

Abstract
Injectable, long-acting drug delivery systems provide effective drug concentrations in the blood for up to 6 months. Naltrexone-loaded poly(lactide-co-glycolide) (PLGA) microparticles were prepared using an in-line homogenization method. It allows the transition from a laboratory scale to scale-up production. This research was designed to understand how the processing parameters affect the properties of the microparticles, such as microparticle size distributions, surface and internal morphologies, drug loadings, and drug release kinetics, and thus, to control them.
The in-line homogenization system was used at high flow rates for the oil- and water-phases, e.g., 100 mL/min and 400 mL/min, respectively, to continuously generate microparticles. A high molecular weight (148 kDa) PLGA at various concentrations was used to generate oil phases with a range of viscosities and also to compare with a 64 and 79 kDa at a single, high concentration. The uniformity of the microparticles was found to be related to the viscosity of the oil phase. As the viscosity of the oil phase increased from 52.6 mPa·s to 4,046 mPa·s, the span value (a measure of uniformity) increased from 1.24 to 3.1 for the microparticles generated at the homogenization speed of 2,000 RPM. Increasing the PLGA concentration from 5.58% to 16.85% showed a corresponding rise in the encapsulation efficiency (EE) from 74.0% to 85.8% and drug loading (DL) from 27.4% to 31.7% for the microparticles made with the homogenization speed of 2,000 RPM. These increases may be due to a faster shell formulation, enabling PLGA microparticles to entrap more naltrexone into the structure. A higher DL, however, shortened the drug release duration from 56 to 42 days. The changes in morphology of the microparticles during different phases of the in vitro release study were also studied for three types of microparticles made with different PLGA concentrations and MWs. As PLGA microparticles went through structural changes, the surface showed raisin-like wrinkled morphologies within the first 10 days. Then, the microparticles swelled to form smooth surfaces. The in-line approach produced PLGA microparticles with a highly reproducible size distribution, DL, and naltrexone release rate.
Keywords: PLGA microparticles, in-line homogenization, naltrexone, oil-phase viscosity, drug loading, encapsulation efficiency
Graphical Abstract
1. Introduction
Opioid overdose continues to cause the majority of drug-related deaths in the United States. In 2017 alone more than 70,000 people died of a drug overdose, and more than 50,000 of those deaths were due to opioids [1]. This number decreased slightly in 2018, but still, about 47,000 people died from opioid overdoses [2]. Two main strategies have been used to overcome the opioid dependence issue and minimize its destructive effects: (i) development of abuse-deterrent formulations (ADFs), such as using physical barriers and aversive agents to hinder opioid extraction and abuse, and (ii) application of antagonists, such as naltrexone, blocking the opioid receptor. The second approach was found to be more effective due to the limited usefulness of current ADFs, which can be cracked readily by various means [3]. Naltrexone has been routinely prescribed to treat opioid dependence for the last few decades [3–5]. Initial treatment with naltrexone relied on daily oral administration, which resulted in low patient compliance. An injectable, one-month naltrexone microparticle formulation has been available clinically to overcome patient compliance issues [6, 7].
Injectable, long-acting depot formulations have been made with biodegradable poly(lactide-coglycolide) (PLGA) polymers [8–10]. Drug-loaded PLGA microparticles have been typically made using an emulsification-solvent extraction approach [10–14]. Drug-loaded PLGA micro- and nano-particles have been studied [15, 16]. For example, the effects of parameters such as polymer and emulsifier concentration, MW, lactide:glycolide (L:G) ratio, and molecular shape of PLGA (linear or branched) were investigated to discover their relationships to the sizes, encapsulation efficiency (EE), DL, and release kinetics of PLGA micro- and nano-particles [10, 17–27].
Despite extensive work on drug-loaded PLGA microparticles, the U.S. Food and Drug Administration (FDA) has approved only about 20 formulations for injectable, long-acting, PLGA-based drug delivery systems since its first use in 1989 [28]. This slow development in controlled drug delivery systems is due to the lack of a clear understanding of the mechanisms of PLGA microparticle formation. In some studies, the drug loading has been low, e.g., <5% [18, 20, 29], but higher drug loadings are preferred as they reduce the number of microparticles to use. Regarding the size characterization of the particles, the size distribution and uniformity of the microparticles were usually not described, and only the mean diameter of the microparticles was reported [18, 19, 24]. The lack of batch-to-batch reproducibility is another commonly cited disadvantage of the conventional PLGA microparticle generation techniques [30, 31]. To address these issues, micro- and nano-particles were made using other approaches, such as microfluidics [32–34], and membrane emulsification [35–37]. Both methods, however, suffer from low production rates, mostly below 10 mL/min, which is a limiting factor for the scale-up production of drug-loaded PLGA micro- and nano-particles. Although some improvements were made to enhance the production rate and compatibility with the scale-up phase, such as parallel microfluidic platforms and pilot-scale membranes [33, 38, 39], they are still not competitive with conventional methods in the industry which produce on the order of liters per minute [40].
This study presents an approach that can potentially shed more light on the mechanisms of the formation of drug-loaded PLGA microparticles and the involved parameters. Naltrexone-loaded PLGA microparticles were made in a continuous manner using an in-line homogenizer with a flow rate of 100 mL/min for the oil phase. The oil-phase was made using PLGA with the concentration range of 5.58–16.85% (w/w) and MW of 148 kDa, and the viscosity ranged from 52.6 mPa·s to 4,046 mPa·s. Additionally, PLGAs, with the concentration of 16.85% and three MWs of 64 kDa, 79 kDa, and 148 kDa, were used to generate the naltrexone-PLGA microparticles, while keeping the L:G ratio constant at 85:15. This study focused on finding dominant parameters controlling the properties of naltrexone-loaded PLGA microparticles, such as size distribution, DL, EE, and drug release kinetics.
2. Materials and methods
2.1. Materials
Two PLGA copolymers molecular weights of 64 kDa and 79 kDa were purchased from LACTEL® Absorbable Polymers (Birmingham, Alabama), and one PLGA of 148 kDa from Evonik industries (Darmstadt, Germany). All three PLGAs were ester end-capped and had the same L:G ratio of 85:15. Naltrexone base anhydrous was purchased from Mallinckrodt Pharmaceuticals (St. Louis, MO). Dichloromethane (DCM), benzyl alcohol, acetonitrile, methanol, ethylene glycol, potassium phosphate monobasic, and sodium azide were obtained from Fisher Scientific Co. (Fair Lawn, NJ, USA). Poly(vinyl alcohol) (PVA) 40–88 (molecular weight ~205,000 g/mol) was purchased from Millipore Sigma (Darmstadt, Germany). Sodium L-ascorbate and phosphate buffered saline Tween 20 (PBST) (pH 7.4) were purchased from Sigma Aldrich (St. Louis, MO).
2.2. Formulation information for naltrexone microparticles
The oil phase included PLGA and naltrexone dissolved in benzyl alcohol and DCM. Benzyl alcohol was used as a co-solvent due to the poor solubility of naltrexone in DCM. In all of the formulations, the theoretical DL was kept constant at 37%. Details about the oil-phase formulations are shown in Table 1. The viscosity of the oil-phase was measured using Brookfield DV2T viscometer with a cone and plate geometry at room temperature (23.4 °C). PLGA and naltrexone were dissolved in benzyl alcohol and DCM using a magnetic stirrer for an hour. Then, the viscosities of the oil-phases made with different Formulations of A-I in Table 1 were measured at 1, 3, 6, and 20 hours after PLGA and naltrexone were in contact with the solvents. For each measurement, 1 mL of the oil phase was transferred to the cup of the viscometer to measure its viscosity. For the in-line homogenization formation of naltrexone microparticles, the oil phase was used after mixing for one hour.
Table 1.
Different oil-phase compositions used for making naltrexone- loaded PLGA microparticles.
| Formulation | PLGA (g) | PLGA MW (kDa) | Naltrexone (g) | Benzyl alcohol (g) | DCM (g) | PLGA Conc. (%)(w/w) |
|---|---|---|---|---|---|---|
| A | 1.0 × 0.625 | 148 | 1.0 × 0.3675 | 1.0 × 0.582 | 10 | 5.58 |
| B | 1.4 × 0.625 | 148 | 1.4 × 0.3675 | 1.4 × 0.582 | 10 | 7.48 |
| C | 2.0 × 0.625 | 148 | 2.0 × 0.3675 | 2.0 × 0.582 | 10 | 10.00 |
| D | 2.4 × 0.625 | 148 | 2.4 × 0.3675 | 2.4 × 0.582 | 10 | 11.63 |
| E | 2.8 × 0.625 | 148 | 2.8 × 0.3675 | 2.8 × 0.582 | 10 | 13.10 |
| F | 3.0 × 0.625 | 148 | 3.0 × 0.3675 | 3.0 × 0.582 | 10 | 13.75 |
| G | 4.0 × 0.625 | 148 | 4.0 × 0.3675 | 4.0 × 0.582 | 10 | 16.85 |
| H | 4.0 × 0.625 | 79 | 4.0 × 0.3675 | 4.0 × 0.582 | 10 | 16.85 |
| I | 4.0 × 0.625 | 64 | 4.0 × 0.3675 | 4.0 × 0.582 | 10 | 16.85 |
MW: molecular weight; DCM: dichloromethane
2.3. In-line homogenization of naltrexone-loaded PLGA microparticles
A Silverson L5M-A homogenizer with an in-line mixing assembly was used to continuously generate the naltrexone-loaded PLGA microparticles at room temperature (23.4 °C). The water phase consisted of 1% (w/v) PVA, as an emulsifier, in deionized water. Briefly, two pumps, a gear pump (Cole-Parmer) and a syringe pump (Harvard Apparatus-PHD 2000), were used to supply the aqueous and oil phases, respectively, to the in-line homogenizer. The flow rates of water and oil phases were kept constant at 400 mL/min and 100 mL/min, respectively, for all formulations. In the homogenizer, the oil was dispersed into oil droplets in the aqueous phase at either 2,000 or 3,000 RPM. A high shear screen with square holes (Silverson) was used to rapidly break oil phase droplets and homogenize them into the aqueous phase.
The outlet of the homogenizer was connected to the aqueous extraction solution for direct transfer of the generated oil/water emulsion to the extraction solution (2 L of deionized water at 4 °C). As the organic solvent diffused into the extraction solution, and naltrexone and PLGA solidified to form microparticles. After 8 hours of extraction, the formed microparticles were collected using a 25 μm sieve and transferred to a vacuum oven for intermediate drying at room temperature for 20 hours. Then, the particles for each formulation were washed using 1 L of 25% (v/v) ethanol for 8 hours at room temperature (23.4 °C). After this step, the particles were collected again using the 25 μm sieve and dried in the vacuum oven for final drying at room temperature for 20 hours. In the next step, a 150 μm sieve was used to collect the dried microparticles. This step helped to separate the particles from each other in case they aggregated during the drying process. The vacuum oven drying and ethanol washing minimized the residual organic solvents.
2.4. Size distribution of the microparticles
The size distribution of the particles was measured using a CILAS 1190 particle size analyzer. The measurement was conducted before collecting the particles using the 25 and 150 μm sieves to capture all of the particles produced with the in-line homogenizer and study their size distribution, uniformity, and the yield of our particle generation approach for different formulations. D-Values (D10, D50, and D90) were measured with the size analyzer, which are the most common metrics to describe particle size distribution, describing the intercepts for 10%, 50%, and 90% of the cumulative volume of microparticles. The span of the size distribution for the microparticles was calculated: Span = (D90-D10)/D50. The span value indicates the distribution width of the microparticles, normalized to the D50. Values closer to zero means narrower distributions of the microparticles. Additionally, since we collected the particles using the 25 and 150 μm sieves, we measured the Loss%, which is the volumetric percentage of the microparticles with diameters smaller than 25 μm and larger than 150 μm.
