

Abstract
African American (AA) smokers are at a higher risk of developing lung cancer compared to whites. The variations in the metabolism of nicotine and tobacco-derived carcinogens in these groups were reported previously with the levels of nicotine metabolites and carcinogen-derived metabolites measured using targeted approaches. While useful, these targeted strategies are not able to detect global metabolic changes for use in predicting the detrimental effects of tobacco use and ultimately lung cancer susceptibility among smokers. To address this limitation, we have performed global untargeted metabolomics profiling in urine of AA and white smokers to characterize the pattern of metabolites, identify differentially regulated pathways, and correlate these profiles with the observed variations in lung cancer risk between these two populations. Urine samples from AA (n = 30) and white (n = 30) smokers were used for metabolomics analysis acquired in both positive and negative electrospray ionization modes. LC-MS data were uploaded onto the cloud-based XCMS online (http://xcmsonline.scripps.edu) platform for retention time correction, alignment, feature detection, annotation, statistical analysis, data visualization, and automated systems biology pathway analysis. The latter identified global differences in the metabolic pathways in the two groups including the metabolism of carbohydrates, amino acids, nucleotides, fatty acids, and nicotine. Significant differences in the nicotine degradation pathway (cotinine glucuronidation) in the two groups were observed and confirmed using a targeted LC-MS/MS approach. These results are consistent with previous studies demonstrating AA smokers with lower glucuronidation capacity compared to whites. Furthermore, the d-glucuronate degradation pathway was found to be significantly different between the two populations, with lower amounts of the putative metabolites detected in AA compared to whites. We hypothesize that the differential regulation of the d-glucuronate degradation pathway is a consequence of the variations in the glucuronidation capacity observed in the two groups. Other pathways including the metabolism of amino acids, nucleic acids, and fatty acids were also identified, however, the biological relevance and implications of these differences across ethnic groups need further investigation. Overall, the applied metabolomics approach revealed global differences in the metabolic networks and endogenous metabolites in AA and whites, which could be used and validated as a new potential panel of biomarkers that could be used to predict lung cancer susceptibility among smokers in population-based studies.
Introduction
Tobacco smoking is the main cause of lung cancer-related mortalities worldwide. Despite the more than 90% lung cancer incidence associated with this lifestyle habit, only a fraction (11–24%) of smokers will develop lung cancer in their lifetime.1−3 This disparity is hypothesized to be due to interindividual genetics differences, which result in variations in the uptake and metabolism of nicotine and tobacco-derived carcinogens leading to differing levels of metabolites in biological fluids.4−6 Epigenetics, behavioral, and environmental factors, including diet and lifestyle, may also contribute to the observed variations in cancer risk and other major causes of mortality across different ethnic groups.7−10 Specifically, AA smokers have been shown to be at a higher risk of developing tobacco-related lung cancer compared to whites.11,12 Thus, the global analysis of biological networks and their associated metabolites in populations with differing lung cancer risk as in African Americans (AA) and whites could identify new potential biomarkers to predict susceptibility to the detrimental effects of tobacco use among smokers. Understanding the differences in the overall metabolic regulation in smokers across different ethnic groups, not only those of nicotine and tobacco-specific carcinogen metabolism, is important in gaining insights into the impact of ethnic and genetic differences on lung cancer susceptibility, which could be used to develop rational strategies for cancer prevention based on targeted surveillance of high-risk and susceptible populations.
Numerous studies have shown differences in genetic backgrounds result in varying capacities to metabolize drugs, nicotine, and tobacco-specific carcinogens.9,13−18 For instance, multiethnic studies demonstrated that AA smokers have a higher risk of developing lung cancer than white smokers due in part to the differing activities of metabolic enzymes in these groups.9,11−13,15,17−21 Interindividual genetic differences can affect nicotine metabolism, which influence smoking behavior, toxicity, and detoxification capacity and thus ultimately impact tobacco-derived carcinogen exposure.9 For example, the gene variants of cytochrome P450 2A6 (CYP2A6) are associated with decreased risk of tobacco smoking-related lung cancer.9,22 CYP2A6 is the main enzyme responsible for metabolizing nicotine. Smokers carrying genetic variants of this gene associated with slower nicotine conversion are more likely to smoke less and have reduced exposure to tobacco smoke carcinogens and thus lower risk of developing lung cancer.9 The reduced activity of CYP2A6 enzyme in Japanese Americans was observed to be associated with lower risk of developing smoking related-lung cancer in this population compared to other ethnic groups.8,9,13,14,23 In addition, the UDP-glucuronosyltransferases (UGT) are another class of enzymes implicated in ethnic differences in the metabolism and detoxification of tobacco-related compounds such as nicotine and tobacco-derived nitrosamines.15,19,24,25 A low glucuronidation capacity in AA compared to whites was observed in several multiethnic studies with the mean urinary cotinine glucuronidation ratio found to be 0.57 in AA over whites.15,18,19,24 AA have a high prevalence of UGT2B10 splice variants resulting in lower cotinine glucuronidation.19,26,27 Similarly, the UGT2B10 splice variants commonly found in AA may increase their exposure to drugs during treatment.26 The predominance of these gene variants including the UGTs and CYP2A6 can lead to differences in the metabolism of nicotine and tobacco-derived carcinogens, which could provide insights into the variations in risk and susceptibility to developing smoking-related cancers in these groups.
Mass spectrometry-based metabolomics have emerged as a powerful tool to investigate global dysregulation of biological networks resulting from specific exposures.28 As more advanced bioinformatics and instrument platforms as well as spectral databases are being developed, its applicability to address various biologically relevant questions is rapidly expanding.29−39 For instance, multiple metabolomics platforms have been applied to probe global changes and patterns in altered metabolites between smokers and nonsmokers.40−42 Metabolomics workflows have also been used in in vitro and in vivo models of tobacco-smoke-induced perturbations to identify tobacco-related biomarkers for lung cancer and other diseases.43−47 In addition, metabolomics analysis has been widely used in human studies to identify biomarkers of smoking habits and assessment of variability in the metabolism of tobacco-derived compounds to understand their contributions in cancer development.40−45,48,49 Although relevant information has been deciphered from these studies, limited information is available on the global metabolic dysregulation in populations with differing genetic backgrounds and in populations from different ethnicities with varying susceptibility to developing lung cancer due to tobacco smoking. To address this limitation, we have performed global untargeted metabolomics profiling in urine of AA and white smokers to characterize the pattern of metabolites, identify potentially dysregulated pathways, and correlate these profiles with the observed variations in lung cancer risk between these two populations. In order to validate the use of these global profiles, the results were compared to those obtained with well-established, traditional targeted MS-based analysis of nicotine-derived metabolites.
Experimental Procedures
Subjects
Urine samples from AA (n = 30) and white (n = 30) smokers were obtained from the study “Ethnic Differences in Tobacco Carcinogen Metabolism” at the University of Minnesota. This study was approved by the University of Minnesota Institutional Review Board: Human Subjects Committee. The detailed 24 h urine sample collection and study design have been previously described.50 The mean age of AA smokers was 46 ± 8 years, while 40 ± 12 years for white smokers. All subjects used in this study were males and were smokers with smoking frequency of more than 10 cigarettes per day (CPD). The total nicotine equivalent (TNE), an established exposure biomarker of cigarette smoke exposure, and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) levels (a biomarker of carcinogen exposure) were measured for the 60 subjects as previously described and reported in the Supporting Table 1.19,51
Caution
NNAL and [13C6]-NNAL are carcinogenic and must be handled with extreme care and proper personal protective equipment and ventilation.
Chemicals and Reagents
NNAL, [13C6]-NNAL, NNAL-O-glucuronide, NNAL-N-glucuronide, NNK-N-oxide, and NNAL-N-oxide were obtained from Toronto Research Chemicals (Ontario, Canada). Oasis HLB (3 cc) and Oasis MCX (2 mg solid phase extraction 96-well plates) were purchased from Waters (Milford, MA). All acids and organic solvents were MS grade.
