

Abstract
Hepatic ischemia-reperfusion injury (IRI), a major risk factor for early allograft dysfunction (EAD) and acute or chronic graft rejection, contributes to donor organ shortage for life-saving orthotopic liver transplantation (OLT). The graft injury caused by local ischemia (warm and/or cold) leads to parenchymal cell death and release of danger-associated molecular patterns (DAMPs), followed by reperfusion-triggered production of reactive oxygen species (ROS), activation of inflammatory cells, hepatocellular damage and ultimate organ failure. Heme oxygenase 1 (HO-1), a heat shock protein-32 induced under IR-stress, is an essential component of the cytoprotective mechanism in stressed livers. HO-1 regulates anti-inflammatory responses and may be crucial in the pathogenesis of chronic diseases, such as arteriosclerosis, hypertension, diabetes and steatosis. An emerging area of study is macrophage-derived HO-1 and its pivotal intrahepatic homeostatic function played in IRI-OLT. Indeed, ectopic hepatic HO-1 overexpression activates intracellular SIRT1/autophagy axis to serve as a key cellular self-defense mechanism in both mouse and human OLT recipients. Recent translational studies in rodents and human liver transplant patients provide novel insights into HO-1 mediated cytoprotection against sterile hepatic inflammation. In this review, we summarize the current bench-to-bedside knowledge on HO-1 molecular signaling and discuss their future therapeutic potential to mitigate IRI in OLT.
Graphical Abstract

Introduction
Liver ischemia-reperfusion injury (IRI), a multifactorial process triggered by transient tissue deprivation of oxygen, followed by reoxygenation, may occur in clinical states such as shock, trauma, hepatic resection, procurement and orthotopic liver transplantation (OLT). Although OLT has become the standard life-saving strategy for patients with end-stage liver failure and those with malignancies, there are currently no therapeutics against or patient-specific diagnostics of peri-transplant hepatic IRI [1].
Although transplantation rates have improved progressively with a concomitant reduction of mortality, the limited supply of donor livers remains a major challenge [2]. According to Organ Procurement and Transplantation Network [US Department of Health and Human Services; http://optn.transplant.hrsa.gov/data/], in 2017, out of 13,239 patients on the liver transplant waiting list, only 8,082 received the transplant; 1,248 died while awaiting for the life-saving organ and another 1,329 individuals were removed from the list because they became too sick. Donor organ shortage has forced the use of extended criteria organs ranging from steatosis, fibrosis, or non-heart beating donors (NHBD), which may also suffer from prolonged periods of warm ischemia. These marginal grafts being highly susceptible to IR-stress, often suffer from primary non-function (PNF), early allograft dysfunction (EAD), as well as undergo more frequent acute and chronic rejection episodes. The incidence of EAD, ranging from 10.8% to 36.3% depending on the facility [3], often closely associates with the overall graft survival rate [4, 5]. As mechanisms that account for liver IRI are not well appreciated, novel biomarkers of hepatocellular graft function as well as strategies to improve clinical OLT outcomes and expand donor pool are warranted.
The mechanism of hepatic tissue IRI encompasses innate immune cell activation and augmented oxidative stress responses. The latter can also be observed during metabolic disorders, such as obesity [6–8] or diabetes [9–11] due to a myriad of environmental stressors, which trigger hepatic sterile inflammation or metabolism disturbances. A key early derivative source of tissue injury is the ischemia phase of IRI [12] when ceasing the blood flow and accompanying lack of oxygen supply triggers cellular glycogen and/or ATP depletion. The resulting initial parenchymal cell death leads to release of danger-associated molecular patterns (DAMPs). During the reperfusion phase, cells recover from the ischemic insult but now generate ROS that lead to further impairment of microcirculation and coagulation disorders with a simultaneous massive influx of inflammatory cells, all contributing to hepatocellular injury and ultimate organ failure. However, as discussed in this review, redox signaling may provide indispensable protection not only in terms of homeostatic maintenance but also for the prevention of several diseases.
The activation of immune cells, such as macrophages, liver-resident Kupffer cells (KCs), neutrophils and T cells leads to the release of pro-inflammatory ROS, cytokines/chemokines, complement activation, and vascular cell adhesion molecular programs, accelerating the hepatocellular damage. Macrophages are central in initiating these inflammatory cascades by activating pattern recognition receptors (PRRs), which further deteriorates the liver function. In particular, macrophage-derived heme oxygenase-1 (HO-1) has been shown to be crucial in the pathophysiology of organ IRI [13–16].
The heme oxygenase (HO) system is the rate-limiting step in the conversion of heme into ferrous iron (Fe2+), biliverdin (BV) and carbon monoxide (CO). HO-1, a heat shock protein 32 (Hsp32), is the inducible HO isoform, the expression of which in mammalian tissues is relatively low under normal physiological conditions, except of the myeloid spleen cells [17]. Our group was the first to report on the protective role of HO-1 induction in the steatotic liver transplant rat model [18], and proposed that HO-1 may serve as a therapeutic target against oxidative stress in OLT recipients [19]. Numerous subsequent studies have confirmed the therapeutic potential of pharmacological or genetic HO-1 modulations against IR-stress in brain, heart, liver, kidney, and intestine [20–25]. Although cytoprotective, antioxidant and anti-inflammatory functions of HO-1 have long been associated with heme catabolism, recent studies have linked HO-1 with the autophagy intracellular pathway [15, 25, 26], a new and potent cytoprotective mechanism against peri-transplant organ damage.
In this review, we first focus on our current understanding of the cellular and molecular mechanisms of HO-1 function, emphasizing the emerging role of HO-1/SIRT1/autophagy in experimental IRI-OLT. Then, we discuss therapeutic prospects for the modulation of HO-1 expression in the clinical settings.
Liver IRI: An innate-dominated local inflammation response
The discovery of Toll-like Receptor 4 (TLR4) and identification of the high mobility group box 1 (HMGB1) and lipopolysaccharide (LPS) as TLR4 endogenous ligands have changed our appreciation of the innate immune system. Functionally, TLR4 is involved in multiple signaling pathways, influencing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) or interferon regulatory factors (IRFs), which then enhance distinct cytokine and chemokine programs. As one of the key mediators in IR-triggered sterile inflammation, disruption of TLR4 prompted high resistance against IR-stress in mouse models of warm as well as extended cold ischemia, followed by OLT [27–29]. Of note, heme, as we discuss later, has been identified as one of the TLR4 ligands to exert pro-inflammatory effects.
HMGB1 is important for the function of transcription factor p53, one of the tumor suppressor genes that regulates cell cycle, apoptosis and DNA repair [30]. Damaged hepatocytes and liver sinusoidal endothelial cells (LSECs) can also release HBGB1 during IRI, which once released outside the cell exerts DAMPs activity via macrophage TLR4 signaling axis. As a result, the expression of cell adhesion molecules, such as ICAM-1 or VCAM-1, increases in IR-stressed liver, coinciding with infiltration by neutrophils, monocyte-derived macrophages and T cells [31, 32]. Such a negative chain of well-orchestrated immune events may prolong IR-triggered acute liver inflammation cascade.
TLR4 and interferon gamma (IFN-γ) signaling polarize liver-sequestered monocyte-derived macrophages into the M1 phentotype. This polarization stimulates the release of proinflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), IL-12, or ROS, which then induce Th1-type immune response. Although M1 macrophages exert strong antibacterial, antiviral and antitumor effects, they promote a pro-inflammatory phenotype in liver IRI [33, 34]. In contrast, macrophages stimulated with IL-4 and IL-13, Th2-type mediators, become M2 macrophages (arginase/mannose receptor positive), promoting tissue repair, angiogenesis and immunosuppressive effects by releasing IL-10. Although KCs are thought to initiate inflammatory responses via TLR4 signaling, they may also produce inhibitory cytokines, such as IL-10 or IL-4. More recently, resveratrol, a SIRT1 activator, and adiponectin have been reported to promote IL-10 secretion from M2-type KCs in alcoholic liver disorders (ALD) and non-alcoholic-steatohepatitis (NASH). Interestingly, IL-10 produced by M2-type KCs promoted selective M1 macrophage death by a mechanism involving arginase activation [35]. Clearly more studies are needed to better understand how polarized macrophages regulate sterile inflammation in liver IRI.
