and results in a negative ∆Tm (as seen with DPA).[12b] It is important to note that while reported thermal shift data have revealed a good correlation between the observed IC50 value and ∆Tm,[30] this correlation has not been observed for inhibitors of NDM-1.[29, 31] In the case of the compounds tested here, ∆Tm did not correlate with IC50 values. All tested compounds displayed a range of positive ∆Tm values, with IDA, 21h, 23c, and 23h yielding ∆Tm on par with, or better than, that of L-captopril. Although no correlation was observed, the small, positive ∆Tm shifts exhibited by all derivatives represents the absence of Zn(II)-chelation and is suggestive evidence for the formation of ternary complexes.
Table 5.
Protein thermal shift of selected compounds against NDM-1.[a]
| Compound | ∆Tm (°C) | Compound | ∆Tm (°C) |
|---|---|---|---|
| l-Captopril | 4.61±0.07 | DPA | −14.5±0.2 |
| IDA | 4.39±0.04 | 1 | 1.9±0.2 |
| 21h | 4.43±0.07 | 21i | 3.4±0.3 |
| 23a | 3.1±0.1 | 23b | 1.6±0.4 |
| 23c | 5.2±0.1 | 23d | 3.4±0.2 |
| 23e | 1.7±0.2 | 23f | 1.5±0.1 |
| 23g | 3.0±0.3 | 23h | 4.2±0.2 |
Values are the average of eight replicates with standard deviations shown. Native NDM-1 was observed to melt at TM = 55.95±0.06 °C.
Native State Electrospray Ionization Mass Spectrometry.
Native state ESI-MS was used to further investigate the formation of an NDM-1 :Zn(II):inhibitor ternary complex of derivatives 1 and 23c – 23f against NDM-1 and VIM-2. This method allows for the rapid determination of the changes in NDM-1:Zn(II):inhibitor stoichiometry.[32]. The major advantage of ESI-MS is in its ability to analyze for the formation of a ternary complex using near physiological concentrations of enzyme and inhibitor. Briefly, NDM-1 and VIM-2 (at 10 μM) were incubated for approximately 5 min with each inhibitor (50 μM) prior to analysis. The control spectra of native NDM-1 revealed a dominant +9 peak at 2,822 m/z, corresponding to the mass of di-Zn(II) NDM-1 (25,385 Da, Table 6, Figure S1). In contrast, incubation of NDM-1 with DPA, a metal chelator, resulted in +9 peaks at 2,807 m/z, 2,814 m/z, and 2,822 m/z, corresponding to the presence of apo-, mono-Zn(II) and di-Zn(II) NDM-1, respectively. These results were similar to the observed spectra of NDM-1 with EDTA, which showed a dominant peak corresponding to metal free NDM-1 (unpublished data). It is important to note that the current native ESI-MS experiment procedures do not generate quantitative results. Higher relative peak intensities (dominant peaks) do not indicate higher concentrations of the solution species, but rather the species that is best ionized by the mass analyzer.[33]
Table 6.
Summary of the native ESI-MS experimental results for NDM-1.[a]
| Sample | NDM-1 + Inhibitor Complex Mass (Da) | Peak Charge (+) | Predicted Peak (m/z) | Observed Peak (m/z) | Complex |
|---|---|---|---|---|---|
| NDM-1 | 25,385 | 9 | 2,821 | 2,822 | 2Zn: NDM-1 |
| 25,385 | 10 | 2,539 | 2,540 | 2Zn: NDM-1 | |
| NDM-1 + DPA | 25,255 | 9 | 2,807 | 2,807 | 0Zn: NDM-1 |
| 25,320 | 9 | 2,814 | 2,814 | 1Zn: NDM-1 | |
| 25,385 | 9 | 2,821 | 2,821 | 2Zn: NDM-1 | |
| NDM-1 + 1 | 25,532 | 9 | 2,838 | 2,837 | 2Zn: NDM-1:1 |
| NDM-1 + 23c | 25,628 | 9 | 2,848 | 2,850 | 2Zn: NDM-1:23c |
| 8 | 3,204 | 3,210 | 2Zn: NDM-1:23c | ||
| NDM-1 + 23d | 25,612 | 9 | 2,847 | 2,846 | 2Zn: NDM-1:23d |
| 8 | 3,202 | 3,202 | 2Zn: NDM-1:23d | ||
| NDM-1 + 23e | 25,628 | 9 | 2,848 | 2,847 | 2Zn: NDM-1:23e |
| 8 | 3,204 | 3,205 | 2Zn: NDM-1:23e | ||
| NDM-1 + 23f | 25,612 | 9 | 2,847 | 2,848 | 2Zn: NDM-1:23f |
| 8 | 3,202 | 3,202 | 2Zn: NDM-1:23f |
Percent error was calculated by subtracting the expected peak value from the actual peak value, dividing by the expected peak value and multiplying by 100 and all observed to be <0.2%.
The spectra of NDM-1 incubated with inhibitors 1 and 23c – 23f all showed the presence of ternary complexes, with the predicted and observed peaks summarized in Table 6 (Figure S1). In all of these experiments, in addition to the dominant di-Zn(II) NDM-1 peak, an additional peak corresponding to the mass of di-Zn(II) NDM-1 plus inhibitor was observed. Protein incubation with derivative 1 revealed a less dominant di-Zn(II) NDM-1:1 +9 peak at 2,837 m/z (25,532 Da). Incubation of protein with 23c yielded +9 and +8 peaks at 2,850 m/z and 3,210 m/z, respectively, corresponding to the di-Zn(II) NDM-1:23c complex (25,628 Da). Similarly, incubation of protein with 23d yielded +9 and +8 peaks at 2,846 m/z and 3,202 m/z, corresponding to the di-Zn(II) NDM-1:23d complex. Inhibitor 23e produced a significantly less intense +9 and +8 peaks at 2,847 m/z and 3,205 m/z, corresponding to the di-Zn(II) NDM-1:23e complex (25,628 Da). Lastly, incubation of NDM-1 with lead inhibitor 23f revealed a less intense +9 peak at 2,848 m/z and a more intense +8 peak at 3,202 m/z, both of which correspond to the mass of di-Zn(II) NDM-1:23f (25,612 Da, Figure 3).
