

Abstract
Cardiac wound healing after myocardial infarction (MI) evolves from pro-inflammatory to anti-inflammatory to reparative responses, and the cardiac fibroblast is a central player during the entire transition. The fibroblast mirrors changes seen in the left ventricle infarct by undergoing a continuum of polarization phenotypes that follow pro-inflammatory, anti-inflammatory, and pro-scar producing profiles. The development of each phenotype transition is contingent upon the MI environment into which the fibroblast enters. In this mini-review, we summarize our current knowledge regarding cardiac fibroblast activation during MI and highlight key areas where gaps remain.
Introduction
Following myocardial infarction (MI), the left ventricle (LV) undergoes a series of wound healing responses that starts with an inflammatory reaction to clear necrotic myocytes and ends with the formation of an infarct scar composed of extracellular matrix (ECM). The infarct scar is dynamic and highly metabolically active, being comprised of cells and vasculature.[1] Long term outcomes are dependent on the quantity and quality of scar formed, as too little or too weak of a scar can generate LV aneurysm, and too much or too stiff of a scar can generate a rigid LV.[2, 3] Too much ECM in the infarct scar can also serve as a conduit for arrhythmias. Therefore, a balance in ECM deposition and cross-linking is needed to minimize infarct expansion while maintaining an optimal tension. Because cardiac fibroblasts are the major source of ECM in the MI LV, their response determines both short and long term outcomes. The purpose of this mini-review is to provide a summary of recent findings made regarding the role of the cardiac fibroblast in the MI LV and detail the changes in fibroblast phenotype over the different phases of cardiac remodeling after MI (Figure 1).
Figure 1. Cardiac fibroblast activation along the time continuum of response to myocardial infarction (MI).
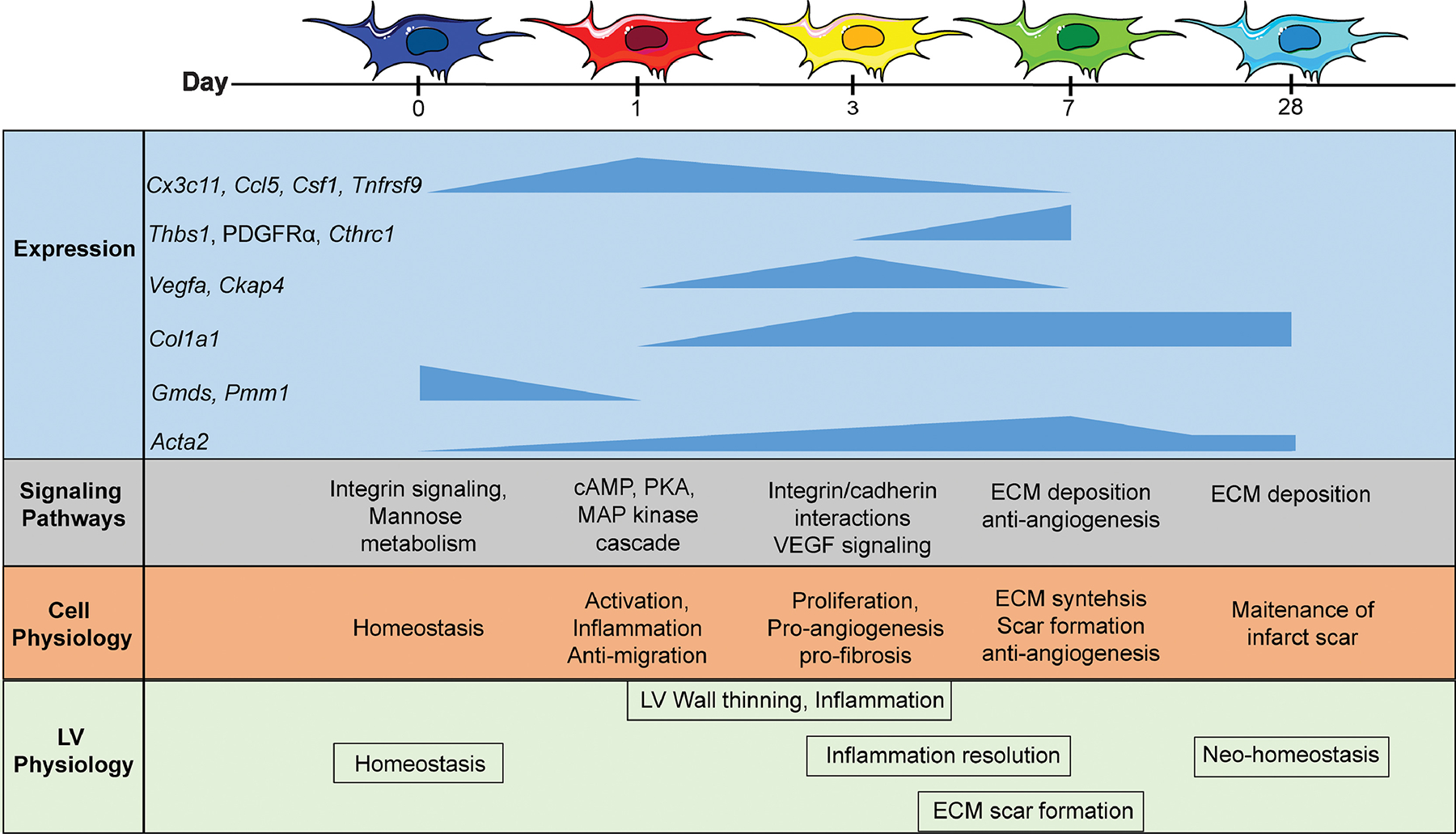
Before MI, fibroblasts are homeostatic and producing extracellular matrix (ECM) to allow normal physiology. At MI day 1, fibroblasts convert to a pro-inflammatory phenotype, signaling activation of inflammatory signaling. At MI day 3, fibroblasts polarize to a proliferative and pro-angiogenesis profile. Peak expression in the genes listed correspond to adaptations in cell and left ventricle physiology (function). At MI day 7, fibroblasts promote ECM synthesis to form the infarct scar and inhibit angiogenic signaling. At MI day 28, fibroblasts return to a state of neo-homeostasis, once again supporting the normal turnover of ECM to support the new infarct environment. Updated from [4].