Hepatic IRI in OLT: A case for innate - adaptive immune interplay
IR-triggered tissue damage in OLT results from two, often overlapping types of cold and warm hepatocellular injury [36]. Cold IRI, characterized by damage to liver sinusoidal endothelial cells (LSECs) following microcirculation disturbance, can occur during the preservation stage after organ procurement [37]. By contrast, warm IRI initiates hepatocellular damage and occurs during the implantation of the cold-preserved liver into the recipient during revascularization. In cases of donation after cardiac death (DCD), donor livers suffer from additional extended periods of warm ischemic injury. In the ischemic stage, lack of oxygen supply leads to the initial hepatocyte cell death, and DAMPs release, such as HMGB1. After reperfusion, activated liver resident KCs produce cytokines and chemokines promoting infiltration of circulating monocyte-derived macrophages, neutrophils and T cells. Tissue-invading inflammatory immune cells further generate ROS and cytokine/chemokine programs, which lead to the increase of adhesion molecule expression and elaboration of other DAMPs.
The adaptive immune system has long been thought to be independent in the pathogenesis of liver IRI. By using adoptive cell transfer system in CD4 T cell-deficient mice, Zwacka et al. first documented the contribution of host T cell immune repertoire for activation and regulation of the hepatic tissue pro-inflammatory response to IR-stress [38]. As CD4 T cells can promote innate immune activation by two mechanisms, i.e., inflammatory cytokines (e.g. IFNγ) or co-stimulatory molecule signaling (e.g. CD28), we have reported that CD4 T cells function in liver IRI via CD154 without de novo antigen-specific activation, while innate immunity-induced CD40 up-regulation may trigger the engagement of CD154-CD40 to facilitate tissue inflammation/injury [38, 39]. Subsequently, we have documented that T cell immunoglobulin mucin family (Tim) and programmed cell death 1 (PD-1), which are currently attracting major attention as immune checkpoint molecules, regulates IR-induced sterile inflammation, as well hepatocellular damage in a murine OLT model [40–44]. Thus, despite the absence of exogenous antigen in hepatic IRI under sterile conditions or in syngeneic liver transplantation, CD4+ T cells may readily promote the pro-inflammatory phenotype of innate immune activation via crucial co-stimulatory signaling. Figure 1 depicts a simplified scheme of intricate innate - adaptive immune interplay in the mechanism of hepatic IRI.
Figure 1:
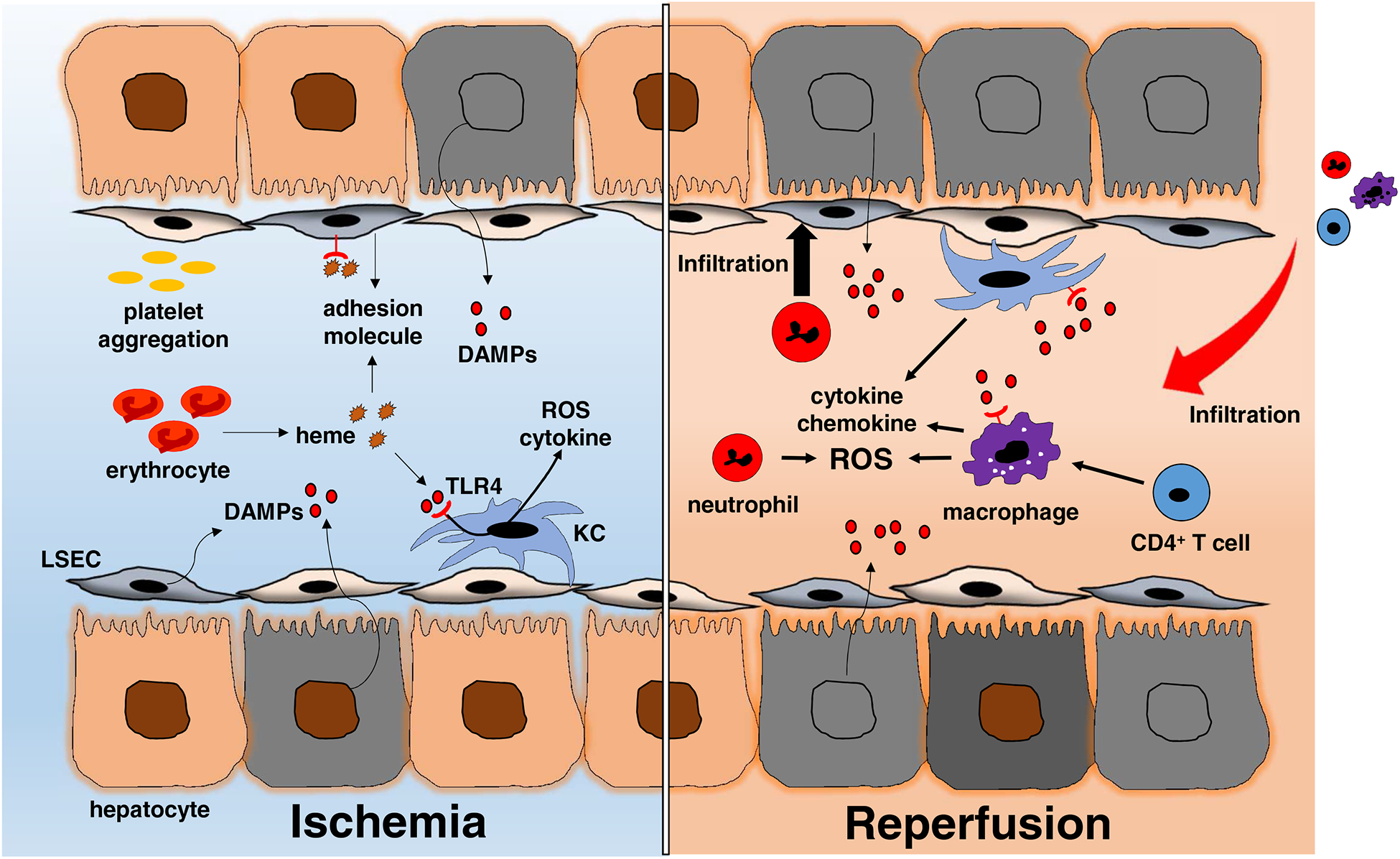
The distinct stages of liver IRI. The ischemia insult, an initial process of hepatic disturbances, results from the lack of oxygen supply. Subsequently, DAMPs released by dead cells up-regulate the expression of adhesion molecules by liver sinusoidal endothelial cells (LSEC) and production of ROS by activated Kupffer cells. Excessive heme released from damaged erythrocytes may trigger inflammatory responses through TLR4-heme signaling axis. Reperfusion injury, triggered by oxygenation, accelerates hepatic IR-immune response via Kupffer cell activation in concert with monocyte-derived macrophage, T cell and neutrophil infiltration. The IRI proinflammatory immune cascade sustains itself by recruiting peripheral immune cells from the circulation.
The “classical” protective functions of HO-1
Heme and its degradation
The biosynthesis of heme, a complex of iron and protoporphyrin IX, occurs in erythroid cells and in the liver [45]. Under physiological conditions, heme provides indispensable cellular functions, but once released into the blood stream, it functions as a cytotoxic protein. In small amounts, free heme promptly binds to haptoglobin and is engulfed by phagocytic cells [46]. The large amounts of heme when released during hemolysis, associate with erythrocyte abnormalities, such as sickle cell disease, sepsis, and ischemia-reperfusion injury [47, 48]. Indeed, excessive heme causes tissue damage due to its ability to produce oxygen free radicals (OFRs), which in turn activate inflammatory cell cascades. Of note, heme may also trigger TLR4 signaling in endothelial cells resulting in the impairment of sinusoidal perfusion through adhesion molecule expression [47]; and may serve as a TLR4 ligand in macrophage proinflammatory function, leading to the secretion of TNF-α in a TLR4, MyD88 and CD14 dependent mechanism [49]. Thus, excessive heme accelerates the inflammatory cascade in TLR4-dependent manner.
HO-1 catabolizes heme complex into its metabolic byproducts (biliverdin, free iron and CO) under the control of the microsomal NADPH-cytochrome P-450 reductase. In the absence of HO-1, heme can cause cell/tissue injury, while heme catabolism exerts cytoprotective functions against oxidative stress in IR-stressed liver [19, 50]. The expression of HO-1 is induced by the several stimuli, such as ROS, heme, loss of cellular glutathione, hydrogen peroxide, LPS and hypoxia [51]. The acute hypoxia also triggers the transcriptional response by hypoxia inducible factor 1 alpha (HIF1α), which in turn induces HO-1 to attenuate IR-mediated cellular damage [52, 53].
Biliverdin
BV, an intermediate produced in the process of heme degradation, is converted into bilirubin (BR) by biliverdin reductase. Both BV and BR have strong antioxidant properties through their ability to scavenge ROS [54]. Several studies have shown that BV may exert potent anti-inflammatory effects against IR-induced cerebral [55], kidney [56], and heart [57] injury. We also reported the protective function of BV in an ex vivo perfusion hepatic IRI model [50, 58], and other attempts have been successfully made to expand the application of BV treatment to large animal hepatic IR models [59]. Notably, BV suppressed T cell proliferation by inhibition of IL-2 synthesis through NFAT/NF-kB pathway [60]. Hence, BV and upstream HO-1 have a prominent protective function against not only innate immune-driven IRI but also host acute rejection adaptive immune-responses. Some even suggest that BV and BR system may modulate late chronic allograft rejection [57].