2.5. Surface morphology and cross-section of the microparticles
A scanning electron microscope (SEM) (Tescan Vega 3) was used to study the effect of different formulations on the surface morphology and internal microstructures of the resulting microparticles. For the sample preparation part, the collected dry microparticles were mounted on carbon taped aluminum stubs. Before transferring the particles into the SEM, they were placed in a −80 °C freezer, which is below their glass transition temperature. Then, the particles were quickly taken out of the freezer and cross-sectioned using a razor blade to study the internal microstructure of the microparticles. This created a glass-like structure and prevented the particles from deformation during cross-sectioning. In addition to SEM imaging of the microparticles before the in vitro release test, we studied the morphology of the microparticles during different phases of the naltrexone release: during the initial release, end of the initial release, end of the zero-order release, and end of the release test. The timeline was selected based on the naltrexone release profile. For each sample, the naltrexone-PLGA microparticles were incubated in the drug release medium at 37 °C with the same weight ratio of microparticles:release media. For SEM imaging, the samples were separated from the release media and desiccated at room temperature (23–24 °C) and atmospheric pressure without vacuum for 48 hours. Then, they were mounted on the carbon taped aluminum stubs.
2.6. In vitro naltrexone release test
The amount of naltrexone, as well as BA, was quantified using high performance liquid chromatography (HPLC) (Agilent 1260 Infinity) with a UV absorbance detector, set at 210 nm. The mobile phase consisted of methanol:10 mM phosphate buffer monobasic (65:35, %v/v), pH=6.6, and its flow rate was at 1 mL/min. As the stationary phase, a Zorbax® SB-C18 column (150 × 4.6 mm, 5 μm; Agilent technologies) was used. The sample injection volume was 2.5 μL for measuring the naltrexone loading and benzyl alcohol residual, and 10 μL for the in vitro naltrexone release rate.
Release media for the naltrexone-loaded PLGA was composed of PBST (10 mM, pH 7.4) containing 0.0625% (w/v) sodium ascorbate and 0.02% (w/v) sodium azide [41]. For each formulation, approximately 5 mg of the naltrexone-loaded PLGA microparticles was accurately weighed and transferred into a 50 mL volumetric flask. 20 mL of release media was added to the flask. All of the flasks were kept in an ethylene glycol-bath (Thermo Scientific Precision Shaking Water Bath SWB 27) where its temperature and shaking speed were set at 37 °C and 40 RPM, respectively. To measure the naltrexone release from the microparticles, 1 mL of each of the samples were withdrawn and replaced with fresh media. The naltrexone concentration in the media was measured using HPLC, and the cumulative release percentage (%) was plotted. The release tests were done in triplicate and the release data for each data point was reported as the mean±standard deviation (SD).
2.7. Naltrexone loading and residual benzyl alcohol
Approximately 5 mg of the naltrexone-loaded PLGA microparticles was accurately weighed and dissolved in 5 mL of acetonitrile and diluted 10 times with the mobile phase. Then, the naltrexone and benzyl alcohol concentrations in the solution were measured with HPLC. The naltrexone loading (%) and benzyl alcohol residual (%) were calculated by dividing the weight of naltrexone or benzyl alcohol by the total weight of PLGA microparticles and multiplied by 100.
3. Results
The schematic of the in-line naltrexone-loaded PLGA microparticle generation using the emulsification-extraction technique is demonstrated in Figure 1-A. The experimental set-up is also shown in Figure 1-B. Two pumps, a gear pump and a syringe pump, were used to flow the aqueous and oil-phases into the line-line homogenizer, respectively. After the oil/water emulsion was formed in the homogenizer chamber, it was flowed into the extraction solution, where the organic solvents, i.e., benzyl alcohol and DCM, diffused out of the oil-phase into the extraction solution, subsequently precipitating naltroxone-PLGA. The flow rates of the oil- and aqueous-phases were kept at 100 mL/min and 400 mL/min, respectively.
Figure 1.
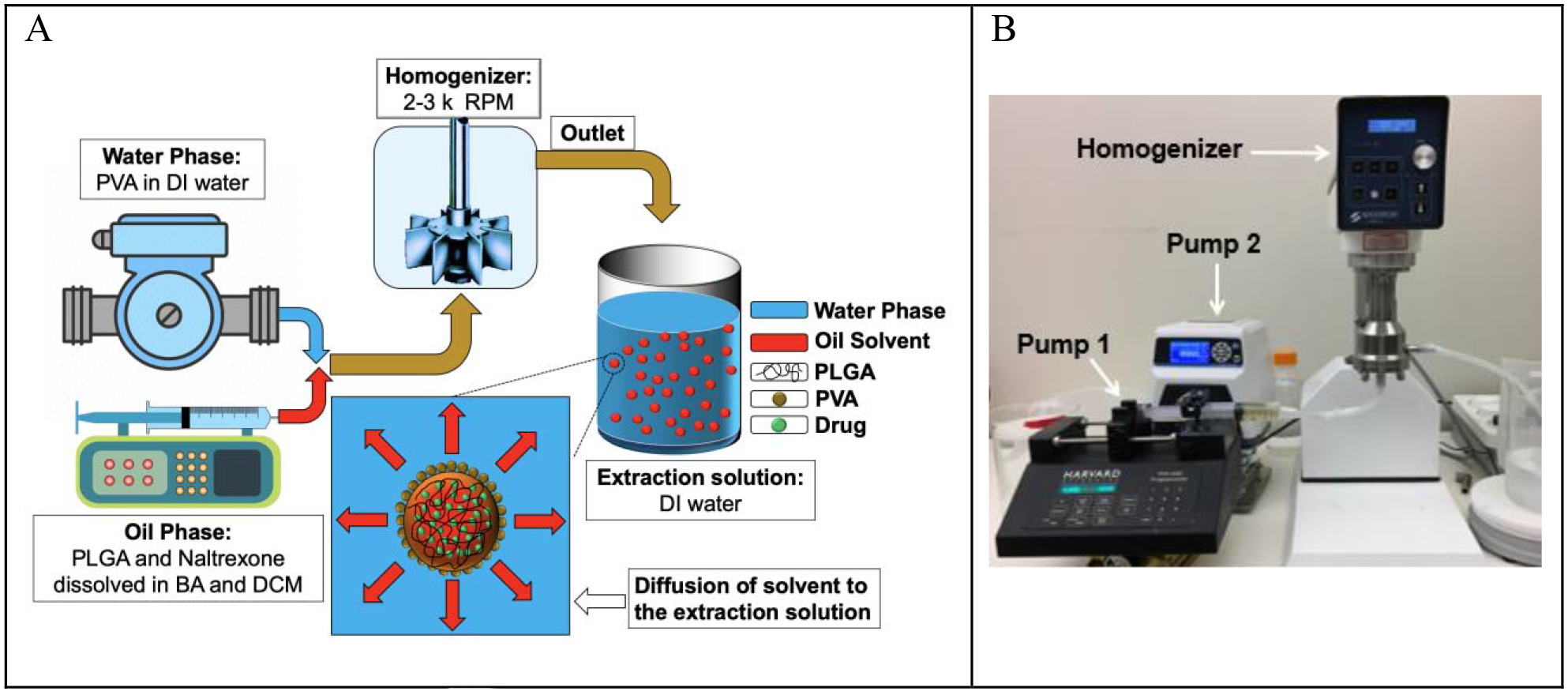
(A) Schematic of naltrexone- loaded PLGA generation using an in-line emulsification-extraction process. (B) Experimental set-up used to generate particles by pumping both oil and aqueous-phases into the homogenizer and transferring the emulsion to the extraction solution.
There are three main advantages of using an in-line homogenization system. First, it is already a scaled-up process over conventional emulsion methods based on manual mixing, where the in-line process can be run continuously versus a batch homogenization process. The current processing conditions can process 1 L of the oil phase in 10 minutes. Depending on the PLGA concentration in the oil phase, this process can generate around 120 g (Formulation A) to 400 g (Formulation G-I) of naltrexone-PLGA microparticles in 10 minutes. The production rate can be increased up to 20 L/min with some minor adjustments. Second, it may minimize the batch-to-batch variability of the microparticles as they are generated under steady state conditions, where all the of the parameters, such as flow rate and geometric factors, are fixed. Third, this platform improves on the properties of current methods, in contrast to simply increasing the size of the container and homogenizer. If the properties of the microparticles, such as size and shape, are not within the desired specification of the product, the process can be modified and brought back within specifications with minimal loss of materials.
Figure 2 and Table 2 show the viscosity of the oil phase of the different formulations (A-I). Figure 2 (a) demonstrates the dependency of the oil phase viscosity on the PLGA concentrations and MWs one hour after PLGA and naltrexone were dissolved in organic solvents, i.e., benzyl alcohol and DCM. It was found that when the concentration of PLGA (with a MW of 148 kDa) increased from 5.58% to 16.85%, the viscosity of the oil phase increased exponentially with a rate of e0.396C from 52.6 mPa·s to 4,046 mPa·s (Figure 2 (A)-red dashed line and Table 2), where C is the concentration of PLGA in the oil phase. Additionally, while keeping the PLGA concentration constant at 16.85%, the increase of the PLGA MW from 64 kDa to 148 kDa enhanced the viscosity of the oil phase with a rate of e0.034MW from 190.7 mPa·s to 4,046 mPa·s (Figure 2 (A)-green solid line and Table 2). Several studies in the area of PLGA microparticle formulations reported the viscosity of the oil phases [29, 42–44]. The range of the oil phase viscosity used in this work was wider than 350 mPa·s which was previously reported in the literature [42].
Figure 2.