Terminology, cell origins, and tissue culture considerations.
Fibroblast is a general term that encapsulates cells in the connective tissue that synthesize collagen and other ECM components.[4] While the term myofibroblast has been used to differentiate a fibroblast cell type that is α smooth muscle actin (Acta2) positive, this terminology is limiting because it does not take into account the non-binary accumulation of Acta2 occurs in fibroblasts of the MI LV and does not take into account other indices of cell activation.[4–6] Fibroblast activation is defined as the transition from a homeostatic fibroblast phenotype over the MI phases of cardiac remodeling. This is a broad term used to generically refer to any increase or change in fibroblast activity. Similar to other cardiac cell types such as macrophages and neutrophils,[7, 8] fibroblast activation in the setting of MI involves polarization to specific shifts in phenotype. Fibroblast activation or polarization cannot be easily assigned based on a single gene or protein marker, as transcriptomics and proteomics examinations have revealed a variety of fibroblast subtype populations. The number of subtypes in part reflect a variety of origins, and cardiac fibroblast origins shift over the MI time course. Fibroblasts from naïve uninjured myocardium reflect for the most part epicardial cells that underwent epithelial-to-mesenchymal transition during development.[9] After MI, cardiac fibroblasts originate from several sources, including resident cardiac fibroblasts, bone marrow derived progenitors, epicardial cells, and endothelial cells.[10]
In tissue culture, cardiac fibroblasts respond to a vast array of stimuli that includes cytokines, growth factors, and hormones.[1] Although in vitro stimulation of naïve fibroblasts provide information on how cells respond to single stimuli challenge, most in vivo responses represent a merging of similar and conflicting cues. Examining cardiac fibroblasts ex vivo actually provides the best means to understand its role in the context of MI. We have shown that cardiac fibroblasts retain their in vivo phenotype through passage 4.[5, 10–14] Two recent articles highlight the ex vivo memory of the MI fibroblast, as the Molkentin lab used freshly isolated fibroblasts and our team used cells to passage 3 for our evaluations.[10, 15] The reproducibility between the two studies was excellent, revealing that isolating fibroblasts and culturing them on plastic in 10% fetal bovine serum provides a means to expand this cell while maintaining in vivo phenotypes. Attempts to recapitulate the perfect in vivo environment, therefore, are not necessary for ex vivo cardiac fibroblast assessments.
Day 0 fibroblasts are homeostatic, continually sensing their environment to provide replacement ECM to maintain normal LV structure and physiology.
In normal LV, cardiac fibroblasts are a major non-cardiomyocyte cell type.[16] Previously, fibroblasts were thought to be quiescent or inactive, and become activated in response to inflammation or other wounding signals. We now know that resident fibroblasts are continually assessing homeostasis in the environment. Enrichment analysis of gene expression for isolated Day 0 no MI fibroblasts from the LV shows high expression for genes involved in integrin signaling and glycolysis. These processes allow fibroblasts to respond to the surrounding environment and replace ECM as needed.[17] In particular, GDP-Mannose 4,6-Dehydratase (Gmds) and Phosphomannomutase 1 (Pmm1) regulate synthesis of substrates in N-glycan biosynthesis, which includes a number of ECM components, and both genes are downregulated with MI.[10] Expression of these genes allows fibroblasts to maintain basal ECM production. Fibroblasts in the uninjured myocardium respond to stretch and are constantly monitoring the environment during the contraction and relaxation cycle.[15, 18] Fibroblast activation induces suppression of sarcolemmal K(ATP) channel opener P-1075 inducing depolarization of the myocardium.[19] While fibroblasts are not excitable, they interact with cardiomyocytes in part through shared expression of connexins 40, 43, and 45 to aid in transduction of electrical potentials throughout the myocardium.[19, 20] Resident cardiac fibroblasts, therefore, have primarily homeostatic roles in the uninjured myocardium to preserve ECM production and assist the cardiomyocyte in providing normal contractile function.