Free Iron and Ferritin
Free iron is toxic to cells due to generated harmful ROS through the Fenton reaction, which prompts rapid expression of the iron-binding proteins [61]. Since Ferritin is responsible for generating pools of iron and can be induced by stress conditions like hypoxia, it is considered an acute reactive protein that is involved in the homeostasis of intracellular concentrations of iron [62, 63]. Indeed, Adenovirus (Ad)-mediated overexpression of the ferritin heavy chain (H-ferritin) protected rat livers from IRI through an anti-apoptotic mechanism [64]. Interestingly, a recent retrospective clinical study documented that high serum ferritin (SF) levels in the donor was associated with increased risk of IRI in OLT recipients, suggesting that SF could be useful as a predictive biomarker of IRI [65]. This may also explain why iron overload might be harmful and result in poor long-term prognosis. Likewise, HO-1 overexpression does not necessarily impose cytoprotection in some disease states, such as neurodegeneration or carcinogenesis [6, 66].
Carbon Monoxide
Carbon monoxide (CO), one of the metabolites of heme degradation by HO-1, is extremely harmful at high doses because of its ability to bind hemoglobin and prevent oxygen supply to the organs. Unlike carbon dioxide, CO is almost insoluble in water. However, one of the beneficial effect of CO has been thought to maintain microcirculation due to its ability to modulate thrombomodulin (TM) or protein C [67–69], as well as endothelium-dependent vasodilatation and inhibition of platelet aggregation [70–72]. We and others have reported the novel anti-apoptotic effects of CO signaling, dependent on the activation of mitogen-activated protein kinase (MAPK) pathway [73, 74]. Recently, Sun et al. have shown that CO treatment mitigated hepatic IRI via inhibition of HMGB1 translocation under the control of SIRT1, referred to as a “longevity gene” [75]. Moreover, CO may also induce immunosuppression by inhibiting T cell proliferation and blocking IL-2 production [76] or by inducing an anti-apoptotic phenotype in regulatory T cells [77]. Importantly, the application of CO has been successfully expanded to large animal models, as evidenced by reduced pulmonary [78] and hepatic IRI [79], as well as prolonged survival of swine lung transplants [80]. Growing body of evidence indicates CO treatment can be one of promising candidates for the clinical use. The development of chemical CO releasing compounds, as well as clinical use of low dose CO therapy, as recently reported in the acute respiratory distress syndrome patients [81], should pave the way to determine the therapeutic usefulness of this gas in human inflammatory liver diseases. Figure 2 illustrates the heme degradation cascade.
Figure 2:
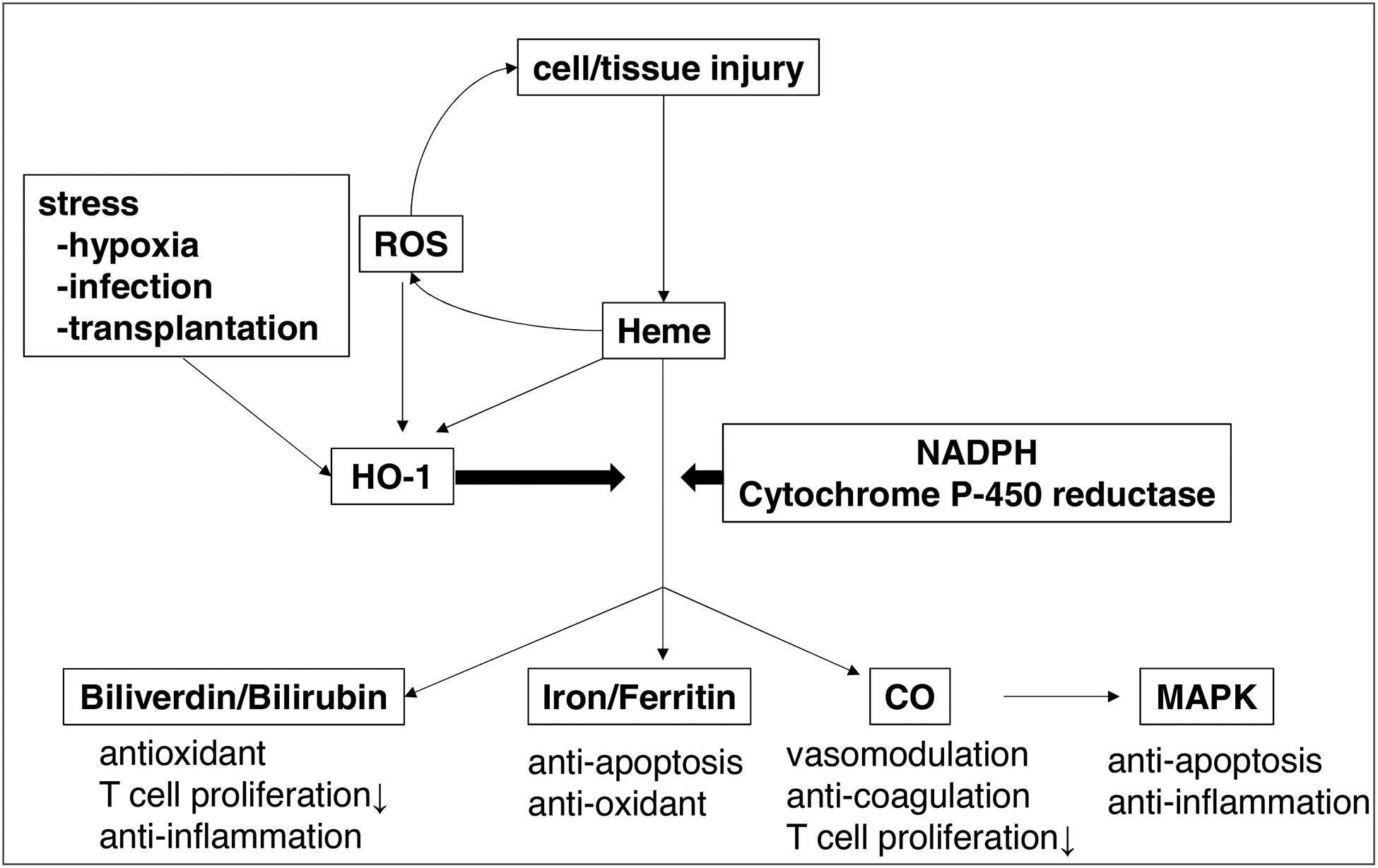
The heme degradation cascade. Heme released from damaged cells produces ROS. HO-1, induced by various stimuli such as heme, ROS, hypoxia, infection or transplantation, catabolizes heme into biliverdin, Fe2+ and CO. Biliverdin scavenges ROS to exert anti-inflammatory effects. Ferritin provides anti-apoptotic effects via its anti-oxidant function. CO promotes anti-apoptosis and anti-inflammatory effects via MAPK activation, modulates the vascular tone and inhibits platelet aggregation, leading to the microcirculatory maintainance.
“Non-classical” HO-1 macrophage pathway
HO-1 cytoprotection in BMDM
The role of HO-1 system as a putative target against innate immune-driven IRI has been extensively studied [19, 82]. Our group was the first to document the cytoprotective functions of exogenous HO-1 induction by pharmacological or gene induction by cobalt-protoporphyrin (CoPP) and adenoviral gene transfer, respectively, in a fatty Zucker rat liver transplantation model [18]. Indeed, HO-1 overexpression was required to maintain tissue integrity, preserve organ function, and improve liver transplant survival, at least in part by sparing graft-infiltrating HO-1 expressing macrophages [83]. We have also shown that HO-1 selectively mitigated infiltration by proinflammatory macrophages, and depressed caspase-3 expression while simultaneously increasing the anti-apoptotic hepatic phenotype [84]. In addition, systemic upregulation of HO-1 by CoPP or gene transfer ameliorated warm hepatic IRI by depressing Type 1 interferon while promoting anti-apoptotic pathway [85–87]. Based on these findings, we proposed that macrophages-derived HO-1 signaling may control the severity of hepatic IRI [13].