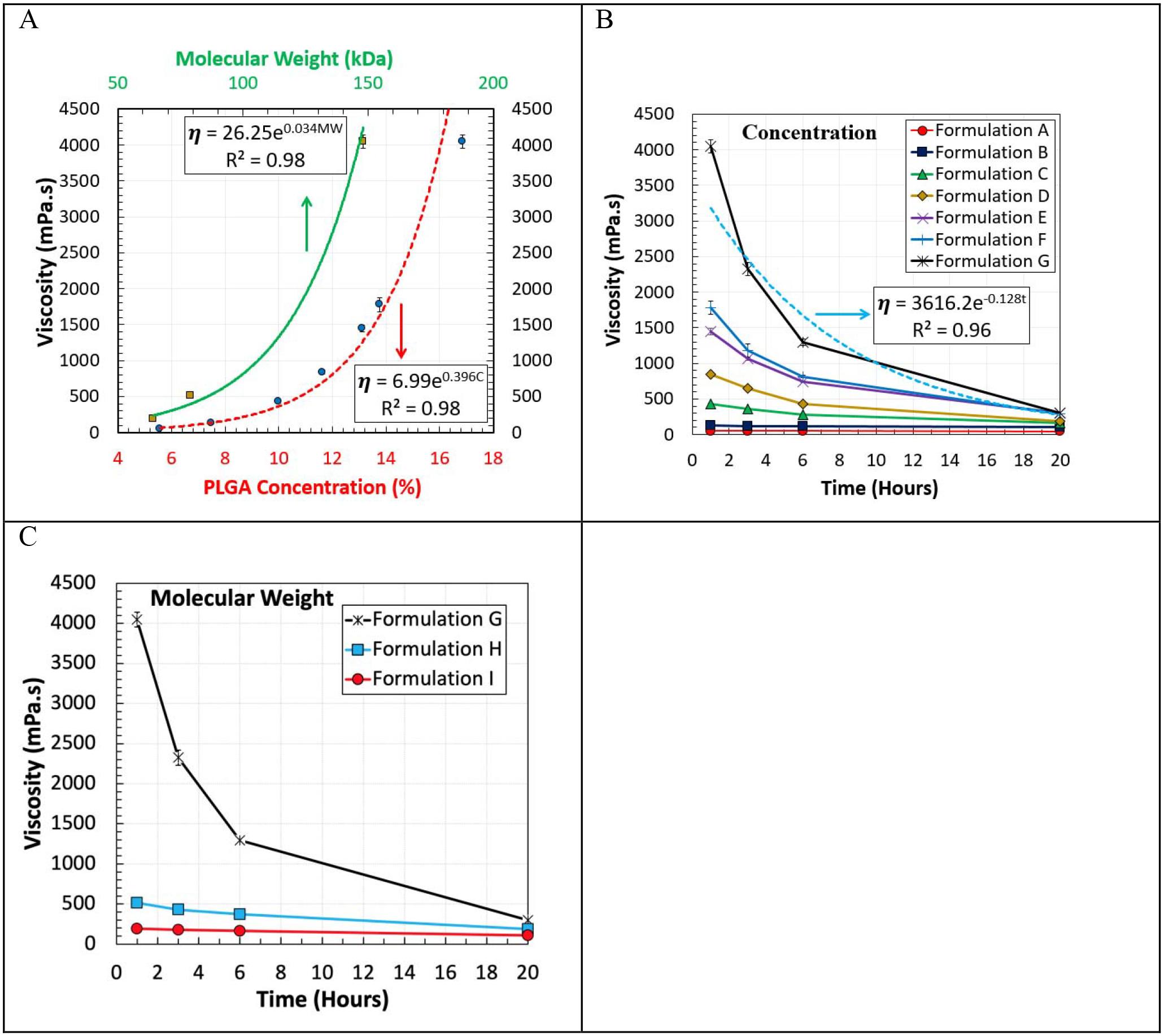
Viscosity of the oil phase at room temperature (23.4 °C) as a function of (A) the PLGA concentration (red dashed line, PLGA MW of 148 kDa), and PLGA MW (green solid line, PLGA concentration of 16.85%) after stirring for one hour. Effect of PLGA/naltrexone contact time with the organic solvents on the viscosity (B and C). All of the viscosity values were reported as mean ± standard deviation. Some of the standard deviations were smaller than the size of the symbols.
Table 2.
Viscosity (mPa·s) and viscosity reduction (%) values of the oil-phase formulations by time at 23.4 °C made with different PLGA concentrations and MWs.
| PLGA Conc. (%) | PLGA MW (kDa) | Viscosity (mPa·s) and Viscosity Reduction (VR) (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Hour 1 | Hour 3 | VR (%) | Hour 6 | VR (%) | Hour 20 | VR (%) | |||
| A | 5.58 | 148 | 52.6 ± 2.3 | 52.5 ± 1.2 | 0.2 | 52.4 ± 1.5 | 0.4 | 47.07 ± 1.86 | 10.5 |
| B | 7.48 | 148 | 125.6 ± 2.3 | 116.6 ± 1.5 | 7.2 | 111.1 ± 1.3 | 11.5 | 107.9 ± 1.55 | 14.1 |
| C | 10.00 | 148 | 426 ± 18.6 | 361.5 ± 6.2 | 15.1 | 282.8 ± 4.65 | 33.6 | 159.7 ± 3.1 | 62.5 |
| D | 11.63 | 148 | 840.3 ± 31.0 | 646.5 ± 15.5 | 23.1 | 433.7 ± 11.6 | 48.4 | 189.8 ± 4.65 | 77.4 |
| E | 13.10 | 148 | 1,442 ± 46.5 | 1,059 ± 18.6 | 26.6 | 740.4 ± 9.3 | 48.6 | 305.4 ± 3 | 78.8 |
| F | 13.75 | 148 | 1,777 ± 93.0 | 1,181 ± 93.0 | 33.5 | 813 ± 18.6 | 54.2 | 290.7 ± 4.65 | 83.6 |
| G | 16.85 | 148 | 4,046 ± 93.0 | 2,325 ± 93.0 | 42.5 | 1,293 ± 46.5 | 68.0 | 298.6 ± 4.65 | 92.6 |
| H | 16.85 | 79 | 515.3 ± 18.6 | 429.7 ± 9.3 | 16.6 | 370.2 ± 4.6 | 28.2 | 189.3 ± 4.65 | 63.3 |
| I | 16.85 | 64 | 190.7 ± 9.3 | 179.5 ± 9.3 | 5.9 | 162.8 ± 4.6 | 14.6 | 110.5 ± 1.86 | 42.0 |
Viscosity reduction (VR) (%) was calculated using the viscosity at Hour 1 as a control.
It was observed that the PLGA solution viscosity decreased over time in the presence of naltrexone. The molecular weights of PLGAs were shown to decrease when they were mixed with nucleophilic drugs, such as naltrexone, risperidone, and oxybutynin, in organic solvents [45–48]. Nucleophilic drugs are capable of cleaving ester bonds of PLGA polymers to cause a molecular weight reduction of the polymer [47]. Here, the degradation of PLGA polymers was examined in more detail by measuring the viscosity changes. Figure 2-B and C show the time-dependent change in the viscosity of the oil phase formulations made with different PLGA concentrations and MWs, respectively. The viscosity of the oil phase drops over time, which can be due to the degradation of PLGA in the presence of naltrexone. Table 2 provides the viscosity decrease over time for 9 different formulations. When the concentration of PLGA (MW of 148 kDa) increased from 5.58% to 16.85%, the viscosity reduction (%) increased from 0.2% to 42.6%, respectively, within two hours. Moreover, at 20 hours the viscosities of the oil phase for Formulations E, F, and G reached about 300 mPa·s. Table 2 demonstrates that the viscosity reduction (%) related to Formulations C and H had similar values by time. Formulation C was made by reducing the PLGA concentration in Formulation G by a factor of 1.68, and Formulation H was created by decreasing the PLGA MW in the Formulation G by a factor of 1.87. This indicates that both PLGA concentrations and MWs were equally important in degrading the dissolved PLGA and reducing the oil phase viscosity by time when the PLGA concentration was above 10%. However, when the PLGA concentration was reduced further to 7.48% and 5.58%, the viscosity reduction (%) significantly decreased to 14.1% and 10.5%, respectively after 20 hours.
Figure 3 shows SEM images of the microparticles made with Formulations C, G, and H in Table 2. The surface morphology (panels Cs, Gs, and Hs) and the cross-section of the particles (panels Cc, Gc, and Hc) were examined. The surface morphology of the particles made with a PLGA concentration of 10% (Formulation C) was found to be smoother compared to the particles made with a PLGA concentration of 16.85% (Formulations G and H). Formulations G and H showed very different surface morphologies with 3 different types of buckling, such as the round dark patch areas (yellow arrows in Figure 3), creased invaginations (green arrows), and deflated buckles (red arrows). Their presence indicates that solvent extraction was not complete in local areas, and while the remaining solvents were removed, the membrane folded inward due to the coalescence process occurring at the skin as a result of the capillary pressure [49–52]. Simply, as the solvent was removed the elastic skin became unstable, leading to shrinking and buckling or invagination [53, 54]. The local buckling phenomena observed with Formulations G and H were due to the high PLGA concentration and high viscosity. The inner morphologies also varied depending on the formulation. The cross-sectional SEM images show that Formulation G has many pores, mainly near the surface, and Formulation H has much fewer pores. The presence of small pores in Formulation G indicates that the viscosity was high preventing smaller water droplets from migrating and merging into bigger water pockets that eventually became empty pores. This is understandable, because the viscosity of Formulation G is about 10 times higher than that of Formulations C and H (Table 2). The relatively smooth inner morphology of Formulations C and H indicates that the skin formation was slower, allowing longer extraction of solvents for homogeneous distribution of PLGA molecules.
Figure 3.
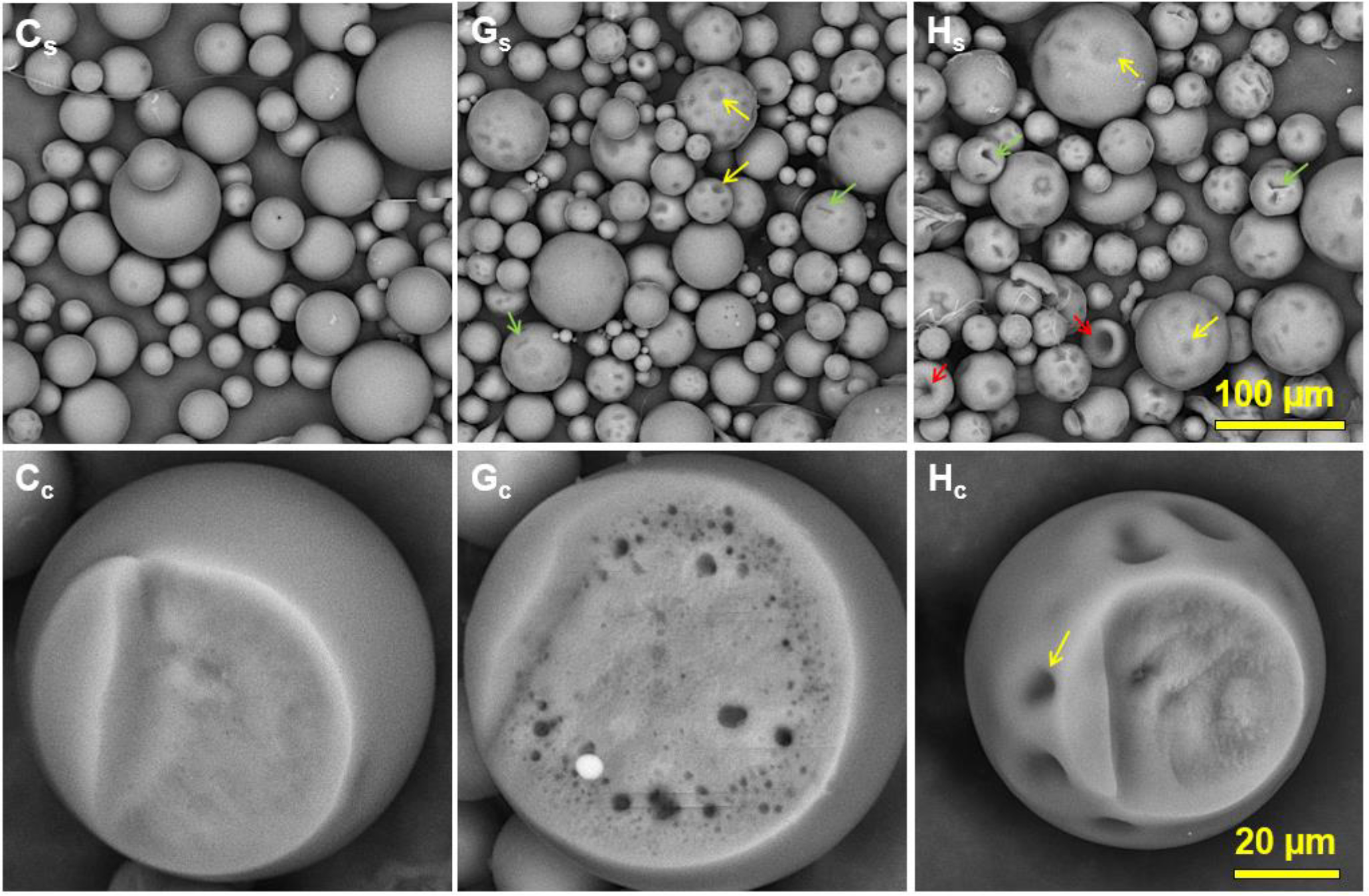
SEM images of the naltrexone- loaded particles of Formulations C, G, and H in Table 2. (Cs-Hs) Surface morphology, scale bar = 100 μm and (Cc-Hc) cross-section of the particles, scale bar = 20 μm. The homogenizer speed for all samples was 3,000 RPM.