Sample Preparation
For the untargeted global metabolomics and targeted metabolite analysis, the same set of samples were used, that is, the same aliquots of urine from each subject were processed and analyzed for both the analyses. Urine samples were centrifuged at 14,000 rpm and 4 °C for 10 min to remove particulates and 500 μL were used for untargeted metabolomics analysis (Figure 1a). The samples were cleaned up using Oasis HLB (3 cc) (Waters, Milford, MA). The cartridge was conditioned with 3 mL of methanol and then with 3 mL of water. The urine samples were loaded into the cartridge and washed with 3 mL of water. After which, the metabolites were eluted with 3 mL of methanol, and the fractions were dried in a refrigerated vacuum centrifuge (T < 10 °C). The fractions were resuspended in acetonitrile/water (20/80, v/v) and the volume (30 μL) normalized based on creatinine concentration. The creatinine concentration (mg/mL) was measured using a colorimetric microplate assay (CRE34-K01) obtained from Eagle Bioscience (http://stores.eaglebio.com/creatinine-microplate-assay-kit). The total nicotine equivalents (TNE: nmol/mL, sum of total nicotine, total cotinine, total 3-hydroxycotinine, and nicotine-N-oxide) were determined for these samples as previously described (Table S1).19 Sample normalization was performed based on the creatinine concentration of each urine sample and the TNE to normalize the levels of specific nicotine metabolites detected in the metabolomics analysis to account for the differences in tobacco smoke exposure.
Figure 1.
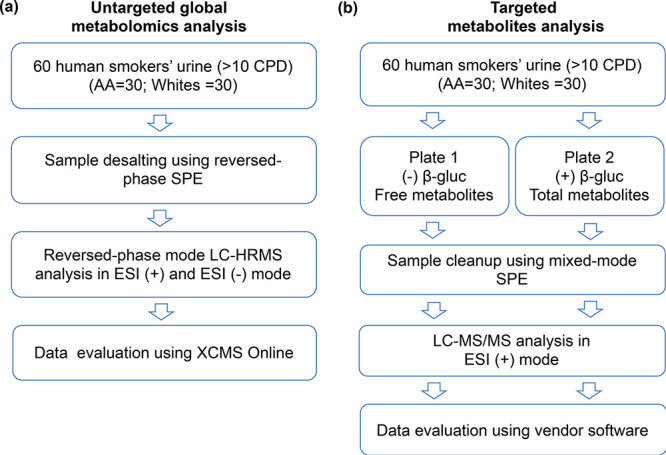
Experimental workflow for global untargeted and targeted metabolomics analyses of smokers’ urine from two ethnic groups. (a) Untargeted approach and (b) targeted approach.
Untargeted LC-HRMS-Based Metabolomics Analysis
LC-HRMS analyses were performed on Agilent 1200 series microflow HPLC (Agilent Technologies, Santa Clara, CA) coupled to a Bruker Impact II quadrupole time-of-flight (Q-TOF) mass spectrometer (Bruker Daltonics, Billerica, MA) in both positive and negative ionization mode. Reversed-phase chromatography was performed on a Waters Atlantis T3 column (3 μm, 1.0 × 150 mm) (Waters, Milford, MA) equipped with a VanGuard precolumn (2.1 × 5 mm). Separation was performed at room temperature and flow rate of 65 μL min–1 using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B. Four μL of the sample was injected on-column. Gradient elution was carried out starting with 2% B for the first 5 min and a linear gradient from 2% to 40% B over 15 min and to 100% B for 7 min followed by a constant 100% B for 4 min. Finally, a linear gradient from 100% to 2% B over 2 min was performed, and the column was re-equilibrated at 2% B for another 6 min. The total run time was 39 min. For the MS analysis, full scan data acquisition was performed with a mass range of m/z 50–1000 with a mass resolving power (fwhm) of 30,000. The Funnel 1 RF was set to 150 Vpp, Funnel 2 RF to 200 Vpp, Hexapole RF to 50 Vpp, quadrupole ion energy was 4.0 eV, and collision energy was 7.0 eV. For data-dependent MS/MS, the isolation width was ±0.5 Da, and 3 ions per full scan were subjected to MS/MS, with exclusion of ions from subsequent analysis for 1 min. Samples were measured in a randomized manner with pooled QC samples injected after every six samples. The QC sample was made by pooling 10 random urine samples from the 60 subjects. A mixture of authentic reference standards (NNAL, [13C6]-NNAL, NNAL-O-glucuronide, NNAL-N-glucuronide, NNK-N-oxide, and NNAL-N-oxide) was utilized as additional QC measure. Internal calibration was performed by injecting sodium formate around 35 min within each run. After the samples were analyzed, they were unblinded and classified into two groups corresponding to AA and whites. LC-HRMS mass spectral data were uploaded to the cloud-based XCMS online platform (http://xcmsonline.scripps.edu) for retention time correction, alignment, feature detection, annotation, and statistical analysis.52 Feature detection parameters include 5 ppm mass tolerance, minimum peak width of 10 s, and maximum peak width of 60 s; obiwarp was used for retention time correction with profStep set to 1, mzwid of 0.015, minfrac of 0.5, bw of 5, allowable retention time deviation of 20 s; unpaired parametric t test (Welch t test) and posthoc analysis for statistical analysis with a p-value threshold of 0.001 for highly significant features, fold change >1.5; adducts considered for the database search include [M + H]+, [M + Na]+, [M + H + Na]2+, [M + NH3+ H]+, [M – NH3+ H]+, and [M + H – H2O]+ for ESI (+) mass spectral data and [M - H]−, [M + Na–2H]− and [M – H2O]− for ESI (−) mass spectral data. Isotopes were searched with m/z absolute error of 0.005 Da, and 5 ppm mass error and the sample biosource were set to human. Systems biology pathway analysis was performed to identify differentially regulated pathways in the two groups of smokers as previously described.54 Putative metabolites were searched against the METLIN and HMDB databases.36,53 Multivariate principal component analysis (PCA) was performed in the XCMS online platform to identify features that show maximum variation in the two groups.55 Finally, autonomous multimodal pathway analysis was performed with a p-value set to 0.01 and 5 ppm mass tolerance on the XCMS online processed data sets (positive and negative modes) as previously described.56 Metabolomics data have been deposited to the EMBL-EBI MetaboLights database (DOI: 10.1093/nar/gkz1019, PMID:31691833) with the identifier MTBLS1705. The complete dataset can be accessed here: https://www.ebi.ac.uk/metabolights/MTBLS1705.71
Targeted LC-MS/MS Quantitation of Nicotine Metabolites
To determine the levels of nicotine metabolites in the urine of the 60 subjects, we performed targeted LC-MS/MS analysis using selected reaction monitoring (SRM) of free and total cotinine and trans-3-hydroxycotinine as previously described.19,57 Briefly, the diluted urine samples (1:10, 10 μL total) from the 60 subjects were combined with 400 μL of 100 mM ammonium acetate, pH 5.0, and methyl-d3 internal standards (1 ng each). The mixture from each sample was added to paired 96-well plates, one for the analysis of free cotinine and trans-3-hydroxycotinine and the second for total (free + glucuronide) cotinine and trans-3-hydroxycotinine (Figure 1b). One of the plates (plate 2) was incubated overnight at 37 °C with β-glucuronidase (500 units) (recombinant β-glucuronidase from overexpressing E. coli BL21). Samples were cleaned up using mixed-mode cation solid-phase extraction 96-well plates using Oasis MCX (2 mg solid-phase extraction 96-well plates) (Waters, Milford, MA). LC-MS/MS analysis was performed on an Agilent 1100 capillary HPLC system coupled to a Thermo Scientific TSQ Vantage mass spectrometer in positive electrospray ionization mode (Thermo Fisher, San Jose, CA). The samples were resuspended in 25 μL of 100 mM ammonium acetate:methanol, and 4 μL was injected on an Atlantis HILIC column (300 μm × 100 mm) (Waters, Milford, MA). Cotinine (tR: 5.03 min) and trans-3-hydroxycotinine (tR: 4.29 min) were eluted with acetonitrile:water:formic acid (95/3.5/1.5). The flow rate was 20 μL min–1. SRM transitions used were m/z 177.1 → 80.1 (confirmation) and m/z 177.1 → 98.1 (quantitation) for d0-cotinine; m/z 180.1 → 80.1 (confirmation) and m/z 180.1 → 101.1 (quantitation) for d3-cotinine; m/z 193.1 → 80.1 (confirmation) and m/z 193.1 → 134.1 (quantitation) for d0-trans-3-hydroxycotinine; m/z 196.1 → 80.1 (confirmation) and m/z 196.1 → 134.1 (quantitation) for d3-trans-3-hydroxycotinine. Peak areas were integrated using Xcalibur 3.0 (Thermo Scientific, Sunnyvale, CA) and the ratio of d0/d3 for each of the metabolites was determined. Total cotinine (free cotinine + cotinine glucuronide) and total trans-3-hydroxycotinine (free trans-3-hydroxycotine + trans-3-hydroxycotinine glucuronide) were measured after β-glucuronidase treatment, while free cotinine and free trans-3-hydroxycotinine glucuronide were measured without β-glucuronidase treatment. The levels of cotinine glucuronide and trans-3-hydroxycotinine glucuronide were obtained by subtraction of the free metabolites from the total metabolites (±β-glucuronidase). Statistical analysis was performed using SigmaPlot 12.5 (Systat Software, Inc. San Jose, CA). Group comparisons using t test and Shapiro–Wilk (P < 0.050) for normality test were performed. Mann–Whitney rank sum test was then performed for data sets that failed the normality test. A p-value < 0.05 was considered statistically significant in the group comparisons.