Day 1 MI fibroblasts are pro-inflammatory and anti-migratory.
MI initiates an inflammatory phase of LV remodeling. Necrotic cardiomyocytes release damage associated molecular patterns such as high motility group box 1 (HMGB1) and complement components that interact with resident non-cardiomycyte cell types to activate the release of pro-inflammatory cytokines and chemokines.[21–23] Resident macrophages recognize HMGB1 through the receptor for advanced glycation end products and toll-like receptor 4 to release pro-inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α).[24, 25] These signals initiate leukocyte infiltration into the infarct region. Leukocytes clear necrotic tissue by releasing extracellular proteases, predominately serine proteases and matrix metalloproteinases (MMPs) that degrade ECM to remove cardiomyocyte debris.[26–29] Fibroblasts respond to and contribute to proinflammatory signaling during the inflammatory phase of MI. Fibroblasts isolated from the infarct region produce pro-inflammatory cytokines in response to MI relevant signaling stimuli such as TNF-α.[30] Stimulation of cultured cardiac myofibroblasts with IL-1α induces secretion of IL-1β, TNF-α, and IL-6, indicating a positive feedback loop is initiated by cytokine stimulation.[31, 32]
In vivo, day 1 MI fibroblasts display a pro-inflammatory phenotype to contribute inflammatory cytokines and MMPs. Ex vivo transcriptomic analysis of day 1 MI fibroblasts (cultured passage 2 cells) show expression of genes contributing to leukocyte infiltration, including Il23a, Ccl5, Csf1, and Cxcl11, and downregulation of anti-inflammatory Il10.[10] Cx3c11, Ccl5, and Csf1 are highly upregulated in day 1 MI fibroblasts and return to day 0 concentrations by day 7.[10] By evaluation of 17 signaling genes, Tnfrsf9 signifies day 1 MI signaling. Pathway analysis of upregulated genes indicate positive regulation of macrophage chemotaxis and regulation of T helper 17 cells by Gene Ontology (GO) biological processes. Downregulation of anti-apoptotic Bcl2 indicate D1 MI fibroblasts have a pro-survival phenotype; this is coupled by an absence of proliferation, such that fibroblast numbers maintain but are not expanded. At MI day 1, cholesterol biosynthesis is the most downregulated GO biological process, indicative of a reduced need for cell membrane synthesis and consistent with the reduction in proliferation.[10] The shift to anti-migratory phenotype is coupled with decreased expression of a number of genes associated with migration, including Cthrc1, Fgf2, and Fzd2.[10] Thus fibroblasts, play a role in the initiation of, and the response to, inflammatory signaling. In response to the damage-induced inflammation, fibroblasts adopt an anti-migratory and anti-proliferative signature.
Day 3 MI fibroblasts are anti-inflammatory and pro-angiogenic.
By MI day 3, fibroblasts in the infarct region enter the proliferative phase of cardiac remodeling. Fibroblast proliferation transcripts become upregulated, along with polarization to an anti-inflammatory and pro-angiogenic phenotype. Polarization to an anti-inflammatory phenotype suggests that fibroblasts play a role in transitioning from the inflammatory phase. Coupled with the increase in proliferation, cholesterol biosynthesis becomes the most upregulated GO biological process.[10] The upregulation in proliferation was due to increased expression of fibroblast proliferation marker cytoskeleton-associated protein 4 (Ckap4) at day 3, as Ckap4 was increased while 9 other proliferation genes were not (Ccnb2, Cenpa, Mcm2, Mik67, Pcna, Top2a, Tubb5, Tuba1a, and Tuba1b).[10, 33] The van Rooij lab used single cell sequencing of freshly isolated cells from day 3 ischemia/reperfused myocardium and also identified Ckap4 as a novel marker for activated fibroblasts.[33] Thus, despite the model (permanent occlusion vs. ischemia/reperfusion), isolation conditions (cultured to passage 1 on plastic vs. freshly used), or approach (RNA-sequencing vs. single cell sequencing), the results were reproducible across labs. Caspase 3 is also downregulated in MI day 3 cardiac fibroblasts, and this inhibition of apoptosis further contributes to an accumulation in fibroblast numbers. Day 3 MI fibroblasts use Il4ra, Mapk7, and Nfkb1 signaling, reflective of the start in shift to anti-inflammation.[10]
Expression of Acta2 is linear in MI cardiac fibroblasts, with MI day 3 fibroblast showing about 50% of the MI day 7 expression.[10] In both ex vivo models and clinical studies, galectin-3 has been identified as a possible activator of the transforming growth factor β1 (TGFβ1)/Acta2/ Collagen I pathway in cardiac fibroblasts, leading to ECM accumulation.[34]
