

Abstract
Inflammation contributes to the development of heart failure (HF) through multiple mechanisms including regulating extracellular matrix (ECM) degradation and deposition. Interactions between cells in the myocardium orchestrates the magnitude and duration of inflammatory cell recruitment and ECM remodeling events associated with HF. More recently, studies have shown T-cells have signficant roles in post-MI wound healing. T-cell biology in HF illustrates the complexity of cross-talk between inflammatory cell types and resident fibroblasts. This review will focus on T-cell recruitment to the myocardium and T-cell specific factors that might influence cardiac wound healing and fibrosis in the heart with consideration of age and sex as important factors in T-cell activity.
Keywords: fibrosis, myocardial infarction, inflammation, T-cell, sex, age
Introduction
Fibrosis is the generic term for accumulation of extracellular matrix (ECM). Excessive ECM accumulation in the heart can result in negative consequences leading to increased morbidity and mortality [1]. An excessive degradation of normal ECM or accumulation of abnormal ECM can also impair ventricular physiology contributing to heart failure (HF) [2–6]. ECM is more than an accumulation of fibrillar collagen but a major reservoir for sequestering signaling molecules, such as inflammatory cytokines and growth factors that not only induce modifications in the ECM, but also influence the overall structure and physiology of the myocardium [7–9].
The cellular and molecular basis for the development and progression of HF are multifactorial and specific to etiology. Inflammation has been proposed as a fundamental mechanism contributing to the development of HF in general and ECM fibrosis in particular.[10–13] However, inflammation after a myocardial infarction (MI) is fluid and therefore difficult to define. The inflammatory environment of the MI left ventricle (LV) at day one is much different from the inflammatory environment seen one week after MI [14]. Key inflammatory cells such as lymphocytes (i.e. T-cells and B-cells), monocytes, and macrophages require precise characterization and definitions to more precisely describe their contribution to the progression to HF in each of its forms.
Importantly specific interactions between types of immune cell in the myocardium likely plays a key role in regulating the magnitude and duration of inflammatory cell recruitment and ECM remodeling events associated with HF. In response to myocardial antigens, recruitment of T-cells can initiate the acute and or propogate the chronic inflammatory processes leading to an impairment of cardiac function by direct cytotoxicity or by enhancing the activity of other cardiac cell types, such as macrophages and fibroblasts [15–17]. As cardiac fibroblasts are the central cell type expressing and depositing fibrillar ECM, an understanding of the interaction of immune cells with cardiac fibroblasts is essential for defining molecular pathways in ECM assembly and/or remodeling. This review will focus on T-cell recruitment to the myocardium and T-cell specific factors that might influence macrophage and fibroblast activity in fibrosis in the heart.
Activation of T-cells Post-MI
T-cells have been shown to play a central role in the immune response in cardiovascular diseases. T-cells are grouped into a series of subsets based on their function and on their protein expression patterns (e.g. CD4 vs CD8). T-cells begin as bone marrow-derived haematopoietic stem cells before circulating to the thymus where they engraft and develop into naive T-cells [18]. In the periphery, CD4+ and CD8+ T-cells undergo further differentiation to specialized cells such as CD4+ helper T-cells (Th1, Th2, and Th17), CD4+ Tregs, and CD8+ (cytotoxic) T-cells further diversifying T-cell specific functional responses (Figure 1).
Figure 1: The post-MI myocardial environment facilitates in T-cell activation and phenotype.
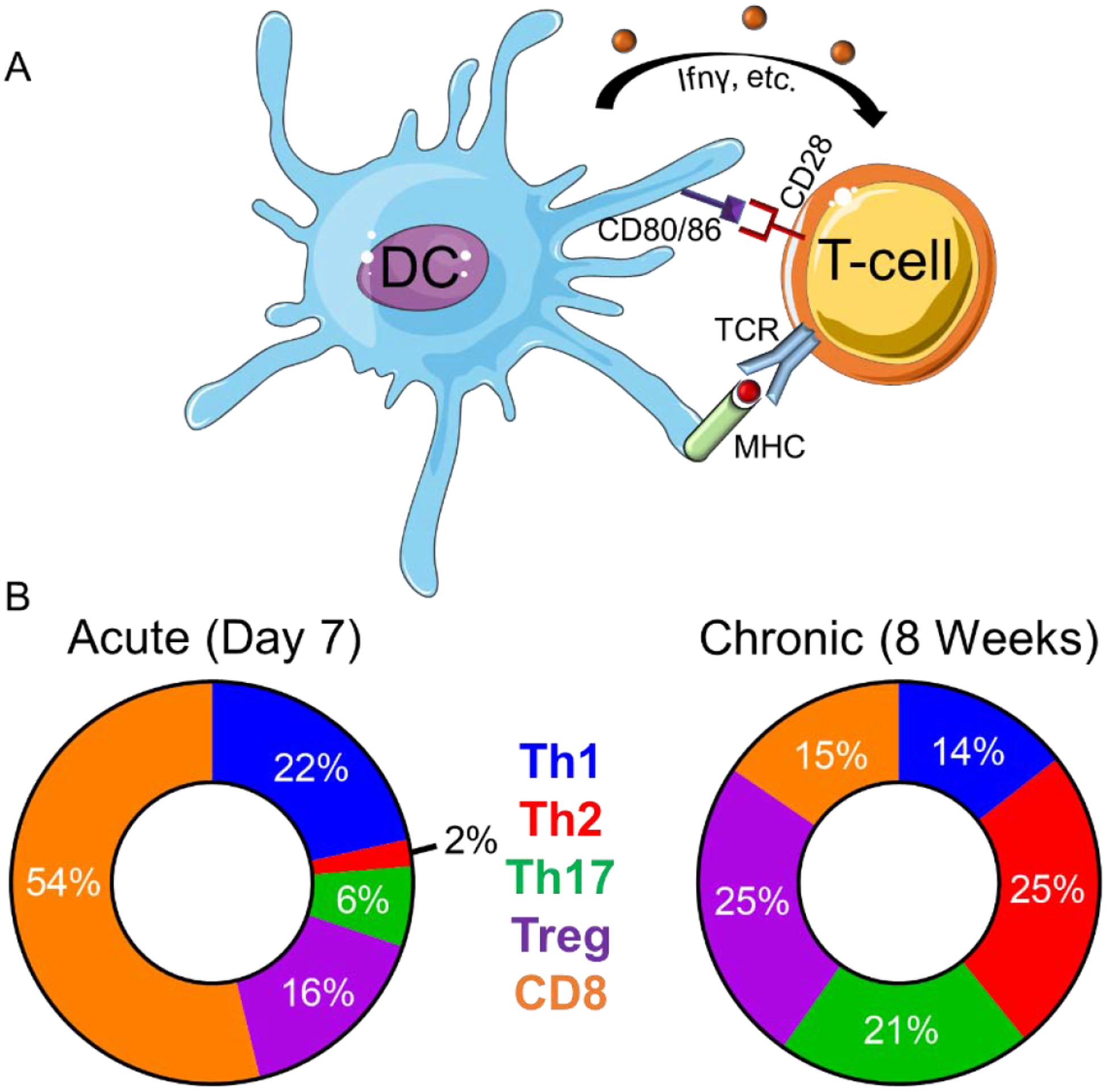
A) Antigen-presenting cells such as dendritic cells (DC) activate T-cells via presentation of cognate peptides expressed on major histocompatibility complex (MHC) to T-cell receptors (TCR), surface expression of co-stimulatory signals (i.e. CD28, CD80, CD86), and production of cytokines (IFNγ, etc.). B) T-cell populations change over time. In the acute post-MI environment Th1 and CD8+ are the predominant T-cells whereby in the chronic post-MI environment Th2, Th17, and Treg become the predominant phenotype. The circle charts reflect averages of T-cell populations (%) reported at acute (7 days) and chronic (8 weeks) post-MI remodeling time points.[14, 19–21]