Consistent with the idea that macrophage HO-1 might regulate innate immune inflammation in OLT, we have shown that adoptive transfer of genetically HO-1 overexpressing BMDMs prior to the transplant, suppressed pro-inflammatory responses and promoted anti-apoptotic effects in a rat syngeneic OLT model [88]. We have identified macrophages (both infiltrating and Kupffer cell resident) in IR-stressed liver grafts as the main HO-1 producers not only in the rat [83] but also in human liver transplant patients [15]. Considering their inherent phagocytic functions, it seems reasonable that macrophages acquire a strong HO-1 production ability for self-protection [89]. These findings confirm the validity of a novel investigative tool in which native macrophages can be transfected ex vivo with cytoprotective HO-1 gene, and then infused, if needed, to prospective recipients to mitigate IR-mediated sterile inflammation, such as during liver transplantation, resection or trauma. As decreasing donor organ quality represents one of the most challenging problems in transplantation, one may then investigate a new and clinically attractive HO-1-based gene targeted strategy to “rejuvenate” extended criteria donor livers and thus improve their function in OLT recipients.
HO-1/SIRT1/Autophagy axis in liver IRI
Activation of silent information regulator factor 2-related enzyme 1 (SIRT1), a NAD+-dependent type III histone/protein deacetylase, is essential for tissue protection against IR-stressI [90]. SIRT1 expression, up-regulated during starvation, under oxidative stress or DNA damage, plays a key role in the autophagy induction [91, 92]. Autophagy, an intracellular “self-cleaning” mechanism that has been in the limelight in recent years, helps to better understand basic hepatic functions, such as glycogenolysis, gluconeogenesis and β-oxidation [93]. Although autophagy was suppressed in hepatocytes under some conditions like metabolic disorders or oxidative stress, pharmacological activation of autophagy alleviated steatosis and hepatic injury in fatty liver mouse models [94, 95].
In our recent studies, macrophage – SIRT1 signaling axis was critical for HO-1 driven anti-inflammatory programs and M2 polarization in IR-stressed livers [15, 16]. Moreover, we have documented that HO-1 enhanced autophagy in IR-stressed livers, both in a clinically relevant mouse model and in human OLT patients [26]. In addition, disruption of SIRT1 depressed the autophagy enhancement in mouse liver grafts subjected to Ad-HO-1 gene transfer, suggesting that HO-1 regulates autophagy in a SIRT1-dependent manner. As a novel anti-inflammatory function of HO-1, we have since elucidated an essential role of macrophage HO-1 in the regulation of IRI-OLT via a SIRT1-autophagy pathway. We have reported that post-reperfusion HO-1 levels in liver grafts positively correlated with microtubule-associated protein light chain 3B (LC3B), one of the established autophagy markers. The expression of LC3B was observed mainly in hepatocytes, while HO-1 was predominantly expressed by infiltrating macrophages in IR-stressed livers. Moreover, bone marrow-derived macrophages overexpressing HO-1 by ex vivo gene transfer significantly increased liver graft LC3B protein levels. As the manipulation of HO-1 expression was limited primarily to non-parenchymal cells, these findings indicate that myeloid cell-derived HO-1 regulates autophagy pathway in IR-stressed hepatocytes. However, how macrophage HO-1 regulates the hepatic autophagy program awaits future investigation. Figure 3 depicts putative function of HO-1/SIRT1/ autophagy axis in liver IRI.
Figure 3:
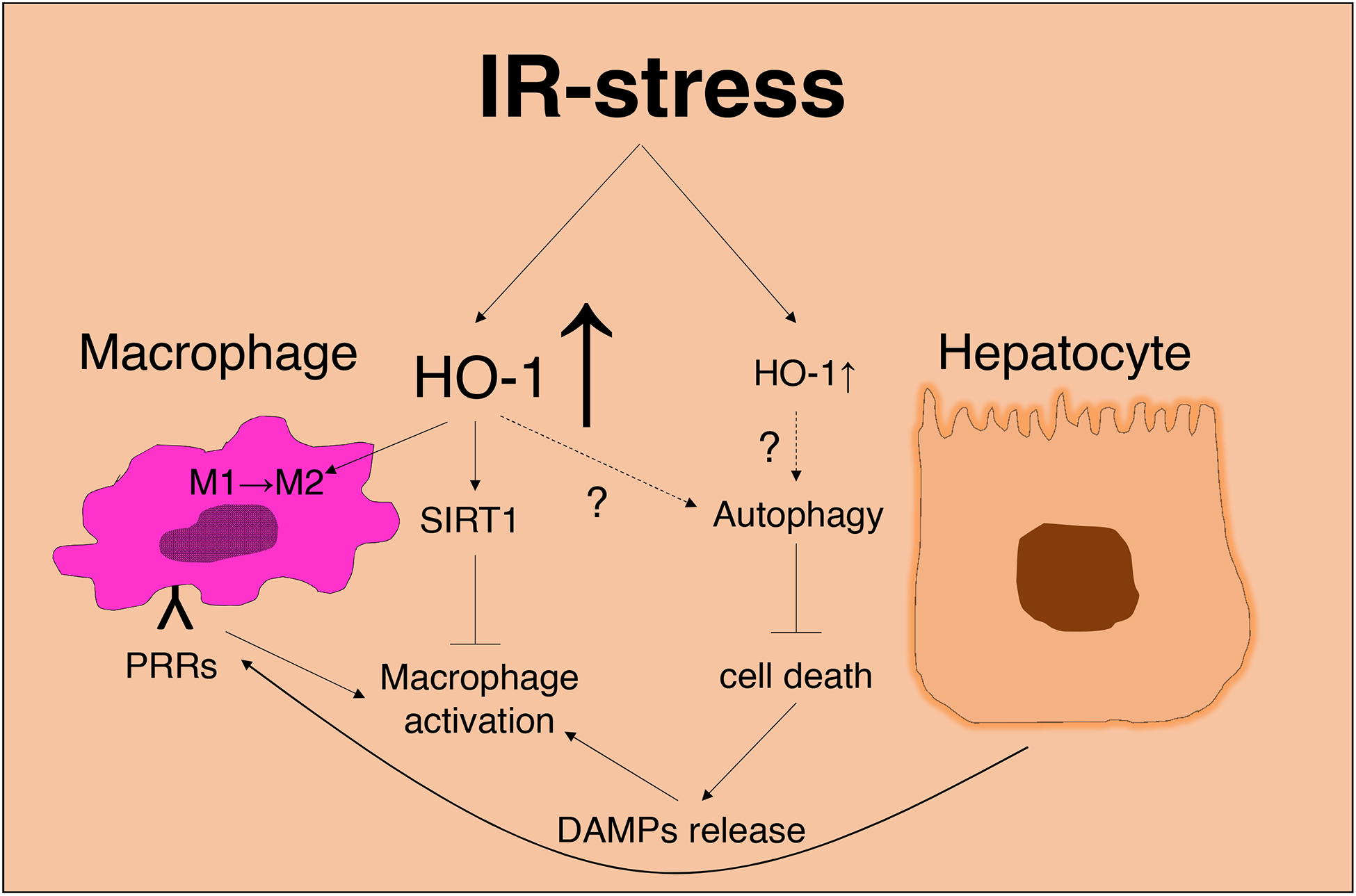
IR-stress induced HO-1 in liver macrophage and hepatocyte function. Stressed hepatocytes release DAMPs, which activate macrophages through PRRs, leading to pro-inflammatory mediator release. By activating SIRT1, myeloid cell-derived HO-1 may also control macrophage polarization towards the anti-inflammatory phenotype. It remains unknown how macrophage HO-1 regulates this “cytoprotective” autophagy pathway in IR-stressed hepatocytes.
HO-1 in LT donor vs. recipient
Our interest in establishing HO-1-based application in clinical organ transplantation has led us to address whether HO-1 should be induced in the donor liver or OLT recipient. We analyzed the hepatic HO-1 expression level in fifty-one adult primary human OLT recipients. Liver biopsies were collected during cold storage (prior to implantation) and 2 hours after portal reperfusion (prior to skin closure). Consistent with our previous experimental data [15], post-operative but not pre-operative liver HO-1 expression negatively correlated with IRI severity in OLT patients. Although a statistically significant difference could not be obtained, when HO-1 expression before and after transplantation was compared between high and low groups in the median, pre-transplant HO-1 low group tended to have a better prognosis, whereas the post-transplant HO-1 low group showed worse clinical outcome. Of note, we found a strong correlation between pre-transplant and post-transplant HO-1 expression levels. In addition, when the post-transplant/pre-transplant HO-1 ratio, representing an index of peri-transplant HO-1 enhancement in the graft was analyzed, we discovered there was a positive correlation with post-transplant HO-1 expression. We hypothesized that pre-transplant HO-1 expression affected post-transplant HO-1 levels, and perioperative HO-1 increases but not basal levels of HO-1 were essential for post-transplant HO-1 as well as hepatoprotection in IR-stressed OLT. In parallel, taking into account that intrahepatic HO-1 expression is derived mostly from macrophages (donor-derived KCs or recipient-derived macrophages), we utilized myeloid-specific HO-1 deficient mice to explore as to whether and how recipient-derived HO-1 may affect liver graft HO-1 levels and hepatic function. Interestingly, recipient myeloid-specific HO-1 deficiency exacerbated the hepatocellular damage and accelerated pro-inflammatory cytokine programs as compared to WT counterparts with proficient myeloid-specific HO-1 expression. Although pre-operative HO-1 expression in the donor liver showed no correlation with the recipient outcome in our study, Geuken et al. reported high expression of HO-1 in the donor liver negatively correlated with recipient outcomes [96]. In addition, Saat et al. demonstrated that both DCD and DBD rat livers were characterized by enhanced hepatic HO-1 gene expression at the time of procurement, while DBD livers exhibited higher pro-inflammatory gene expression, such as IL-1β, TNF-α or IL-6. Interestingly, after 12 hours of cold storage, HO-1 expression in the DCD donor livers returned to that seen in the control group, whereas DBD donor livers still exhibited higher gene expression [97]. As for the effect of pre-transplant HO-1 on clinical OLT outcomes, further in-depth investigations are warranted.