TGFβ1 is a known pro-fibrotic growth factor and a key paracrine signal to induce fibroblast activation.[35–37] TGFβ1 enhances ECM protein synthesis and is well- documented for its role in activating fibroblasts and cardiac repair following MI.[38–42] Activated fibroblasts further signal ECM accumulation through multiple signaling pathways, using platelet derived growth factor receptor-alpha and beta (PDGFRα and β) in particular.[43, 44] TGFβ1 and vascular endothelial growth factor (VEGF), released by both anti-inflammatory macrophages and cardiac fibroblasts, increase connective tissue growth factor (CTGF) secretion by fibroblasts.[4, 45–49] CTGF is also downregulated by IL-1 to yield lower activity in the inflammatory phase.[50] CTGF mAb treatment in MI mice at day 3 improved ejection fraction and survival at day 7. Long term treatment with CTGF blocking antibody limited fibrosis and cardiac hypertrophy.[51] The increase in VEGF in MI D3 fibroblasts also signals a stimulus from fibroblasts to initiate angiogenesis.[52] VEGF interacts with fibroblasts to promote angiogenesis in endothelial cells through both autocrine and paracrine mechanisms. VEGF can be induced by fibroblasts through fibroblast growth factor-2 (FGF-2) to stimulate endothelial tube formation. [52–55]
Day 3 MI fibroblasts isolated from the infarct region of mice deficient in secreted protein acidic and rich in cysteine (SPARC) showed reduced expression of 22 ECM genes out of 84 measured.[14] Fibronectin, connective tissue growth factor, MMP-3, and tissue inhibitor of metalloproteinase-2 (TIMP-2) were all lower in expression (measured in passage 2–4 cells) compared to wild type day 3 MI fibroblasts. Fibroblast gene expression mirrored LV tissue protein for fibronectin, CTGF, and MMP-3 but not TIMP-2.[14] Day 3 fibroblasts are proliferative, pro-angiogenic, show an increase in activation, and upregulate pro-fibrotic pathways that will continue to be upregulated in the maturation phase.
Day 7 MI fibroblasts are ECM-producing and anti-angiogenic.
The maturation phase of cardiac remodeling occurs at day 7 and is characterized by ECM accumulation and scar formation. Fibroblasts are the major contributors of ECM during the maturation phase. Day 7 MI fibroblasts show reduced migration rates and a shift in adhesion preference from laminin to collagen IV.[5] Collagen synthesis increases 169% in fibroblasts isolated from the MI region, reflecting a shift to producing ECM to support the generation of the infarct scar. Collagen I and III are predominant ECM proteins in the infarct scar.[1] There is a gradual increase in collagen in the infarct region for first 6 days of MI followed by its subsequent gradual condensation into a scar with a steady increase in complexity.[56] Fibronectin (Fn1) is also upregulated in MI fibroblast gene expression.[10] Collagen complexity over the infarct scar further increases from day 7 to day 21 producing a robust contractile scar.[15, 56]
The expression of Acta2 is linear over the first week, peaking at MI day 7 and returning towards day 0 values by MI day 28.[15, 57] Acta2 expression indicates that fibroblasts have acquired a contractile phenotype. This phenotype is necessary to maintain structural integrity during the process of scar formation. Periostin is another marker of contractile fibroblasts that increases in the infarct region in the first week of MI.[10, 58] Treatment with periostin locally improves cardiac physiology after MI but causes increased cardiac fibrosis suggesting periostin increases ECM production after MI.[59] At MI day 7, Acta2 increases in cardiac fibroblasts isolated from the infarct zone compared to fibroblasts isolated from the remote zone, and remote zone fibroblasts show more Acta2 expression than fibroblasts isolated from day 0 no MI hearts.[5] Of note, markers of origin shift with MI time. Wt1 and Twist1 are increased at MI day 7 compared to day 0 no MI and day 1 or 3 MI. Tbx18 is increased at both MI days 3 and 7 compared to MI day 1, while Tcf21 shows uniform expression across the time continuum.[10] Day 7 MI fibroblasts use Pik3r3 and Fgfr2 signaling. Pdgfra and Cthrc1 denote the day 7 MI fibroblast, showing highest expression at this time. Cthrc1 has been associated with tumor invasion and metastasis, working through upregulation of MMPs −7 and −9.[60] While Cthrc1 reverses collagen synthesis in keloid fibroblasts, its role in cardiac fibroblasts remains to be divulged.[61]
Fibroblasts convert from a pro-angiogenic state at MI day 3 to an anti-angiogenic profile at MI day 7.[10] Fibroblast-mediated inhibition of angiogenesis is modulated by thrombospondin-1 (Thbs1). Treatment of cultured microvascular endothelial cells showed reduced endothelial cell tube formation with treatment of day 7 fibroblast secretome. Thbs1 is the highest upregulated fibroblast gene showing the most linear increase over MI time, and secreted Thbs1 increases dramatically at day 7. Treatment with a Thbs1 Ab significantly attenuates the decrease in endothelial cell tube formation. The increase in Thbs1 could be related to the decrease in fibroblast-derived VEGF at day 7, as VEGF is a known negative regulator of Thbs1.[10, 62] Like Thbs1, VEGF also negatively regulates Thbs2 through hypoxia inducible factor-1α, which would serve as an additional mechanism to prevent angiogenesis.[63]
Secretomes collected from day 7 LV infarct fibroblast cultures show increased release of collagen I alpha 1 and 2 from cardiac fibroblasts, along with increases in the cross-linkers SPARC and lysyl oxidase (LOX) concentrations. Thus, the MI fibroblast secretome is a rich source of both ECM and its cross-linkers, and the fibroblast retains its secretome memory in culture. Secretome fractions collected under serum free conditions are also useful stimuli of other MI-relevant cell types, including inflammatory and endothelial cells, to examine cell crosstalk.