Figure 4 shows the uniformity of the naltrexone-loaded PLGA microparticles generated using different PLGA concentrations (A and B) and MWs (C and D). The microparticles were made with two different homogenizer speeds of 2,000 (A and C) and 3,000 RPM (B and D)). Table 3 provides the D-Values (D10, D50, D90), span, and Loss% to have a better understanding of the effects of the viscosity and homogenization speed have on the size distribution of the microparticles. Figure 4 (A and B) and Table 3 demonstrate that the uniformity of microparticles gradually decreased as the viscosity increased at both homogenization speeds. The same behavior was observed in Figure 4 C and D, where the uniformity of the microparticles was improved by decreasing the viscosity of the oil-phase. Table 3 demonstrates that the decrease of the viscosity from 4,046 mPa·s to 426 mPa·s, due to the decrease of PLGA concentration and MW, improved the uniformity of the microparticles. However, by reducing the viscosity further to 52.6 mPa·s, no significant change was observed on the size distribution of the resulting microparticles. Table 3 also shows that Formulations C and H have very similar size distributions. Both Formulations C and H have similar viscosities, although they were made with different PLGA concentrations and MWs. Thus, it appears that the viscosity of the oil phase is a significant factor affecting the size distribution of the microparticles, in comparison to the PLGA MW or concentration. The same trend was observed for Formulations B and I, having similar viscosities but with different PLGA concentrations and MWs. The total loss (%) and the volume percentage (V%) of the microparticles with the diameters of <25 μm and >150 μm was increased from 14.1% to 53% when the oil phase viscosity increased from 52.6 mPa·s to 4,046 mPa·s. The information on the total loss (%) is an important parameter for scale-up production, especially for expensive drugs, such as peptides and proteins. This is because an oil phase with high viscoelasticity (as in Formulation G) retards the thread breakup process dramatically, as it stretches into a long slender liquid thread, resulting in much higher proportions of smaller droplets [55]. The size distribution of Formulation G in Figure 4 clearly shows increased volume percentages of microparticles < 25 μm. As the homogenizer speed increased from 2,000 RPM to 3,000 RPM, the D50 decreased from 22.2 μm to 18.8 μm, as expected.
Figure 4.
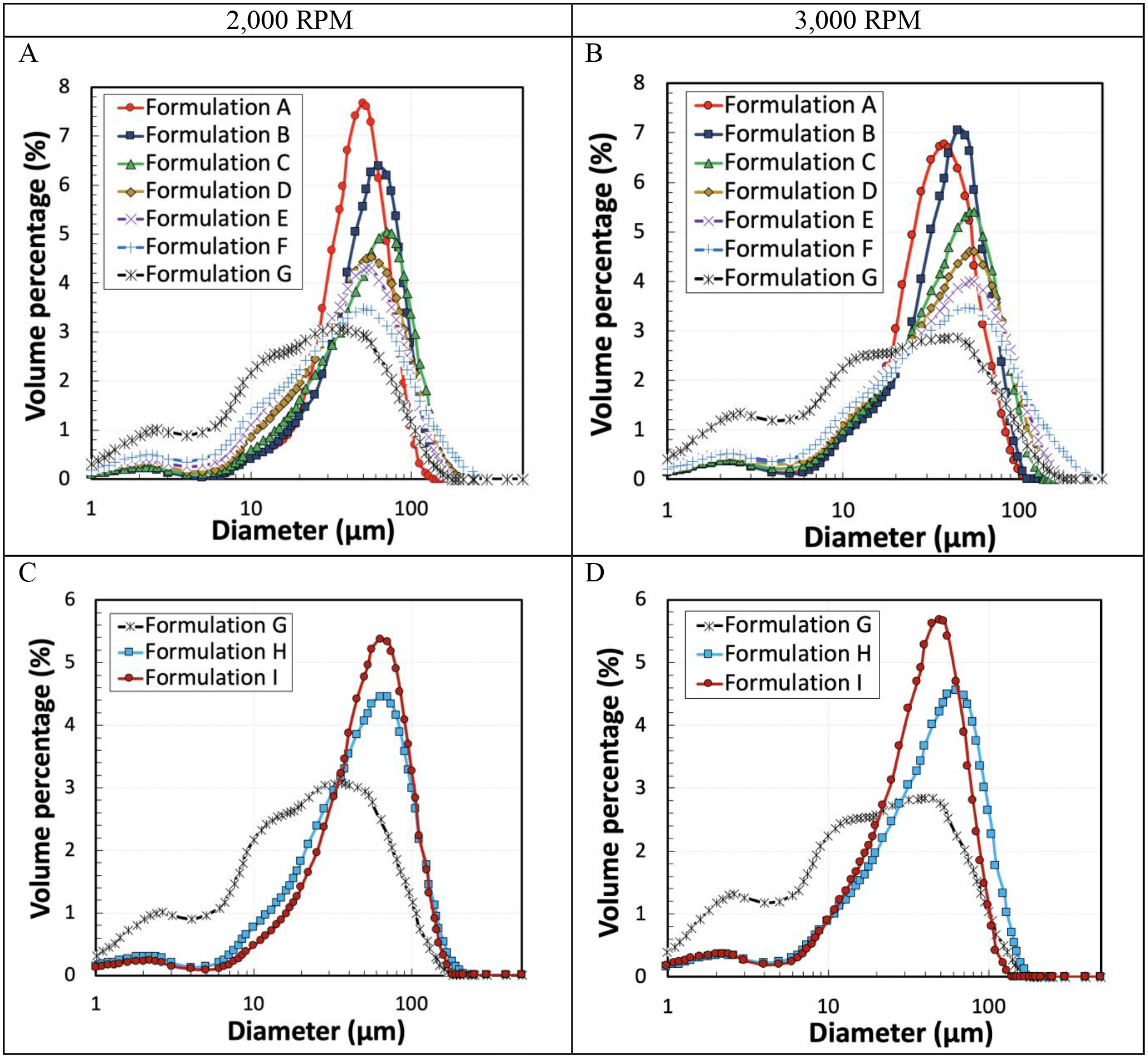
Size distribution of the particles made with different formulations prepared with a homogenizer speed of 2,000 RPM (A and C) and 3,000 RPM (B and D).
Table 3.
Size distribution metrics of the microparticles made with different oil-phase formulations and homogenizer speeds of 2,000 and 3,000 RPM.
| Homogenizer Speed = 2000 RPM | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Formulation | Viscosity (mPa·s) | D10 (μm) | D50 (μm) | D90 (μm) | DMax (μm) | Span | V% (<25 μm) | V% (>150 μm) | Total loss (%) |
| A | 52.6 ± 2.3 | 18.5 | 46.4 | 76.7 | 130 | 1.24 | 12.9 | 0 | 12.9 |
| B | 125.6 ± 2.3 | 18.2 | 54.8 | 94.7 | 170 | 1.39 | 12.6 | 0.15 | 12.8 |
| C | 426 ± 18.6 | 13.8 | 53.0 | 104.6 | 170 | 1.7 | 17.1 | 0.51 | 17.6 |
| D | 840.3 ± 31.0 | 10.6 | 44.9 | 97.9 | 212 | 1.9 | 23.1 | 1.01 | 24.1 |
| E | 1,442 ± 46.5 | 8.4 | 40.0 | 92.0 | 170 | 2.1 | 28.2 | 0.33 | 28.5 |
| F | 1,777 ± 93.0 | 6.2 | 36.7 | 103.9 | 250 | 2.7 | 31.8 | 3.22 | 35.0 |
| G | 4,046 ± 93.0 | 2.9 | 22.2 | 72.5 | 170 | 3.1 | 49.4 | 0.16 | 49.5 |
| H | 515.3 ± 18.6 | 11.6 | 48.6 | 105.2 | 212 | 1.9 | 20.2 | 1.34 | 21.6 |
| I | 190.7 ± 9.3 | 16.1 | 54.1 | 102.9 | 170 | 1.60 | 14.3 | 0.47 | 14.8 |
| Homogenizer Speed = 3000 RPM | |||||||||
| Formulation | Viscosity (mPa·s) | D10 (μm) | D50 (μm) | D90 (μm) | DMax (μm) | Span | V% (<25 μm) | V% (>150 μm) | Total loss (%) |
| A | 52.6 ± 2.3 | 11.2 | 34.4 | 62.1 | 106 | 1.48 | 26.4 | 0 | 26.4 |
| B | 125.6 ± 2.3 | 11.9 | 40.2 | 69.8 | 106 | 1.44 | 22.2 | 0 | 22.2 |
| C | 426 ± 18.6 | 9.6 | 40.0 | 78.7 | 130 | 1.7 | 26.6 | 0 | 26.6 |
| D | 840.3 ± 31.0 | 7.8 | 38.6 | 85.0 | 170 | 2.0 | 29.2 | 0.14 | 29.4 |
| E | 1,442 ± 46.5 | 6.0 | 36.4 | 89.3 | 170 | 2.3 | 32.1 | 0.28 | 32.3 |
| F | 1,777 ± 93.0 | 2.9 | 20.4 | 69.6 | 170 | 3.3 | 52.5 | 0.1 | 52.6 |
| G | 4,046 ± 93.0 | 2.4 | 18.8 | 69.3 | 170 | 3.5 | 53.4 | 0.15 | 53.5 |
| H | 515.3 ± 18.6 | 9.7 | 45.0 | 96.5 | 170 | 1.9 | 23.6 | 0.35 | 23.9 |
| I | 190.7 ± 9.3 | 10.5 | 39.4 | 75.6 | 130 | 1.65 | 25.9 | 0 | 25.9 |
The EE (%), DL (%), and residual benzyl alcohol (%) in the microparticles generated with two homogenization speeds of 2,000 and 3,000 RPM are shown in Table 4. The theoretical naltrexone loading in the particles was 37% for all 9 formulations. Figure 5 demonstrates the EE (%) of naltrexone and DL (%) of the microparticles made with different oil-phase formulations and a homogenizer speed of 3,000 RPM. The PLGA concentration was found to be the main factor for improving the EE and increase of the organic solvent residual. For instance, at the homogenizer speed of 3,000 RPM, when the PLGA concentration increased from 5.58% to 16.85%, the EE (%), DL (%), and residual benzyl alcohol (%) all increased by 13.1%, 4.9%, and 0.8%, respectively. At the lower PLGA concentration, the skin is formed more slowly, resulting in a longer time for the drug to leave the particles during the extraction process. Thus, the EE, DL, and residual benzyl alcohol were reduced at lower PLGA concentrations. Comparison of Formulations G, H, and I, indicates that the MW and the viscosity of the oil phase had a negligible impact on the EE, DL, and residual benzyl alcohol in the microparticles. Similar behavior was observed in the literature in regards to the effects of the PLGA concentration and MW on the EE of different drugs [19, 56, 57].