Figure 3.
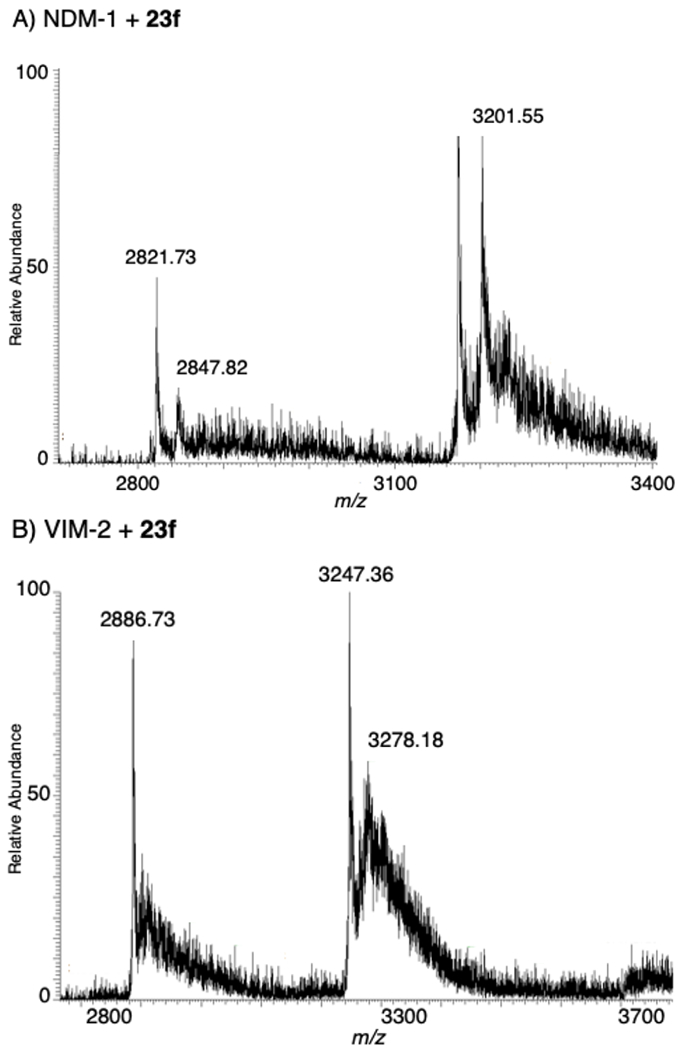
Native state ESI-MS of lead inhibitor 23f with: NDM-1 (top) and VIM-2 (bottom).
Previous work has shown that different mechanisms of inhibition can be observed for the same inhibitors against varying MBLs (unpublished data). To verify that the IDA inhibitors are able to form ternary complexes with other MBLs and have the potential to be developed into pan-MBL inhibitors, additional ESI-MS experiments were performed for inhibitors 1 and 23c – 23f with VIM-2. Control spectra of VIM-2 revealed dominant +9 and +8 peaks at 2,886 m/z and 3,247 m/z, respectively, corresponding to the mass of di-Zn(II) VIM-2 (25,972 Da, Table 7, Figure S2). VIM-2 incubated with DPA revealed dominant +9 and +8 peaks at 2,891 m/z and 3,253 m/z, respectively, corresponding most closely with the mass of apo-VIM-2 with 1 equivalent of DPA bound (26,010 Da).
Table 7.
Summary of the native ESI-MS experimental results for VIM-2.[a]
| Sample | NDM-1 + Inhibitor Complex Mass (Da) | Peak Charge (+) | Predicted Peak (m/z) | Observe d Peak (m/z) | Complex |
|---|---|---|---|---|---|
| VIM-2 | 25,972 | 9 | 2,887 | 2,886 | 2Zn: VIM-2 |
| 25,972 | 8 | 3,247 | 3,247 | 2Zn: VIM-2 | |
| VIM-2 + DPA | 26,010 | 9 | 2,891 | 2,891 | 0Zn: VIM-2:DPA |
| 26,010 | 8 | 3,252 | 3,253 | 0Zn: VIM-2:DPA | |
| VIM-2 + 1 | 26,119 | 9 | 2,903 | 2,902 | 2Zn: VIM-2:1 |
| VIM-2 + 23c | 26,215 | 8 | 3,278 | 3,277 | 2Zn: VIM-2:23c |
| VIM-2 + 23d | 26,199 | 8 | 3,276 | 3,276 | 2Zn: VIM-2:23d |
| VIM-2 + 23e | 26,215 | 8 | 3,278 | 3,277 | 2Zn: VIM-2:23e |
| VIM-2 + 23f | 26,199 | 8 | 3,276 | 3,278 | 2Zn: VIM-2:23f |
Percent error was calculated by subtracting the expected peak value from the actual peak value, dividing by the expected peak value and multiplying by 100 and all observed to be <0.07%.
Similar to the previously reported NDM-1:inhibitor complexes, spectra of VIM-2 incubated with inhibitors 1 and 23c – 23f all revealed the presence of ternary complexes. In addition to the dominant di-Zn(II) VIM-2 peak, additional di-Zn(II) VIM-2:inhibitor peaks were observed. The predicted and observed peaks are summarized in Table 7 (Figure S2). Incubation of VIM-2 with 1 revealed a less dominant +9 peak at 2,902 m/z, corresponding to di-Zn(II) VIM-2:1 complex (26,119 Da). The native MS of VIM-2 incubated with inhibitors 23c – 23f displayed similar secondary peaks at 3,277 m/z (26,215 Da), 3,276 m/z (26,199 Da), 3,277 m/z (26,215 Da), and 3,278 m/z (26,199 Da) representing the formation of the di-Zn(II) VIM-2:23c, di-Zn(II) VIM-2:23d, di-Zn(II) VIM-2:23e, and di-Zn(II) VIM-2:23f (Figure 3) ternary complex, respectively. This data is evident that the representative IDA inhibitors form ternary complexes with VIM-2, and represents a promising scaffold for future development against other MBLs.
