

Abstract
Extracellular matrix (ECM) is the foundation on which all cells and organs converge to orchestrate normal physiological functions. In the setting of pathology, the ECM is modified to incorporate additional roles, with modifications including turnover of existing ECM and deposition of new ECM. The fibroblast is center stage is coordinating both normal tissue homeostasis and response to disease. Understanding how fibroblasts work under normal conditions and are activated in response to injury or stress will provide mechanistic insight that triggers discovery of new therapeutic treatments for a wide range of disease. We highlight here fibroblast roles in the cancer, lung, and heart as example systems where fibroblasts are major contributors to homeostasis and pathology.
Introduction
The extracellular matrix (ECM) is defined as all extracellular constituents that combined provide structure, function, and signaling to the cells and organs it surrounds. ECM is comprised of a number of varied components, ranging from fibrillar molecules that provide structure to small molecules that refine structure to membrane proteins that coordinate signaling across cells. For example, cell shape is directed by basement membrane ligands that initiate distinct signaling cascades.[1] As such, the ECM is the foundation on which cells align to form organ systems. The ECM provides cues to maintain homeostasis under normal conditions and to respond to signaling induced under disease states.[2] Of note, both transforming growth factor β (Tgfb) and Wnt signaling are common across conditions where ECM accumulates.[3–7]
The fibroblast is the predominant producer of ECM across all organs. The ECM is necessary for cell and tissue maintenance and for responses to injury, and too little or too much ECM production can yield severe dysfunction. In addition to ECM abundance, variations in ECM composition and quality can alter the ECM microenvironment in dramatic ways. The fibroblast is a key cell type that oversees ECM quantity and quality. Because of this, much attention has been given to fibroblast roles in fibrotic disorders. The term fibroblast activation is used to denote a fibroblast response to any stimuli, whether it be to immune cues, mechanical stretch, or other causes.
The goal of this review is to summarize recent knowledge advancements in the field of fibroblast biology, highlighting the articles that comprise this special issue (Figure 1). We focus our review on 3 major avenues, namely fibroblast roles in cancer, lung, and heart, as well as discussion on how these separate areas can inform each other. Taken together, this overview provides the current state of knowledge regarding fibroblast functions and highlights the gaps that remain to be filled.
Figure 1.
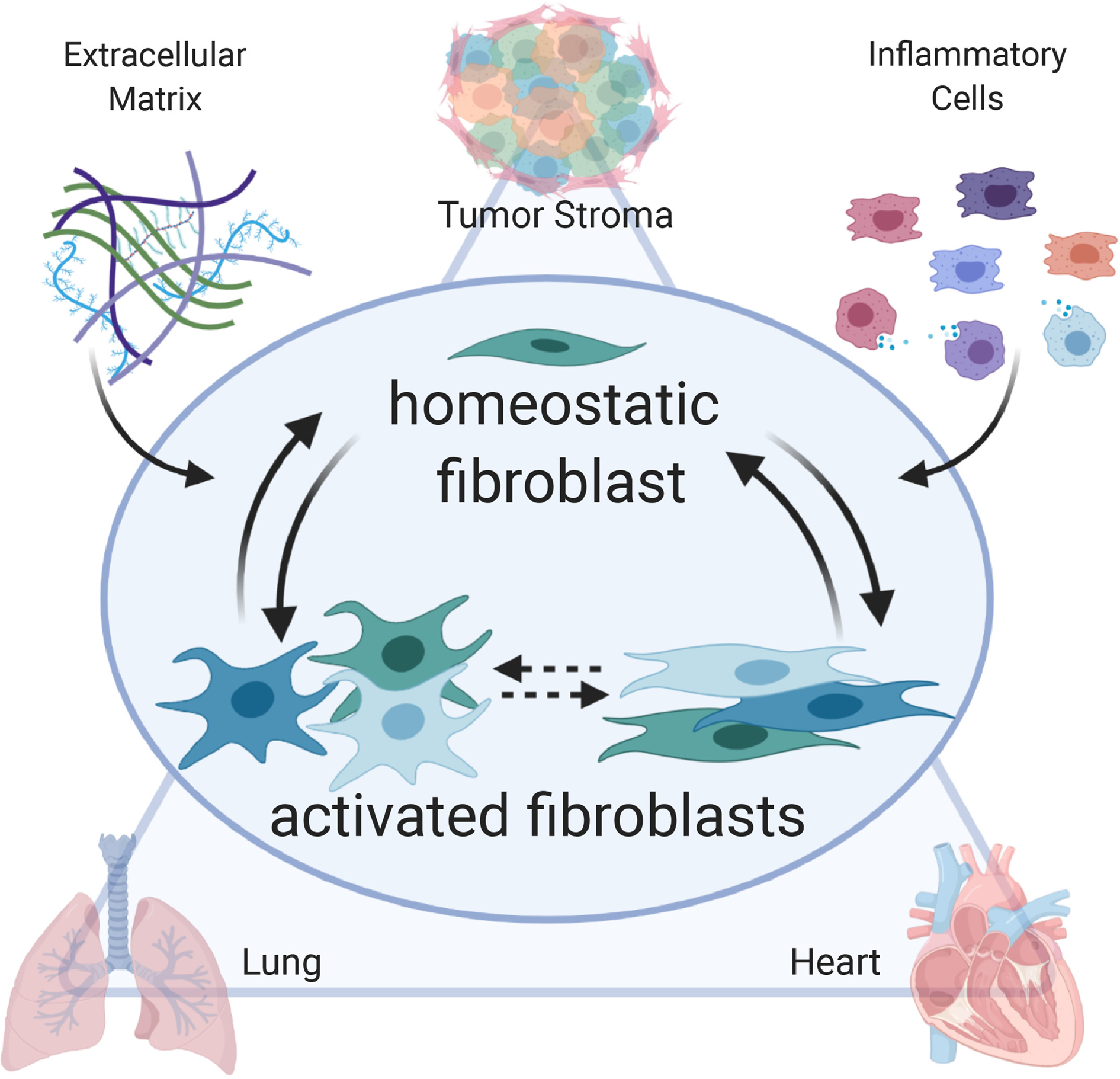
In a variety of systems (e.g., tumor stroma, lung, and heart), the fibroblast is the central arbiter of homeostasis and response to injury. The fibroblast integrates cues from the environment, including signals from inflammatory cells and the extracellular matrix (ECM), to coordinate breakdown of existing ECM and production of new ECM. During response to injury, the fibroblast becomes activated in a continuum of states ranging from pro-inflammatory to anti-inflammatory to pro-reparative. The culmination of the fibroblast response is a return to homeostasis, albeit often within a newly defined scar environment, or continued activation to generate pathological fibrosis.