A number of modifications alter fibroblast responses to MI. MMP-28 deletion decreases collagen deposition and yields fewer myofibroblasts in the day 7 MI LV.[13] Collagen cross-linking is impaired in the absence of MMP-28 as a result of decreased expression and activation of lysyl oxidase. Porphyromonas gingivalis lipopolysaccharide pre-treatment exacerbates MI induction of Ccl12 in mice, which inhibits fibroblast activation, measured in vivo and ex vivo, to reduce scar formation and increase rupture rates.[11] Likewise, infusion of anti-inflammatory IL-10 starting at one day after MI reduces LV dilation and improves ejection fraction without affecting infarct area or mortality.[12] The change in cardiac physiology resulted from increased M2 anti-inflammatory expression in macrophages. Ex vivo, fibroblasts from the in vivo IL-10 treated group showed more fibroblast activation (increased proliferation, migration, and collagen production) that was the result of both direct and indirect effects on macrophage polarization. Importantly, the fibroblasts examined ex vivo in these studies were isolated, cultured in media with 10% fetal bovine serum, and used through passage 4. The major role of fibroblasts on day 7 is to deposit ECM to build a scar. Fibroblasts regulate angiogenesis by switching a pro-angiogenic phenotype at MI day 3 to an anti-angiogenic phenotype at MI day 7. Fibroblast activity, therefore, has direct effects on cardiac physiology through regulation of ECM and has indirect effects through regulation of other cardiac cell types.
Long-term cardiac fibroblast activation to maintain the MI reset in homeostasis.
Beginning at MI day 7, fibroblasts begin to de-activate and return to their homeostatic roles sensing the ECM needs of the myocardium. Because there is a large replacement of myocytes with infarct scar, the ECM needs are greater than in the pre-MI state. Differences in cell physiology between cardiac fibroblasts before MI and long-term fibroblasts after MI is reflected primarily by the reset in cardiac homeostasis that occurs, including increased ECM production needed to maintain the infarct scar structure. The maintenance of the infarct requires constant sentinel feedback to the fibroblast. As late as 18 years after MI in humans, fibroblasts have been observed to be present and functional in the infarct region.[64] The Blankestein lab has put forth the concept that well-healed infarcts require the continued presence of a minimum number of fibroblasts to prevent adverse remodeling that progresses to heart failure.[6, 64] Thus, strategies to eliminate fibroblasts from healed myocardium will likely prove detrimental.
Conclusions
Fibroblasts are major players in scar formation following MI. Fibroblasts phenotypes shift through the different phases of remodeling (Figure 1). In the control setting, fibroblasts interact with the extracellular environment to maintain cardiac homeostasis. Following MI, fibroblasts immediately transition to an inflammatory phenotype, corresponding to the inflammatory phase of MI. During the proliferative phase, fibroblasts are pro-angiogenic. During the reparative phase, fibroblasts are responsible for laying down new extracellular matrix and downregulation of angiogenesis. Establishing the role of fibroblasts through different phases of cardiac remodeling has implications in targeting fibroblasts to improve scar formation.
There remains several gaps in our understanding of cardiac fibroblast roles after MI. Future studies are needed to understand what mechanisms induce fibroblast to polarize and whether polarization is bi-directional and reversible. Understanding the regulators of each fibroblast cell state will be important for both mechanistic insight and to develop targets that can adjust the system in controlled ways.[65] Computational studies will be informative in allowing a broad number of scenarios to be rapidly evaluated.[66, 67] Cell to cell interactions play important roles in polarizing macrophages, and similar mechanisms likely work for fibroblasts.[68] Using spatial-omics approaches will help to define cell heterogeneity and its effect on ECM regulation by the fibroblast will provide a greater depth of knowledge.[69] While the cardiac phenotypes across the MI time continuum are now better defined, elucidating signaling patterns and cell interactions will allow us to modulate these cells, to assess if long-term MI outcomes can be predicted based on early fibroblast phenotypes. For example, in vitro renin-angiotensin-aldosterone signaling effects on cardiac fibroblasts are well understood, and a better understanding of its relationship with other signaling pathways such as Tgfb signaling is needed to predict how modulation will alter overall ECM accumulation in vivo.[70] In addition, more information is needed on the role of post-translational modifications. For example, fibronectin is citrullinated at cell-binding domain sites to alter fibroblast migration rates and support wound healing.[71] Thus, ECM records exposure to inflammation through post-translational modifications that are then read by fibroblasts to alter their cell physiology. How these mechanisms may work in cardiac fibroblasts has not been examined. Limiting the inflammatory phenotype and promoting the reparative phenotype could potentially improve the process of scar formation. In contrast, enhancing the reparative phenotype could cause excess collagen deposition and fibrosis, leading to reduced cardiac function. Thus, the right balance in ECM deposition is needed for the happy medium of providing enough structure. Acquiring new knowledge of fibroblast phenotypes in relation to MI time will allow us to more accurately target fibroblasts to improve long-term outcomes.