Conclusion
Since the discovery of AMA as an effective inhibitor against NDM-1 capable of restoring the efficacy of antibiotic meropenem in mouse models, the synthesis and development of this compound into a more potent and selective inhibitor have been of interest.[19e, 22a, 22b] AMA, similar to EDTA, is a non-competitive inhibitor that deactivates NDM-1 via active site Zn(II) metal sequestration.[20a] Inhibitor development of AMA through modification of the carboxylic acid functional groups has been unsuccessful, as that motif is necessary for metal-chelation.[21a] Herein, we report the FBDD of IDA as a novel MBP for NDM-1 inhibitor development. IDA is a simplified analogue of AMA and EDTA, allowing for more facile inhibitor derivatization to probe the NDM-1 active site pocket. Reducing the number of carboxylates should also reduce the affinity of these compounds for free Zn(II) ions, thereby reducing their metal-stripping propensity. From a preliminary screen of a small library of MBPs, IDA and 1 were verified as novel hits for inhibitor development. Upon rounds of library design, synthesis, and mechanistic analysis IDA (IC50 = 122 μM) was developed into inhibitor 23f (IC50 = 8.6 μM). To study the mode of inhibition, protein thermal shift and native state ESI-MS were utilized. Both experiments revealed 23f and related derivatives inhibited NDM-1 via the formation of stable ternary complexes. Additional studies are currently underway to elucidate the precise protein-inhibitor binding interactions; however, similarities are observed with optimized dipicolinic acid derivatives.[19d] Each scaffold is optimized through the addition of a central hydrophobic substituent that includes a hydrogen-bond partner that appears to require precise positioning, presumably reflecting the binding interactions made with the beta-hairpin loop neighboring the di-nuclear Zn(II) ion site of NDM-1. While lead compound 23f displayed an inhibitory value similar to that of AMA (IC50 = 4 – 9 μM), rational inhibitor design integrated with detailed mechanistic studies has allowed for the development of an AMA-inspired alternative that displays the formation of a NDM-1:Zn(II):inhibitor ternary complex with a Ki of 2.6 μM. This work represents the benefit of performing mechanistic analysis hand-in-hand with inhibitor derivation for the development of inhibitors with a mode of inhibition more suitable for drug development. By utilizing a novel IDA MBP scaffold, traditional metal chelators (such as AMA and EDTA) not viewed as candidates for inhibitor development can be elaborated into potent inhibitors that form favorable ternary complexes. Our findings provide a path for the development of IDA-based inhibitors against NDM-1 and other clinically relevant MBLs. Upon the development of advance inhibitors with greater potency and selectivity, detailed spectroscopy and microbiology studies can be performed to further validate the mechanism of action.
Experimental Section
Inhibitors 1 – 11, IDA, reagents, and solvents were obtained from commercial sources and used without further purification. All reactions, unless otherwise stated, were performed at room temperature under a nitrogen atmosphere. Flash column chromatography was performed using a Teledyne ISCO CombiFlash Rf system using hexanes, ethyl acetate, dichloromethane, and methanol as eluents with prepacked silica cartridges. Reverse phase column chromatography was performed on the same instrument using methanol and water (w/ 0.1% formic acid) as eluents with high-performance Gold C18 columns. Column separation was monitored via Teledyne ISCO RF+ Purlon ESI-MS. 1H and 13C NMR spectra were recorded at ambient temperature on a 400 Varian Mercury Plus or 500 Varian VX NMR instrument located in the Department of Chemistry and Biochemistry at the University of California, San Diego. Mass spectra were obtained at the Molecular Mass Spectrometry Facility (MMSF) in the Department of Chemistry and Biochemistry at the University of California, San Diego. The purity of all compounds used in assays was determined to be ≥95% by high-performance liquid chromatography. Enzymatic assays were performed via monitoring the hydrolysis of substrate meropenem on Synergy H4 Hybrid Microplate Reader using 96-well UV-transparent microplates #3635 (Corning) according to previously published procedures.[25] Thermal shift assays were performed on QuantStudio 3 real-time PCR machines (Applied Biosystems) using 96-well 0.2 mL optical MicroAmp thermocycler plates and SYPRO orange Thermal Shift dye (ThermoFisher). Native state ESI-MS experiments were performed on a LTQ Orbitrap XL hybrid ion trap-orbitrap mass spectrometer (ThermoScientific).
Synthesis
tert-Butyl 3-((2-(tert-butoxy)-2-oxoethyl)(methyl)amino)propanoate (12a). Clear colorless oil, yield: 90% (676 mg, 2.47 mmol).
3-((Carboxymethyl)(methyl)amino)propanoic acid (13a). White solid, quantitative yield (102 mg, 0.40 mmol).
tert-Butyl 3-(benzyl(2-(tert-butoxy)-2-oxoethyl)amino)propanoate (12b). Clear colorless oil, yield: 43% (239 mg, 0.67 mmol).
3-(Benzyl(carboxymethyl)amino)propanoic acid (13b). White solid, quantitative yield (168 mg, 0.47 mmol).
Ethyl N-benzyl-N-(2-(diethoxyphosphoryl)ethyl)glycinate (14). Clear colorless oil, yield: 40% (150 mg, 0.42 mmol).
N-Benzyl-N-(2-phosphonoethyl)glycine (15). White solid, yield: 99% (114 mg, 0.42 mmol).
Ethyl N-benzyl-N-(cyanomethyl)glycinate (16). Clear colorless oil, yield: 74% (447 mg, 1.92 mmol).
N-((1H-tetrazol-5-yl)methyl)-N-benzylglycine (17). Yellow solid, yield: 18% (40.0 mg, 0.16 mmol).