Results
To identify variations in biological pathways and their associated putative metabolites in smokers from two ethnic groups with differing lung cancer risk, 60 24 h urine samples (AA = 30 and whites = 30) were used for the LC-HRMS-based metabolomics analysis acquired in both positive and negative modes. The subjects were all males and smoking at least 10 cigarettes per day (CPD). The TNE, which is the sum of smokers’ urinary nicotine, cotinine, 3-hydroxycotinine, their corresponding glucuronides, plus nicotine N-oxide accounts for >85% of the nicotine dose consumed and considered as an excellent biomarker of cigarette smoke exposure, was measured for these subjects.58,59 The TNE levels for AA ranged from 30 to 159 nmol/mL, while for whites, it ranged from 20 to 173 nmol/mL (Table S1). In addition, the level of urinary NNAL, a biomarker of tobacco-specific nitrosamine exposure with a longer half-life compared to TNE, was measured for the 60 subjects. The average concentration of urinary NNAL in AA was 218 pg mL–1 (±119) and 224 pg mL–1 (±110) for whites. Both TNE and urinary NNAL levels showed no significant differences (p = 0.587) between the two populations, indicating that the cohort was exposed to similar levels of tobacco smoke and tobacco-specific carcinogens (Figure S1 and Table S1).
Metabolic Profiles of Smokers’ Urine from AA and Whites
The XCMS online platform identified features that are significantly different between the two groups. The metabolic cloud plot in positive mode showed 114 features with fold change ≥1.5 and p-value ≤0.001 (Figure 2a). Likewise, in negative mode, 36 features were detected with fold change ≥1.5 and p-value ≤0.001 (Figure 2b). Minimal retention time shifts (<0.6 min in both modes) were observed in the chromatographic runs of the 60 samples, which indicate good run-to-run reproducibility (Figure S2). The multivariate principal component analysis (PCA) in both positive and negative modes showed modest separation between the two groups (Figure 2c,d). This modest clustering was expected as both groups included only smokers, the sample size was relatively small, and other factors such as diet/lifestyle factors were not matched. Overall, a robust and reproducible LC-HRMS-based metabolomics analysis of the urine samples from AA and whites yielded features or putative metabolites associated with differentially regulated metabolic pathways, with the majority of these pathways being down-regulated and resulting in lower levels of associated metabolites in AA compared to whites.
Figure 2.

Metabolic cloud plots showing significantly upregulated features (green circles) and down-regulated features (red circles) between the two groups in (a) positive mode and (b) negative mode. PCA analysis in both (c) positive mode and (d) negative mode showing modest separation between the two groups.
Putative Pathway Analysis for Identifying Differentially Regulated Biological Networks
To identify differences in metabolic pathways in the two groups of smokers, an automated pathway analysis tool was performed as previously described.54 Significant and differentially regulated features identified by XCMS online were mapped onto known biological pathways, and differentially regulated biological networks were identified using the Fisher’s exact test based on the processed accurate mass spectral data.54 The systems biology results show metabolic pathways differentially regulated between the two populations in both positive and negative modes (Table 1). In addition, an autonomous multimodal pathway analysis was performed using both the positive and negative mode-acquired and processed data set as previously described.56 Using this integrated approach, metabolic pathways involving carbohydrate/sugar metabolism, amino acids, nucleic acids, fatty acids, and nicotine were identified (Tables S3 and S4). The nicotine degradation (p-value = 0.0002) and d-glucuronate degradation (p-value = 0.0002) pathways were significantly and differentially regulated between the two groups with reduced amounts of the putative metabolites detected in AA compared to whites. The metabolites implicated in the nicotine degradation pathway using the multimodal pathway analysis were 3-pyridylacetate, 4-(3-pyridyl)-butanoate, cotinine methonium ion, cotinine glucuronide, and trans-3-hydroxycotinine glucuronide (Table 2, Table S4, and Figure S6). The metabolites, cotinine glucuronide, and trans-3-hydroxycotinine glucuronide, were down-regulated in AA compared to whites (Table 2). To confirm the results obtained by our untargeted metabolomics analysis, the levels of cotinine glucuronide and trans-3-hydroxycotinine glucuronide were quantified by LC-MS/MS (Table S2).
Table 1. Differentially Regulated Metabolic Pathways Identified in Positive and Negative Modes.
| pathway | |||
|---|---|---|---|
| overlapping putative metabolites | all metabolites | p-value | |
| (+) Mode | |||
| d-glucuronate degradation | 4 | 4 | 1.10 × 10–8 |
| lysine degradation I (saccharopine pathway) | 3 | 6 | 4.20 × 10–8 |
| lactose degradation III | 2 | 2 | 1.60 × 10–7 |
| d-galactose degradation V (Leloir pathway) | 2 | 2 | 1.60 × 10–7 |
| trehalose degradation | 2 | 3 | 5.20 × 10–7 |
| bupropion degradation | 3 | 4 | 1.50 × 10–6 |
| sucrose degradation | 3 | 5 | 4.20 × 10–6 |
| lysine degradation II (pipecolate pathway) | 2 | 8 | 5.50 × 10–5 |
| tRNA charging | 2 | 11 | 4.10 × 10–4 |
| nicotine degradation V | 2 | 18 | 9.80 × 10–3 |
| (−) Mode | |||
| d-glucuronate degradation | 4 | 4 | 3.60 × 10–3 |
| tryptophan degradation via tryptamine | 4 | 4 | 3.60 × 10–3 |
| gluconeogenesis | 2 | 2 | 3.10 × 10–2 |
| sorbitol degradation I | 2 | 2 | 3.10 × 10–2 |
| taurine biosynthesis | 2 | 2 | 3.10 × 10–2 |
| lactose degradation III | 2 | 2 | 3.10 × 10–2 |
| d-galactose degradation V (Leloir pathway) | 2 | 2 | 3.10 × 10–2 |
| trehalose degradation | 2 | 2 | 3.10 × 10–2 |
| urate biosynthesis/inosine 5-phosphate degradation | 2 | 2 | 3.10 × 10–2 |
| adenosine nucleotides degradation | 2 | 2 | 3.10 × 10–2 |
| glycolysis | 2 | 2 | 3.10 × 10–2 |
| putrescine degradation III | 2 | 2 | 3.10 × 10–2 |
| lysine degradation II (pipecolate pathway) | 3 | 6 | 4.60 × 10–2 |
Table 2. Representative Metabolites Associated with the Nicotine Degradation and d-Glucuronate Degradation Pathways in Multimodal Pathway Analysis.
| pathway/metabolite | METLIN ID | dysregulationa | fold change | p-value | m/z | tR (min) | adduct type |
|---|---|---|---|---|---|---|---|
| Nicotine Degradation V | |||||||
| 3-pyridylacetate | NA | down | 2.1 | 4.50 × 10–3 | 121.0279 | 12.9 | [M – NH3 + H]+1 |
| 4-(3-pyridyl)-butanoate | NA | up | 1.7 | 6.60 × 10–3 | 166.0855 | 13.7 | [M + H]+1 |
| cotinine-gluc | NA | down | 1.5 | 7.20 × 10–3 | 373.1022 | 14.6 | [M + Na – 2H]−1 |
| cotinine methonium ion | NA | down | 1.5 | 5.00 × 10–3 | 209.1516 | 18.8 | [M + NH3 + H]+1 |
| trans-3-hydroxycotinine-gluc | NA | down | 2.1 | 6.00 × 10–3 | 196.0593 | 12.8 | [M + H + Na]+2 |
| Nicotine Degradation IV | |||||||
| 3-pyridylacetate | NA | down | 2.1 | 4.50 × 10–3 | 121.0279 | 12.9 | [M – NH3 + H]+1 |
| 4-(3-pyridyl)-butanoate | NA | up | 1.7 | 6.60 × 10–3 | 166.0855 | 13.7 | [M + H]+1 |
| d-Glucuronate Degradation Pathway | |||||||
| l-xylulose | 139 | down | 2.2 | 3.00 × 10–3 | 151.0604 | 9.1 | [M + H]+1 |
| l-gulonate | 63,144 | down | 2.4 | 2.40 × 10–3 | 197.0652 | 12.3 | [M + H]+1 |
| l-gulonate | 63,144 | down | 3 | 1.50 × 10–4 | 195.0503 | 12.0 | [M – H]−1 |
| aldehydo-d-glucuronate | NA | down | 2 | 1.70 × 10–3 | 177.0392 | 13.2 | [M – H2O + H]+1 |
| 3-keto-l-gulonate | 58,394 | down | 2 | 1.70 × 10–3 | 177.0392 | 13.2 | [M – H2O + H]+1 |
Dysregulation (fold change) relative to whites; NA: not applicable.