Highlights.
Cardiac fibroblasts undergo temporal pattern changes throughout the wound healing response.
Resident fibroblasts work to maintain cardiac homeostasis, producing sufficient extracellular matrix (ECM) to coordinate normal turnover.
Myocardial infarction (MI) day 1 fibroblasts are pro-inflammatory, day 3 fibroblasts are proliferative and pro-angiogenic, and day 7 fibroblasts are scar producing and anti-angiogenic.
By MI day 28, fibroblasts have established a neo-homeostasis, de-activating while continuing to produce sufficient ECM to coordinate the new kinetics of turnover.
Acknowledgements.
Dr. Lindsey is a Stokes-Shackleford Professor at UNMC. We acknowledge funding from National Institutes of Health under Award Numbers HL075360, HL129823, and HL137319, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Numbers 5I01BX000505. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies. All authors have reviewed and approved the article. All authors have read the journal authorship agreement and policy on disclosure of potential conflicts of interest and have nothing to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ma Y, Halade GV, Lindsey ML, Extracellular matrix and fibroblast communication following myocardial infarction, Journal of cardiovascular translational research 5(6) (2012) 848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Turner NA, Porter KE, Function and fate of myofibroblasts after myocardial infarction, Fibrogenesis Tissue Repair 6(1) (2013) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li L, Zhao Q, Kong W, Extracellular matrix remodeling and cardiac fibrosis, Matrix Biology 68–69 (2018) 490–506. [DOI] [PubMed] [Google Scholar]
- [4].Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML, Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps, Trends Pharmacol Sci 38(5) (2017) 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, Mains IM, Mingoia JT, Flack EC, Lindsey ML, Altered fibroblast function following myocardial infarction, J Mol Cell Cardiol 39(4) (2005) 699–707. [DOI] [PubMed] [Google Scholar]
- [6].Van Den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J, Myocardial remodeling after infarction: the role of myofibroblasts, Nature Reviews Cardiology 7(1) (2010) 30. [DOI] [PubMed] [Google Scholar]
- [7].Daseke MJ 2nd, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML, Neutrophil proteome shifts over the myocardial infarction time continuum, Basic Res Cardiol 114(5) (2019) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML, Mapping macrophage polarization over the myocardial infarction time continuum, Basic Res Cardiol 113(4) (2018) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ivey MJ, Tallquist MD, Defining the Cardiac Fibroblast, Circ J 80(11) (2016) 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ 2nd, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, Lindsey ML, Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis, Basic Res Cardiol 114(2) (2019) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML, Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing, JCI Insight 2(18) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML, IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation, Basic Res Cardiol 112(3) (2017) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML, Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation, Circ Res 112(4) (2013) 675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML, SPARC mediates early extracellular matrix remodeling following myocardial infarction, Am J Physiol Heart Circ Physiol 301(2) (2011) H497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J Clin Invest 128(5) (2018) 2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Camelliti P, Borg TK, Kohl P, Structural and functional characterisation of cardiac fibroblasts, Cardiovascular Research 65(1) (2005) 40–51. [DOI] [PubMed] [Google Scholar]
- [17].Manso AM, Kang SM, Ross RS, Integrins, focal adhesions, and cardiac fibroblasts, J Investig Med 57(8) (2009) 856–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].van Putten S, Shafieyan Y, Hinz B, Mechanical control of cardiac myofibroblasts, J Mol Cell Cardiol 93 (2016) 133–42. [DOI] [PubMed] [Google Scholar]
- [19].Miragoli M, Salvarani N, Rohr S, Myofibroblasts induce ectopic activity in cardiac tissue, Circ Res 101(8) (2007) 755–8. [DOI] [PubMed] [Google Scholar]
- [20].Vasquez C, Benamer N, Morley GE, The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts, J Cardiovasc Pharmacol 57(4) (2011) 380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED, Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart, J Thorac Cardiovasc Surg 128(2) (2004) 170–9. [DOI] [PubMed] [Google Scholar]
- [22].Frangogiannis NG, Regulation of the inflammatory response in cardiac repair, Circ Res 110(1) (2012) 159–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turner NA, Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs), J Mol Cell Cardiol 94 (2016) 189–200. [DOI] [PubMed] [Google Scholar]