Fibroblasts in Cancer
Cancer develops when a temporary wound healing process is not resolved, resulting in perpetually inappropriate ECM that establishes co-dependency between tissue and tumor.[8] There are dynamic reciprocal systems in cancer that continually evolve both in response to the initial perturbation as well as in response to the complex adaptations of the response and to treatment.[9] The ECM is a fundamental component of cancer, as tumor invasion and progression involve continual ECM synthesis and turnover.[10] The ECM fluctuates to become more permissive for tumor invasion, and ECM is produced by both tumor and tissue.[11] As such, tissue architecture dictates regulatory controls, and fibroblasts influence tissue structure in multiple ways.[12, 13] Cancer-associated fibroblasts (CAFs) are defined as chronically activated fibroblasts within the tumor topography and serve as the primary source of ECM, as well as primary cell type regulating ECM remodeling and coordinating angiogenesis.[14, 15] Extrinsic modulation of the environment by substrate rigidity influences the intrinsic forces of the CAF, which in turn affects CAF production of ECM.[15] CAFs have two major polarizing roles: 1) inflammatory CAFs secrete cytokines (e.g., IL-6 and IL-8) and 2) Tgfb-dependent CAFs produce ECM and are force generating.[12] It is important to note that binary characterization is simplistic and limiting, as CAFs also contribute immunosuppression components and can serve a range of roles that span the full heterogeneity gamut from pro- to anti-tumor.[12]
CAF heterogeneity is derived from the shifting of phenotypes that occurs within the tissue-tumor environment, coincident with the shifting of the environment. Sources of CAFs include endothelial-to-mesenchymal transition and bone marrow derived progenitors. Regardless of source, CAFs respond similarly to the local microenvironment by expression a common gene signature. Indeed, CAF phenotype is determined by the environment into which they were activated and the environment is which they currently reside.[10] Cell lineage tracing experiments, therefore, may not be important if present environment rather than source dictates phenotype. While the concept of heterogeneity has been put forward, fibroblast specific protein 1 and alpha smooth muscle actin are the two frequently used markers of CAF. There is no current consensus on how many fibroblast subtypes there are, their biological properties and signaling, and whether there is plasticity, which impedes our understanding of the mechanisms of fibrosis.[16] This lack of defined markers to differentiate heterogeneity limits our current understanding.[8] More details on the gene and protein markers that distinguish CAFs, as well as differences in secretome patterns, would help to define expression differences that translation to differences in CAF physiology.