In addition, the d-glucuronate degradation pathway was the top pathway identified being differentially regulated in AA in both positive (p = 1.10 × 10–8) and negative modes (p = 3.60 × 10–3) as well as in the multimodal pathway analysis (p = 2.0 × 10–4) (Table 2 and Table S3). Figure 3 shows the pathway of the degradation of d-glucuronate in humans including the enzymes responsible for each step of the process. The metabolites associated in this pathway are 3-keto-l-gulonate, aldehydo-d-glucuronate, l-gulonate, and l-xylulose. The levels of these metabolites were lower in AA compared to whites (Table 2, Table S4, and Figure S6). Figure 4 shows a representative EIC, zoomed precursor ion MS spectrum, and box plot for l-gulonate. Overall, metabolic pathways involving carbohydrate/sugar metabolism that are differentially regulated in AA compared to whites were identified.
Figure 3.

d-Glucuronate degradation pathway in Homo sapiens illustrating the different metabolites associated with the pathway (https://biocyc.org). The metabolites implicated in the pathway are down-regulated in AA compared to whites (https://humancyc.org/HUMAN/NEW-IMAGE?type=PATHWAY=PWY-5525=3).
Figure 4.

Representative (a) extracted ion chromatogram (EIC), (b) zoomed precursor ion full scan MS spectrum in positive mode, and (c) box plot of the putative metabolite, l-gulonate (fold change = 2.4; p-value = 0.0024).
Targeted LC-MS Analysis of Nicotine Metabolites
To confirm the results of our metabolomics analysis, in particular when considering the metabolites implicated in the nicotine degradation pathway, the levels of cotinine glucuronide and trans-3-hydroxycotine glucuronide were measured in the same samples used for the global untargeted metabolomics analysis. Total cotinine, total trans-3-hydroxycotinine, free cotinine, free trans-3-hydroxycotinine, cotinine glucuronide, and trans-3-hydroxycotinine glucuronide were measured in the 60 samples. Figure 5 shows the levels of nicotine metabolites (cotinine glucuronide, trans-3-hydroxycotinine glucuronide, free cotinine, free trans-3-hydroxycotinine, total cotinine, and total trans-3-hydroxycotinine) in the urine samples of AA and whites. Figures S3a,b and S4a,b show representative chromatograms of the LC-MS/MS analysis of total cotinine and total trans-3-hydroxycotinine detected in the urine of a heavy smoker with their corresponding d3-labeled internal standard. The levels of cotinine glucuronide are significantly lower (p < 0.001) in AA compared to whites, while the levels of free cotinine were significantly higher (p = 0.026) in AA compared to white smokers. These results are consistent with previous studies where the levels of cotinine glucuronide are lower and the free cotinine higher in the urine of AA compared to white smokers.15 The other metabolites did not show any significant difference in the levels between the two groups.
Figure 5.
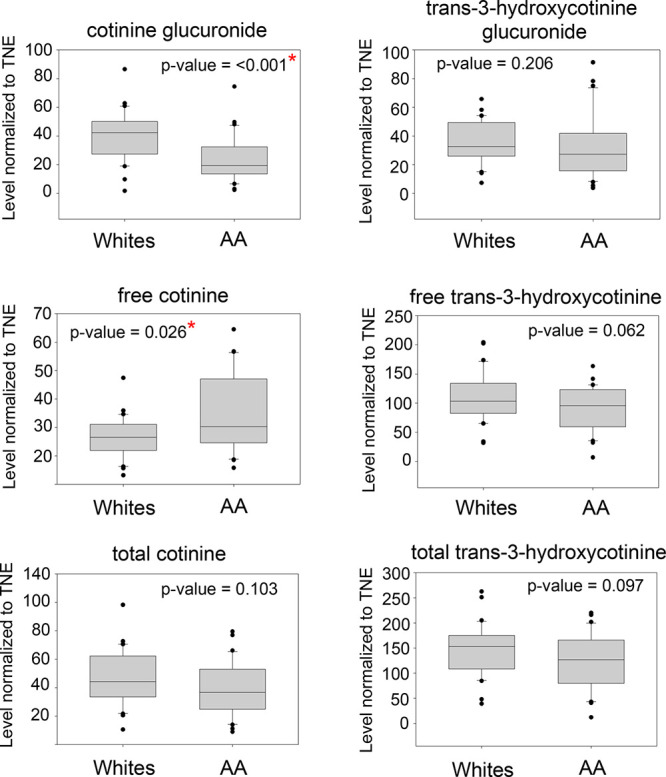
Quantitation of nicotine metabolites (ng nmol–1) in the 60 subjects using targeted LC-MS approach.
Discussion
The overall goal of this work is to identify differentially regulated metabolic pathways and their associated metabolites using global MS-based metabolomics in a cohort with established differences in the levels of tobacco-related metabolites previously measured by targeted approaches. The identification of other pathways and biomarkers that may be relevant in cancer development may be used and validated as new potential panel of biomarkers to better understand and therefore potentially predict cancer susceptibility in smokers in future population-based studies. Previous studies demonstrated that AA smokers are at a higher risk of developing lung cancer compared to whites.11,12 The metabolic pathways of nicotine and tobacco-specific carcinogens are well studied in these two groups, providing an ideal cohort to test the use of untargeted metabolomics methods to uncover other biologically relevant and novel pathways that are valuable in understanding the association of genetics, ethnic differences, and environmental and lifestyle factors such as smoking in disease development. The use of untargeted mass spectrometry-based metabolomics has been widely used to identify differentially regulated biological pathways resulting from tobacco exposure. For example, metabolomics analyses were used to compare the global metabolic profiles between smokers and nonsmokers and current smokers before and after smoking, and using mentholated cigarettes.40,42−45 These metabolomics investigations serve as proof-of-principle for using metabolomics to identify novel tobacco-exposure biomarkers and provide important information on the dysregulated pathways and associated metabolites in smokers, which maybe relevant in cancer development while confirming the ability to detect the known differences in these groups.45 Hsu et al. reported the identification of unique metabolites in smokers’ plasma that are affected by acute smoking including menthol glucuronide, the reduction of glutamate, oleamide, and 13 glycerophospholipids.45 However, global metabolomics studies comparing the variations in metabolic pathways in current smokers from different ethnic groups are limited or non-existent. There are metabolomics studies comparing the differences between nonsmokers and smokers of the same ethnic background, but limited studies on the combined effects of smoking and race/ethnic group. Furthermore, because these studies have only used methods focusing on specific pathways related to a few toxicants, the overall changes in an individual’s metabolic pathways could not be explored. Here, we present a unique study that relates global metabolic changes in smokers’ urine from two populations of different ethnic backgrounds using untargeted metabolomics to test the possibility of identifying a panel of biomarkers, including those traditionally measured in relation to tobacco smoke that may enable identification and stratification of highly susceptible population to the detrimental effects of tobacco use.