Table 4.
DL (%), EE (%), and residual benzyl alcohol (%) in the microparticles made with different formulations and two homogenizer speeds of 2,000 and 3,000 RPM.
| Homogenizer Speed = 2,000 RPM | ||||||
|---|---|---|---|---|---|---|
| Formulation | PLGA Conc. (%) | PLGA Mw (kDa) | Viscosity (mPa·s) | DL (%) | EE (%) | Residual BA (%) |
| A | 5.58 | 148 | 52.6 ± 2.3 | 27.36 ± 0.35 | 73.96 ± 0.94 | 0.48 ± 0.06 |
| B | 7.48 | 148 | 125.6 ± 2.3 | 28.52 ± 0.18 | 77.09 ± 0.48 | 0.71 ± 0.03 |
| C | 10.0 | 148 | 426 ± 18.6 | 29.63 ± 0.24 | 80.08 ± 0.66 | 0.89 ± 0.02 |
| D | 11.63 | 148 | 840.3 ± 31.0 | 30.31 ± 0.30 | 81.93 ± 0.81 | 0.91 ± 0.01 |
| E | 13.10 | 148 | 1,442 ± 46.5 | 30.98 ± 0.18 | 83.72 ± 0.48 | 1.40 ± 0.03 |
| F | 13.75 | 148 | 1,777 ± 93.0 | 30.97 ± 0.35 | 83.71 ± 0.96 | 1.29 ± 0.02 |
| G | 16.85 | 148 | 4,046 ± 93.0 | 31.75 ± 0.44 | 85.81 ± 1.19 | 1.76 ± 0.02 |
| H | 16.85 | 79 | 515.3 ± 18.6 | 31.01 ± 0.24 | 83.82 ± 0.65 | 1.71 ± 0.03 |
| I | 16.85 | 64 | 190.7 ± 9.3 | 31.61 ± 0.47 | 85.43 ± 1.28 | 1.39 ± 0.02 |
| Homogenizer Speed = 3,000 RPM | ||||||
| Formulation | PLGA Conc. (%) | PLGA Mw (kDa) | Viscosity (mPa·s) | DL (%) | EE (%) | Residual BA (%) |
| A | 5.58 | 148 | 52.6 ± 2.3 | 27.63 ± 0.63 | 74.68 ± 1.69 | 0.47 ± 0.03 |
| B | 7.48 | 148 | 125.6 ± 2.3 | 28.45 ± 0.47 | 76.90 ± 1.28 | 0.79 ± 0.16 |
| C | 10.0 | 148 | 426 ± 18.6 | 30.28 ± 0.12 | 81.84 ± 0.32 | 0.84 ±0.001 |
| D | 11.63 | 148 | 840.3 ± 31.0 | 30.21 ± 0.46 | 81.65 ± 1.24 | 1.14 ± 0.02 |
| E | 13.10 | 148 | 1,442 ± 46.5 | 30.88 ± 0.36 | 83.47 ± 0.99 | 1.36 ± 0.06 |
| F | 13.75 | 148 | 1,777 ± 93.0 | 31.40 ± 0.31 | 84.87 ± 0.83 | 1.11 ± 0.08 |
| G | 16.85 | 148 | 4,046 ± 93.0 | 32.49 ± 0.24 | 87.80 ± 0.66 | 1.24 ± 0.04 |
| H | 16.85 | 79 | 515.3 ± 18.6 | 31.30 ± 0.11 | 84.59 ± 0.29 | 1.52 ± 0.06 |
| I | 16.85 | 64 | 190.7 ± 9.3 | 31.64 ± 0.28 | 85.50 ± 0.75 | 0.96 ± 0.06 |
BA: Benzyl alcohol
Figure 5.
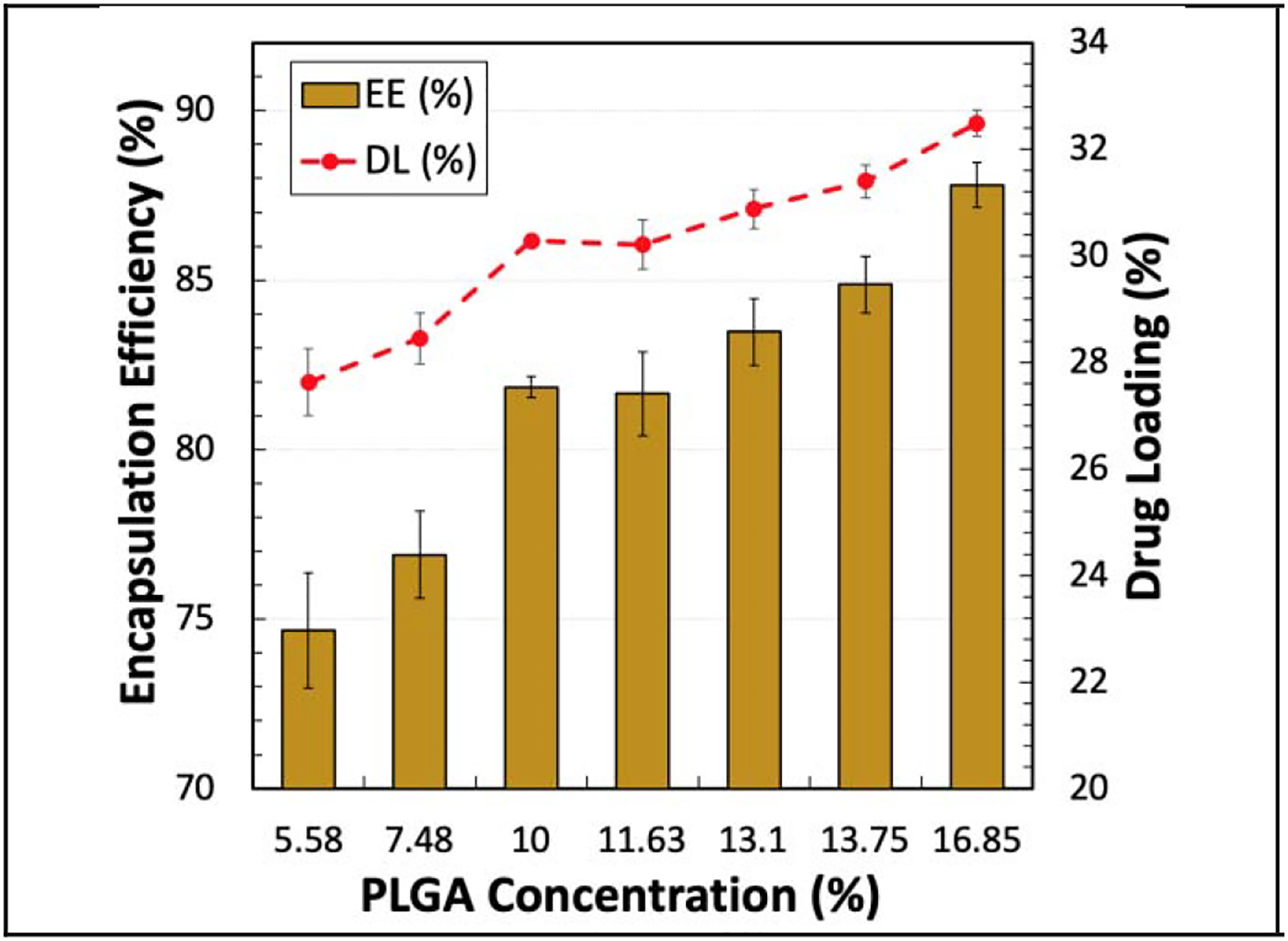
EE (%) and DL (%) of naltrexone in the microparticles made with different PLGA concentrations at a homogenizer speed of 3,000 RPM. The targeted DL for all of the formulations was 37%.
The increase of the homogenizer speed from 2,000 RPM to 3,000 RPM had minor effects on the EE and DL, and only some minor effects on residual benzyl alcohol. Caution is necessary, however, to conclude that the homogenizer speed does not have a major impact on the entrapment efficiency, because the particles were normalized by collecting them only in the range of 25~150 μm. The results suggest that the EE, DL, and residual benzyl alcohol from the microparticles made at different shear rates will be the same as long as their size distributions are the same. In other words, increasing the shear force at the fluid/fluid interface had negligible impact on the EE, DL, and residual benzyl alcohol as long as the size distribution remained the same.
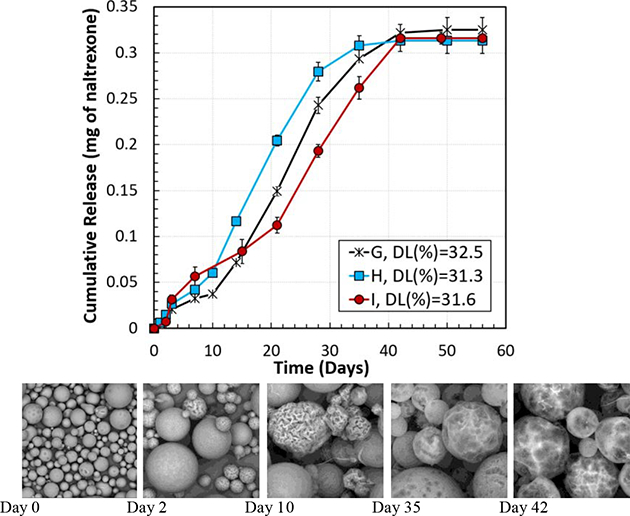
Figure 6 demonstrates the in vitro cumulative drug release percentage (Figure 6 (A and C)) and the actual amount (mg) of naltrexone (Figure 6 (B and D)) released per mg PLGA microparticles with different formulations. Figure 6 (A and B) shows the effect of PLGA concentration, which changed from 5.58% to 16.85% in the initial oil phase, with a constant PLGA MW of 148 kDa. The lower PLGA concentration in the oil phase resulted in the lower DL and the lower naltrexone release rate. Thus, the PLGA concentration affects the drug release kinetics through altering the DL. When the DL was 27.6% (Formulation A), the naltrexone release was extended to 56 days, as compared with 42 days for Formulation G with a drug loading of 32.5%. However, Figure 6 (B) shows that the total amount of the released naltrexone from Formulation A was 52 ng/mg microparticles, less than that from Formulation G.
Figure 6.