Tgfb and Wnt signaling are common denominators between cancer and ECM accumulation, and understanding how changes in signaling influence CAF phenoytpes will help to elucidate the connections between wound healing and cancer.[4] Likewise, the connections between inflammation, cancer, and fibrosis are also very strong, and understanding how the macrophage coordinates across processes will be informative.[17] In addition to genomics technologies, quantitative proteomics will reveal translational and post-translational effects of inflammation and how it influences ECM in cancer.[18]
Fibroblasts in the Lung
The lung is responsible for a number of functions, including filtering and delivering inhaled and exhaled air through the conducting airways, serving as conduits of air, blood, and lymphatics through bronchovascular bundles, and exchanging gas in the alveolar region.[19] Resident fibroblasts are important for maintaining normal homeostasis in the lung.[19, 20] Continual low secretion of ECM, collagens in particular, provide structural integrity to maintain distinct anatomic and physiological features. Fibroblasts continually secrete ECM to maintain the normal pulmonary environment.
Fibrosis in the lung tends to follow the general wound healing paradigm (i.e. hemostasis, inflammation, proliferation and remodeling), with a few additional mechanisms of fibrosis unique to the lung.[20] The lung contains of a variety of cell types including type I and II alveolar epithelial cells (AECs), endothelial cells, and fibroblasts. Type I AECs repair minor damage by initiating proliferation and differentiation of the type II AECs to reestablish the epithelial barrier. This allows the lung to heal without scarring.[21] In contrast, if damage is more severe, cells such as macrophages and T-cells are recruited often resulting in lung fibrosis. Mild repeated injury can prolong strain on the alveolar unit which in turn activates both fibroblasts and immune cells leading to enhancement of injury and sustained and self-propagating fibrosis.[22] As a result, instead of a resolving the injury response, the lung enters a vicious cycle, with increased matrix stiffness, enhanced local injury, and accumulation of fibroblasts and epithelial cells.[23]
Karina-Perl et al. highlight the complexity of lung fibroblasts and suggest that while some lineages exist, most of what we observe are variations in functional stage along degrees of plasticity.[24] Computational and mathematical models have been developed and used to track and monitor different fibroblast phenotypic states in real time and have provided a way to more deeply and thoroughly investigate the underlying drivers of lung fibrosis in different disease states.[20] Analysis of dissociated non-fibrotic lungs samples identified two major fibroblast populations that were conserved across mouse, rat, pig, and human pulmonary tissues: Col13a1+ fibroblasts and Col14a1+ fibroblasts.[25] Col13a1+ fibroblasts display genes involved in type II AEV cell regulation and may represent a regenerative population of lipofibroblasts.[24, 25] Less is known about Col14a1+ fibroblast other than they harbor a unique set of significant genes, including Pi16, Mmp3, Cygb, and Rtp4.[16]
Clustering of data generated from single cell RNA-seq reveal that fibroblasts from bleomycin-induced fibrotic lungs show considerable heterogeneity within the fibroblast population [26]. Peyser et al. illustrated that fibroblasts exhibit widely varying degrees of activation including a mix of markers involved in ECM development (Timp1, Col1a1, Col1a2, Col5a1, Cthrc1, Eln), muscle contraction (Acta2, Actg2, Tagln, Tpm2, Des), cell–matrix interaction (Spp1, Thbs1, Tnc, Dcn), and cell signaling (Serpine1, Spp1, Postn, Ltbp2, Tnc, Dcn, Igfbp2) [26]. Xie et al. identified Pdgfrbhi fibroblasts as newly emerging cell population in response to the bleomycin-induced fibrotic injury.[16] Pdgfrb was highly expressed by both Col13a1 and Col14a1 matrix fibroblasts.