Multiple studies have shown that T-cell infiltration of the myocardium is both necessary and sufficient to produce LV fibrosis and dysfunction indicating a functional role for T-cells in cardiac fibrosis [15]. However, the complex nature of T-cell types and context-dependent activity render the task of a concise summary of T-cell function in the heart a difficult one. T-cell biology in HF illustrates the complexity of cross-talk between inflammatory cell types and resident fibroblasts (Table 1). A growing consensus is that T-cells are involved in induction of cardiac fibrosis and can act through direct adhesion and actions on cardiac fibroblasts [15]. T-cells can also synthesize and export a variety of inflammatory molecules (e.g. tumor necrosis factor (Tnf)α and IL-1β) suggesting they have an indirect role in the regulation of fibroblast ECM deposition and proliferation by facilitating polarization of macrophages as well as fibroblast activation [22, 23]. Below we discuss the activation of T-cells post-MI and the role they play on cardiac fibrosis.
Table 1:
Summary of T-cell subsets involved in myocardial infarction (MI).
| T-cell classification | Secretion profile | Function |
|---|---|---|
| Th1 | Cxcl10, Ifnγ, IL-2, Tnfβ | |
| Th2 | IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, | |
| Th17 | IL-17, IL-21, IL-22, Ccl20 | |
| Treg | Tgfβ, IL-10 | |
| Cytotoxic | Performin, Granzyme B |
bFGF- basic fibroblast growth factor; IFN-interferon; IL-interleukin; MCP- monocyte chemotactic protein; TGF-transforming growth factor; TNF-tumor necrosis factor
Antigen-presenting cells:
Antigen-presenting cells such as monocytes and dendritic cells are potent key immunoregulators that direct specification of multiple types of inflammatory cells including T-cells. After necrotic injury, antigen-presenting cells internalize damage-associated molecular patterns released from the necrotic tissue and migrate into the lymph nodes [30]. In the lymph nodes, antigen-presenting cells activate T-cells into tissue-homing effector cells via a multistep activation process that includes upregulation of major histocompatibility complexes class I and II to display cognate peptides to T-cell receptors, surface expression of co-stimulatory signals (e.g CD28, CD80/86), and production of cytokines (e.g. interferon (IFN)γ) for effector T-cell activation and polarization [40–42].
Hofmann et al. demonstrated that antigen-mediated activation was vital for the protective function of CD4+ T-cells after MI using a transgenic mouse model (OT-II) [40]. OT-II mice have CD4+ T cells that primarily recognize an irrelevant chicken ovalbumin-derived peptide via their T-cell receptor thus inhibiting T-cell receptor-ligand interactions, cross-presentation of antigens, and central and peripheral T-cell tolerance and induction [43]. The OT-II mice had more loosely distributed and disarrayed collagen on post-MI day 7 compared to WT mice. Interestingly, no differences compared to WT mice were observed in terms of collagen mRNA expression or metalloproteinase (MMP) activity indicating impaired post-transcriptional processing of matrix proteins most likely accounts for the observed phenotype [40].
Memory T-cells:
A subset of the effector T-cells differentiate into memory T-cells, which are defined by their re-circulation characteristics [44]. In addition to circulating memory T-cells, cardiac resident memory T-cells have been identified under-steady state conditions (e.g. with increased age) [45]. In patients with acute MI (59±10 years; 75% males), circulating effector and memory T-cells decreased 90 min after reperfusion returning to levels comparable to pre reperfusion by 24 hours [46]. The initial loss in peripheral blood CD8+ memory T-cell subsets has been suggested to be caused by entrapment in the coronary microcirculation and possible be due to infiltration of cells into the reperfused myocardium [17]. In addition, expansion of splenic memory CD4+ T-cells has been detected in mice 8 weeks after permanent occlusion. Although the biological significance of memory T-cells in myocardial homeostasis and disease is not fully understood, changes in levels and distribution suggest an active role for this subset in cardiac remodeling [19].