General procedures for the synthesis of 19a – 19d
To a solution of the corresponding amine (1.1 equivalents) and TEA (2 equivalents) in DMF (10 mL) was added t-butyl 2-bromoacetate (1 equivalent) dropwise at 0°C. The reaction was allowed to warm to 25 °C and stirred for additional 16 h. After completion of the reaction, the salts were removed via vacuum filtration. The collected filtrate was concentrated in vacuo and the residue was purified via flash column chromatograph to afford the desired intermediates 18a – 18d. Intermediates were dissolved in TFA:CH2Cl2 (4:1 mL) and the reaction mixture was stirred at 25 °C for 16 h. The excess TFA removed via coevaporation with copious amounts of MeOH under reduced pressure. The product was purified via reverse phase column chromatography using MeOH in H2O (w/ 0.1% formic acid) as eluent to afford the title compounds 19a – 19d.
tert-Butyl (2-(tert-butoxy)-2-oxoethyl)-D-alaninate (18a). Clear oil, yield: 40% (175 mg, 0.68 mmol).
(Carboxymethyl)-D-alanine (19a). Clear oil, yield: 99% (93 mg, 0.63 mmol).
tert-Butyl (2-(tert-butoxy)-2-oxoethyl)-L-alaninate (18b). Clear oil, yield: 40% (176 mg, 0.68 mmol).
(Carboxymethyl)-L-alanine (19b). White solid, yield: 99% (72 mg, 0.49 mmol).
tert-Butyl (2-(tert-butoxy)-2-oxoethyl)-D-phenylalaninate (18c). Clear oil, yield: 52% (296 mg, 0.88 mmol).
(Carboxymethyl)-D-phenylalanine (19c). White solid, yield: 98% (116 mg, 0.52 mmol).
tert-Butyl (2-(tert-butoxy)-2-oxoethyl)-L-phenylalaninate (18d). Clear oil, yield: 54% (308 mg, 0.92 mmol).
(Carboxymethyl)-L-phenylalanine (19d). White solid, yield: 98% (110 mg, 0.49 mmol).
General procedures for the synthesis of 21a – 21m and 23a – 23h
The synthesis of compounds 21a – 21m and 23a – 23h were adapted from literature reported procedures.[34] To a solution of the corresponding amine (1 equivalent) and KHCO3 (4 equivalents) in THF or DMF (10 mL) was added t-butyl 2-bromoacetate (2.25 equivalents), and the reaction was stirred at 25 °C for 16 h. After completion of the reaction, as indicated by TLC, the salts were removed via vacuum filtration. The collected filtrate was concentrated in vacuo and the residue was purified via flash column chromatograph using hexane/ethyl acetate as eluent to afford the desired intermediates 20a – 20m and 22a – 22h. Compounds 21a – 21m and 23a – 23h were obtained through the following deprotection procedures: A) Intermediate was dissolved in a mixture of TFA:CH2CI3 (4:1 mL) and the reaction was stirred at 25 °C for 16 h. The excess TFA was removed under reduced pressure and co-evaporated with copious amounts of MeOH. The acid product was purified via reverse phase column chromatography with MeOH and H2O (w/ 0.1% formic acid) as eluent to afford the desired products; or B) Intermediate was dissolved in 1M NaOH:THF:MeOH (3:1:1 mL) and the reaction mixture was stirred at 65 °C for 16 h. THF and MeOH was removed under reduced pressure and the aqueous solution was acidified to pH 5 with 4M HCl. The precipitate was collected via vacuum filtration.
Di-tert-butyl 2,2’-(phenethylazanediyl)diacetate (20a). Viscous clear colorless oil, yield: 84% (708 mg, 2.03 mmol).
2,2’-(Phenethylazanediyl)diacetic acid (21a). Deprotection procedure A. White solid, yield: 99% (104 mg, 0.44 mmol).
Di-tert-butyl 2,2’-(([1,1’-biphenyl]-4-ylmethyl)azanediyl)diacetate (20b). White fluffy solid, yield: 69% (680 mg, 1.65 mmol).
2,2’-(([1,1’-Biphenyl]-4-ylmethyl)azanediyl)diacetic acid (21b). Deprotection procedure A. White solid, yield: 99% (72 mg, 0.24 mmol).
Di-tert-butyl 2,2’-(([1,1’-biphenyl]-3-ylmethyl)azanediyl)diacetate (20c). Clear colorless oil, yield: 98% (967 mg, 2.35 mmol).
2,2’-(([1,1’-Biphenyl]-3-ylmethyl)azanediyl)diacetic acid (21c). Deprotection procedure A. White solid, yield: 98% (141 mg, 0.47 mmol).
Di-tert-butyl 2,2’-((4-hydroxybenzyl)azanediyl)diacetate (20d). White solid, yield: 63% (530 mg, 1.51 mmol).
2,2’-((4-Hydroxybenzyl)azanediyl)diacetic acid (21d). Deprotection procedure A. White solid, yield: 99% (105 mg, 0.44 mmol).
Di-tert-butyl 2,2’-((4-chlorobenzyl)azanediyl)diacetate (20e). White crystalline solid, yield: 82% (727 mg, 1.97 mmol).
2,2’-((4-Chlorobenzyl)azanediyl)diacetic acid (21e). Deprotection procedure A. White solid, yield: 98% (120 mg, 0.48 mmol).
Di-tert-butyl 2,2’-((4-cyanobenzyl)azanediyl)diacetate (20f). White crystalline solid, yield: 75% (1.30 g, 3.60 mmol).
2,2’-((4-Cyanobenzyl)azanediyl)diacetic acid (21f). White solid, yield: 94% (67 mg, 0.27 mmol).
Di-tert-butyl 2,2’-((4-(1H-tetrazol-5-yl)benzyl)azanediyl)diacetate (20g). Pale yellow solid, yield: 46% (130 mg, 0.32 mmol).
2,2’-((4-(1H-Tetrazol-5-yl)benzyl)azanediyl)diacetic acid (21g). Deprotection procedure B. Beige solid, yield: 54% (50 mg, 0.17 mmol).