Tsukui and colleagues identified a subset of Cthrc1 (collagen triple helix repeat containing 1)+ fibroblasts which were independent from Col13a1 and Col14a1+ fibroblast in fibrotic lungs of both mice and humans. Cthrc1+ fibroblasts demonstrated an increase in migratory capacity compared to other subsets of collagen-producing cells within the lungs of bleomycin-treated mice. Using spatially-resolved proteomics, lungs from both bleomycin-induced lung fibrosis and lung adenocarcinoma share a common ECM signature not observed in healthy lung tissues [18]. What distinguished these diseases from one another was the presence of specific ECM components such as fibrinogen which is elevated in lung fibrosis compared to lung adenocarcinoma samples. The diversity of genes identified highlights the complex regulation of matrix production and turnover and cell–matrix interaction involved in the early stages of lung fibrosis.
Computational models can also be used to test the effects of different treatments in a high-throughput and cost-efficient manner.[20] Hao et al. used a simplified model of a lung as a cube that contained spatially distributed smaller cubes representative of alveoli [27]. Using this model system they tested five potential treatments: anti-TNF-α, anti-PDGF, anti-IL-13, anti-TGF-β and a combination treatment of anti-IL-13 and anti-TGF-β. Treatment was simulated for both mild and severe cases of IPF and concentration of ECM over time was measured as a means to test for treatment efficacy. Both the mild and severe cases responded to anti-TGF-β however the combination treatment of anti-IL-13 and anti-TGF-β proved to be the most effective [27]. As techniques to simplify the complexity of the fibroblast population are developed, our understanding of the multiple subpopulations and their contribution to the production and remodeling of the ECM in healthy and diseased tissues improves.
Better understanding of how changes in the distribution of lung ECM are both the response to fibrosis as well as the initiator could identify novel therapeutic targets for chronic lung disease.[23] Circadian rhythm needs to be considered when evaluating lung pathological processes involving ECM accumulation, as lungs exhibit diurnal variations in immune response, similar to skin and heart.[28] Differentiating common and distinct process across different types of lung injury will reveal targets that may have more generalizable applications.
Fibroblasts in the Heart
Resident cardiac fibroblasts are critical for maintaining normal homeostasis in the heart. Continual low secretion of basement membrane ECM provides structure on which cardiomyocytes contract and the coronary vasculature is maintained. While previously considered an inert cell in the absence of pathology, we now know that fibroblasts continually secrete ECM to maintain the normal cardiac environment. The two cardiovascular pathologies that contribute most to mortality rates are pressure overload induced by hypertension and myocardial infarction (MI). The cardiac fibroblast responds to these pathologies in very different ways, which highlights that information from one pathology does not easily translate over to inform the other.[29–31]
The heart responds to hypertension induced pressure overload by undergoing hypertrophic growth of the cardiac myocytes along with initial perivascular fibrosis. With prolonged pressure overload, the hypertrophy response becomes insufficient and heart failure can ensue. During this transition, interstitial fibrosis is observed. The renin-angiotensin-aldosterone signaling (RAAS) system has been very well studied in pressure overload and cardiac fibroblasts, in particular fibroblasts in the in vitro setting.[30] A better understanding of how cardiac fibroblasts respond to pressure overload in vivo is needed, along with more depth of details regarding interrelationship among signaling pathways, such as RAAS and transforming growth factor β signaling, is needed to predict how modulation will adjust net ECM accumulation.[32] A major driving component in the progression from pressure overload compensated hypertrophy to heart failure is the activation of an abnormal fibroblast phenotype that is resistant to apoptosis and degrades normal ECM to replace it with a fibrotic matrix structure.[33] The Spinale lab has made the observation that this fibroblast phenotype mimics the CAF that responds to chemotherapeutic agents.[33] By this analogy, activated fibroblasts in the heart likely arise primarily from bone marrow precursors during initial stages where perivascular fibrosis dominates, and from a mesenchymal source during later stages where interstitial fibrosis dominates. Use of such agents targeting the pressure overload activated cardiac fibroblast might attenuate the pathophysiological fibrosis that is a hallmark of heart failure. A note of caution in that these agents must be designed to target abnormal fibroblasts with high specificity without also inducing damage to the cardiomyocyte that would negate benefit.