Although there is no doubt about hepatoprotective effects of macrophage-derived HO-1, recent studies showed LSECs and hepatocytes have the ability to generate HO-1 against oxidative stress [98–100]. Indeed, HO-1 down-regulation by siRNA inhibited autophagy leading to hepatocyte apoptosis in vitro [101, 102], while HO-1 up-regulation by gene transfer protected hepatocytes from TNF-α induced cell death after ethanol stimulation [103]. Thus, despite relatively low expression levels in IR-stressed livers, HO-1 produced by hepatocytes or LSECs may certainly contribute to hepatoprotection in IR-stressed liver. To date, however, no HO-1 specific conditional knockout mice (e.g. hepatocyte-specific or LSECs-specific) has been tested in IRI-OLT settings.
Future prospects: From bench to bedside
The accruing clinical data supports the idea that selectively inducing HO-1 in host’s macrophages may mitigate peri-transplant hepatic IR-stress and improve OLT outcomes. Other factors, such as HO-1 overexpression level and its duration should be carefully considered though, based on the pathophysiological condition as well as the recipient background. Indeed, HO-1 overexpression did not result in cytoprotection in some clinical settings [6]; while the toxicity of excessive HO-1 overexpression has been described in hamster fibroblasts [102]. In nerve cells, neurodegeneration can occur if HO-1 is overexpressed independent of the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway [104]. One needs to keep in mind that HO-1 expression may be causatively associated with carcinogenesis/tumorigenesis or induce cytochrome P-450-centered hepatic dysfunction [105]. In our own experiments, successful chemical induction of HO-1 in the rat livers after a single dose of CoPP, could still be observed more than 100 days after OLT [18]. In the clinical setting, application of HO-1 still poses significant limitations because there is no evidence of its optimal expression levels or duration. Recently, promising autologous cell therapies with ex vivo gene engineered macrophages devoid of viral gene modulation and few side effects have been carried out to treat a variety of human tumors [106]. Experimental HO-1 macrophage-targeted therapies using Hb-Hp complex, which binds to a scavenger receptor CD163; or Heme-Hx complex, which is taken-up through a receptor mediated endocytosis [107] await confirmation in clinical trials.
Diverse HO-1 functions
A major obstacle to the clinical application of HO-1 is its diverse biological function. For instance, in liver transplant patients due to terminal malignant disease, anti-inflammatory HO-1 effects against IR-stress may prove detrimental and counterproductive as HO-1 overexpression can spoil the anti-tumor immunity, as shown by suppressing ICAM-1 signaling in colorectal carcinoma cells [108], inhibit apoptosis in renal cell carcinoma [109] or prompt cell proliferation in pancreatic cancers [110]. On the other hand, Zhou et al. has recently reported stable HO-1 expression suppressed the progression of hepatocellular carcinoma [111]. Moreover, Norberto et al. documented HO-1 expression negatively correlated with poor prognosis of breast cancer and its cytoplasm rather than nucleus localization being crucial for HO-1 mediated antitumor effects [112].
Common underlying mechanisms in liver IRI and pandemic metabolic disorders
The number of OLTs in Hepatitis C virus (HCV) patients is decreasing, while alcoholic liver disorders (ALD) and nonalcoholic-steatohepatitis (NASH) have been dramatically increasing [113]. The major problem with NASH, characterized by macrovesicular steatosis and lobular hepatitis with necrosis or ballooning degeneration and fibrosis, is that there are no subjective symptoms until the end stage, and when diagnosed, no effective treatment except for liver transplantation exists. Hence, there is an unmet need for novel preventive and treatment strategies against fatty liver disease in human patients. In a limited patient cohort, the HO-1 expression was significantly increased in NASH patients and the increase reflected the severity of the disease [114]. In addition, Jais et al. demonstrated that mice with hepatocyte or macrophage conditional HO-1 deletion evoked resistance to diet-induced insulin resistance and inflammation, dramatically reducing secondary disease such as steatosis and liver toxicity [9]. Surprisingly, in matched biopsies from “healthy” versus insulin-resistant obese human subjects, HO-1 expression was among the strongest positive predictors of metabolic disease [9]. Consistent with the latter, livers suffering from cirrhosis showed higher HO-1 expression in both animals and humans [115–117]. To the contrary, others have shown that CoPP-induced HO-1 prompted resistance to high fat diet and prevented liver steatosis in mice [118]. Collectively, these conflicting data provide insights as to how environmental stresses in human donor livers with increased HO-1 levels may dictate the donor liver tissue quality.
As discussed, HO-1 therapeutic effects in liver transplant inflammation are mediated/controlled by TLR4, SIRT1 and/or autophagy signaling pathways. However, TLR4/HMGB1 signaling, as well as SIRT1 gene and autophagy functions are closely interrelated to the pathogenesis of metabolic disorders, including liver steatosis following steatohepatitis [93, 119, 120]. Indeed, Li et al. demonstrated that liver specific SIRT1 deficient mice fed with high fat diet (HFD) exhibited severe steatosis with increased expression of genes regulating fatty acid oxidation, as compared with wild-type littermates [121]; while Pedersen et al. reported that SIRT1 mRNA levels were lower in adipose tissue of obese patients as compared with normal-weight controls [122]. It remains to be determined whether hepatic IR-inflammation, and NASH do indeed share a common underlying etiology via a putative HO-1 dependent mechanism.
Unlike in animal models, ‘normal’ human donor livers do not necessarily reflect a ‘healthy’ hepatic status. In other words, the human liver is often stressed (alcohol, high-fat diet, various environmental factors, etc.) with inconsistent basal HO-1 levels, making it difficult to define what is truly ‘normal’. As discussed [123], relative changes of HO-1 expression, rather than the hepatic HO-1 steady state, may be crucial to exert ultimate cytoprotection against IR-stress in OLT. Moreover, one needs to keep in mind most experiments include HO-1 induction in ‘normal’ livers of inbred animals. This may contribute to apparent dissociation between human and murine centered HO-1 studies. For personalized and precise organ matching to reduce risks of IRI in OLT, HO-1 induction should be tailored to fit an individual’s immune status.
Concluding remarks
Recent bench-to-bedside translational studies provide new insights into HO-1 signaling in the mechanism of macrophage activation and hepatocellular damage in IRI-OLT. The immune phenotypes and pathways reviewed here are likely to be applicable to other types of liver inflammatory diseases, such as NASH, an emerging global epidemic projected to become the leading indication for liver transplantation. Thus, better understanding of the crosstalk between HO-1 signaling in the context of host innate – adaptive immune interface should open infinite possibilities to regulate redox signaling beyond the IR-stress in OLT.
HIGHLIGHTS.
Hepatic ischemia-reperfusion injury (IRI) is a major risk factor in orthotopic liver transplantation (OLT), which affects early and late clinical outcomes.
Heme oxygenase 1 (HO-1), induced under IR-stress, exerts potent cytoprotective and homeostatic functions in IRI-OLT.
By activating intracellular SIRT1/autophagy molecular signaling axis, HO-1 overexpression is essential for self-defense mechanism in experimental and clinical OLT settings.
Bench-to-bedside translational approach on the crosstalk between HO-1 signaling in the context of host innate – adaptive immune interface may offer novel insights to regulate redox signaling beyond the IRI in OLT.