Di-tert-butyl 2,2’-((thiophen-3-ylmethyl)azanediyl)diacetate (20h). Yellow oil, yield: 62% (506 mg, 1.48 mmol).
2,2’-((Thiophen-3-ylmethyl)azanediyl)diacetic acid (21h). Deprotection procedure A. White solid, yield: 80% (118 mg, 0.52 mmol).
Di-tert-butyl 2,2’-((furan-3-ylmethyl)azanediyl)diacetate (20i). Clear colorless oil, yield: 29% (230 mg, 0.71 mmol).
2,2’-((Furan-3-ylmethyl)azanediyl)diacetic acid (21i). Deprotection procedure A. White solid, yield: 99% (179 mg, 0.84 mmol).
Di-tert-butyl 2,2’-((phenylsulfonyl)azanediyl)diacetate (20j). White crystalline solid, yield: 70% (273 mg, 0.71 mmol).
2,2’-((Phenylsulfonyl)azanediyl)diacetic acid (21j). Deprotection Procedure A. White solid, yield: 100% (115 mg, 0.42 mmol).
Di-tert-butyl 2,2’-((benzylsulfonyl)azanediyl)diacetate (20k). White solid, yield: 76% (310 mg, 0.78 mmol).
2,2’-((Benzylsulfonyl)azanediyl)diacetic acid (21k). Deprotection Procedure A. White solid, yield: 99% (90 mg, 0.31 mmol).
Di-tert-butyl 2,2’-((thiophen-2-ylsulfonyl)azanediyl)diacetate (20l). White crystalline solid, yield: 77% (308 mg, 0.79 mmol).
2,2’-((Thiophen-2-ylsulfonyl)azanediyl)diacetic acid (21l). Deprotection Procedure A. Off-white solid, yield: 90% (105 mg, 0.38 mmol).
Di-tert-butyl 2,2’-((benzo[b]thiophen-2-ylsulfonyl)azanediyl)diacetate (20m). Off-white crystalline solid, yield: 70% (312 mg, 0.71 mmol).
2,2’-((Benzo[b]thiophen-2-ylsulfonyl)azanediyl)diacetic acid (21m). Deprotection Procedure A. Yellow solid, yield: 99% (101 mg, 0.31 mmol).
Di-tert-butyl 2,2’-((thiophen-2-ylmethyl)azanediyl)diacetate (22a). Clear colorless oil, yield: 87% (717 mg, 2.10 mmol).
2,2’-((Thiophen-2-ylmethyl)azanediyl)diacetic acid (23a). Deprotection Procedure A. White solid, yield: 70% (130 mg, 0.57 mmol).
Di-tert-butyl 2,2’-((furan-2-ylmethyl)azanediyl)diacetate (22b). Yellow oil, yield: 82% (646 mg, 1.99 mmol).
2,2’-((Furan-2-ylmethyl)azanediyl)diacetic acid (23b). Deprotection Procedure A. Yellow oil, yield: 99% (160 mg, 0.75 mmol).
Di-tert-butyl 2,2’-(((5-methylthiophen-2-yl)methyl)azanediyl)diacetate (22c). Clear colorless oil, yield: 82% (700 mg, 1.97 mmol).
2,2’-(((5-Methylthiophen-2-yl)methyl)azanediyl)diacetic acid (23c). Deprotection Procedure A. Yellow solid, yield: 97% (113 mg, 0.46 mmol).
Di-tert-butyl 2,2’-(((5-methylfuran-2-yl)methyl)azanediyl)diacetate (22d). Clear colorless oil, yield: 64% (522 mg, 1.54 mmol).
2,2’-(((5-Methylfuran-2-yl)methyl)azanediyl)diacetic acid (23d). Deprotection Procedure B. Yellow solid, yield: 53% (70 mg, 0.31 mmol).
Di-tert-butyl 2,2’-((2-(thiophen-2-yl)ethyl)azanediyl)diacetate (22e). Light yellow oil, yield: 85% (726 mg, 2.04 mmol).
2,2’-((2-(Thiophen-2-yl)ethyl)azanediyl)diacetic acid (23e). Deprotection Procedure A. White solid, yield: 75% (109 mg, 0.45 mmol).
Di-tert-butyl 2,2’-((2-(furan-2-yl)ethyl)azanediyl)diacetate (22f). Yellow oil, yield: 84% (683 mg, 2.01 mmol).
2,2’-((2-(Furan-2-yl)ethyl)azanediyl)diacetic acid (23f). Deprotection Procedure A. Yellow solid, yield: 33% (24 mg, 0.11 mmol).
Di-tert-butyl 2,2’-(((5-bromothiophen-2-yl)methyl)azanediyl)diacetate (22g). Yellow crystalline solid, yield: 90% (908 mg, 2.16 mmol).
2,2’-(((5-Bromothiophen-2-yl)methyl)azanediyl)diacetic acid (23g). Deprotection Procedure A. White crystalline solid, yield: 98% (108 mg, 350 mmol).
Di-tert-butyl 2,2’-(((5-chlorothiophen-2-yl)methyl)azanediyl)diacetate (22h). Yellow solid, yield: 80% (510 mg, 1.36 mmol).
2,2’-(((5-Chlorothiophen-2-yl)methyl)azanediyl)diacetic acid (23h). Deprotection Procedure A. Dark yellow solid, yield: 98% (107 mg, 0.41 mmol).
Inhibition screening and IC50 determination
A soluble truncation of NDM-1 was over-expressed and purified as described previously.[35] IC50 values were determined using 11 concentrations of compound that span the IC50 value, and were assayed using meropenem as described previously[25] except that total assay volumes were increased to 200 μL. Final DMSO concentrations (derived from compound stock solutions) were 1% (v/v). For initial screening of compounds, the percent inhibition at 200 μM and 250 μM for each compound was determined using a similar procedure. The NDM-1 catalyzed hydrolysis rate in the absence of added inhibitor (adjusted for constant DMSO concentration) was set at 100% activity (0% inhibition), and the relative rates determined in the presence of inhibitors were used to calculate percent inhibition with respect to that control (e.g. 90% activity is reported as 10% inhibition). Briefly, each compound (357 μM) was incubated with NDM-1 (3.6 nM) for 20 min at 25 °C and diluted upon addition of the meropenem substrate to initiate the reaction. Final concentrations: NDM-1 (2.5 nM), compound (200 μM and 250 μM), meropenem (180 μM), CHAPS (2 mM), HEPES (50 mM), DMSO (0.5 %) at pH 7. Assays were completed as described for the IC50 determinations above.