Th1:
Th1 cells increase in the circulation of patients with acute MI (26 men/7 women) and unstable angina (17 men/5 women) within 24 hours after the onset of symptoms [47]. In patients with unstable angina, levels of Th1 cells returned to baseline after 1 week, whereas in post-MI patients Th1 cells remained elevated 1 month after hospitalization. This prolonged Th1 response after MI was also observed in a mouse model where Th1 cells remained elevated in the LV and in the circulation 8 weeks post-MI [14, 19].
Th1 polarization is associated with increased cardiomyocyte apoptosis, imbalanced ECM turnover and decreased myofibroblast differentiation leading to cardiac rupture [30]. Activation of Th1 cells through T-cell receptor peptide administration increased pro-fibrotic signals leading to an increase in interstitial LV fibrosis.[28] IFNγ is one of the primary markers of Th1-mediated inflammation and has been shown to inhibit fibrosis by blocking the pro-fibrotic activity of transforming growth factor-β (TGF-β) [24]. IFNγ can also directly inhibit fibroblast proliferation and expression of collagen-I and -III mRNA. In addition, IFNγ inhibits Th2-mediated fibroblast activation and indirectly regulates fibrosis through activation of macrophages [25, 26, 29]. The secretome of macrophages from post-MI day 3 hearts suppressed TGF-β induced α-smooth muscle actin (αSMA) expression, a marker of activated fibroblasts, in cultured cells [30]. Addition of an IFNγ neutralizing antibody reversed the TGF-β induction of α-SMA expression demonstrating Th1 cells may be indirectly regulating fibrosis through macrophage-dependent fibroblast activation.
The Th1 ligand, Cxcl10, increases in the LV during the first 24 hours post-MI and inhibits disproportionate fibrotic remodeling most likely through proteoglycan signaling [48]. In vitro, Cxcl10 did not modulate cardiac fibroblast proliferation and apoptosis, but significantly inhibited basic fibroblast growth factor-induced fibroblast migration and enhanced growth factor-mediated wound contraction in fibroblast-populated collagen lattices [27]. Overall, the presence of Th1 cells in the post-MI heart is predicted to dampen fibroblast activation and limit fibrosis through both direct and indirect mechanisms.
Th17:
Little is known about the post-MI role of Th17 cells nevertheless, 8 weeks after occlusion Th17 cells become one of the predominant T-cell phenotypes in the LV implying they may have a major role in the chronic cardiac remodeling process [19]. Deletion of IL17, a primary Th17 secreted factor, improved survival and attenuated LV dilation at post-MI day 28 by reducing cardiomyocyte apoptosis and neutrophil infiltration [49]. It must be noted however, Th17 cells are not the only source of IL-17 in the post-MI setting and therefore this study may not truly describe the effect of Th17 cells [49, 50].
Th2:
In a mouse model of MI, the expression levels of Th2-related transcriptional factor GATA3 does not increase in the LV acutely (first 7 days) however, 8 weeks after occlusion Th2 cells become the predominant T-cell phenotype in the LV and are key in regulation cardiac fibrosis and hypertrophy [14, 19]. Patients at baseline (no previous cardiovascular events; 65±1 years old; 53% men) with elevated numbers of circulating Th2 cells also showed reduced risk of future coronary events (log rank test P for trend=0.004) [51].
Th2 cells counteract the Th1 response by secreting several cytokines including IL-10, IL-4, IL-5, and IL-13. IL-4 induces the expression of GATA-3 in a STAT-6-dependent mechanism, which leads to a further increase in IL-4 and IL-5, and inhibits the production of IFN-γ [52]. Secretion of Th2 cytokines IL-4 and IL13 also promote recruitment of monocyte-derived M2 macrophages thus indirectly regulating cardiac fibroblasts [31–33]. While data suggests that Th2 cells are pro-fibrotic, our understanding of the role of this cell type requires further evaluation.
Tregs:
Myocardial regulatory T (Treg) cells increase gradually over the first week following MI. Tregs co-cultured with cardiac fibroblasts led to decreased αSMA and MMP-3 expression and attenuated fibroblast mediated-contraction of collagen pads. This data suggests direct contact may be necessary for Tregs to stimulate fibroblast and preserve the matrix [38]. Tregs potentially have a dual role in contributing to cardiac fibrosis by releasing the pro-fibrotic molecule TGF-β and by inhibiting Th17-mediated fibrosis through IL-10 secretion [36, 37].
Treg activation also induces an M2-like macrophage differentiation within the healing myocardium, associated with fibroblast activation, and increased expression of macrophage-derived proteins fostering wound healing [38, 41]. Depletion of Tregs increased pro-inflammatory M1 macrophages and impaired M2-like differentiation [41]. Treg expansion induced by CD28 antibodies significantly improved survival and fewer cardiac ruptures during the first 7 days after MI. This was primarily due to decreased MMP-mediated degradation of collagen within the infarct [41]. In vitro, co-culture experiments of Tregs with macrophages support the notion that Tregs facilitate cardiac fibrosis by increasing the expression of genes associated with healing such as osteopontin and arginase-1 [41]. Hence, increases in Tregs after MI is likely to promote ECM deposition and scar formation.