Supported in part by:
NIH Grants P01 AI120944, R01 DK062357, DK107533, DK102110 (JWKW)
Abbreviations:
- Ad
Adenovirus
- BMDM
bone marrow-derived macrophage
- BR
bilirubin
- BV
biliverdin
- CO
carbon monoxide
- DAMPs
danger-associated molecular patterns
- DBD
donation after brain death
- DCD
donation after cardiac death
- EAD
early allograft dysfunction
- Hb
hemoglobin
- HMGB1
high-mobility group box 1
- HO-1
heme oxygenase-1
- Hp
haptoglobin
- Hsp
heat shock protein
- Hx
hemopexin
- IRI
ischemia-reperfusion injury
- KCs
Kupffer cells
- LPS
lipopolysaccharide
- LSECs
liver sinusoidal endothelial cells
- MAPK
mitogen-activated protein kinase
- NHBD
non-heart beating donor
- NPC
non parenchymal cell
- OLT
orthotopic liver transplantation
- PNF
primary non-function
- PRRs
pattern recognition receptors
- ROS
reactive oxygen species
- SF
serum ferritin
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have nothing to disclose
REFERENCES
- [1].Wertheim JA, Petrowsky H, Saab S, Kupiec-Weglinski JW, Busuttil RW. Major challenges limiting liver transplantation in the United States. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2011;11:1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim WR, Lake JR, Smith JM, Skeans MA, Schladt DP, Edwards EB, et al. Liver. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2016;16 Suppl 2:69–98. [DOI] [PubMed] [Google Scholar]
- [3].Hudcova J, Scopa C, Rashid J, Waqas A, Ruthazer R, Schumann R. Effect of early allograft dysfunction on outcomes following liver transplantation. Clin Transplant 2017;31. [DOI] [PubMed] [Google Scholar]
- [4].Olthoff KM, Kulik L, Samstein B, Kaminski M, Abecassis M, Emond J, et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl 2010;16:943–949. [DOI] [PubMed] [Google Scholar]
- [5].Deschenes M Early allograft dysfunction: causes, recognition, and management. Liver Transpl 2013;19 Suppl 2:S6–8. [DOI] [PubMed] [Google Scholar]
- [6].Drummond GS, Baum J, Greenberg M, Lewis D, Abraham NG. HO-1 overexpression and underexpression: Clinical implications. Arch Biochem Biophys 2019;673:108073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carmona-Montesinos E, Velazquez-Perez R, Pichardo Aguirre E, Rivas-Arancibia S. Obesity, Oxidative Stress, and Their Effect on Serum Heme Oxygenase-1 Concentrations and Insulin in Children Aged 3 to 5 Years in a Pediatric Hospital of the Ministry of Health CDMX. Child Obes 2016;12:474–481. [DOI] [PubMed] [Google Scholar]
- [8].Cao J, Singh SP, McClung JA, Joseph G, Vanella L, Barbagallo I, et al. EET intervention on Wnt1, NOV, and HO-1 signaling prevents obesity-induced cardiomyopathy in obese mice. Am J Physiol Heart Circ Physiol 2017;313:H368–h380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jais A, Einwallner E, Sharif O, Gossens K, Lu TT, Soyal SM, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 2014;158:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xin G, Du J, Wang YT, Liang TT. Effect of oxidative stress on heme oxygenase-1 expression in patients with gestational diabetes mellitus. Exp Ther Med 2014;7:478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rochette L, Zeller M, Cottin Y, Vergely C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol Metab 2018;29:74–85. [DOI] [PubMed] [Google Scholar]
- [12].Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2011;11:1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ke B, Shen XD, Gao F, Ji H, Qiao B, Zhai Y, et al. Adoptive transfer of ex vivo HO-1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Molecular therapy : the journal of the American Society of Gene Therapy 2010;18:1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J, et al. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Molecular therapy : the journal of the American Society of Gene Therapy 2010;18:1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, et al. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J Hepatol 2017;67:1232–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang M, Nakamura K, Kageyama S, Lawal AO, Gong KW, Bhetraratana M, et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol 1996;15:9–19. [DOI] [PubMed] [Google Scholar]
- [18].Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest 1999;104:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Katori M, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 system in organ transplantation. Transplantation 2002;74:905–912. [DOI] [PubMed] [Google Scholar]
- [20].Farmer DG, Anselmo D, Da Shen X, Ke B, Carmody IC, Gao F, et al. Disruption of P-selectin signaling modulates cell trafficking and results in improved outcomes after mouse warm intestinal ischemia and reperfusion injury. Transplantation 2005;80:828–835. [DOI] [PubMed] [Google Scholar]
- [21].Wasserberg N, Pileggi A, Salgar SK, Ruiz P, Ricordi C, Inverardi L, et al. Heme oxygenase-1 upregulation protects against intestinal ischemia/reperfusion injury: a laboratory based study. Int J Surg 2007;5:216–224. [DOI] [PubMed] [Google Scholar]
- [22].Yang Y, Wang J, Li Y, Fan C, Jiang S, Zhao L, et al. HO-1 Signaling Activation by Pterostilbene Treatment Attenuates Mitochondrial Oxidative Damage Induced by Cerebral Ischemia Reperfusion Injury. Mol Neurobiol 2016;53:2339–2353. [DOI] [PubMed] [Google Scholar]
- [23].Akamatsu Y, Haga M, Tyagi S, Yamashita K, Graca-Souza AV, Ollinger R, et al. Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. Faseb j 2004;18:771–772. [DOI] [PubMed] [Google Scholar]
- [24].Rossi M, Thierry A, Delbauve S, Preyat N, Soares MP, Roumeguere T, et al. Specific expression of heme oxygenase-1 by myeloid cells modulates renal ischemia-reperfusion injury. Sci Rep 2017;7:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakamura K, Zhang M, Kageyama S, Ke B, Araujo JA, Kupiec-Weglinski JW. Reply to: “Protective effects of heme oxygenase 1 during ischemia-reperfusion injury: Hepatocytes or non parenchymal cells?”. J Hepatol 2018;69:753–755. [DOI] [PubMed] [Google Scholar]
- [26].Nakamura K, Kageyama S, Yue S, Huang J, Fujii T, Ke B, et al. Heme oxygenase-1 regulates sirtuin-1-autophagy pathway in liver transplantation: From mouse to human. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2018;18:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2005;5:1793–1800. [DOI] [PubMed] [Google Scholar]
- [28].Shen XD, Ke B, Zhai Y, Gao F, Tsuchihashi S, Lassman CR, et al. Absence of toll-like receptor 4 (TLR4) signaling in the donor organ reduces ischemia and reperfusion injury in a murine liver transplantation model. Liver Transpl 2007;13:1435–1443. [DOI] [PubMed] [Google Scholar]
- [29].Ke B, Shen XD, Tsuchihashi S, Gao F, Araujo JA, Busuttil RW, et al. Viral interleukin-10 gene transfer prevents liver ischemia-reperfusion injury: Toll-like receptor-4 and heme oxygenase-1 signaling in innate and adaptive immunity. Hum Gene Ther 2007;18:355–366. [DOI] [PubMed] [Google Scholar]
- [30].Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 2013;201:613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. The Journal of experimental medicine 2007;204:2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology 2000;32:169–173. [DOI] [PubMed] [Google Scholar]
- [33].Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature reviews Immunology 2005;5:953–964. [DOI] [PubMed] [Google Scholar]
- [34].Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 2013;495:524–528. [DOI] [PubMed] [Google Scholar]
- [35].Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014;59:130–142. [DOI] [PubMed] [Google Scholar]
- [36].Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol 2013;10:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Datta N, Devaney SG, Busuttil RW, Azari K, Kupiec-Weglinski JW. Prolonged Cold Ischemia Time Results in Local and Remote Organ Dysfunction in a Murine Model of Vascularized Composite Transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2017;17:2572–2579. [DOI] [PubMed] [Google Scholar]