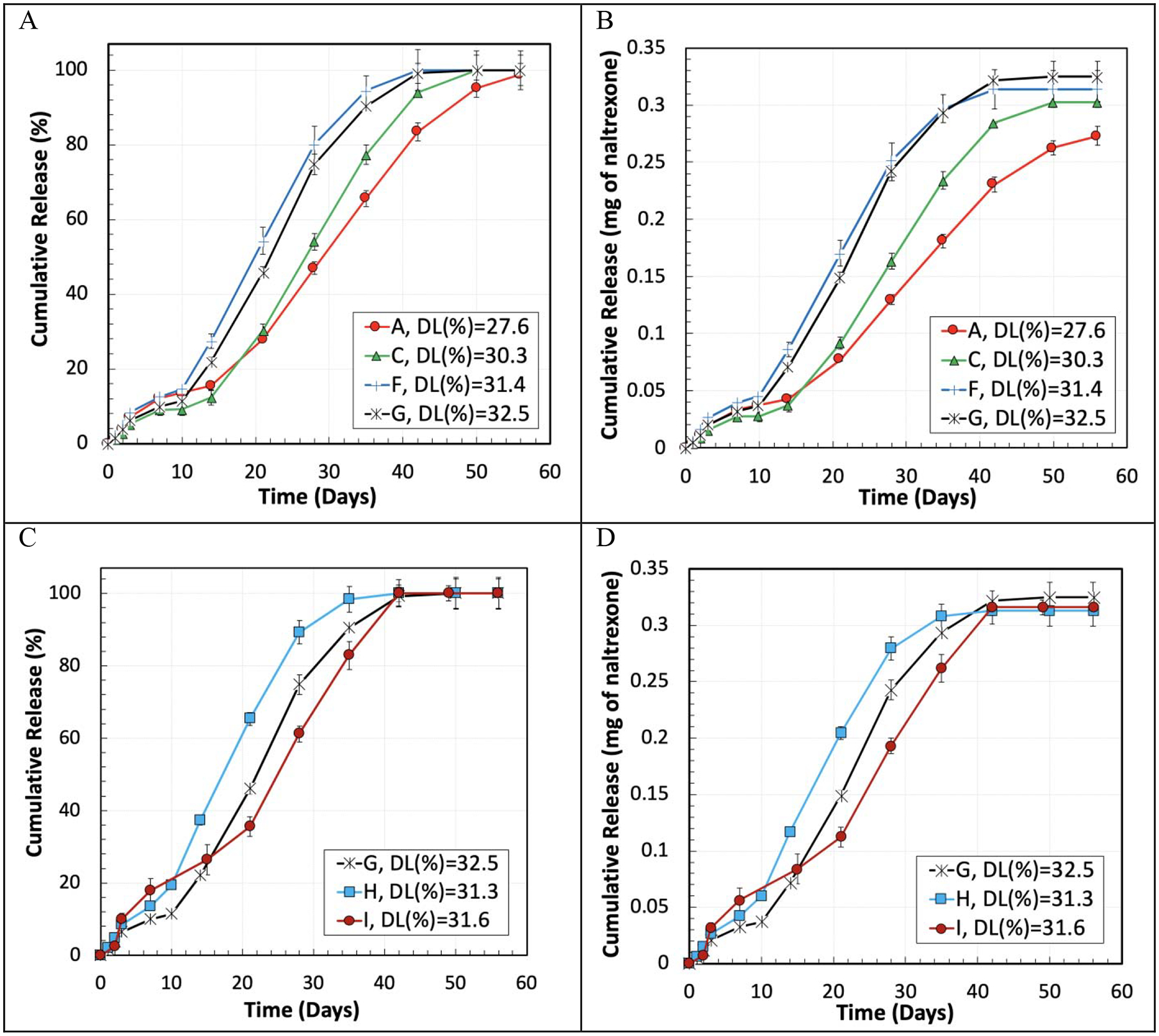
In vitro cumulative drug release in % (A and C) and in the absolute amount (B and D) per mg of naltrexone-PLGA microparticles made with different PLGA concentrations (A and B) in the oil-phase and PLGA MWs (C and D). The homogenizer speed was 3,000 RPM.
In addition to the PLGA concentration, the effect of the PLGA MW was also examined. Microparticles were made with PLGA MWs of 64 kDa (Formulation I), 79 kDa (Formulation H), and 148 kDa (Formulation G) at the PLGA concentration of 16.85% (Figure 6 C and D). When the PLGA MW was decreased from 148 kDa (Formulation H) to 79 kDa (Formulation G), the drug release duration was reduced from 42 to 35 days, although the DL of the particles with Formulation G was 1.2% higher than Formulation H. Reducing the PLGA MW further from 79 kDa to 64 kDa (Formulation I), led to the increase of the naltrexone release to 42 days, similar to Formulation G, with a DL of 31.6%, which is 0.9% less than the one in Formulation G. Figure 6 D demonstrates that the highest difference between the total naltrexone release per mg of naltrexone-PLGA microparticles was 12 ng (between Formulations H and G), which can be negligible as it is 3.2% of the theoretical drug content per mg of the microparticles. Furthermore, the rate of the cumulative drug release in the linear region of these three formulations, i.e., G, H, and I, were 11.5 μg/day, 12.1 μg/day, and 9.7 μg/day, respectively, which are close to each other, indicating the fact that the effect of the PLGA MW factor was not as significant as the DL on the release rate of the naltrexone from the microparticles. Other parameters such as change of the oil phase viscosity had minor effects on the drug release rate. The same behavior was observed for the microparticles made with the Formulations A-H and a homogenizer speed of 2,000 RPM.
Based on the release kinetics shown in Figure 6, at Day 2 all formulations remained in the initial release phase. Then, between days 10–14, as the initial release neared completion, the linear release (zero-order release) started. Between days 35–42, zero-order release ended, and the drug release started to plateau. Figure 7 demonstrates SEM images of the microparticles made with Formulations C, G, and H, incubated in the release media. Days 2, 10, 35, and 42 were selected to study the morphology of the microparticles at time points where the release rate changed, which can be due to the change in the release mechanism. After 2 days of in vitro release, smaller microparticles in all three formulations showed corrugation or wrinkles on the surface. Formulation C, which had a smooth surface, started showing the wrinkled morphology, indicating non-isotropic swelling. Formulations G and H which had buckled morphologies also started showing more extensive wrinkled surfaces for small microparticles. All formulations maintained spherical shaped microparticles indicating that the microparticles were in the swollen state. At Day 10, most of the smaller microparticles appeared as “deflated balls” [58] and larger microparticles started to have corrugation. The results show that within the first 10 days, the release mechanism was diffusion from monolithic naltrexone-PLGA microparticles, where the drug molecules were distributed throughout the PLGA matrix. The naltrexone release from the microparticles was not zero-order during the first 0–10 days, mainly because the drug molecules located near the surface of the microparticles were initially released. Then, the drug residing in the interior parts of the microparticles had to migrate longer distances and diffuse out at slower rates relative to the molecules close to the surface, altering the observed naltrexone release rate from the microparticles. After this region (days 10–14), zero-order release kinetics was observed for all formulations. The morphology of the microparticles at the end of the zero-order release region (Day 35) shows that the wrinkled surface morphed into a smooth surface with occasional large craters. This change in the surface morphology is likely due to swelling of the microparticles resulting from the presence of the saturated naltrexone and degradation of PLGA producing acid groups, ultimately increasing the osmotic pressure inside the microparticles [59, 60]. In this region, the naltrexone release rate from the microparticles was constant (zero-order release). As most naltrexone is released, the concentration gradient decreases, resulting in reduced naltrexone release. The surface morphologies of PLGA microparticles at day 42 remained similar to those at day 35, as the PLGA with a L:G ratio of 85:15 degrades slowly beyond 42 days.
Figure 7.

SEM images of the naltrexone- loaded microparticles made with Formulations C, G, and H during the in vitro test at days 0 (before the in vitro test), 2, 10, 35, and 42.
Determining the batch-to-batch consistency in the properties of the microparticles was another important objective in this study. Formulation C was selected to evaluate the reproducibility of the results. The results of size distribution, DL, and in vitro cumulative drug release from PLGA microparticles made with the homogenization speeds of 2,000 RPM and 3,000 RPM for two replicates showed minor differences between the two replicates (Figure 8). For instance, the difference in span and DL values in the microparticles made with the homogenization speed of 3,000 RPM was around 1.91% and 0.5%, respectively. Additionally, the drug release profiles had the same slope and the release duration was the same for both of the replicates.
Figure 8.
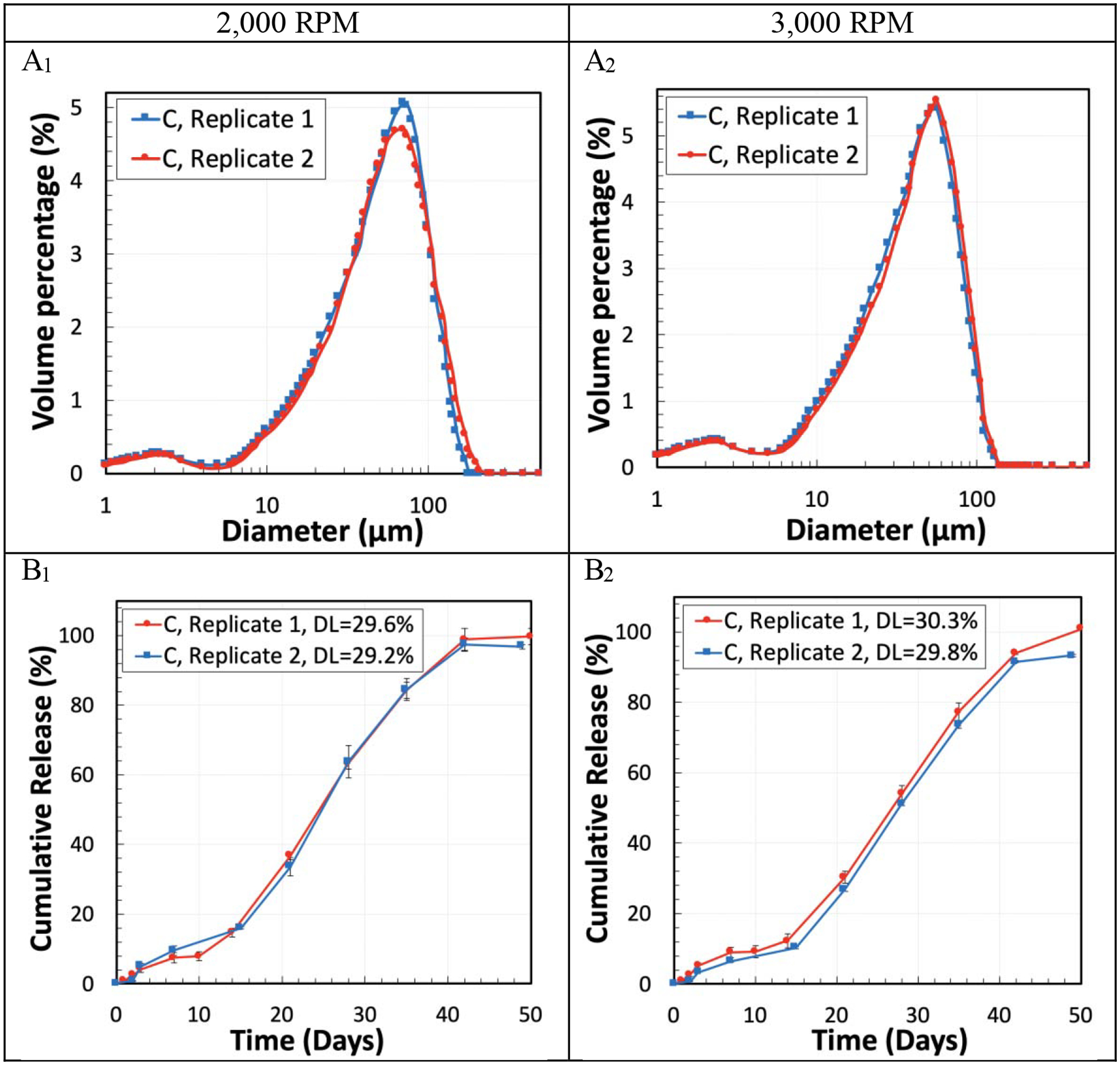
Reproducibility test using Formulation C for the size distribution (A1, A2), and DL and in vitro cumulative drug release (B1, B2) from two replicates of PLGA microparticles. The homogenization speed was either 2,000 RPM (A1, B1) or 3,000 RPM (A2, B2).