- [24].Kwak MS, Lim M, Lee YJ, Lee HS, Kim YH, Youn JH, Choi JE, Shin JS, HMGB1 Binds to Lipoteichoic Acid and Enhances TNF-alpha and IL-6 Production through HMGB1-Mediated Transfer of Lipoteichoic Acid to CD14 and TLR2, J Innate Immun 7(4) (2015) 405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML, Temporal neutrophil polarization following myocardial infarction, Cardiovasc Res 110(1) (2016) 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].DeCoux A, Lindsey ML, Villarreal F, Garcia RA, Schulz R, Myocardial matrix metalloproteinase-2: inside out and upside down, J Mol Cell Cardiol 77 (2014) 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML, Matrix metalloproteinase-9: Many shades of function in cardiovascular disease, Physiology (Bethesda) 28(6) (2013) 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y, Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling, J Mol Cell Cardiol 91 (2016) 134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Iyer RP, Jung M, Lindsey ML, MMP-9 signaling in the left ventricle following myocardial infarction, Am J Physiol Heart Circ Physiol 311(1) (2016) H190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jacobs M, Staufenberger S, Gergs U, Meuter K, Brandstatter K, Hafner M, Ertl G, Schorb W, Tumor necrosis factor-alpha at acute myocardial infarction in rats and effects on cardiac fibroblasts, J Mol Cell Cardiol 31(11) (1999) 1949–59. [DOI] [PubMed] [Google Scholar]
- [31].Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE, Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts, Am J Physiol Heart Circ Physiol 297(3) (2009) H1117–27. [DOI] [PubMed] [Google Scholar]
- [32].van Nieuwenhoven FA, Hemmings KE, Porter KE, Turner NA, Combined effects of interleukin-1α and transforming growth factor-β1 on modulation of human cardiac fibroblast function, Matrix Biology 32(7) (2013) 399–406. [DOI] [PubMed] [Google Scholar]
- [33].Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, Lacraz GPA, Huibers MMH, van Oudenaarden A, van Rooij E, Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation, Circulation 138(2) (2018) 166–180. [DOI] [PubMed] [Google Scholar]
- [34].Shen H, Wang J, Min J, Xi W, Gao Y, Yin L, Yu Y, Liu K, Xiao J, Zhang Y, Wang Z, Activation of TGF-β1/α-SMA/Col I Profibrotic Pathway in Fibroblasts by Galectin-3 Contributes to Atrial Fibrosis in Experimental Models and Patients, Cellular Physiology and Biochemistry 47(2) (2018) 851–863. [DOI] [PubMed] [Google Scholar]
- [35].Dobaczewski M, Chen W, Frangogiannis NG, Transforming growth factor (TGF)-β signaling in cardiac remodeling, Journal of molecular and cellular cardiology 51(4) (2011) 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cellular and molecular life sciences 71(4) (2014) 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Györfi AH, Matei A-E, Distler JHW, Targeting TGF-β signaling for the treatment of fibrosis, Matrix Biology 68–69 (2018) 8–27. [DOI] [PubMed] [Google Scholar]
- [38].Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn W-P, Schaper J, Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms, Circulation 107(7) (2003) 984–991. [DOI] [PubMed] [Google Scholar]
- [39].Talman V, Ruskoaho H, Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration, Cell and tissue research 365(3) (2016) 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Frangogiannis NG, The role of transforming growth factor (TGF)-beta in the infarcted myocardium, J Thorac Dis 9(Suppl 1) (2017) S52–s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hanna A, Frangogiannis NG, The Role of the TGF-beta Superfamily in Myocardial Infarction, Front Cardiovasc Med 6 (2019) 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Avery D, Govindaraju P, Jacob M, Todd L, Monslow J, Puré E, Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts, Matrix Biology 67 (2018) 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mechanical regulation of cardiac fibroblast profibrotic phenotypes, Molecular Biology of the Cell 28(14) (2017) 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG, The Role of Platelet-Derived Growth Factor Signaling in Healing Myocardial Infarcts, Journal of the American College of Cardiology 48(11) (2006) 2315–2323. [DOI] [PubMed] [Google Scholar]
- [45].Sonnylal S, Shi-Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, Leask A, Abraham D, Bou-Gharios G, de Crombrugghe B, Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis, Arthritis Rheum 62(5) (2010) 1523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shi-Wen X, Leask A, Abraham D, Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis, Cytokine & Growth Factor Reviews 19(2) (2008) 133–144. [DOI] [PubMed] [Google Scholar]
- [47].Zhao L, Eghbali-Webb M, Release of pro- and anti-angiogenic factors by human cardiac fibroblasts: effects on DNA synthesis and protection under hypoxia in human endothelial cells, Biochim Biophys Acta 1538(2–3) (2001) 273–82. [DOI] [PubMed] [Google Scholar]