CD8:
In a mouse model of permanent ischemia, levels of CD8+ T-cell increase as early as day 1 and remain elevated at day 14 post-MI [14, 40]. Plasma levels of Granzyme B, a CD8+ T-cell secreted protein, correlated with left ventricular end-diastolic diameter, suggesting CD8+ T-cells may be involved in adverse post-MI LV remodeling [53]. In contrast, a drop in circulating CD8+ T-cells within an hour following percutaneous coronary intervention was associated with poor prognosis suggesting effects from CD8+ T-cells are temporally-dependent [46].
Mice lacking functional CD8+ T-cells exhibit improved cardiac physiology and less mortality compared to wild-type animals 7 days post-MI [21]. Despite better overall survival, animals that died did so due to cardiac rupture. This was linked to delayed removal of the necrotic debris. In contrast, depletion of CD8+ T-cells by administration of monoclonal antibodies markedly increased both wound-breaking strength and collagen synthesis [23]. One difference between the two studies is the genetic knockdown of CD8+ T-cell activation resulted in an increase in CD4+ T-cells whereas antibody depletion of CD8+ cells resulted in only a slight decrease in CD4+ T-cells. These observations suggest that CD8+ T-cell regulation of CD4+ T-cell recruitment might be a critical player in collagen scar formation.
The frequency of CD8+CD57+ T-cells, a T-cell subset involved in replicative senescence, correlated with cardiovascular mortality 6 months after acute MI [54]. CD57+ T-cells fail to proliferate after in vitro antigen-specific stimulation and are highly vulnerable to activation-induced apoptosis [55]. This specific subset of CD8+ T-cells most likely regulates acute coronary events via their pro-inflammatory and high cytotoxic capacities. In contrast, a subset of CD8+ T-cells expressing the angiotensin type 2 receptor (AT2R) has been shown to have no cytotoxic activity and are believed to have a cardioprotective effect after MI by infiltrating the peri-infarct zone and downregulating pro-inflammatory cytokine expression [39]. These studies suggest that a balance in the CD8+ T-cell response is needed for beneficial cardiac wound healing and highlight the complexity of CD8+ T-cell biology in post-MI remodeling.
Effect of Confounding Variables on T-cell function
Sex:
After menopause, women lose the protective effect of female hormones and experience increased risk of adverse cardiac remodeling following MI. Hormone replacement therapy (HRT) was initially thought to lower cardiac death incidence however, two randomized trials, the Women’s Health Initiative and the Heart and Estrogen/Progestin Replacement Study, showed that HRT may actually increase the risk and events of cardiovascular disease in postmenopausal women [56–58]. Interestingly, secondary analysis of the Women’s Health Initiative data set indicated that when HRT was started closer to start of menopause, risk of cardiovascular disease was reduced [59]. One possible reason why the timing of HRT is crucial may be due to changes in the immune system with menopause [60].
Hormonal changes during menopause results in stimulation of the pro-inflammatory and T-cell-dependent responses [61]. In a study that screened for circulating T-cell populations in patients between 63–68 years (mean age 65) without previous cardiovascular events, Th1 and Th2 cells were significantly higher in women (930±720 Th1 cells/μL; 54±47 Th2 cells/μL) compared with men (710±640 Th1 cells/μL and 41±38 Th2 cells/μL) [51]. In women, elevated Th2 cells were also independently associated with a reduced risk of MI (hazard ratio, 0.19; 95% confidence interval, 0.06–0.56; p=0.002 for the highest versus the lowest tertile of Th2 cells) [51]. In mice, old females post MI (23.3 ± 0.1 months) had an increase in acute phase response and a decrease in the Th1/Th2 and the liver X receptors/retinoid X receptor (LXR/RXR) pathways compared to young female mice (5.4 ± 0.1 months) [62]. In vitro stimulation of LXR/RXR in T-cells enhanced Tregs and attenuated Th1 and Th17 differentiation denoting the loss of LXR/RXR signaling in old females may be facilitating adverse remodeling through T-cell mediated mechanisms [63, 64].
In addition to hormonal regulation, genes expressed by the X-chromosomes also affect T-cell activity. In vitro studies have shown in mammalian female T-cells, the inactive X-chromosome is predisposed to reactivate, resulting in the overexpression of immune-related genes including CD40LG, Cxcr3, and Foxp3 [65, 66]. In a mouse model of stroke, the X chromosome exacerbates ischemic injury in old but not young females [67, 68]. In addition, a novel population of memory T cells increase with age in the brain and appear to be primed to potentiate inflammation and leukocyte recruitment following ischemic injury [66].
Age:
The incidence and severity of cardiovascular disease are heightened with advanced age. Studies have shown with increased age, both animals and humans present increased afterload and impaired vasodilation, which increased wall stress in the LV and led to cardiomyocyte hypertrophy [69]. After ischemia/reperfusion, mice greater than 2 years old have: 1) impaired phagocytosis of dead cardiomyocytes, 2) decreased neutrophil infiltration, 3) delayed macrophage recruitment, and 4) markedly decreased collagen deposition in the scar compared to young mice (2–3 months old) [70]. Beyond macrophage quantity, increased M1 and decreased M2 macrophage polarization correlated with age in mice [71].
T-cells become senescent with increased age resulting in a decrease in the number of naive T-cells along with an expansion of memory and effector subpopulations [72, 73]. The functional capacity of senescent T-cells is linked to decreased responsiveness to stimuli and impaired differentiation [74]. Old mice (12–15 month old) have more T-cells that exhibit cardiotropism compared to cells isolated from 2–3 month old mice [45]. Adoptive transfer of T-cells from old mice into young (2–3 months old) Rag−/− mice (mice lacking mature B and T lymphocytes) resulted in a slight yet significant decrease in cardiac function signifying with age there may be a heart-directed immune responses even in the absence of myocardial tissue damage [45].