4. Discussion
In making PLGA microparticles, there are many variables that need to be controlled to produce them with reproducible properties. Some of the parameters are shown in Figure 9. Additional processing parameters that are not shown include the processing temperature, solvent type, and post treatment (i.e., additional treatment of fully dried microparticles). In this study, the 4 parameters examined (highlighted in red in Figure 9) were the PLGA amount (concentration), PLGA MW, viscosity of PLGA-dissolved in solvents, and the homogenizer speed. These aspects were varied and then examined for their impacts on the resultant microparticle properties, including drug release kinetics. Highlighted in blue are the results of varying those parameters, i.e., microparticle properties, such as the drug loading, drug encapsulation efficiency, microparticle size distribution, surface and internal morphology, and drug release kinetics.
Figure 9.
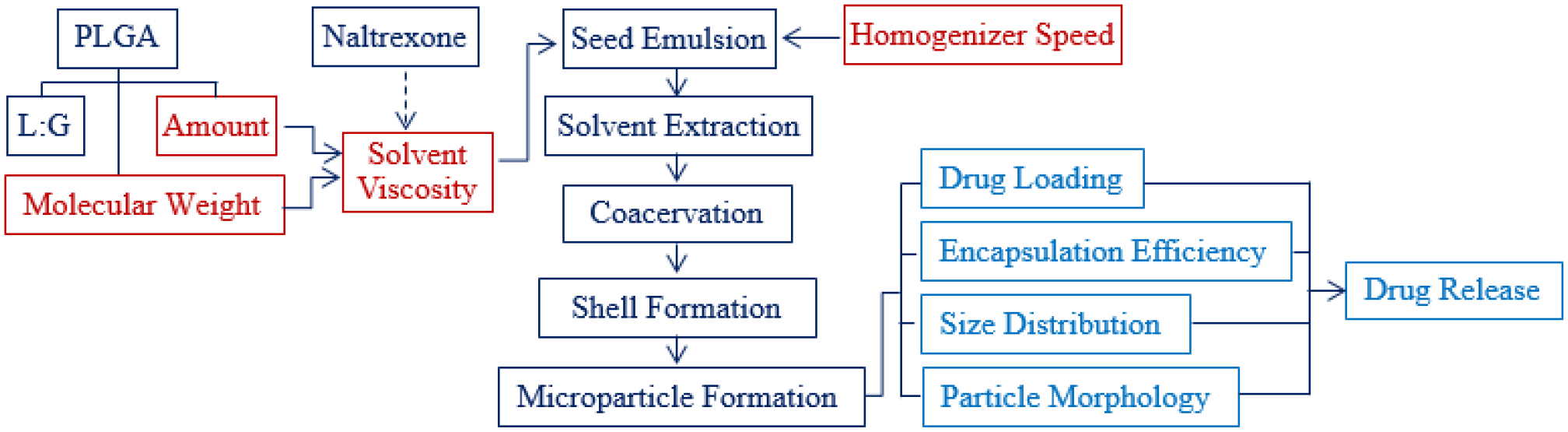
Parameters and processing steps of making PLGA microparticles. The parameters examined are shown in red, and the resulting microparticle properties are in blue.
Naltrexone, a nucleophilic drug, is known to cleave ester bonds and reduce the MW, as observed before by other investigators [45–48]. Our study also shows that the viscosity of PLGAs dissolved in DCM and benzyl alcohol decreased over time in the presence of naltrexone. The decrease in the viscosity was found to be significant when the PLGA concentrations were above 10% for all the PLGA MWs which were used in this study, i.e. 64 kDa, 79 kDa, and 148 kDa. For instance, the oil phase viscosity dropped by 42.5% only three hours after PLGA, with the concentration of 16.85% and MW of 148 kDa, and naltrexone, were in contact with the organic solvents. Naltrexone and PLGA can be dissolved in separate solvents and mixed together before emulsification, but it still takes time for homogeneous mixing and emulsification. Thus, it is important to consider that the MW in the final PLGA microparticles may be significantly different from the initial MW.
When PLGA microparticles are prepared, the information presented is usually the PLGA lactide:glycolide (L:G) ratio, molecular weight, and concentration. Many studies have examined the effects of PLGA concentration on the uniformity and mean diameter of the PLGA microparticles [17, 19, 56, 57, 61, 62]. The results, however, have not been straightforward. Depending on the studies, the uniformity of the microparticles was enhanced [56], reduced [61, 62], or remained the same [17]. Regardless of the results, simply focusing on the PLGA concentration may not provide useful information, because different molecular weights of the same concentration may have very different solution properties. Thus, the viscosity may be a better parameter to examine. The effects of the oil phase viscosity, PLGA MW and concentration, and L:G ratio on the properties of the salmon calcitonin-loaded PLGA microparticles were studied [29]. The viscosity of the organic phase was changed from 2 mPa·s to 10 , and within this viscosity range, the mean size of the microparticles was found to have a linear relationship with the viscosity of the organic phase. However, the viscosity range was small to obtain a deeper understanding of its effect on the properties of the microparticles. In addition to the mean microparticle size, measurement of the size distribution of the microparticles would also have helped to understand the effect of viscosity on the size range and polydispersity of the PLGA microparticles. In our study, the PLGA concentration was varied to obtain a wide range of viscosities, 52.6~4,046 mPa·s (Table 2). Our results in Figure 4 shows that the size distribution of the microparticles was highly dependent on the viscosity of the organic phase. Formulations C (148 kDa at 10%) and H (79 kDa at 16.85%) were found to have the same size distribution because their viscosity was similar, although the PLGA concentration was different. As shown by Formulations H and I in Table 4, the increase in the PLGA concentration resulted in higher drug loading and encapsulation efficiency, even with the low viscosity. The decrease in the viscosity enhanced the size uniformity of the microparticles, and the span value was reduced from 3.1 to 1.24 at the homogenization speed of 2,000 RPM when the oil phase viscosity was reduced from 4,046 mPa·s to 52.6 mPa·s. Controlling the viscosity of the oil phase can play a significant role on the reproducibility of the results as well as drug release kinetics.
The effect of homogenizer speed was studied in the literature. For instance, haloperidol-loaded nanoparticles made of PLGA and poly(L-lactide) (PLA) were prepared using an emulsification-solvent evaporation approach with two agitation techniques of homogenization and sonication [19]. The study showed that the particle size was reduced with the increased shear stress by either increasing the homogenizer speed or decreasing the polymer concentration. The size distribution is an important parameter, but it is commonly not reported [18, 19, 24, 29]. As shown in Figure 4, the microparticles formed from different formulations have different size distributions, and such differences may not be obvious if only the mean particle size is presented. The viscosity has a significant effect on the size distribution of microparticles. When microparticles of a certain size range are used in a study (such as 25 μm ~150 μm in this study), the size distribution is an important factor to report as it may influence the drug loading, encapsulation efficiency, and reproducibility of results.
More studies are necessary to further understand the mechanisms of the formation of drug-loaded PLGA microparticles. In this study, naltrexone was used, and other similar nucleophilic drugs, such as naltrexone, risperidone, and oxybutynin [45–47] are expected to have similar results in decreasing the viscosity over time. While other drugs may not cleave ester bonds of PLGA, the effect of the viscosity on the properties of PLGA microparticles remains the same, although the exact impact may vary. Other parameters to study include the solvent extraction kinetics which affect the change in the glass transition temperature while PLGA solidifies [11, 63], processing temperature, solvent type, and post treatment (i.e., additional treatment of fully dried microparticles). There are a large number of articles on PLGA microparticles presenting valuable information. Through careful, systematic studies, we will be able to obtain the mechanisms that govern the formation of PLGA microparticles. Such a thorough understanding will allow us to design PLGA microparticles having predetermined drug release properties with a minimal trial-and-error approach.
Acknowledgments
This study was supported by the grant UG3 DA048774 from the National Institute of Drug Abuse (NIDA), the Chong Kun Dang Pharmaceutical Corp., and the Showalter Trust Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths - United States, 2013–2017. MMWR Morb Mortal Wkly Rep 67 (2019) 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].CDC. Opioid overdose. (2019) https://www.cdc.gov/drugoverdose/index.html.
- [3].Park K, Otte A. Prevention of opioid abuse and treatment of opioid addiction: Current status and future possibilities. Ann. Rev. Biomed. Eng 21 (2019) 61–84. [DOI] [PubMed] [Google Scholar]
- [4].Morgan JR, Schackman BR, Leff JA, Linas BP, Walley AY. Injectable naltrexone, oral naltrexone, and buprenorphine utilization and discontinuation among individuals treated for opioid use disorder in a United States commercially insured population. Journal of Substance Abuse Treatment 85 (2018) 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Comer SD, Sullivan MA, Yu E, Rothenberg JL, Kleber HD, Kampman K, Dackis C, O’Brien CP. Injectable, sustained-release naltrexone for the treatment of opioid dependence: A randomized, placebo-controlled trial. Archives of General Psychiatry 63 (2006) 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Alkermes. VIVITROL (naltrexone for extended-release injectable suspension), for intramuscular use. (1984) https://www.vivitrol.com/content/pdfs/prescribing-information.pdf.