Metabolic information can be derived from a number of biological sources such as saliva, urine, and blood. However, urine provides a non-invasive and accessible biofluid for longitudinal studies. In addition, urine is a rich source of cellular metabolites and has been extensively used for diagnostic and clinical applications. So far, there are about 4500 metabolites detected in urine associated with approximately 600 human diseases/conditions such as obesity, cancer, inflammation, neurodegeneration, infectious disease, and diet to name a few.60 Therefore, it is an ideal biofluid for global metabolomics studies as it reflects the overall metabolic network regulation of an individual resulting from specific and complex exposures. Using global LC-HRMS-based metabolomics, we have identified differentially regulated biological pathways and metabolites in urine of AA and white smokers. One pathway we have identified is the nicotine degradation pathway, which is different in AA compared to whites. The metabolites associated with the pathway are down-regulated in AA compared to whites according to our analysis. We have confirmed the low cotinine glucuronidation in AA using a targeted LC-MS/MS approach in the same aliquots of urine and is consistent with previous multiethnic studies.8,13,15,19 In addition, we also found that trans-3-hydroxycotinine glucuronide is significantly lower in AA compared to whites in our metabolomics analysis. However, we found no significant difference in the levels of trans-3-hydroxycotinine glucuronide in AA using a targeted approach (Figure 5). Previous studies have shown that trans-3-hydroxycotinine glucuronide is not different within these groups.22,57 When the levels of cotinine glucuronide and trans-3-hydroxycotine glucuronide were adjusted to TNE, only cotinine glucuronide (p = 0.027) showed significantly lower amounts, while trans-3-hydroxycotinine glucuronide (p = 0.061) was not significant anymore in the metabolomics analysis. These results are consistent with our targeted analysis and previous studies and support the need to normalize the levels of nicotine metabolites to TNE, rather than to CPD, to account for the differences in the exposure levels or nicotine dose in smokers.15,22 We have used the indirect measurement of glucuronide metabolites of nicotine such as cotinine glucuronide and trans-3-hydoxycotinine glucuronide (β-glucuronidase treatment), as reported previously by others, to allow appropriate comparison of our current results with previous studies that used such indirect approaches.15,57 The targeted analysis of nicotine metabolites in the two groups confirmed the results of our metabolomics analysis and therefore provided more confidence in the identification of the other differentially regulated pathways even those not directly related to nicotine. The biological relevance of these pathways, however, is still unknown and warrants further investigation.
The low levels of cotinine glucuronidation in AA are associated with a relatively higher frequency of UGT2B10 splice variants in this population.19,26,27 For instance, UGT2B10 is the enzyme responsible for cotinine glucuronidation.19,61 The majority (mean = 78% in two studies) of the UGT2B10-null individuals were AA in a multi-ethnic study based on the racial/ethnic-specific frequency of the UGT2B10 splice variant.15 Genetic variations in UGT enzyme activities are associated with increased risk of developing solid cancers including colon, GI, lung, liver, oral, orolaryngeal, and prostate, which indicates that these variants are likely involved in detoxification of various carcinogens.62 Furthermore, the UGT2B10 splice variants common in AA have been shown to greatly increase drug exposure in this population and therefore should be considered in the treatment regimen.26
In addition to the nicotine degradation pathway/cotinine glucuronidation, we have identified other pathways differentially regulated in the two groups. The d-glucuronate degradation pathway was highly and significantly dysregulated in AA in both positive and negative ESI modes and in the multimodal pathway analysis. The majority of the metabolites in the d-glucuronate degradation pathway were down-regulated in AA. We hypothesize that the altered d-glucuronate degradation pathway could be influenced by the low glucuronidation in AA, or it could be due to differentially regulated carbohydrate metabolic pathways. The upstream pathways such as the sugar/carbohydrate degradation influence the level of the metabolites associated with downstream pathways such as the d-glucuronate degradation pathway. d-Glucuronic acid is important in cellular processes including glucuronidation in xenobiotic metabolism and as a precursor for vitamin C biosynthesis.63,64 These results demonstrate that even without using the nicotine or tobacco-specific biomarkers to stratify these two groups with differing lung cancer risk, the variations in the pattern and profile of metabolites as a result of differentially regulated biological networks between AA and whites can still be ascertained. For instance, excluding the nicotine metabolites in the metabolomics analysis, unique patterns of metabolites in smokers were observed and were used to differentiate this population from nonsmokers based on the global profiles and patterns of metabolites implicated in other affected pathways.40 The differential regulation of the d-glucuronate degradation pathway is not likely smoke induced as both groups of smokers showed no significant differences in TNE, an established biomarker of tobacco smoke exposure. We speculate that analysis of populations of AA and white nonsmokers or healthy controls using our metabolomics approach would allow for the observation of the same metabolic alterations as observed for smokers, except for the nicotine degradation pathway, which is specific to tobacco use. Further studies are warranted to elucidate and understand the consequences of the differentially regulated d-glucuronate degradation pathway as it relates to ethnic and genetics differences and potentially to lung cancer susceptibility among smokers.
Variations in other pathways including the metabolism of amino acids, nucleic acids, and fatty acids were also identified (Table 1 and Table S3–S4). The fatty acid biosynthesis pathway was found to be significantly decreased in AA compared to whites with putative metabolites, palmitate and oleate, being lower in AA. In addition, the amino acid degradation pathways (lysine degradation I and II; saccharopine and pipecolate pathways) were differentially regulated with lower amounts of putative metabolites in AA compared to whites. Furthermore, the carbohydrate degradation and biosynthesis pathways (sucrose degradation, lactose degradation, d-galactose degradation, trehalose degradation, sorbitol degradation, myo-inositol de novo biosynthesis, d-myo-inositol (1,4,5)-trisphosphate biosynthesis, glycogenolysis, UDP-N-acetyl-d-galactosamine biosynthesis II) were also decreased in AA compared to whites, with levels of putative metabolites implicated in the pathways being lower in AA (Table S4). Because the majority of the putative metabolites associated with the carbohydrate metabolic pathways are isobaric and associated with other pathways as well, it is difficult to decipher the exact nature of the differentially regulated metabolic pathway(s) without synthetic standards. For example, β-d-glucose is implicated in multiple pathways including the sucrose degradation, lactose degradation, trehalose degradation, glycogenolysis, and UDP-N-acetyl-d-galactosamine biosynthesis II (Table S4). Further studies are warranted to investigate the biological relevance and implications of these differences between the two groups.
This study has its strengths and limitations. This work, for the first time, provides a comprehensive and global urinary metabolome of smokers from two ethnic groups with differing lung cancer risk. Because of the well-established differences in the metabolism of a few specific nicotine metabolites and tobacco-derived carcinogens in AA and whites, we were able to further confirm these variations in the metabolomics analysis by using a targeted approach to quantify selected nicotine metabolites in the same samples. In addition, the multimodal pathway analysis (positive and negative mode data acquisition and data processing) identified other potentially relevant and novel pathways and putative metabolites not related to smoking, which may still be important in understanding differences in susceptibility to the detrimental effects of tobacco use. Although we were able to confirm the levels and structures of a few nicotine metabolites in the same samples using a targeted MS-based approach, other significant and differentially regulated metabolites detected need further confirmation in the future using synthetic standards. In addition, highly curated and annotated metabolic pathways should be developed. For instance, the metabolic pathways of several tobacco-derived carcinogens such as NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone), NNN (N′-nitrosonornicotine), PAHs (polyaromatic hydrocarbons) are yet to be integrated into online pathway databases such as BioCyc (https://biocyc.org). Previous work of our colleagues and others have comprehensively characterized the metabolism of these tobacco-derived carcinogens as well as the metabolism of nicotine in both human and animal models.9,15,19,65−67 The results from this extensive body of work need to be incorporated into pathway databases or in-house metabolomics workflows to enable more comprehensive “tobacco-focused” metabolomics studies in population-based settings to assess exposure and effects of tobacco use. Because the metabolomics analysis used here measures global metabolite profiles and typically the abundant ones in the sample, trace level compounds due to carcinogen-specific compounds such as NNK, NNN, and PAHs could be missed during the analysis. Furthermore, a minimal cleanup (desalting) step was performed before the metabolomics analysis to capture most of the metabolites in urine, resulting however in being less likely to detect trace level metabolites, which need thorough sample cleanup. In fact, we have previously developed a “focused” metabolomics analysis of known and unknown NNK metabolites in rat urine and the workflow involved extensive cleanup to detect all known and novel NNK metabolites.65 We plan to evaluate this “focused” metabolomics workflow in human smokers’ urine to investigate the NNK metabolite profiles across ethnic groups.