Tserel et al. showed that older patients (73–84 years) have more DNA methylation changes in both CD8+ and CD4+ T-cells compared to young patients (age 22–34), with increased methylation variation in CD8+ T-cells compared to CD4+ T-cells [75]. Methylation status was negatively correlated with expression levels in genes associated with T-cell mediated immune response and differentiation. For example, in older patients (73–84 years) there was hypermethylation of central genes that regulated CD8+ T-cell differentiation such as SATB-1 (special AT-rich sequence-binding protein-1) that resulted in terminally differentiated effector CD8+ T cells and increased expression of galectin 1 gene (LGALS1), interferon gamma, CCL5, and granzyme H [75]. The study suggested impaired age-dependent function in T-cell activity, particularly in CD8+ cells. However, this study did not mention the sex of the patients nor were the subsets separated by memory vs. naive T-cells, which may be contributing to some of the differences observed as methylation status may vary depending on sex or T-cell phenotype.
Conclusion
The role of T-cells in cardiovascular pathology is a new emerging field with much more to learn. Clearly, T-cells contribute both directly and indirectly to post-MI ECM remodeling and fibrosis (Figure 2). Direct binding of T-cells to cardiac fibroblasts and secretion of cytokines, such as the potent fibroblast activation factor TGF-β, undoubtedly play a role in production and degradation of myocardial ECM.
Figure 2: T-cells regulate fibrosis through direct and indirect mechanisms.
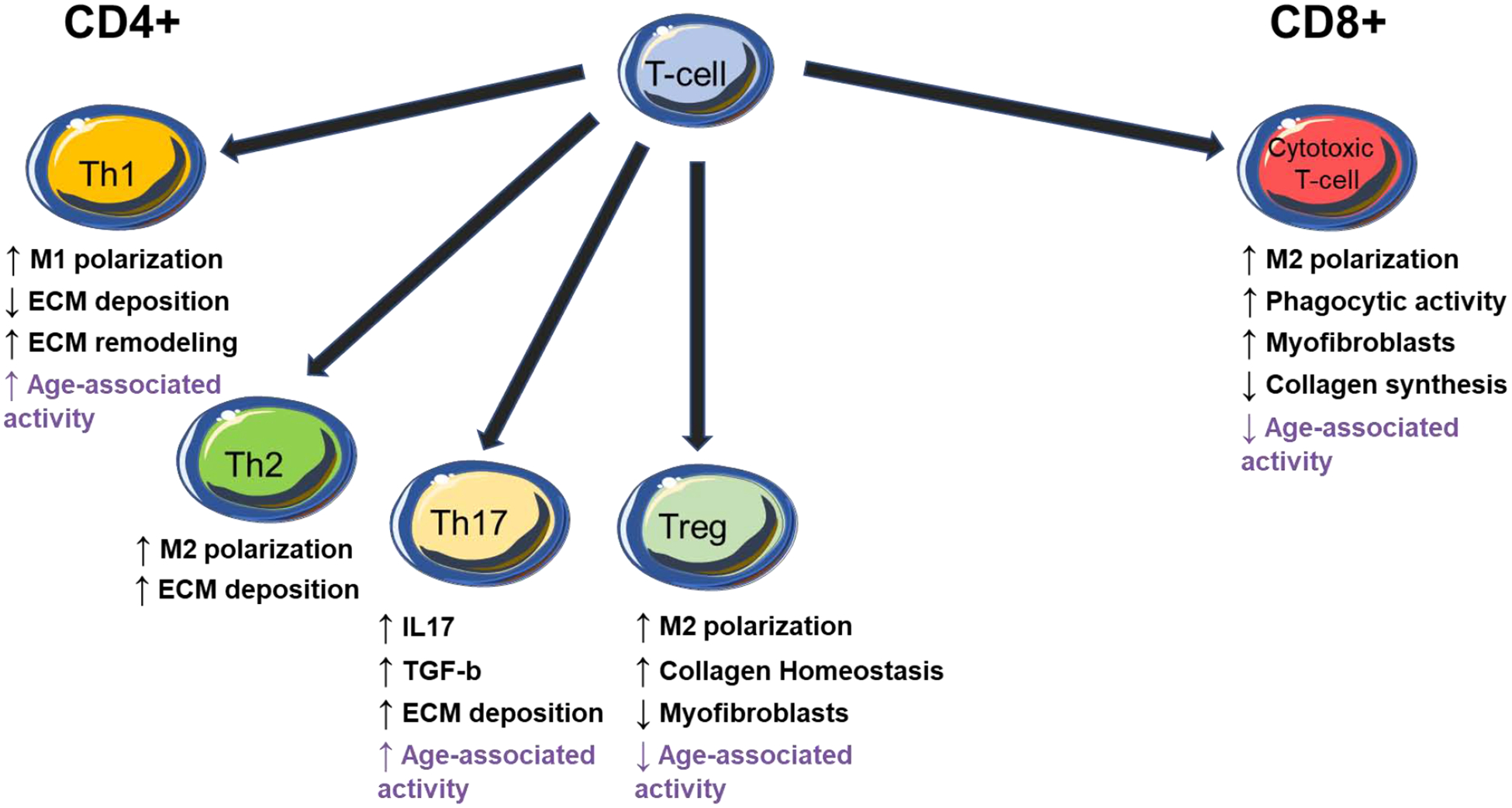
CD4+ T-cells produce multiple factors that influence macrophage (M1 versus M2) polarization which in turn influence fibroblast activity and fibrotic deposition of collagen. T-reg cells influence fibroblast activity through direct cellular interaction in tissues. CD8+ cells are critical at early and late times after ischemic injury. T-cell activity with known age-dependent changes in activity are shown in purple. IL- interleukin; TGFβ- tissue growth factor β.