Thermal Shift Assay
To each well of a 96-well 0.2 mL optical MicroAmp (ThermoFisher) thermocycler plate was added 9.5 μL of buffer (50 mM HEPES pH 7.5), 4 μL of NDM-1 in buffer (25 μM), 4 μL of inhibitor in buffer (1 mM), and 2.5 μL of SYPRO orange Thermal Shift dye (ThermoFisher) in buffer. Each well contained a final concentration of 5 μM NDM-1 and 200 μM inhibitor. Thermocycler plate wells were sealed prior to analysis, and the plate was then heated in a thermocycler from 25 to 99 °C at a ramp rate of 0.05 °C/sec. Each thermal shift measurement was taken in eight replicates. Fluorescence was read using the ROX filter channel (λx = 580 nm; λem = 623 nm), and the TM was determined by plotting the first derivative of the fluorescence emission as a function of temperature (-dF/dT) to identify TM via Applied Biosystems® Protein Thermal Shift™ Software. Native NDM-1was observed to melt with a TM = 55.95±0.06 °C.
Native state electrospray ionization mass spectrometry
NanoESI-MS was used to analyze the mechanism of inhibition of some of the inhibitors in this study. Expression and purification of NDM-1 and VIM-2 were performed according to literature reported procedures.[12a, 36] Samples (50 μM of VIM-2 and NDM-1) were incubated for 1 h and dialyzed overnight against 100 mM ammonium acetate, pH 7.5, after addition of tris(2-carboxyethyl) phosphine hydrochloride (TCEP, final concentration 2 mM) and 3 equivalents (VIM-2) or 2 equivalents (NDM-1) of Zn(II) (from a 0.1 M ZnCl2 stock). To analyze samples, a nano-electrospray ionization (n-ESI) probe (ThermoFisher Scientific, San Jose, CA, USA) with positive mode protein detection was used on a Thermo Scientific LTQ Orbitrap XL™ hybrid ion trap-orbitrap mass spectrometer. The major parameters were set as follows: capillary temperature, 180 °C; sheath gas, 0; auxiliary gas, 0; sweep gas, 0; spray voltage, 1.1-1.9kV; tube-lens, 150 V; capillary voltage, 35 V; full scan ranging 1000-4000 (m/z); and resolution set to 30,000. The automated gain control was set as follows: full scan, 3×104; SIM, 1×104; and MSn 1×105 for Fourier-transform. Making slight modifications, the nESI source was equipped with an offline unite (Catalog number ES260) which was constructed based on previously published material.[37] To construct the source, a platinum white (0.25 mm diameter) was inserted into the center of the offline unit. The glass capillaries (inner tip diameter 0.8 mm, outer tip diameter 1.5 mm) were produced in-house using a micropipette puller (model P-87 Flaming/Brown Micropipette Puller, Sutter Instrument Inc., USA), 5 μL of sample was loaded into the pulled glass capillary. The platinum wire was inserted into the capillary and the capillary position was adjusted approximately 3 mm from the MS inlet.
Supplementary Material
Scheme 2.
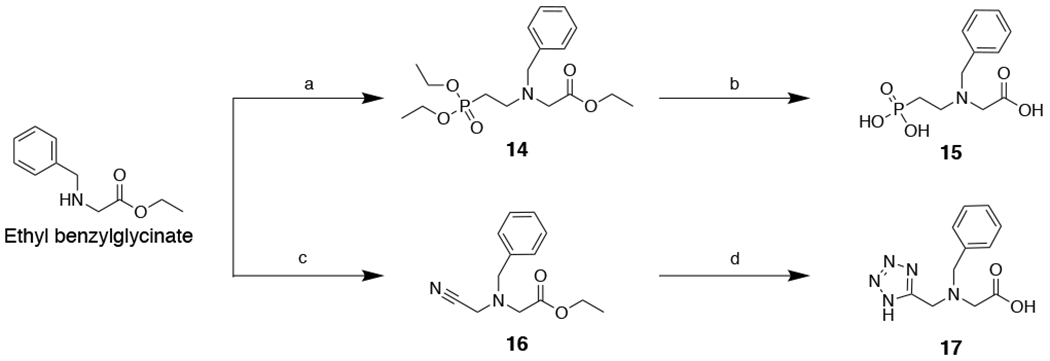
Synthesis of IDA inhibitors 15 and 17. Reagents and conditions: (a) diethyl(2-bromoethyl)phosphonate, K2CO3, KI, ACN, 82 °C, 16 h, 40%; (b) HCl, 100 °C, 16 h, ~99%; (c) 2-bromoacetonitrile, K2CO3, KI, CH3CN, 25 °C, 16 h, 74%; (d) NaN3, NH4Cl, DMF, 110 °C, 16 h; then 1:1:1 MeOH:THF:1 M NaOH, 60 °C, 16 h; two steps 19%.