- [48].Twardowski RL, Black LD 3rd, Cardiac fibroblasts support endothelial cell proliferation and sprout formation but not the development of multicellular sprouts in a fibrin gel co-culture model, Ann Biomed Eng 42(5) (2014) 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L, Levtchenko E, Kuypers D, Goldschmeding R, Connective tissue growth factor (CTGF) from basics to clinics, Matrix Biology 68–69 (2018) 44–66. [DOI] [PubMed] [Google Scholar]
- [50].Maqbool A, Hemmings KE, O’Regan DJ, Ball SG, Porter KE, Turner NA, Interleukin-1 has opposing effects on connective tissue growth factor and tenascin-C expression in human cardiac fibroblasts, Matrix Biology 32(3) (2013) 208–214. [DOI] [PubMed] [Google Scholar]
- [51].Vainio LE, Szabó Z, Lin R, Ulvila J, Yrjölä R, Alakoski T, Piuhola J, Koch WJ, Ruskoaho H, Fouse SD, Seeley TW, Gao E, Signore P, Lipson KE, Magga J, Kerkelä R, Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction, JACC: Basic to Translational Science 4(1) (2019) 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tille JC, Pepper MS, Mesenchymal cells potentiate vascular endothelial growth factor-induced angiogenesis in vitro, Exp Cell Res 280(2) (2002) 179–91. [DOI] [PubMed] [Google Scholar]
- [53].Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P, Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis, J Cell Biol 141(7) (1998) 1659–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gutpell KM, Hoffman LM, VEGF induces stress fiber formation in fibroblasts isolated from dystrophic muscle, J Cell Commun Signal 9(4) (2015) 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Humeres C, Frangogiannis NG, Fibroblasts in the Infarcted, Remodeling, and Failing Heart, JACC Basic Transl Sci 4(3) (2019) 449–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML, Han HC, Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction, J Mol Cell Cardiol 85 (2015) 229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD, Resident fibroblast expansion during cardiac growth and remodeling, Journal of molecular and cellular cardiology 114 (2018) 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD, Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat Commun 7 (2016) 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ladage D, Yaniz-Galende E, Rapti K, Ishikawa K, Tilemann L, Shapiro S, Takewa Y, Muller-Ehmsen J, Schwarz M, Garcia MJ, Sanz J, Hajjar RJ, Kawase Y, Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis, PLoS One 8(5) (2013) e59656–e59656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wu Q, Yang Q, Sun H, Role of collagen triple helix repeat containing-1 in tumor and inflammatory diseases, J Cancer Res Ther 13(4) (2017) 621–624. [DOI] [PubMed] [Google Scholar]
- [61].Zhao M-J, Chen S-Y, Qu X-Y, Abdul-Fattah B, Lai T, Xie M, Wu S.-d., Zhou Y-W, Huang C-Z, Increased Cthrc1 Activates Normal Fibroblasts and Suppresses Keloid Fibroblasts by Inhibiting TGF-β/ mad Signal Pathway and Modulating YAP Subcellular Location, Curr Med Sci 38(5) (2018) 894–902. [DOI] [PubMed] [Google Scholar]
- [62].Lawler PR, Lawler J, Molecular basis for the regulation of angiogenesis by thrombospondin-1 and −2, Cold Spring Harb Perspect Med 2(5) (2012) a006627–a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].MacLauchlan SC, Calabro NE, Huang Y, Krishna M, Bancroft T, Sharma T, Yu J, Sessa WC, Giordano F, Kyriakides TR, HIF-1α represses the expression of the angiogenesis inhibitor thrombospondin-2, Matrix Biology 65 (2018) 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hermans KCM, Daskalopoulos EP, Blankesteijn WM, The Janus face of myofibroblasts in the remodeling heart, Journal of Molecular and Cellular Cardiology 91 (2016) 35–41. [DOI] [PubMed] [Google Scholar]
- [65].Bretherton R, Bugg D, Olsewski E, Davis J, Regulators of Cardiac Fibroblast Cell State, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ, A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation, J Mol Cell Cardiol 94 (2016) 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zeigler AC, Nelson AR, Chandrabhatla AS, Brazhkina O, Holmes JW, Saucerman JJ, Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction, Matrix Biology in press (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S, Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype, Eur Heart J 38(3) (2017) 187–197. [DOI] [PubMed] [Google Scholar]
- [69].Bingham GC, Lee F, Naba A, Barker TH, Spatial-omics: Novel Approaches to Probe Cell Heterogeneity and ECM Biology, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].AlQudah M, Hale TM, Czubryt MP, Targeting the Renin-Angiotensin-Aldosterone System in Fibrosis, Matrix Biology (in press) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Stefanelli VL, Choudhury S, Hu P, Liu Y, Schwenzer A, Yeh C-R, Chambers DM, Pesson K, Li W, Segura T, Midwood KS, Torres M, Barker TH, Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion, MatrixBiology 82 (2019) 86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]