MI occurs when the myocardium is exposed to ischemia of longer than 30 min duration, usually due to an occluded coronary artery preventing blood flow to the left ventricle. In response to MI, the left ventricle reacts with a wound healing response that involves a robust upregulation of inflammation that stimulates protease induced removal of necrotic myocytes followed by scar formation to replace the myocytes. Fibroblasts are major players in scar formation following MI. Daseke et al reveal how fibroblast phenotypes shift through the different phases of MI remodeling.[34] Early after MI, fibroblasts transition to an inflammatory phenotype, followed by an anti-inflammatory and pro-angiogenic transition and later by a pro-reparative and anti-angiogenic phenotype.[35] The culmination of fibroblast activation is production of sufficient scar in the infarct zone to replace necrotic cardiomyocytes. The final fibroblast phenotype reverts back to homeostasis, however this is under new baseline conditions as the infarct scar contains a lot more ECM than the normal myocardium. While the cardiac fibroblast transitions across the MI time continuum are now better defined, how to modify to influence long-term outcomes is the next arena.
For both pressure overload and MI, recognizing the regulator nodes of each fibroblast cell state will allow us to dial the system up or down in controlled ways.[36] Computational studies provide a way to rapidly inform on responses from a wide number of scenarios.[37] Understanding cell to cell cross-talk and using spatial-omics approaches can reveal effects on cell heterogeneity.[38] For example, there are a number of inflammatory cells including macrophages and T cells that co-reside within the infarcted myocardium that both regulate and are regulated by the fibroblast.[39, 40] In addition, more information on post-translational modifications (identification and roles) is needed.[41] In particular, understanding how glycosylation influences ECM structure and function are needed.[42, 43] Limiting the inflammatory phenotype and promoting the reparative phenotype could potentially improve the process of scar formation. In contrast, enhancing the reparative phenotype could cause excess collagen deposition and fibrosis, leading to reduced cardiac function. Of note, strategies that are effective for pressure overload induced pathological cardiac ECM accumulation will likely be much different from effective strategies to modify infarct scar. While this review focused on pressure overload and MI as two predominant stimuli of fibroblast activation, use of cancer treatment also triggers cardiac fibroblast activation. This is a growing arena of investigation into fibroblast roles. Several have shown that doxorubicin treatment leads to cardiac fibroblast activation consistent with impaired inflammation resolution.[44, 45]
It is also important to understand the role of the fibroblasts in different regions of the heart. While not studied extensively, there are noted differences in behaviors and regulations of ventricular vs. atrial fibroblasts. Understanding the role of the atrium in atrial fibrillation will show how cross-talk within the heart is influenced by pathologies that activate fibroblasts.[46] Likewise, atrial fibroblasts are likely differentially activated based on pathological stimulus. Thus, the right balance between ECM degradation and synthesis is needed for a balanced medium of providing enough structure without creating an overly stiff environment. How this is defined will vary by pathology. Acquiring new knowledge of fibroblast phenotypes in relation to pathology stage and region will allow us to more accurately target fibroblasts to improve long-term outcomes.
Conclusions
There are a number of areas in the fibroblast research arena that are fruitful for investigation (Table 1). Patient response to disease are not uniform, and this diversity of response may involve heterogeneity in fibroblast activation. In addition, patient responses change as a factor of age or in the presence of other modifying processes, such that individual responses may change depending on the context under which the fibroblast is activated. Understanding how fibroblasts activate to yield differences in responses may inform clinic decisions, including the need for transient vs. continual treatment. It would be interesting to know if patients have similar fibroblast responses to injury across their different organ systems, such that one system could inform and predict response by another organ system.
Table 1.