Acknowledgements
We thank Dr. Yongxuan Su for mass spectrometry sample analysis at The Molecular Mass Spectrometry Facility (MMSF) at UC San Diego. This work was supported by the National Institutes of Health (Grant GM111926, GM098435, and GM134454) and by the Robert A. Welch Foundation (Grant F-1572 to W.F.). S.M.C. is a cofounder of and has an equity interest in Cleave Therapeutics and Forge Therapeutics, companies that may potentially benefit from the research results. S.M.C. also serves on the Scientific Advisory Board for these companies. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
References
- [1].Centers for Disease Control and Prevetion 2019, p. 148. [Google Scholar]
- [2].a) Bonomo RA, Cold Spring Harbor Perspect. Med 2017, 7, a025239; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fisher JF, Meroueh SO, Mobashery S, Chem. Rev 2005, 105, 395–424; [DOI] [PubMed] [Google Scholar]; c) Munita JM, Arias CA, Microbiol. Spectr 2016, 4, VMBF-0016–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bush K, Bradford PA, Cold Spring Harb Perspect. Med 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bush K, Jacoby GA, Antimicrob. Agents Chemother 2010, 54, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sabath LD, Abraham EP, Biochem. J 1966, 98, 11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Meini MR, Llarrull LI, Vila AJ, FEBS Lett. 2015, 589, 3419–3432; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mojica MF, Bonomo RA, Fast W, Curr. Drug Targets 2016, 17, 1029–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Ju L-C, Cheng Z, Fast W, Bonomo RA, Crowder MW, Trends Pharmacol. Sci 2018, 39, 635–647; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bush K, Bradford PA, Nat. Rev. Microbio 2019, 17, 295–306; [DOI] [PubMed] [Google Scholar]; c) Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, Spencer J, J. Mol. Biol 2019, 431, 3472–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Drawz SM, Bonomo RA, Clin. Microbiol. Rev 2010, 23, 160–201; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crowder MW, Spencer J, Vila AJ, Acc. Chem. Res 2006, 39, 721–728; [DOI] [PubMed] [Google Scholar]; c) Nordmann P, Naas T, Poirel L, Emerging Infect. Dis 2011, 17, 1791–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR, Antimicrob. Agents Chemother 2009, 53, 5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zou D, Huang Y, Liu W, Yang Z, Dong D, Huang S, He X, Ao D, Liu N, Wang S, Wang Y, Tong Y, Yuan J, Huang L, Sci. Rep 2017, 7, 9405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Gonzalez LJ, Bahr G, Nakashige TG, Nolan EM, Bonomo RA, Vila AJ, Nat. Chem. Biol 2016, 12, 516–522; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) King D, Strynadka N, Protein Sci. 2011, 20, 1484–1491; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bahr G, Vitor-Horen L, Bethel CR, Bonomo RA, Gonzalez LJ, Vila AJ, Antimicrob. Agents Chemother 2018, 62, e01849–01817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Cheng Z, Thomas PW, Ju L, Bergstrom A, Mason K, Clayton D, Miller C, Bethel CR, VanPelt J, Tierney DL, Page RC, Bonomo RA, Fast W, Crowder MW, J. Biol. Chem 2018, 293, 12606–12618; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stewart AC, Bethel CR, VanPelt J, Bergstrom A, Cheng Z, Miller CG, Williams C, Poth R, Morris M, Lahey O, Nix JC, Tierney DL, Page RC, Crowder MW, Bonomo RA, Fast W, Acs Infect. Dis 2017, 3, 927–940; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI, J. Enzyme Inhib. Med. Chem 2017, 32, 917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kang JS, Zhang AL, Faheem M, Zhang CJ, Ai N, Buynak JD, Welsh WJ, Oelschlaeger P, J. Chem. Inf. Model 2018, 58, 1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim Y, Tesar C, Mire J, Jedrzejczak R, Binkowski A, Babnigg G, Sacchettini J, Joachimiak A, PLoS One 2011, 6, e24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Sun Z, Hu L, Sankaran B, Prasad BVV, Palzkill T, Nat. Commun 2018, 9, 4524; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Hao Q, FASEB J. 2011, 25, 2574–2582. [DOI] [PubMed] [Google Scholar]
- [16].Linciano P, Cendron L, Gianquinto E, Spyrakis F, Tondi D, ACS Infect. Dis 2018, 5, 9–34. [DOI] [PubMed] [Google Scholar]
- [17].a) Li N, Xu Y, Xia Q, Bai C, Wang T, Wang L, He D, Xie N, Li L, Wang J, Zhou HG, Xu F, Yang C, Zhang Q, Yin Z, Guo Y, Chen Y, Bioorg. Med. Chem. Lett 2014, 24, 386–389; [DOI] [PubMed] [Google Scholar]; b) King DT, Worrall LJ, Gruninger R, Strynadka NC, J. Am. Chem. Soc 2012, 134, 11362–11365; [DOI] [PubMed] [Google Scholar]; c) Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, Rao Z, Protein Cell 2011, 2, 384–394; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cain R, Brem J, Zollman D, McDonough MA, Johnson RM, Spencer J, Makena A, Abboud MI, Cahill S, Lee SY, McHugh PJ, Schofield CJ, Fishwick CWG, J. Med. Chem 2018, 61, 1255–1260. [DOI] [PubMed] [Google Scholar]