- [38].Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest 1997;100:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology 2009;50:1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Uchida Y, Ke B, Freitas MC, Ji H, Zhao D, Benjamin ER, et al. The emerging role of T cell immunoglobulin mucin-1 in the mechanism of liver ischemia and reperfusion injury in the mouse. Hepatology 2010;51:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology 2010;139:2195–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ji H, Shen X, Gao F, Ke B, Freitas MC, Uchida Y, et al. Programmed death-1/B7-H1 negative costimulation protects mouse liver against ischemia and reperfusion injury. Hepatology 2010;52:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu Y, Ji H, Zhang Y, Shen XD, Gao F, Nguyen TT, et al. Negative CD4 + TIM-3 signaling confers resistance against cold preservation damage in mouse liver transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2015;15:954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu Y, Ji H, Zhang Y, Shen X, Gao F, He X, et al. Recipient T cell TIM-3 and hepatocyte galectin-9 signalling protects mouse liver transplants against ischemia-reperfusion injury. J Hepatol 2015;62:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ponka P Cell biology of heme. The American journal of the medical sciences 1999;318:241–256. [DOI] [PubMed] [Google Scholar]
- [46].Jeney V Pro-Inflammatory Actions of Red Blood Cell-Derived DAMPs. Exp Suppl 2018;108:211–233. [DOI] [PubMed] [Google Scholar]
- [47].Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014;123:377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, et al. A central role for free heme in the pathogenesis of severe sepsis. Science translational medicine 2010;2:51ra71. [DOI] [PubMed] [Google Scholar]
- [49].Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. The Journal of biological chemistry 2007;282:20221–20229. [DOI] [PubMed] [Google Scholar]
- [50].Tsuchihashi S, Fondevila C, Kupiec-Weglinski JW. Heme oxygenase system in ischemia and reperfusion injury. Ann Transplant 2004;9:84–87. [PubMed] [Google Scholar]
- [51].Immenschuh S, Ramadori G. Gene regulation of heme oxygenase-1 as a therapeutic target. Biochemical pharmacology 2000;60:1121–1128. [DOI] [PubMed] [Google Scholar]
- [52].Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Experimental & molecular medicine 2019;51:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. The Journal of biological chemistry 1997;272:5375–5381. [PubMed] [Google Scholar]
- [54].Asad SF, Singh S, Ahmad A, Khan NU, Hadi SM. Prooxidant and antioxidant activities of bilirubin and its metabolic precursor biliverdin: a structure-activity study. Chem Biol Interact 2001;137:59–74. [DOI] [PubMed] [Google Scholar]
- [55].Li JJ, Zou ZY, Liu J, Xiong LL, Jiang HY, Wang TH, et al. Biliverdin administration ameliorates cerebral ischemia reperfusion injury in rats and is associated with proinflammatory factor downregulation. Exp Ther Med 2017;14:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Adin CA, Croker BP, Agarwal A. Protective effects of exogenous bilirubin on ischemia-reperfusion injury in the isolated, perfused rat kidney. Am J Physiol Renal Physiol 2005;288:F778–784. [DOI] [PubMed] [Google Scholar]
- [57].Ollinger R, Wang H, Yamashita K, Wegiel B, Thomas M, Margreiter R, et al. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid Redox Signal 2007;9:2175–2185. [DOI] [PubMed] [Google Scholar]
- [58].Fondevila C, Katori M, Lassman C, Carmody I, Busuttil RW, Bach FH, et al. Biliverdin protects rat livers from ischemia/reperfusion injury. Transplantation proceedings 2003;35:1798–1799. [DOI] [PubMed] [Google Scholar]
- [59].Andria B, Bracco A, Attanasio C, Castaldo S, Cerrito MG, Cozzolino S, et al. Biliverdin protects against liver ischemia reperfusion injury in swine. PloS one 2013;8:e69972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yamashita K, McDaid J, Ollinger R, Tsui TY, Berberat PO, Usheva A, et al. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. Faseb j 2004;18:765–767. [DOI] [PubMed] [Google Scholar]
- [61].Carocci A, Catalano A, Sinicropi MS, Genchi G. Oxidative stress and neurodegeneration: the involvement of iron. Biometals 2018;31:715–735. [DOI] [PubMed] [Google Scholar]
- [62].Ozkan AK, Yemisci OU, Saracgil Cosar SN, Oztop P, Turhan N. Can high-sensitivity C-reactive protein and ferritin predict functional outcome in acute ischemic stroke? A prospective study. Top Stroke Rehabil 2013;20:528–536. [DOI] [PubMed] [Google Scholar]
- [63].Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: Past, present and future. Biochim Biophys Acta 2010;1800:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Berberat PO, Katori M, Kaczmarek E, Anselmo D, Lassman C, Ke B, et al. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. Faseb j 2003;17:1724–1726. [DOI] [PubMed] [Google Scholar]
- [65].Wakiya T, Sanada Y, Urahashi T, Ihara Y, Yamada N, Okada N, et al. Impact of the serum ferritin concentration in liver transplantation. Liver Transpl 2015;21:1419–1427. [DOI] [PubMed] [Google Scholar]
- [66].Waza AA, Hamid Z, Ali S, Bhat SA, Bhat MA. A review on heme oxygenase-1 induction: is it a necessary evil. Inflamm Res 2018;67:579–588. [DOI] [PubMed] [Google Scholar]
- [67].Fei D, Meng X, Kang K, Nan C, Zhao M, Pan S, et al. Heme oxygenase-1 modulates thrombomodulin and activated protein C levels to attenuate lung injury in cecal ligation and puncture-induced acute lung injury mice. Exp Lung Res 2012;38:173–182. [DOI] [PubMed] [Google Scholar]
- [68].Fei D, Meng X, Zhao M, Kang K, Tan G, Pan S, et al. Enhanced induction of heme oxygenase-1 suppresses thrombus formation and affects the protein C system in sepsis. Transl Res 2012;159:99–109. [DOI] [PubMed] [Google Scholar]
- [69].Kang K, Nan C, Fei D, Meng X, Liu W, Zhang W, et al. Heme oxygenase 1 modulates thrombomodulin and endothelial protein C receptor levels to attenuate septic kidney injury. Shock 2013;40:136–143. [DOI] [PubMed] [Google Scholar]
- [70].Peng L, Mundada L, Stomel JM, Liu JJ, Sun J, Yet SF, et al. Induction of heme oxygenase-1 expression inhibits platelet-dependent thrombosis. Antioxid Redox Signal 2004;6:729–735. [DOI] [PubMed] [Google Scholar]
- [71].Lindenblatt N, Bordel R, Schareck W, Menger MD, Vollmar B. Vascular heme oxygenase-1 induction suppresses microvascular thrombus formation in vivo. Arterioscler Thromb Vasc Biol 2004;24:601–606. [DOI] [PubMed] [Google Scholar]
- [72].Desbuards N, Rochefort GY, Schlecht D, Machet MC, Halimi JM, Eder V, et al. Heme oxygenase-1 inducer hemin prevents vascular thrombosis. Thromb Haemost 2007;98:614–620. [PubMed] [Google Scholar]
- [73].Amersi F, Shen XD, Anselmo D, Melinek J, Iyer S, Southard DJ, et al. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology 2002;35:815–823. [DOI] [PubMed] [Google Scholar]
- [74].Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. The Journal of experimental medicine 2000;192:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sun J, Guo E, Yang J, Yang Y, Liu S, Hu J, et al. Carbon monoxide ameliorates hepatic ischemia/reperfusion injury via sirtuin 1-mediated deacetylation of high-mobility group box 1 in rats. Liver Transpl 2017;23:510–526. [DOI] [PubMed] [Google Scholar]
- [76].Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. Journal of immunology (Baltimore, Md : 1950) 2004;172:4744–4751. [DOI] [PubMed] [Google Scholar]
- [77].Choi BM, Pae HO, Jeong YR, Oh GS, Jun CD, Kim BR, et al. Overexpression of heme oxygenase (HO)-1 renders Jurkat T cells resistant to fas-mediated apoptosis: involvement of iron released by HO-1. Free Radic Biol Med 2004;36:858–871. [DOI] [PubMed] [Google Scholar]
- [78].Sahara H, Shimizu A, Setoyama K, Okumi M, Oku M, Samelson-Jones E, et al. Carbon monoxide reduces pulmonary ischemia-reperfusion injury in miniature swine. The Journal of thoracic and cardiovascular surgery 2010;139:1594–1601. [DOI] [PubMed] [Google Scholar]
- [79].Murokawa T, Sahara H, Sekijima M, Pomposelli T, Iwanaga T, Ichinari Y, et al. The Protective Effects of Carbon Monoxide Against Hepatic Warm Ischemia-Reperfusion Injury in MHC-Inbred Miniature Swine. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract 2019. [DOI] [PubMed] [Google Scholar]
- [80].Sahara H, Shimizu A, Setoyama K, Oku M, Okumi M, Nishimura H, et al. Beneficial effects of perioperative low-dose inhaled carbon monoxide on pulmonary allograft survival in MHC-inbred CLAWN miniature swine. Transplantation 2010;90:1336–1343. [DOI] [PubMed] [Google Scholar]
- [81].Fredenburgh LE, Perrella MA, Barragan-Bradford D, Hess DR, Peters E, Welty-Wolf KE, et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury--a fresh look. Exp Mol Pathol 2003;74:86–93. [DOI] [PubMed] [Google Scholar]
- [83].Kato H, Amersi F, Buelow R, Melinek J, Coito AJ, Ke B, et al. Heme oxygenase-1 overexpression protects rat livers from ischemia/reperfusion injury with extended cold preservation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2001;1:121–128. [PubMed] [Google Scholar]