Another limitation of the present work is that we only used male subjects. This gender-specific cohort was used to avoid confounding factors that might complicate the metabolomics data analysis. Since previous studies have shown differences in lung cancer susceptibility between gender, females being at higher risk than males of the same ethnic group.68,69 Furthermore, the differences in the diet between the two groups of smokers could also affect the observed variations in the urinary metabolite profiles. For instance, in a randomized, controlled, crossover trial on healthy volunteers, taking four different types of diets, differences in the urinary metabolic profiles were observed between the four groups.70 While it is possible that the variations we observed in the untargeted metabolomics analysis could be an indirect or direct effects of diet on the overall global urinary metabolome in the two groups of smokers, we did not detect diet-specific metabolites such as hippurate (a marker of fruit and vegetable consumption), (N-acetyl)-S-methyl-l-cysteine-sulfoxide (cruciferous vegetables), dimethylamine and TMAO (fish), and 1-methylhistidine and 3-methyl- histidine (oily fish and chicken) or observed differences in the levels of these compounds in smokers’ urine.70 Future investigation is warranted to evaluate these metabolomics results in a large cohort of subjects including females, influence of dietary intake, as well as in other ethnic groups where disparities in the levels of urinary metabolites are not consistent with the observed cancer risk for these populations. Finally, the metabolomics analysis was performed on a relatively small number of subjects (60 subjects total; AA, n = 30, and white, n = 30), resulting in reduced ability to perform any stratification. Nevertheless, the significant differences in the levels of the metabolites detected in such a small cohort and the consistency of these findings with previous studies performed on selected metabolites in larger cohorts support the robustness of the method and the need to use this approach on a larger study.
Conclusions
The LC-HRMS-based metabolomics analysis of smokers’ urine from AA and whites with differing lung cancer risk identified differentially regulated biological pathways and metabolites, which may be used and validated in future studies as potential biomarkers for predicting the detrimental effects of tobacco use and ultimately contribute in the development of bioanalytical tools to predict lung cancer susceptibility among smokers in population-based studies. We have identified differentially regulated pathways including decreased nicotine degradation, in particular cotinine glucuronidation in AA, and the d-glucuronate degradation. Other pathways including the metabolism of amino acids, nucleic acids, and fatty acids were also identified. Further studies are warranted to investigate the biological relevance and implications of these differences in the metabolic pathway regulations between the two groups. Finally, our metabolomics analysis provides an alternative approach of characterizing the global patterns of metabolites in various ethnic groups where the disparities and differences in risks are not explained by the metabolites currently being measured and illustrate the importance of global profiling of all metabolites to identify other relevant biomarkers and dysregulated biological networks to ultimately develop tools to identify susceptible populations to smoking-related lung cancer.
Acknowledgments
The authors would like to thank the American Society for Mass Spectrometry Postdoctoral Career Development Award to R.D., Prof. Gary Siuzdak and group members (Aries Aisporna, Tao Han, Erica Forsberg, H. Paul Benton, Xavi Domingo, Rafael Montenegro, Carlos Guijas, Brian Hilmers, Bill Web, Linh Hoang, Winnie Heim Duane Rinehart, and Bill Webb) at the Scripps Center for Metabolomics and Mass Spectrometry, The Scripps Research Institute, CA for hosting R.D. to conduct this work, Prof. Sharon E. Murphy and group members, and Laura A. Maertens and Anshu Jain for the sample inventory. B.W. acknowledges the Austrian Science Fund (FWF) for funding (Erwin-Schrödinger Fellowship J-3808). P.V. acknowledges funding from grant NCI R50 CA-211256. Targeted mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support grant P30 CA-077598. This work was supported by grant CA-138338 from the National Cancer Institute.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.0c00064.
Figures S1–S6 and Tables S1–S4 with targeted LC-MS/MS data for NNAL and nicotine metabolites, TNE, creatinine, demographics of the 60 subjects, multimodal pathway analysis results, and extracted ion chromatograms for the associated putative metabolites (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hecht S. S. (2003) Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 3 (10), 733–44. 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- Hecht S. S. (2008) Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol. 21 (1), 160–71. 10.1021/tx7002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Jemal A. (2017) Cancer Statistics, 2017. Ca-Cancer J. Clin. 67 (1), 7–30. 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Antona C.; Gomez A.; Karlgren M.; Sim S. C.; Ingelman-Sundberg M. (2010) Molecular genetics and epigenetics of the cytochrome P450 gene family and its relevance for cancer risk and treatment. Hum. Genet. 127 (1), 1–17. 10.1007/s00439-009-0748-0. [DOI] [PubMed] [Google Scholar]
- Schwartz A. G.; Prysak G. M.; Bock C. H.; Cote M. L. (2006) The molecular epidemiology of lung cancer. Carcinogenesis 28 (3), 507–18. 10.1093/carcin/bgl253. [DOI] [PubMed] [Google Scholar]
- Taioli E. (2008) Gene-environment interaction in tobacco-related cancers. Carcinogenesis 29 (8), 1467–74. 10.1093/carcin/bgn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett W. C. (2002) Balancing life-style and genomics research for disease prevention. Science 296 (5568), 695–8. 10.1126/science.1071055. [DOI] [PubMed] [Google Scholar]
- Murphy S. E.; Park S. L.; Balbo S.; Haiman C. A.; Hatsukami D. K.; Patel Y.; Peterson L. A.; Stepanov I.; Stram D. O.; Tretyakova N.; Hecht S. S.; Le Marchand L. (2018) Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. npj Precis. Oncol. 2, 17. 10.1038/s41698-018-0057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. E. (2017) Nicotine Metabolism and Smoking: Ethnic Differences in the Role of P450 2A6. Chem. Res. Toxicol. 30 (1), 410–419. 10.1021/acs.chemrestox.6b00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland A.; Miners J. O.; Mackenzie P. I. (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 45 (6), 1121–32. 10.1016/j.biocel.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Le Marchand L.; Wilkens L. R.; Kolonel L. N. (1992) Ethnic differences in the lung cancer risk associated with smoking. Cancer Epidemiol. Biomarkers Prev. 1 (2), 103–7. [PubMed] [Google Scholar]
- Haiman C. A.; Stram D. O.; Wilkens L. R.; Pike M. C.; Kolonel L. N.; Henderson B. E.; Le Marchand L. (2006) Ethnic and racial differences in the smoking-related risk of lung cancer. N. Engl. J. Med. 354 (4), 333–42. 10.1056/NEJMoa033250. [DOI] [PubMed] [Google Scholar]
- Wang H.; Park S. L.; Stram D. O.; Haiman C. A.; Wilkens L. R.; Hecht S. S.; Kolonel L. N.; Murphy S. E.; Le Marchand L. (2015) Associations Between Genetic Ancestries and Nicotine Metabolism Biomarkers in the Multiethnic Cohort Study. Am. J. Epidemiol. 182 (11), 945–51. 10.1093/aje/kwv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. L.; Tiirikainen M. I.; Patel Y. M.; Wilkens L. R.; Stram D. O.; Le Marchand L.; Murphy S. E. (2016) Genetic determinants of CYP2A6 activity across racial/ethnic groups with different risks of lung cancer and effect on their smoking intensity. Carcinogenesis 37 (3), 269–279. 10.1093/carcin/bgw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. E.; Sipe C. J.; Choi K.; Raddatz L. M.; Koopmeiners J. S.; Donny E. C.; Hatsukami D. K. (2017) Low Cotinine Glucuronidation Results in Higher Serum and Saliva Cotinine in African American Compared to white Smokers. Cancer Epidemiol., Biomarkers Prev. 26 (7), 1093–1099. 10.1158/1055-9965.EPI-16-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht S. S.; Carmella S. G.; Murphy S. E.; Akerkar S.; Brunnemann K. D.; Hoffmann D. (1993) A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N. Engl. J. Med. 329 (21), 1543–6. 10.1056/NEJM199311183292105. [DOI] [PubMed] [Google Scholar]