Areas for future investigation
|
A number of current technologies aid in filling these gaps. RNA-sequencing (single cell and global) will provide information on transcriptional effects. For example, Xie and colleagues have recently provided a collection of single-cell transcriptomes and distinct classification of fibroblast subsets in the lung of normal and bleomycin treated mice.[16] They classified six mesenchymal cell types in normal lung and seven in fibrotic lung. A similar atlas of collagen-producing cells has also been generated for human lungs, both under normal conditions and from patients with idiopathic pulmonary fibrosis and scleroderma.[19] Experiments such as these provide a roadmap for studying the unique fibroblast populations and provide new resources for understanding the fibroblast landscape its roles in disease. Coupling these with proteomic approaches will allow translation and post-translational effects to be understood.[18, 47] Adding in temporal and spatial considerations will provide hints on cause and effect mechanisms by assigning which events occur first and in proximity with outcome measurements. A better understanding of how exosomes coordinate cell-ECM signaling is needed.[13, 48] Multiplex spatial omics will help resolve the cellular complexities that contribute to overall cell phenotype/state, cell-cell interactions, and tissue architecture. Although beneficial, spatial omics will require advances such as developing techniques for live cell imaging to increase the resolution and throughput. For all of these approaches, we continue to develop statistics and visualization tools to allow us to digest our results. In addition, there is a need to standardize within the field how we present and analyze omics results.
As shown in this review and by the articles in this special issue, fibroblasts share a common name, yet have a wide range of activities depending on the stimulus to which they are responding. The fibroblast research community has exploded over the last two decades, and we look forward to seeing how the field continues to evolve.
Highlights.
Fibroblasts are the arbiters of extracellular matrix remodeling.
Fibroblasts undergo temporal activation changes throughout the wound healing response.
Fibroblasts share homeostatic and wound healing roles across systems.
Acknowledgements.
Dr. Lindsey is a Stokes-Shackleford Professor at UNMC. We acknowledge funding from National Institutes of Health under Award Numbers R13HL149435, U54DA016511, HL075360, HL127283, HL129823, HL132585, and HL137319, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Numbers 5I01BX000505 and IK2BX003922. This work was also financially supported, in part, by the 2019 S&R Foundation Ryuji Ueno Award that was bestowed upon KYD-P by the American Physiological Society. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies.
Abbreviations
- CAF
cancer-associated fibroblast
- ECM
extracellular matrix
- IL
interleukin
- MI
myocardial infarction
- RAAS
renin-angiotensin-aldosterone signaling
- Tgfb
transforming growth factor beta
- Wnt
wingless and int-1 signaling transduction pathway
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Randles MJ, Lausecker F, Humphries JD, Byron A, Clark SJ, Miner JH, Zent R, Humphries MJ, Lennon R, Basement membrane ligands initiate distinct signalling networks to direct cell shape, Matrix Biol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Karamanos NK, Theocharis AD, Neill T, Iozzo RV, Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases, Matrix Biol 75–76 (2019) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Siljamaki E, Rappu P, Riihila P, Nissinen L, Kahari VM, Heino J, H-Ras activation and fibroblast-induced TGF-beta signaling promote laminin-332 accumulation and invasion in cutaneous squamous cell carcinoma, Matrix Biol 87 (2020) 26–47. [DOI] [PubMed] [Google Scholar]
- [4].Burgy O, Konigshoff M, The WNT signaling pathways in wound healing and fibrosis, Matrix Biol 68–69 (2018) 67–80. [DOI] [PubMed] [Google Scholar]
- [5].Gyorfi AH, Matei AE, Distler JHW, Targeting TGF-beta signaling for the treatment of fibrosis, Matrix Biol 68–69 (2018) 8–27. [DOI] [PubMed] [Google Scholar]
- [6].Rebolledo DL, Gonzalez D, Faundez-Contreras J, Contreras O, Vio CP, Murphy-Ullrich JE, Lipson KE, Brandan E, Denervation-induced skeletal muscle fibrosis is mediated by CTGF/CCN2 independently of TGF-beta, Matrix Biol 82 (2019) 20–37. [DOI] [PubMed] [Google Scholar]