- [18].a) Cahill ST, Cain R, Wang DY, Lohans CT, Wareham DW, Oswin HP, Mohammed J, Spencer J, Fishwick CW, McDonough MA, Schofield CJ, Brem J, Antimicrob. Agents Chemother 2017, 61; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brem J, Cain R, Cahill S, McDonough MA, Clifton IJ, Jimenez-Castellanos JC, Avison MB, Spencer J, Fishwick CW, Schofield CJ, Nat. Commun 2016, 7, 12406; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Krajnc A, Brem J, Hinchliffe P, Calvopina K, Panduwawala T, Lang PA, Kamps J, Tyrell JM, Widlake E, Saward BG, Walsh TR, Spencer J, Schofield CJ, J. Med. Chem 2019, 62, 8544–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Zhang E, Wang MM, Huang SC, Xu SM, Cui DY, Bo YL, Bai PY, Hua YG, Xiao CL, Qin S, Bioorg. Med. Chem. Lett 2018, 28, 214–221; [DOI] [PubMed] [Google Scholar]; b) Schnaars C, Kildahl-Andersen G, Prandina A, Popal R, Radix S, Le Borgne M, Gjoen T, Andresen AMS, Heikal A, Okstad OA, Frohlich C, Samuelsen O, Lauksund S, Jordheim LP, Rongved P, Astrand OAH, ACS Infect. Dis 2018, 4, 1407–1422; [DOI] [PubMed] [Google Scholar]; c) Yu ZJ, Liu S, Zhou S, Li H, Yang F, Yang LL, Wu Y, Guo L, Li GB, Bioorg. Med. Chem. Lett 2018, 28, 1037–1042; [DOI] [PubMed] [Google Scholar]; d) Chen AY, Thomas PW, Stewart AC, Bergstrom A, Cheng ZS, Miller C, Bethel CR, Marshal SH, Credille CV, Riley CL, Page RC, Bonomo RA, Crowder MW, Tierney DL, Fast W, Cohen SM, J. Med. Chem 2017, 60, 7267–7283; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD, Nature 2014, 510, 503–506; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu S, Zhou Y, Niu X, Wang T, Li J, Liu Z, Wang J, Tang S, Wang Y, Deng X, Cell Death Discovery 2018, 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Bergstrom A, Katko A, Adkins Z, Hill J, Cheng Z, Burnett M, Yang H, Aitha M, Mehaffey MR, Brodbelt JS, Tehrani K, Martin NI, Bonomo RA, Page RC, Tierney DL, Fast W, Wright GD, Crowder MW, ACS Infect. Dis 2018, 4, 135–145; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang J, Wang S, Wei Q, Guo Q, Bai Y, Yang S, Song F, Zhang L, Lei X, Bioorg. Med. Chem 2017, 25, 5133–5141. [DOI] [PubMed] [Google Scholar]
- [21].a) Albu SA, Koteva K, King AM, Al-Karmi S, Wright GD, Capretta A, Angew. Chem., Int. Ed. Engl 2016, 55, 13259–13262; [DOI] [PubMed] [Google Scholar]; b) Tehrani K, Fu H, Bruchle NC, Mashayekhi V, Prats Lujan A, van Haren MJ, Poelarends GJ, Martin NI, Chem. Commun 2020, [Online early access], DOI: 10.1039/D1030CC00356E; [DOI] [PubMed] [Google Scholar]; c) Proschak A, Kramer J, Proschak E, Wichelhaus TA, J. Antimicrob. Chemother 2018, 73, 425–430. [DOI] [PubMed] [Google Scholar]
- [22].a) Liao D, Yang S, Wang J, Zhang J, Hong B, Wu F, Lei X, Angew. Chem., Int. Ed. Engl 2016, 55, 4291–4295; [DOI] [PubMed] [Google Scholar]; b) Koteva K, King AM, Capretta A, Wright GD, Angew. Chem., Int. Ed. Engl 2016, 55, 2210–2212; [DOI] [PubMed] [Google Scholar]; c) Zhang J, Wang S, Bai Y, Guo Q, Zhou J, Lei X, J. Org. Chem 2017, 82, 13643–13648. [DOI] [PubMed] [Google Scholar]
- [23].Ni LB, Zhang RH, Liu QX, Xia WS, Wang H, Zhou ZH, Solid State Chem J. 2009, 182, 2698–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Raczynska JE, Shabalin IG, Minor W, Wlodawer A, Jaskolski M, Drug Resist. Updat 2018, 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen AY, Thomas PW, Cheng Z, Xu NY, Tierney DL, Crowder MW, Fast W, Cohen SM, ChemMedChem 2019, 14, 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Krezel A, Maret W, Arch. Biochem. Biophys 2016, 611, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheng Y, Prusoff WH, Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- [28].Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR, J. Biomol. Screen 2001, 6, 429–440. [DOI] [PubMed] [Google Scholar]
- [29].Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Muller HF, Sjuts H, Gottig S, Koenigs A, Pos KM, Pogoryelov D, Proschak E, J. Med. Chem 2015, 58, 3626–3630. [DOI] [PubMed] [Google Scholar]
- [30].Credille CV, Morrison CN, Stokes RW, Dick BL, Feng Y, Sun J, Chen Y, Cohen SM, J. Med. Chem 2019, 62, 9438–9449. [DOI] [PubMed] [Google Scholar]
- [31].Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G, Anal. Biochem 2004, 332, 153–159. [DOI] [PubMed] [Google Scholar]
- [32].a) Selevsek N, Tholey A, Heinzle E, Lienard BMR, Oldham NJ, Schofield CJ, Heinz U, Adolph H-W, Frere J-M, J. Am. Soc. Mass Spectrom 2006, 17, 1000–1004; [DOI] [PubMed] [Google Scholar]; b) Heck AJR, Nat. Methods 2008, 5, 927–933. [DOI] [PubMed] [Google Scholar]
- [33].Stokvis E, Rosing H, Beijnen JH, Rapid Commun. Mass Spectrom 2005, 19, 401–407. [DOI] [PubMed] [Google Scholar]
- [34].a) Paramella D, Cantel S, Enjalbal C, Amblard M, Forest E, Heymann M, Geourjon C, Martinez J, Subra G, Proteomics 2009, 9, 5384–5388; [DOI] [PubMed] [Google Scholar]; b) Rolland O, Turrin C-O, Baquet G, Poupot R, Poupot M, Caminade A-M, Majoral J-P, Tet. Lett 2009, 50, 2078–2082. [Google Scholar]
- [35].Yang H, Aitha M, Hetrick AM, Richmond TK, Tierney DL, Crowder MW, Biochemistry 2012, 51, 3839–3847. [DOI] [PubMed] [Google Scholar]
- [36].Cheng Z, Shurina BA, Bethel CR, Thomas PW, Marshall SH, Thomas CA, Yang K, Kimble RL, Montgomery JS, Orischak MG, Miller CM, Tennenbaum JL, Nix JC, Tierney DL, Fast W, Bonomo RA, Page RC, Crowder MW, mBio 2019, 10, e02412–02419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harvey SR, Porrini M, Stachl C, MacMillan D, Zinzalla G, Barran PE, J. Am. Chem. Soc 2012, 134, 19384–19392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.