The complexity of T-cell subsets combined with injury and time-dependent effects of T-cell activity complicate simple categorical conclusions about whether T-cells might be beneficial and/or detrimental in different cardiac milieus. Nonetheless, some patterns are emerging that suggest, for example, that CD4+ Th1 cells are recruited earlier to the MI myocardium whereas the Th2 population emerges at later time points to overtake the Th1 cells. Th1 secreted factors are implicated in matrix remodeling and pro-inflammatory responses. Factors secreted by Th2, on the other hand, favor ECM deposition and wound repair. CD8+ cells recruited at both early times after MI and at later time points facilitate clearance of cellular debris and aid in ECM remodeling and repair thus necessitating activity both during the initial inflammatory wound healing phase and during scar formation. Clearly, both age and sex influence T-cell function and activity and represent important areas of research for future studies. Future work should also focus on characterizing both naïve and memory T-cells in patients with MI to determine how these cells contribute to post-ischemic heart dilation and dysfunction. Therapeutic strategies targeting the adaptive immune system by either stimulating protective immune functions or attenuating the activity of immune pathogenic effectors should be assessed.
Highlights.
T-cells contribute both directly and indirectly to ECM remodeling and fibrosis
T-cell activity influences monocyte recruitment and macrophage activation
Direct binding of T-cells to cardiac fibroblasts can lead to activation of collagen producing fibroblasts
Secretion of T-cell related cytokines play a role in production and degradation of myocardial ECM
Funding Sources.
This work was supported by the National Institute of Health (NIDA) U54DA016511 to KYD-P and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award IK2BX003922 to KYD-P and I01CX001608 to ADB. This work was also financially supported, in part, by the 2019 S&R Foundation Ryuji Ueno Award that was bestowed upon KYD-P by the American Physiological Society. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Veterans Administration, or the American Physiological Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Frangogiannis NG, The Extracellular Matrix in Ischemic and Nonischemic Heart Failure, Circ Res 125(1) (2019) 117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nattel S, Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation, JACC Clin Electrophysiol 3(5) (2017) 425–435. [DOI] [PubMed] [Google Scholar]
- [3].Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde AV, Conway SJ, Graff J, Frangogiannis NG, Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program, Circ Res 124(8) (2019) 1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rubart M, Tao W, Lu XL, Conway SJ, Reuter SP, Lin SF, Soonpaa MH, Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart, Cardiovascular research 114(3) (2018) 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nielsen SH, Mouton AJ, DeLeon-Pennell KY, Genovese F, Karsdal M, Lindsey ML, Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes, Matrix biology : journal of the International Society for Matrix Biology 75–76 (2019) 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Christensen G, Herum KM, Lunde IG, Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease, Matrix biology : journal of the International Society for Matrix Biology 75–76 (2019) 286–299. [DOI] [PubMed] [Google Scholar]
- [7].McDonald LT, Zile MR, Zhang Y, Van Laer AO, Baicu CF, Stroud RE, Jones JA, LaRue AC, Bradshaw AD, Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis, Am J Physiol Heart Circ Physiol 315(1) (2018) H92–H100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Bras LE, A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis, J Am Coll Cardiol 66(12) (2015) 1364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kim SH, Turnbull J, Guimond S, Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor, J Endocrinol 209(2) (2011) 139–51. [DOI] [PubMed] [Google Scholar]
- [10].Mack M, Inflammation and fibrosis, Matrix biology : journal of the International Society for Matrix Biology 68–69 (2018) 106–121. [DOI] [PubMed] [Google Scholar]
- [11].DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML, Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing, JCI insight 2(18) (2017) e94207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].van Nieuwenhoven FA, Hemmings KE, Porter KE, Turner NA, Combined effects of interleukin-1alpha and transforming growth factor-beta1 on modulation of human cardiac fibroblast function, Matrix biology : journal of the International Society for Matrix Biology 32(7–8) (2013) 399–406. [DOI] [PubMed] [Google Scholar]
- [13].Maqbool A, Hemmings KE, O’Regan DJ, Ball SG, Porter KE, Turner NA, Interleukin-1 has opposing effects on connective tissue growth factor and tenascin-C expression in human cardiac fibroblasts, Matrix biology : journal of the International Society for Matrix Biology 32(3–4) (2013) 208–14. [DOI] [PubMed] [Google Scholar]
- [14].Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M, Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction, J Mol Cell Cardiol 62 (2013) 24–35. [DOI] [PubMed] [Google Scholar]
- [15].Blanton RM, Carrillo-Salinas FJ, Alcaide P, T-cell recruitment to the heart: friendly guests or unwelcome visitors?, Am J Physiol Heart Circ Physiol 317(1) (2019) H124–H140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hofmann U, Frantz S, Role of Lymphocytes in Myocardial Injury, Healing, and Remodeling After Myocardial Infarction, Circ Res 116(2) (2015) 354–367. [DOI] [PubMed] [Google Scholar]
- [17].Hofmann U, Frantz S, Role of T-cells in myocardial infarction, European heart journal 37(11) (2016) 873–9. [DOI] [PubMed] [Google Scholar]
- [18].Zlotoff DA, Bhandoola A, Hematopoietic progenitor migration to the adult thymus, Annals of the New York Academy of Sciences 1217 (2011) 122–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD, Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure, Circ Heart Fail 10(3) (2017) e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD, Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy, Circulation 139(2) (2019) 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, DeLeon-Pennell KY, CD8(+) T-cells negatively regulate inflammation post-myocardial infarction, Am J Physiol Heart Circ Physiol 317(3) (2019) H581–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Diaz A, Munoz E, Johnston R, Korn JH, Jimenez SA, Regulation of human lung fibroblast alpha 1(I) procollagen gene expression by tumor necrosis factor alpha, interleukin-1 beta, and prostaglandin E2, J Biol Chem 268(14) (1993) 10364–71. [PubMed] [Google Scholar]