- [7].Valle-Tenney R, Rebolledo DL, Lipson KE, Brandan E, Role of hypoxia in skeletal muscle fibrosis: Synergism between hypoxia and TGF-beta signaling upregulates CCN2/CTGF expression specifically in muscle fibers, Matrix Biol 87 (2020) 48–65. [DOI] [PubMed] [Google Scholar]
- [8].Kurie J, Fibroblast heterogeneity and its impact on extracellular matrix and immune landscape remodeling in cancer, Matrix biology (in press) (2020). [DOI] [PubMed] [Google Scholar]
- [9].Xiao W, Wang S, Zhang R, Sohrabi A, Yu Q, Liu S, Ehsanipour A, Liang J, Bierman RD, Nathanson DA, Seidlits SK, Bioengineered scaffolds for 3D culture demonstrate extracellular matrix-mediated mechanisms of chemotherapy resistance in glioblastoma, Matrix Biol 85–86 (2020) 128–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Avery D, Govindaraju P, Jacob M, Todd L, Monslow J, Pure E, Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts, Matrix Biol 67 (2018) 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gotte M, Kovalszky I, Extracellular matrix functions in lung cancer, Matrix Biol 73 (2018) 105–121. [DOI] [PubMed] [Google Scholar]
- [12].Cukierman E, Cancer Associated Fibroblast: Mediators of Tumorigenesis, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lu H, Bowler N, Harshyne LA, Craig Hooper D, Krishn SR, Kurtoglu S, Fedele C, Liu Q, Tang HY, Kossenkov AV, Kelly WK, Wang K, Kean RB, Weinreb PH, Yu L, Dutta A, Fortina P, Ertel A, Stanczak M, Forsberg F, Gabrilovich DI, Speicher DW, Altieri DC, Languino LR, Exosomal alphavbeta6 integrin is required for monocyte M2 polarization in prostate cancer, Matrix Biol 70 (2018) 20–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hutchenreuther J, Vincent K, Norley C, Racanelli M, Gruber SB, Johnson TM, Fullen DR, Raskin L, Perbal B, Holdsworth DW, Postovit LM, Leask A, Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma, Matrix Biol 74 (2018) 52–61. [DOI] [PubMed] [Google Scholar]
- [15].Malik R, Luong T, Cao X, Han B, Shah N, Franco-Barraza J, Han L, Shenoy VB, Lelkes PI, Cukierman E, Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2, Matrix Biol 81 (2019) 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, Liu N, Kulur V, Yao C, Chen P, Liu Z, Stripp B, Tang J, Liang J, Noble PW, Jiang D, Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis, Cell Rep 22(13) (2018) 3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].El-Nadi M, Hassan H, Saleh ME, Nassar E, Ismail YM, Amer M, Greve B, Götte M, El-Shinawi M, Ibrahim SA, Induction of heparanase via IL-10 correlates with a high infiltration of CD163+ M2-type tumor-associated macrophages in inflammatory breast carcinomas, Matrix Biology Plus (2020) 100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gocheva V, Naba A, Bhutkar A, Guardia T, Miller KM, Li CM, Dayton TL, Sanchez-Rivera FJ, Kim-Kiselak C, Jailkhani N, Winslow MM, Del Rosario A, Hynes RO, Jacks T, Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival, Proc Natl Acad Sci U S A 114(28) (2017) E5625–E5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, Kaminski N, Matthay MA, Wolters PJ, Sheppard D, Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis, Nat Commun 11(1) (2020) 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leonard-Dukea J, Evans S, Hannanc RT, Barker TH, Bates JHT, Bonham CA, Moore BB, Kirschner DE, Peirce SM, Multi-scale Models of Lung Fibrosis, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, Amselem S, Clement A, Alveolar epithelial cells: master regulators of lung homeostasis, Int J Biochem Cell Biol 45(11) (2013) 2568–73. [DOI] [PubMed] [Google Scholar]
- [22].Yu G, Ibarra GH, Kaminski N, Fibrosis: Lessons from OMICS analyses of the human lung, Matrix Biol 68–69 (2018) 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gu BH, Madison MC, Corry D, Kheradmand F, Matrix remodeling in chronic lung diseases, Matrix Biol 73 (2018) 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Perl A-K, The Elephant in the Lung: An attempt to decipher overlap of fibroblast populations by integrating known lineage-tracing, molecular markers, and single cell sequencing data, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Raredon MSB, Adams TS, Suhail Y, Schupp JC, Poli S, Neumark N, Leiby KL, Greaney AM, Yuan Y, Horien C, Linderman G, Engler AJ, Boffa DJ, Kluger Y, Rosas IO, Levchenko A, Kaminski N, Niklason LE, Single-cell connectomic analysis of adult mammalian lungs, Sci Adv 5(12) (2019) eaaw3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, Zhang W, Devalaraja-Narashimha K, Grindley J, Halasz G, Morton L, Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing, Am J Respir Cell Mol Biol 61(1) (2019) 74–85. [DOI] [PubMed] [Google Scholar]