- Benowitz N. L.; Dains K. M.; Dempsey D.; Wilson M.; Jacob P. (2011) Racial differences in the relationship between number of cigarettes smoked and nicotine and carcinogen exposure. Nicotine Tob. Res. 13 (9), 772–83. 10.1093/ntr/ntr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar C. A.; Conti D. V.; Das S.; Chen P.; Cook E. H.; Ratain M. J.; Benowitz N. L.; Tyndale R. F. (2015) UGT1A and UGT2B genetic variation alters nicotine and nitrosamine glucuronidation in european and african American smokers. Cancer Epidemiol., Biomarkers Prev. 24 (1), 94–104. 10.1158/1055-9965.EPI-14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. E.; Park S. S.; Thompson E. F.; Wilkens L. R.; Patel Y.; Stram D. O.; Le Marchand L. (2014) Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 35 (11), 2526–33. 10.1093/carcin/bgu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. L.; Kotapati S.; Wilkens L. R.; Tiirikainen M.; Murphy S. E.; Tretyakova N.; Le Marchand L. (2014) 1,3-Butadiene exposure and metabolism among Japanese American, Native Hawaiian, and white smokers. Cancer Epidemiol., Biomarkers Prev. 23 (11), 2240–9. 10.1158/1055-9965.EPI-14-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz N. L.; St Helen G.; Dempsey D. A.; Jacob P.; Tyndale R. F. (2016) Disposition kinetics and metabolism of nicotine and cotinine in African American smokers: impact of CYP2A6 genetic variation and enzymatic activity. Pharmacogenet. Genomics 26 (7), 340–350. 10.1097/FPC.0000000000000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derby K. S.; Cuthrell K.; Caberto C.; Carmella S. G.; Franke A. A.; Hecht S. S.; Murphy S. E.; Le Marchand L. (2008) Nicotine metabolism in three ethnic/racial groups with different risks of lung cancer. Cancer Epidemiol., Biomarkers Prev. 17 (12), 3526–35. 10.1158/1055-9965.EPI-08-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Y. M.; Park S. L.; Han Y.; Wilkens L. R.; Bickeböller H.; Rosenberger A.; Caporaso N.; Landi M. T.; Brüske I.; Risch A.; Wei Y.; Christiani D. C.; Brennan P.; Houlston R.; McKay J.; McLaughlin J.; Hung R.; Murphy S.; Stram D. O.; Amos C.; Le Marchand L. (2016) Novel Association of Genetic Markers Affecting CYP2A6 Activity and Lung Cancer Risk. Cancer Res. 76 (19), 5768–5776. 10.1158/0008-5472.CAN-16-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. E.; Spina D. A.; Nunes M. G.; Pullo D. A. (1995) Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol. 8 (5), 772–9. 10.1021/tx00047a018. [DOI] [PubMed] [Google Scholar]
- Murphy S. E.; Nunes M. G.; Hatala M. A. (1997) Effects of phenobarbital and 3-methylcholanthrene induction on the formation of three glucuronide metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, NNK. Chem.-Biol. Interact. 103 (3), 153–66. 10.1016/S0009-2797(96)03756-8. [DOI] [PubMed] [Google Scholar]
- Fowler S.; Kletzl H.; Finel M.; Manevski N.; Schmid P.; Tuerck D.; Norcross R. D.; Hoener M. C.; Spleiss O.; Iglesias V. A. (2015) A UGT2B10 splicing polymorphism common in african populations may greatly increase drug exposure. J. Pharmacol. Exp. Ther. 352 (2), 358–67. 10.1124/jpet.114.220194. [DOI] [PubMed] [Google Scholar]
- Patel Y. M.; Stram D. O.; Wilkens L. R.; Park S. S.; Henderson B. E.; Le Marchand L.; Haiman C. A.; Murphy S. E. (2015) The contribution of common genetic variation to nicotine and cotinine glucuronidation in multiple ethnic/racial populations. Cancer Epidemiol., Biomarkers Prev. 24 (1), 119–27. 10.1158/1055-9965.EPI-14-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth B.; Spangler S.; Fang M.; Johnson C. H.; Forsberg E. M.; Granados A.; Martin R. L.; Domingo-Almenara X.; Huan T.; Rinehart D.; Montenegro-Burke J. R.; Hilmers B.; Aisporna A.; Hoang L. T.; Uritboonthai W.; Benton H. P.; Richardson S. D.; Williams A. J.; Siuzdak G. (2017) Exposome-Scale Investigations Guided by Global Metabolomics, Pathway Analysis, and Cognitive Computing. Anal. Chem. 89 (21), 11505–11513. 10.1021/acs.analchem.7b02759. [DOI] [PubMed] [Google Scholar]
- Forsberg E. M.; Huan T.; Rinehart D.; Benton H. P.; Warth B.; Hilmers B.; Siuzdak G. (2018) Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 13 (4), 633–651. 10.1038/nprot.2017.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R.; Patti G. J.; Rinehart D.; Siuzdak G. (2012) XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem. 84 (11), 5035–9. 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F.; Pon A.; Wilson M.; Greiner R.; Wishart D. (2014) CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 42, W94–W99. 10.1093/nar/gku436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S.; Schmidt S.; Müller-Hannemann M.; Neumann S. (2010) In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinf. 11, 148. 10.1186/1471-2105-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Xing X.; Chen L.; Yang L.; Su X.; Rabitz H.; Lu W.; Rabinowitz J. D. (2019) Peak Annotation and Verification Engine for Untargeted LC-MS Metabolomics. Anal. Chem. 91 (3), 1838–1846. 10.1021/acs.analchem.8b03132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaženović I.; Kind T.; Sa M. R.; Ji J.; Vaniya A.; Wancewicz B.; Roberts B. S.; Torbašinović H.; Lee T.; Mehta S. S.; Showalter M. R.; Song H.; Kwok J.; Jahn D.; Kim J.; Fiehn O. (2019) Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Anal. Chem. 91 (3), 2155–2162. 10.1021/acs.analchem.8b04698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H.; Cajka T.; Kind T.; Ma Y.; Higgins B.; Ikeda K.; Kanazawa M.; VanderGheynst J.; Fiehn O.; Arita M. (2015) MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12 (6), 523–6. 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijas C.; Montenegro-Burke J. R.; Domingo-Almenara X.; Palermo A.; Warth B.; Hermann G.; Koellensperger G.; Huan T.; Uritboonthai W.; Aisporna A. E.; Wolan D. W.; Spilker M. E.; Benton H. P.; Siuzdak G. (2018) METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 90 (5), 3156–3164. 10.1021/acs.analchem.7b04424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djoumbou-Feunang Y.; Pon A.; Karu N.; Zheng J.; Li C.; Arndt D.; Gautam M.; Allen F.; Wishart D. S. (2019) CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 9 (4), 72. 10.3390/metabo9040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-De-La-Fuente A.; Godzien J.; Saugar S.; Garcia-Carmona R.; Badran H.; Wishart D. S.; Barbas C.; Otero A. (2019) CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 18 (2), 797–802. 10.1021/acs.jproteome.8b00720. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Marcu A.; Guo A. C.; Liang K.; Vázquez-Fresno R.; Sajed T.; Johnson D.; Li C.; Karu N.; Sayeeda Z.; Lo E.; Assempour N.; Berjanskii M.; Singhal S.; Arndt D.; Liang Y.; Badran H.; Grant J.; Serra-Cayuela A.; Liu Y.; Mandal R.; Neveu V.; Pon A.; Knox C.; Wilson M.; Manach C.; Scalbert A. (2018) HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46 (D1), D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez I.; Lindon J. C.; Minet E. (2014) Application of CE-MS to a metabonomics study of human urine from cigarette smokers and non-smokers. Bioanalysis 6 (20), 2733–49. 10.4155/bio.14.136. [DOI] [PubMed] [Google Scholar]
- Kaluarachchi M. R.; Boulangé C. L.; Garcia-Perez I.; Lindon J. C.; Minet E. F. (2016) Multiplatform serum metabolic phenotyping combined with pathway mapping to identify biochemical differences in smokers. Bioanalysis 8 (19), 2023–43. 10.4155/bio-2016-0108. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P.; Nair S.; Rangiah K. (2016) A method for comparative metabolomics in urine using high resolution mass spectrometry. J. Chromatogr A 1443, 83–92. 10.1016/j.chroma.2016.02.080. [DOI] [PubMed] [Google Scholar]