- [23].Barbul A, Breslin RJ, Woodyard JP, Wasserkrug HL, Efron G, The effect of in vivo T helper and T suppressor lymphocyte depletion on wound healing, Annals of surgery 209(4) (1989) 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ulloa L, Doody J, Massague J, Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway, Nature 397(6721) (1999) 710–3. [DOI] [PubMed] [Google Scholar]
- [25].Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D, Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation, Journal of leukocyte biology 83(6) (2008) 1323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gurujeyalakshmi G, Giri SN, Molecular mechanisms of antifibrotic effect of interferon gamma in bleomycin-mouse model of lung fibrosis: downregulation of TGF-beta and procollagen I and III gene expression, Exp Lung Res 21(5) (1995) 791–808. [DOI] [PubMed] [Google Scholar]
- [27].Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG, Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction, Circ Res 105(10) (2009) 973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu Q, Vazquez R, Zabadi S, Watson RR, Larson DF, T-lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice, Matrix biology : journal of the International Society for Matrix Biology 29(6) (2010) 511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Panduro M, Benoist C, Mathis D, Treg cells limit IFN-gamma production to control macrophage accrual and phenotype during skeletal muscle regeneration, Proceedings of the National Academy of Sciences of the United States of America 115(11) (2018) E2585–E2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yan X, Zhang H, Fan Q, Hu J, Tao R, Chen Q, Iwakura Y, Shen W, Lu L, Zhang Q, Zhang R, Dectin-2 Deficiency Modulates Th1 Differentiation and Improves Wound Healing After Myocardial Infarction, Circ Res 120(7) (2017) 1116–1129. [DOI] [PubMed] [Google Scholar]
- [31].Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K, IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice, Scientific reports 7(1) (2017) 6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Han J, Kim YS, Lim MY, Kim HY, Kong S, Kang M, Choo YW, Jun JH, Ryu S, Jeong HY, Park J, Jeong GJ, Lee JC, Eom GH, Ahn Y, Kim BS, Dual Roles of Graphene Oxide To Attenuate Inflammation and Elicit Timely Polarization of Macrophage Phenotypes for Cardiac Repair, ACS Nano 12(2) (2018) 1959–1977. [DOI] [PubMed] [Google Scholar]
- [33].Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S, Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction, Circ Heart Fail 7(5) (2014) 822–30. [DOI] [PubMed] [Google Scholar]
- [34].Wang K, Wen S, Jiao J, Tang T, Zhao X, Zhang M, Lv B, Lu Y, Zhou X, Li J, Nie S, Liao Y, Wang Q, Tu X, Mallat Z, Xia N, Cheng X, IL-21 promotes myocardial ischaemia/reperfusion injury through the modulation of neutrophil infiltration, Br J Pharmacol 175(8) (2018) 1329–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY, Zhu ZF, Xia N, Nie SF, Zhang M, Zeng ZP, Lv BJ, Jiao J, Liu H, Xian ZS, Yang XP, Hu Y, Liao YH, Wang Q, Tu X, Mallat Z, Huang Y, Shi GP, Cheng X, Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction, Theranostics 8(16) (2018) 4552–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Van Linthout S, Miteva K, Tschope C, Crosstalk between fibroblasts and inflammatory cells, Cardiovascular research 102(2) (2014) 258–69. [DOI] [PubMed] [Google Scholar]
- [37].Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, Lai A, Ghahary A, Broady R, Levings MK, Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells, Journal of immunology 185(6) (2010) 3199–208. [DOI] [PubMed] [Google Scholar]
- [38].Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG, Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function, Am J Physiol Heart Circ Physiol 307(8) (2014) H1233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifro W, Miteva K, Kaschina E, Thiel A, Imboden H, Wang J, Steckelings U, Steinhoff G, Unger T, Li J, Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury, Journal of immunology 185(10) (2010) 6286–93. [DOI] [PubMed] [Google Scholar]
- [40].Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S, Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice, Circulation 125(13) (2012) 1652–63. [DOI] [PubMed] [Google Scholar]
- [41].Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S, Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation, Circ Res 115(1) (2014) 55–67. [DOI] [PubMed] [Google Scholar]
- [42].Van der Borght K, Scott CL, Nindl V, Bouche A, Martens L, Sichien D, Van Moorleghem J, Vanheerswynghels M, De Prijck S, Saeys Y, Ludewig B, Gillebert T, Guilliams M, Carmeliet P, Lambrecht BN, Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells, Cell Rep 18(12) (2017) 3005–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barnden MJ, Allison J, Heath WR, Carbone FR, Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements, Immunol Cell Biol 76(1) (1998) 34–40. [DOI] [PubMed] [Google Scholar]
- [44].Jameson SC, Masopust D, Understanding Subset Diversity in T Cell Memory, Immunity 48(2) (2018) 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abesser M, Heinze KG, Schuh K, Beyersdorf N, Kerkau T, Demengeot J, Frantz S, Hofmann U, Myocardial aging as a T-cell-mediated phenomenon, Proceedings of the National Academy of Sciences of the United States of America 114(12) (2017) E2420–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N, Bennaceur K, Zaman A, Keavney B, Spyridopoulos I, T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients, J Clin Invest 125(8) (2015) 3063–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cheng X, Liao YH, Ge H, Li B, Zhang J, Yuan J, Wang M, Liu Y, Guo Z, Chen J, Zhang J, Zhang L, TH1/TH2 functional imbalance after acute myocardial infarction: coronary arterial inflammation or myocardial inflammation, J Clin Immunol 25(3) (2005) 246–53. [DOI] [PubMed] [Google Scholar]