- [27].Hao W, Marsh C, Friedman A, A Mathematical Model of Idiopathic Pulmonary Fibrosis, PLoS One 10(9) (2015) e0135097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sherratt MJ, Hopkinson L, Naven M, Hibbert SA, Ozols M, Eckersley A, Newton VL, Bell M, Meng QJ, Circadian rhythms in skin and other elastic tissues, Matrix Biol 84 (2019) 97–110. [DOI] [PubMed] [Google Scholar]
- [29].Soliman H, Rossi FMV, Cardiac fibroblast diversity in health and disease, Matrix biology (in press) (2020). [DOI] [PubMed] [Google Scholar]
- [30].Frangogiannis NG, The Extracellular Matrix in Ischemic and Nonischemic Heart Failure, Circ Res 125(1) (2019) 117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li L, Zhao Q, Kong W, Extracellular matrix remodeling and cardiac fibrosis, Matrix Biol 68–69 (2018) 490–506. [DOI] [PubMed] [Google Scholar]
- [32].AlQudah M, Hale TM, Czubryt MP, Targeting the Renin-Angiotensin-Aldosterone System in Fibrosis, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oatmen KE, Cull E, Spinale FG, Heart failure as interstitial cancer: emergence of a malignant fibroblast phenotype, Nat Rev Cardiol (2019). [DOI] [PubMed] [Google Scholar]
- [34].Daseke II MJ, Tenkorang MA, Chalise U, Konfrst SR, Lindsey ML, Cardiac Fibroblast Activation during Myocardial Infarction Wound Healing, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ 2nd, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, Lindsey ML, Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis, Basic Res Cardiol 114(2) (2019) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bretherton R, Bugg D, Olsewski E, Davis J, Regulators of Cardiac Fibroblast Cell State, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zeigler AC, Nelson AR, Chandrabhatla AS, Brazhkina O, Holmes JW, Saucerman JJ, Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bingham GC, Lee F, Naba A, Barker TH, Spatial-omics: Novel Approaches to Probe Cell Heterogeneity and ECM Biology, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bradshaw AD, DeLeon-Pennell KY, T-cell regulation of fibroblast and cardiac fibrosis, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML, Mapping macrophage polarization over the myocardial infarction time continuum, Basic Res Cardiol 113(4) (2018) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brás L.E.d.C., Frangogiannis NG, Extracellular matrix-derived peptides in tissue remodeling and fibrosis, Matrix biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Christensen G, Herum KM, Lunde IG, Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease, Matrix Biol 75–76 (2019) 286–299. [DOI] [PubMed] [Google Scholar]
- [43].Contessotto P, Ellis BW, Jin C, Karlsson NG, Zorlutuna P, Kilcoyne M, Pandit A, Distinct glycosylation in membrane proteins within neonatal versus adult myocardial tissue, Matrix Biol 85–86 (2020) 173–188. [DOI] [PubMed] [Google Scholar]
- [44].Jadapalli JK, Wright GW, Kain V, Sherwani MA, Sonkar R, Yusuf N, Halade GV, Doxorubicin triggers splenic contraction and irreversible dysregulation of COX and LOX that alters the inflammation-resolution program in the myocardium, Am J Physiol Heart Circ Physiol 315(5) (2018) H1091–h1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lindsey ML, Lange RA, Parsons H, Andrews T, Aune GJ, The tell-tale heart: molecular and cellular responses to childhood anthracycline exposure, Am J Physiol Heart Circ Physiol 307(10) (2014) H1379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Reese-Petersen AL, Olesen MS, Karsdal M, Svendsen JH, Genovese F, Atrial Fibrillation and Cardiac Fibrosis: A Review on the Potential of Extracellular Matrix Proteins as Biomarkers, Matrix biology (in press) (2020). [DOI] [PubMed] [Google Scholar]
- [47].Stefanelli VL, Choudhury S, Hu P, Liu Y, Schwenzer A, Yeh CR, Chambers DM, Pesson K, Li W, Segura T, Midwood KS, Torres M, Barker TH, Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion, Matrix Biol 82 (2019) 86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sanderson RD, Bandari SK, Vlodavsky I, Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling, Matrix Biol 75–76 (2019) 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]