- Hsu P. C.; Zhou B.; Zhao Y.; Ressom H. W.; Cheema A. K.; Pickworth W.; Shields P. G. (2013) Feasibility of identifying the tobacco-related global metabolome in blood by UPLC-QTOF-MS. J. Proteome Res. 12 (2), 679–91. 10.1021/pr3007705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. C.; Lan R. S.; Brasky T. M.; Marian C.; Cheema A. K.; Ressom H. W.; Loffredo C. A.; Pickworth W. B.; Shields P. G. (2017) Menthol Smokers: Metabolomic Profiling and Smoking Behavior. Cancer Epidemiol., Biomarkers Prev. 26 (1), 51–60. 10.1158/1055-9965.EPI-16-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. C.; Lan R. S.; Brasky T. M.; Marian C.; Cheema A. K.; Ressom H. W.; Loffredo C. A.; Pickworth W. B.; Shields P. G. (2017) Metabolomic profiles of current cigarette smokers. Mol. Carcinog. 56 (2), 594–606. 10.1002/mc.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulimiri S. V.; Misra M.; Hamm J. T.; Mitchell M.; Berger A. (2009) Effects of mainstream cigarette smoke on the global metabolome of human lung epithelial cells. Chem. Res. Toxicol. 22 (3), 492–503. 10.1021/tx8003246. [DOI] [PubMed] [Google Scholar]
- Barupal D. K.; Pinkerton K. E.; Hood C.; Kind T.; Fiehn O. (2016) Environmental Tobacco Smoke Alters Metabolic Systems in Adult Rats. Chem. Res. Toxicol. 29 (11), 1818–1827. 10.1021/acs.chemrestox.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross A. J.; Boca S.; Freedman N. D.; Caporaso N. E.; Huang W. Y.; Sinha R.; Sampson J. N.; Moore S. C. (2014) Metabolites of tobacco smoking and colorectal cancer risk. Carcinogenesis 35 (7), 1516–22. 10.1093/carcin/bgu071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.; Zhang J.; Fu X.; Ma S.; Wang C.; Wang J.; Tian S.; Liu S.; Zhao B.; Wang X. (2016) LC-MS based metabolomics identification of novel biomarkers of tobacco smoke-induced chronic bronchitis. Biomed. Chromatogr. 30 (1), 68–74. 10.1002/bmc.3620. [DOI] [PubMed] [Google Scholar]
- Stepanov I.; Upadhyaya P.; Carmella S. G.; Feuer R.; Jensen J.; Hatsukami D. K.; Hecht S. S. (2008) Extensive metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers. Cancer Epidemiol., Biomarkers Prev. 17 (7), 1764–73. 10.1158/1055-9965.EPI-07-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht S. S.; Carmella S. G.; Chen M.; Dor Koch J. F.; Miller A. T.; Murphy S. E.; Jensen J. A.; Zimmerman C. L.; Hatsukami D. K. (1999) Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. 59 (3), 590–6. [PubMed] [Google Scholar]
- Warth B.; Levin N.; Rinehart D.; Teijaro J.; Benton H. P.; Siuzdak G. (2017) Metabolizing Data in the Cloud. Trends Biotechnol. 35 (6), 481–483. 10.1016/j.tibtech.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; O’Maille G.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. (2005) METLIN: a metabolite mass spectral database. Ther. Drug Monit. 27 (6), 747–751. 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Huan T.; Forsberg E. M.; Rinehart D.; Johnson C. H.; Ivanisevic J.; Benton H. P.; Fang M.; Aisporna A.; Hilmers B.; Poole F. L.; Thorgersen M. P.; Adams M. W. W.; Krantz G.; Fields M. W.; Robbins P. D.; Niedernhofer L. J.; Ideker T.; Majumder E. L.; Wall J. D.; Rattray N. J. W.; Goodacre R.; Lairson L. L.; Siuzdak G. (2017) Systems biology guided by XCMS Online metabolomics. Nat. Methods 14 (5), 461–462. 10.1038/nmeth.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda H.; Ivanisevic J.; Johnson C. H.; Kurczy M. E.; Benton H. P.; Rinehart D.; Nguyen T.; Ray J.; Kuehl J.; Arevalo B.; Westenskow P. D.; Wang J.; Arkin A. P.; Deutschbauer A. M.; Patti G. J.; Siuzdak G. (2014) Interactive XCMS Online: simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal. Chem. 86 (14), 6931–9. 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T.; Palermo A.; Ivanisevic J.; Rinehart D.; Edler D.; Phommavongsay T.; Benton H. P.; Guijas C.; Domingo-Almenara X.; Warth B.; Siuzdak G. (2018) Autonomous Multimodal Metabolomics Data Integration for Comprehensive Pathway Analysis and Systems Biology. Anal. Chem. 90 (14), 8396–8403. 10.1021/acs.analchem.8b00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. E.; Wickham K. M.; Lindgren B. R.; Spector L. G.; Joseph A. (2013) Cotinine and trans 3′-hydroxycotinine in dried blood spots as biomarkers of tobacco exposure and nicotine metabolism. J. Exposure Sci. Environ. Epidemiol. 23 (5), 513–8. 10.1038/jes.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer G.; Engl J.; Urban M.; Gilch G.; Janket D.; Riedel K. (2007) Relationship between machine-derived smoke yields and biomarkers in cigarette smokers in Germany. Regul. Toxicol. Pharmacol. 47 (2), 171–83. 10.1016/j.yrtph.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liang Q.; Mendes P.; Sarkar M. (2011) Is 24h nicotine equivalents a surrogate for smoke exposure based on its relationship with other biomarkers of exposure?. Biomarkers 16 (2), 144–54. 10.3109/1354750X.2010.536257. [DOI] [PubMed] [Google Scholar]
- Miller I. J.; Peters S. R.; Overmyer K. A.; Paulson B. R.; Westphall M. S.; Coon J. J. (2019) Real-time health monitoring through urine metabolomics. NPJ. Digit Med. 2, 109. 10.1038/s41746-019-0185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J. Z.; von Weymarn L. B.; Thompson E. A.; Wickham K. M.; Weisensel N. A.; Hatsukami D. K.; Murphy S. E. (2010) UGT2B10 genotype influences nicotine glucuronidation, oxidation, and consumption. Cancer Epidemiol., Biomarkers Prev. 19 (6), 1423–31. 10.1158/1055-9965.EPI-09-0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie P. I.; Hu D. G.; Gardner-Stephen D. A. (2010) The regulation of UDP-glucuronosyltransferase genes by tissue-specific and ligand-activated transcription factors. Drug Metab. Rev. 42 (1), 99–109. 10.3109/03602530903209544. [DOI] [PubMed] [Google Scholar]
- Mazerska Z.; Mróz A.; Pawłowska M.; Augustin E. (2016) The role of glucuronidation in drug resistance. Pharmacol. Ther. 159, 35–55. 10.1016/j.pharmthera.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Linster C. L.; Van Schaftingen E. (2007) Vitamin C Biosynthesis, recycling and degradation in mammals. FEBS J. 274 (1), 1–22. 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- Dator R.; von Weymarn L. B.; Villalta P. W.; Hooyman C. J.; Maertens L. A.; Upadhyaya P.; Murphy S. E.; Balbo S. (2018) In Vivo Stable-Isotope Labeling and Mass-Spectrometry-Based Metabolic Profiling of a Potent Tobacco-Specific Carcinogen in Rats. Anal. Chem. 90 (20), 11863–11872. 10.1021/acs.analchem.8b01881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht S. S.; Young R.; Chen C. B. (1980) Metabolism in the F344 rat of 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Cancer Res. 40 (11), 4144–50. [PubMed] [Google Scholar]
- Hecht S. S. (1997) Approaches to cancer prevention based on an understanding of N-nitrosamine carcinogenesis. Exp. Biol. Med. 216 (2), 181–91. 10.3181/00379727-216-44168. [DOI] [PubMed] [Google Scholar]
- Dresler C. M.; Fratelli C.; Babb J.; Everley L.; Evans A. A.; Clapper M. L. (2000) Gender differences in genetic susceptibility for lung cancer. Lung Cancer 30 (3), 153–60. 10.1016/S0169-5002(00)00163-X. [DOI] [PubMed] [Google Scholar]
- Zang E. A.; Wynder E. L. (1996) Differences in lung cancer risk between men and women: examination of the evidence. J. Natl. Cancer Inst 88 (3–4), 183–192. 10.1093/jnci/88.3-4.183. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez I.; Posma J. M.; Gibson R.; Chambers E. S.; Hansen T. H.; Vestergaard H.; Hansen T.; Beckmann M.; Pedersen O.; Elliott P.; Stamler J.; Nicholson J. K.; Draper J.; Mathers J. C.; Holmes E.; Frost G. (2017) Objective assessment of dietary patterns by use of metabolic phenotyping: a randomised, controlled, crossover trial. Lancet Diabetes Endocrinol. 5 (3), 184–195. 10.1016/S2213-8587(16)30419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huag K.; Cochrane K.; Nainala V. C.; Williams M.; Chang J.; Jayaseelan K. V.; O'Donovan C. (2020) MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444. 10.1093/nar/gkz1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.