- [84].Coito AJ, Buelow R, Shen XD, Amersi F, Moore C, Volk HD, et al. Heme oxygenase-1 gene transfer inhibits inducible nitric oxide synthase expression and protects genetically fat Zucker rat livers from ischemia-reperfusion injury. Transplantation 2002;74:96–102. [DOI] [PubMed] [Google Scholar]
- [85].Tsuchihashi S, Zhai Y, Fondevila C, Busuttil RW, Kupiec-Weglinski JW. HO-1 upregulation suppresses type 1 IFN pathway in hepatic ischemia/reperfusion injury. Transplantation proceedings 2005;37:1677–1678. [DOI] [PubMed] [Google Scholar]
- [86].Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation 2007;83:1628–1634. [DOI] [PubMed] [Google Scholar]
- [87].Ke B, Shen XD, Gao F, Qiao B, Ji H, Busuttil RW, et al. Small interfering RNA targeting heme oxygenase-1 (HO-1) reinforces liver apoptosis induced by ischemia-reperfusion injury in mice: HO-1 is necessary for cytoprotection. Hum Gene Ther 2009;20:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shen XD, Ke B, Uchida Y, Ji H, Gao F, Zhai Y, et al. Native macrophages genetically modified to express heme oxygenase 1 protect rat liver transplants from ischemia/reperfusion injury. Liver Transpl 2011;17:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Virag L, Jaen RI, Regdon Z, Bosca L, Prieto P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol 2019;26:101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Nakamura K, Kageyama S, Ke B, Fujii T, Sosa RA, Reed EF, et al. Sirtuin 1 attenuates inflammation and hepatocellular damage in liver transplant ischemia/Reperfusion: From mouse to human. Liver Transpl 2017;23:1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ou X, Lee MR, Huang X, Messina-Graham S, Broxmeyer HE. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014;32:1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sathyanarayan A, Mashek MT, Mashek DG. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell reports 2017;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ueno T, Komatsu M. Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol 2017;14:170–184. [DOI] [PubMed] [Google Scholar]
- [94].Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 2013;58:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gupta NA, Kolachala VL, Jiang R, Abramowsky C, Shenoi A, Kosters A, et al. Mitigation of autophagy ameliorates hepatocellular damage following ischemia-reperfusion injury in murine steatotic liver. Am J Physiol Gastrointest Liver Physiol 2014;307:G1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Geuken E, Buis CI, Visser DS, Blokzijl H, Moshage H, Nemes B, et al. Expression of heme oxygenase-1 in human livers before transplantation correlates with graft injury and function after transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2005;5:1875–1885. [DOI] [PubMed] [Google Scholar]
- [97].Saat TC, Susa D, Kok NF, van den Engel S, Roest HP, van der Laan LJ, et al. Inflammatory genes in rat livers from cardiac- and brain death donors. J Surg Res 2015;198:217–227. [DOI] [PubMed] [Google Scholar]
- [98].Peralta C, Jimenez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol 2013;59:1094–1106. [DOI] [PubMed] [Google Scholar]
- [99].Qu S, Yuan B, Zhang H, Huang H, Zeng Z, Yang S, et al. Heme Oxygenase 1 Attenuates Hypoxia-Reoxygenation Injury in Mice Liver Sinusoidal Endothelial Cells. Transplantation 2018;102:426–432. [DOI] [PubMed] [Google Scholar]
- [100].Zhang H, Yuan B, Huang H, Qu S, Yang S, Zeng Z. Gastrodin induced HO-1 and Nrf2 up-regulation to alleviate H2O2-induced oxidative stress in mouse liver sinusoidal endothelial cells through p38 MAPK phosphorylation. Braz J Med Biol Res 2018;51:e7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011;53:2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Suttner DM, Dennery PA. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. Faseb j 1999;13:1800–1809. [DOI] [PubMed] [Google Scholar]
- [103].Bakhautdin B, Das D, Mandal P, Roychowdhury S, Danner J, Bush K, et al. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J Hepatol 2014;61:1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Nitti M, Piras S, Brondolo L, Marinari UM, Pronzato MA, Furfaro AL. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Muhoberac BB, Hanew T, Halter S, Schenker S. A model of cytochrome P-450-centered hepatic dysfunction in drug metabolism induced by cobalt-protoporphyrin administration. Biochemical pharmacology 1989;38:4103–4113. [DOI] [PubMed] [Google Scholar]
- [106].Lee S, Kivimae S, Dolor A, Szoka FC. Macrophage-based cell therapies: The long and winding road. J Control Release 2016;240:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Vijayan V, Wagener F, Immenschuh S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochemical pharmacology 2018;153:159–167. [DOI] [PubMed] [Google Scholar]
- [108].Seo GS, Jiang WY, Chi JH, Jin H, Park WC, Sohn DH, et al. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015;6:19792–19806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Balan M, Chakraborty S, Flynn E, Zurakowski D, Pal S. Honokiol inhibits c-Met-HO-1 tumor-promoting pathway and its cross-talk with calcineurin inhibitor-mediated renal cancer growth. Sci Rep 2017;7:5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Han L, Jiang J, Ma Q, Wu Z, Wang Z. The inhibition of heme oxygenase-1 enhances the chemosensitivity and suppresses the proliferation of pancreatic cancer cells through the SHH signaling pathway. Int J Oncol 2018;52:2101–2109. [DOI] [PubMed] [Google Scholar]
- [111].Zou C, Zou C, Cheng W, Li Q, Han Z, Wang X, et al. Heme oxygenase-1 retards hepatocellular carcinoma progression through the microRNA pathway. Oncol Rep 2016;36:2715–2722. [DOI] [PubMed] [Google Scholar]
- [112].Gandini NA, Alonso EN, Fermento ME, Mascaro M, Abba MC, Colo GP, et al. Heme Oxygenase-1 Has an Antitumor Role in Breast Cancer. Antioxid Redox Signal 2019;30:2030–2049. [DOI] [PubMed] [Google Scholar]
- [113].Cholankeril G, Wong RJ, Hu M, Perumpail RB, Yoo ER, Puri P, et al. Liver Transplantation for Nonalcoholic Steatohepatitis in the US: Temporal Trends and Outcomes. Dig Dis Sci 2017;62:2915–2922. [DOI] [PubMed] [Google Scholar]
- [114].Malaguarnera L, Madeddu R, Palio E, Arena N, Malaguarnera M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J Hepatol 2005;42:585–591. [DOI] [PubMed] [Google Scholar]
- [115].Matsumi M, Takahashi T, Fujii H, Ohashi I, Kaku R, Nakatsuka H, et al. Increased heme oxygenase-1 gene expression in the livers of patients with portal hypertension due to severe hepatic cirrhosis. J Int Med Res 2002;30:282–288. [DOI] [PubMed] [Google Scholar]
- [116].Wei CL, Lee KH, Khoo HE, Hon WM. Expression of haem oxygenase in cirrhotic rat liver. J Pathol 2003;199:324–334. [DOI] [PubMed] [Google Scholar]
- [117].Goh BJ, Tan BT, Hon WM, Lee KH, Khoo HE. Nitric oxide synthase and heme oxygenase expressions in human liver cirrhosis. World J Gastroenterol 2006;12:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Sacerdoti D, Singh SP, Schragenheim J, Bellner L, Vanella L, Raffaele M, et al. Development of NASH in Obese Mice is Confounded by Adipose Tissue Increase in Inflammatory NOV and Oxidative Stress. Int J Hepatol 2018;2018:3484107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chenxu G, Minxuan X, Yuting Q, Tingting G, Jing F, Jinxiao L, et al. Loss of RIP3 initiates annihilation of high-fat diet initialized nonalcoholic hepatosteatosis: A mechanism involving Toll-like receptor 4 and oxidative stress. Free Radic Biol Med 2019;134:23–41. [DOI] [PubMed] [Google Scholar]
- [120].Liu X, Gao Y, Li M, Geng C, Xu H, Yang Y, et al. Sirt1 mediates the effect of the heme oxygenase inducer, cobalt protoporphyrin, on ameliorating liver metabolic damage caused by a high-fat diet. J Hepatol 2015;63:713–721. [DOI] [PubMed] [Google Scholar]
- [121].Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014;146:539–549.e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Pedersen SB, Olholm J, Paulsen SK, Bennetzen MF, Richelsen B. Low Sirt1 expression, which is upregulated by fasting, in human adipose tissue from obese women. International journal of obesity (2005) 2008;32:1250–1255. [DOI] [PubMed] [Google Scholar]
- [123].Kageyama S, Hirao H, Nakamura K, Ke B, Zhang M, Ito T, et al. Recipient HO-1 inducibility is essential for posttransplant hepatic HO-1 expression and graft protection: From bench-to-bedside. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2019;19:356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]