- [48].Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez-Quesada C, Lu B, Gerard C, Frangogiannis NG, CXCR3-independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans, Cardiovascular research 103(2) (2014) 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X, Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration, J Am Coll Cardiol 59(4) (2012) 420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yan X, Shichita T, Katsumata Y, Matsuhashi T, Ito H, Ito K, Anzai A, Endo J, Tamura Y, Kimura K, Fujita J, Shinmura K, Shen W, Yoshimura A, Fukuda K, Sano M, Deleterious effect of the IL-23/IL-17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction, J Am Heart Assoc 1(5) (2012) e004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Engelbertsen D, Andersson L, Ljungcrantz I, Wigren M, Hedblad B, Nilsson J, Bjorkbacka H, T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke, Arteriosclerosis, thrombosis, and vascular biology 33(3) (2013) 637–44. [DOI] [PubMed] [Google Scholar]
- [52].Maier E, Duschl A, Horejs-Hoeck J, STAT6-dependent and -independent mechanisms in Th2 polarization, Eur J Immunol 42(11) (2012) 2827–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kondo H, Hojo Y, Tsuru R, Nishimura Y, Shimizu H, Takahashi N, Hirose M, Ikemoto T, Ohya K, Katsuki T, Yashiro T, Shimada K, Elevation of plasma granzyme B levels after acute myocardial infarction, Circ J 73(3) (2009) 503–7. [DOI] [PubMed] [Google Scholar]
- [54].Tae Yu H, Youn JC, Lee J, Park S, Chi HS, Lee J, Choi C, Park S, Choi D, Ha JW, Shin EC, Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction, Cell Mol Immunol 12(4) (2015) 466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA, Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells, Blood 101(7) (2003) 2711–20. [DOI] [PubMed] [Google Scholar]
- [56].Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing I Group for the Women’s Health Initiative, Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial, JAMA 288(3) (2002) 321–33. [DOI] [PubMed] [Google Scholar]
- [57].Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, Trevisan M, Black HR, Heckbert SR, Detrano R, Strickland OL, Wong ND, Crouse JR, Stein E, Cushman M, I. Women’s Health Initiative, Estrogen plus progestin and the risk of coronary heart disease, N Engl J Med 349(6) (2003) 523–34. [DOI] [PubMed] [Google Scholar]
- [58].Xu Y, Arenas IA, Armstrong SJ, Davidge ST, Estrogen modulation of left ventricular remodeling in the aged heart, Cardiovascular research 57(2) (2003) 388–94. [DOI] [PubMed] [Google Scholar]
- [59].Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, Ko M, LaCroix AZ, Margolis KL, Stefanick ML, Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause, JAMA 297(13) (2007) 1465–77. [DOI] [PubMed] [Google Scholar]
- [60].Yang XP, Reckelhoff JF, Estrogen, hormonal replacement therapy and cardiovascular disease, Curr Opin Nephrol Hypertens 20(2) (2011) 133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pollow DP Jr., Uhlorn JA, Sylvester MA, Romero-Aleshire MJ, Uhrlaub JL, Lindsey ML, Nikolich-Zugich J, Brooks HL, Menopause and FOXP3(+) Treg cell depletion eliminate female protection against T cell-mediated angiotensin II hypertension, Am J Physiol Heart Circ Physiol 317(2) (2019) H415–H423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].DeLeon-Pennell KY, Mouton AJ, Ero OK, Ma Y, Padmanabhan Iyer R, Flynn ER, Espinoza I, Musani SK, Vasan RS, Hall ME, Fox ER, Lindsey ML, LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling, Basic research in cardiology 113(5) (2018) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Takeuchi H, Yokota-Nakatsuma A, Ohoka Y, Kagechika H, Kato C, Song SY, Iwata M, Retinoid X receptor agonists modulate Foxp3(+) regulatory T cell and Th17 cell differentiation with differential dependence on retinoic acid receptor activation, Journal of immunology 191(7) (2013) 3725–33. [DOI] [PubMed] [Google Scholar]
- [64].Herold M, Breuer J, Hucke S, Knolle P, Schwab N, Wiendl H, Klotz L, Liver X receptor activation promotes differentiation of regulatory T cells, PloS one 12(9) (2017) e0184985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC, Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X, Proceedings of the National Academy of Sciences of the United States of America 113(14) (2016) E2029–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, Jellison ER, McCullough LD, Age-Associated Resident Memory CD8 T Cells in the Central Nervous System Are Primed To Potentiate Inflammation after Ischemic Brain Injury, Journal of immunology 196(8) (2016) 3318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].McCullough LD, Mirza MA, Xu Y, Bentivegna K, Steffens EB, Ritzel R, Liu F, Stroke sensitivity in the aged: sex chromosome complement vs. gonadal hormones, Aging (Albany NY) 8(7) (2016) 1432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Manwani B, Bentivegna K, Benashski SE, Venna VR, Xu Y, Arnold AP, McCullough LD, Sex differences in ischemic stroke sensitivity are influenced by gonadal hormones, not by sex chromosome complement, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 35(2) (2015) 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML, The impact of aging on cardiac extracellular matrix, GeroScience 39(1) (2017) 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG, Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction, J Am Coll Cardiol 51(14) (2008) 1384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, Zouein F, Lindsey ML, Jin YF, Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence, Cardiovascular research 106(3) (2015) 421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ, The influence of age on T cell generation and TCR diversity, Journal of immunology 174(11) (2005) 7446–52. [DOI] [PubMed] [Google Scholar]
- [73].Ponnappan S, Ponnappan U, Aging and immune function: molecular mechanisms to interventions, Antioxid Redox Signal 14(8) (2011) 1551–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Maue AC, Yager EJ, Swain SL, Woodland DL, Blackman MA, Haynes L, T-cell immunosenescence: lessons learned from mouse models of aging, Trends Immunol 30(7) (2009) 301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, Saare M, Vilo J, Metspalu A, Milani L, Peterson P, Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes, Scientific reports 5 (2015) 13107. [DOI] [PMC free article] [PubMed] [Google Scholar]