- [7].Brewer C, Krupitsky E. Antagonists for the treatment of opioid dependence, in: Miller PM (Ed.) Interventions for Addiction, Academic Press, San Diego, CA: 2013, pp. 427–438 (Chapter 445). [Google Scholar]
- [8].Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm 364 (2008) 298–327. [DOI] [PubMed] [Google Scholar]
- [9].Gasmi H, Danede F, Siepmann J, Siepmann F. Does PLGA microparticle swelling control drug release? New insight based on single particle swelling studies. J. Control. Release 213 (2015) 120–127. [DOI] [PubMed] [Google Scholar]
- [10].Park K, Skidmore S, Hadar J, Garner J, Park H, Otte A, Soh BK, Yoon G, Yu D, Yun Y, Lee BK, Jiang X, Wang Y. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 304 (2019) 125–134. [DOI] [PubMed] [Google Scholar]
- [11].Vay K, Frieß W, Scheler S. A detailed view of microparticle formation by in-process monitoring of the glass transition temperature. Eur. J. Pharm. Biopharm 81 (2012) 399–408. [DOI] [PubMed] [Google Scholar]
- [12].Baena-Aristizábal CM, Fessi H, Elaissari A, Mora-Huertas CE. Biodegradable microparticles preparation by double emulsification—Solvent extraction method: A Systematic study. Colloids and Surfaces A: Physicochemical and Engineering Aspects 492 (2016) 213–229. [Google Scholar]
- [13].Fang Y, Zhang N, Li Q, Chen J, Xiong S, Pan W. Characterizing the release mechanism of donepezil-loaded PLGA microspheres in vitro and in vivo. J. Drug Del. Sci. Tech 51 (2019) 430–437. [Google Scholar]
- [14].Ding S, Serra CA, Vandamme TF, Yu W, Anton N. Double emulsions prepared by two–step emulsification: History, state-of-the-art and perspective. Journal of Controlled Release 295 (2019) 31–49. [DOI] [PubMed] [Google Scholar]
- [15].Acharya G, Shin CS, Vedantham K, McDermott M, Rish T, Hansen K, Fu Y, Park K. A study of drug release from homogeneous PLGA microstructures. J. Control. Release 146 (2010) 201–206. [DOI] [PubMed] [Google Scholar]
- [16].Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. European Journal of Pharmaceutical Sciences 15 (2002) 355–366. [DOI] [PubMed] [Google Scholar]
- [17].Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int. J. Pharm 290 (2005) 137–144. [DOI] [PubMed] [Google Scholar]
- [18].Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm 314 (2006) 198–206. [DOI] [PubMed] [Google Scholar]
- [19].Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm 336 (2007) 367–375. [DOI] [PubMed] [Google Scholar]
- [20].Song X, Zhao Y, Hou S, Xu F, Zhao R, He J, Cai Z, Li Y, Chen Q. Dual agents loaded PLGA nanoparticles: Systematic study of particle size and drug entrapment efficiency. Eur. J. Pharm. Biopharm 69 (2008) 445–453. [DOI] [PubMed] [Google Scholar]
- [21].Tsukada Y, Hara K, Bando Y, Huang CC, Kousaka Y, Kawashima Y, Morishita R, Tsujimoto H. Particle size control of poly(dl-lactide-co-glycolide) nanospheres for sterile applications. International Journal of Pharmaceutics 370 (2009) 196–201. [DOI] [PubMed] [Google Scholar]
- [22].Kwon H-Y, Lee J-Y, Choi S-W, Jang Y, Kim J-H. Preparation of PLGA nanoparticles containing estrogen by emulsification–diffusion method. Colloids and Surfaces A: Physicochemical and Engineering Aspects 182 (2001) 123–130. [Google Scholar]
- [23].Araújo J, Vega E, Lopes C, Egea MA, Garcia ML, Souto EB. Effect of polymer viscosity on physicochemical properties and ocular tolerance of FB-loaded PLGA nanospheres. Colloids and Surfaces B: Biointerfaces 72 (2009) 48–56. [DOI] [PubMed] [Google Scholar]
- [24].Rawat A, Burgess DJ. Effect of ethanol as a processing co-solvent on the PLGA microsphere characteristics. Int. J. Pharm 394 (2010) 99–105. [DOI] [PubMed] [Google Scholar]
- [25].Shen J, Choi S, Qu W, Wang Y, Burgess DJ. In vitro-in vivo correlation of parenteral risperidone polymeric microspheres. J. Control. Release 218 (2015) 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang T, Xue P, Wang A, Yin M, Han J, Tang S, Liang R. Pore change during degradation of octreotide acetate-loaded PLGA microspheres: The effect of polymer blends. Eur. J. Pharm. Sci 138 (2019) 104990. [DOI] [PubMed] [Google Scholar]
- [27].Skidmore S, Hadar J, Garner J, Park H, Park K, Wang Y, Jiang X. Complex sameness: Separation of mixed poly(lactide-co-glycolide)s based on the lactide:glycolide ratio. J. Control. Release 300 (2019) 174–184. [DOI] [PubMed] [Google Scholar]
- [28].Takeda. LUPRON DEPOT (leuprolide acetate for depot suspension). Initial U.S. Approval: 1989. (2014) https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020517s036_019732s041lbl.pdf.
- [29].Jeyanthi R, Mehta RC, Thanoo BC, Deluca PP. Effect of processing parameters on the properties of peptide-containing PLGA microspheres. Journal of Microencapsulation 14 (1997) 163–174. [DOI] [PubMed] [Google Scholar]
- [30].Jahn A, Reiner JE, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Preparation of nanoparticles by continuous-flow microfluidics. Journal of Nanoparticle Research 10 (2008) 925–934. [Google Scholar]
- [31].Valencia PM, Farokhzad OC, Karnik R, Langer R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nature Nanotechnology 7 (2012) 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Keohane K, Brennan D, Galvin P, Griffin BT. Silicon microfluidic flow focusing devices for the production of size-controlled PLGA based drug loaded microparticles. Int. J. Pharm 467 (2014) 60–69. [DOI] [PubMed] [Google Scholar]
- [33].Min K-I, Im DJ, Lee H-J, Kim D-P. Three-dimensional flash flow microreactor for scale-up production of monodisperse PEG–PLGA nanoparticles. Lab on a Chip 14 (2014) 3987–3992. [DOI] [PubMed] [Google Scholar]
- [34].Rezvantalab S, Keshavarz Moraveji M. Microfluidic assisted synthesis of PLGA drug delivery systems. RSC Advances 9 (2019) 2055–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Albisa A, Piacentini E, Sebastian V, Arruebo M, Santamaria J, Giorno L. Preparation of drug-loaded PLGA-PEG nanoparticles by membrane-assisted nanoprecipitation. Pharm. Res 34 (2017) 1296–1308. [DOI] [PubMed] [Google Scholar]
- [36].Qi F, Wu J, Hao D, Yang T, Ren Y, Ma G, Su Z. Comparative studies on the influences of primary emulsion preparation on properties of uniform-sized exenatide-loaded PLGA microspheres. Pharm. Res 31 (2014) 1566–1574. [DOI] [PubMed] [Google Scholar]
- [37].Qi F, Wu J, Fan Q, He F, Tian G, Yang T, Ma G, Su Z. Preparation of uniform-sized exenatide-loaded PLGA microspheres as long-effective release system with high encapsulation efficiency and bio-stability. Colloids and Surfaces B: Biointerfaces 112 (2013) 492–498. [DOI] [PubMed] [Google Scholar]
- [38].Lim J-M, Bertrand N, Valencia PM, Rhee M, Langer R, Jon S, Farokhzad OC, Karnik R. Parallel microfluidic synthesis of size-tunable polymeric nanoparticles using 3D flow focusing towards in vivo study. Nanomedicine: Nanotechnology, Biology and Medicine 10 (2014) 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Joscelyne SM, Trägårdh G. Membrane emulsification- a literature review. J. Memb. Sci 169 (2000) 107–117. [Google Scholar]
- [40].Lyons SL, Wright SG, Apparatus and method for preparing microparticles using in-line solvent extraction. US 2008/0054220 A1. (2008) US Patent 2008/0054220 A1.
- [41].Andhariya JV, Choi S, Wang Y, Zou Y, Burgess DJ, Shen J. Accelerated in vitro release testing method for naltrexone loaded PLGA microspheres. Int. J. Pharm 520 (2017) 79–85. [DOI] [PubMed] [Google Scholar]
- [42].Dillen K, Vandervoort J, Van den Mooter G, Ludwig A. Evaluation of ciprofloxacin-loaded Eudragit® RS100 or RL100/PLGA nanoparticles. Int. J. Pharm 314 (2006) 72–82. [DOI] [PubMed] [Google Scholar]
- [43].Gaignaux A, Réeff J, Siepmann F, Siepmann J, De Vriese C, Goole J, Amighi K. Development and evaluation of sustained-release clonidine-loaded PLGA microparticles. Int. J. Pharm 437 (2012) 20–28. [DOI] [PubMed] [Google Scholar]
- [44].Bahl Y, Sah H. Dynamic changes in size distribution of emulsion droplets during ethyl acetatebased microencapsulation process. AAPS PharmSciTech 1 (2000) E5–E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wright SG, Rickey ME, Ramstack JM, Lyons SL, Hotz JM, Method for preparing microparticles having a selected polymer molecular weight. (2003) US Patent 6,534,092.
- [46].D’Souza S, Faraj JA, Dorati R, DeLuca PP. Enhanced degradation of lactide-co-glycolide polymer with basic nucleophilic drugs. Advances in Pharmaceutics 2015 (2015) 10pages. [Google Scholar]
- [47].Thanoo BC, Murtagh J, Johns G, Prevention of molecular weight reduction of the polymer, impurity formation and gelling in polymer composition. (2015) US Patent 9,017,715.
- [48].Kohno M, Andhariya JV, Wan B, Bao Q, Rothstein S, Hezel M, Wang Y, Burgess DJ. The effect of PLGA molecular weight differences on risperidone release from microspheres. Int. J. Pharm 582 (2020) 119339. [DOI] [PubMed] [Google Scholar]
- [49].Brown GL. Formation of films from polymer dispersion. J. Polym. Sci 22 (1956) 423–434. [Google Scholar]
- [50].Pinnau I, Koros WJ. A qualitative skin layer formation mechanism for membranes made by dry/wet phase inversion. J. Polym. Sci. B: Polym. Phys 31 (1993) 419–427. [Google Scholar]
- [51].Giorgiutti-Dauphine F, Pauchard L. Drying drops containing solutes: From hydrodynamical to mechanical instabilities. Eur. Phys. J. E 41 (2018) 32(15 pages). [DOI] [PubMed] [Google Scholar]
- [52].Vo TT, Nezamabadi S, Mutabaruka P, Delenne J-Y, Izard E, Radja PR,F. Agglomeration of wet particles in dense granular flows. Eur. Phys. J. E 42 (2019) 127(112 pages). [DOI] [PubMed] [Google Scholar]
- [53].Giorgiutti-Dauphiné F, Pauchard L. Drying drops containing solutes: From hydrodynamical to mechanical instabilities. Eur. Phys. J. E 41 (2018) 32(15 pages). [DOI] [PubMed] [Google Scholar]
- [54].Bacchin P, Brutin D, Davaille A, Di Giuseppe E, Chen XD, Gergianakis I, Giorgiutti-Dauphiné F, Goehring L, Hallez Y, Heyd R, Jeantet R, Le Floch-Fouéré C, Meireles M, Mittelstaedt E, Nicloux C, Pauchard L, Saboungi M-L. Drying colloidal systems: Laboratory models for a wide range of applications. Eur. Phys. J. E 41 (2018) 94(34 pages). [DOI] [PubMed] [Google Scholar]
- [55].Janssen JMH. Polymer melt mixing: Liquid-liquid mixing, in: Jürgen Buschow KH, Cahn RW, Flemings MC, Ilschner B, Kramer EJ, Mahajan S, Veyssière P (Eds.) Encyclopedia of Materials: Science and Technology, Elsevier; 2001. [Google Scholar]
- [56].Ito F, Fujimori H, Makino K. Incorporation of water-soluble drugs in PLGA microspheres. Colloids and Surfaces B: Biointerfaces 54 (2007) 173–178. [DOI] [PubMed] [Google Scholar]
- [57].Song X, Zhao Y, Wu W, Bi Y, Cai Z, Chen Q, Li Y, Hou S. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: Systematic study of particle size and drug entrapment efficiency. Int. J. Pharm 350 (2008) 320–329. [DOI] [PubMed] [Google Scholar]
- [58].Mylonaki I, Allémann E, Delie F, Jordan O. Imaging the porous structure in the core of degrading PLGA microparticles: The effect of molecular weight. J. Control. Release 286 (2018) 231–239. [DOI] [PubMed] [Google Scholar]
- [59].Gu B, Sun X, Papadimitrakopoulos F, Burgess DJ. Seeing is believing, PLGA microsphere degradation revealed in PLGA microsphere/PVA hydrogel composites. J. Control. Release 228 (2016) 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Brunner A, Mäder K, Göpferich A. pH and osmotic pressure inside biodegradable microspheres during erosion. Pharm. Res 16 (1999) 847–853. [DOI] [PubMed] [Google Scholar]
- [61].Huang W, Zhang C. Tuning the size of poly(lactic-co-glycolic acid) (PLGA) nanoparticles fabricated by nanoprecipitation. Biotechnol. J 13 (2018) 10.1002/biot.201700203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Thomasin C, Nam-Trȃn H, Merkle HP, Gander B. Drug microencapsulation by PLA/PLGA coacervation in the light of thermodynamics. 1. Overview and theoretical considerations. J. Pharm. Sci 87 (1998) 259–268. [DOI] [PubMed] [Google Scholar]
- [63].Vay K. Analysis and control of the manufacturing process and the release properties of PLGA microparticles for sustained delivery of a poorly water-soluble drug (Ph.D. Thesis, 148pages). (2012) https://edoc.ub.uni-muenchen.de/15049/1/Vay_Kerstin.pdf. [Google Scholar]