

Abstract
Background
Maple sirup urine disease (MSUD) is an autosomal recessive inherited metabolic disorder. The disease‐causing mutations can affect the BCKDHA, BCKDHB, and DBT genes encoding for the E1α, E1β, and E2 subunits of the multienzyme branched‐chain α‐keto acid dehydrogenase (BCKDH) complex. In the present study, novel pathogenic variants in BCKDHB and DBT genes were identified in three Vietnamese families with MSUD.
Methods
Three newborn patients from three unrelated Vietnamese families were diagnosed with MSUD at the Metabolic Clinic, National Hospital of Pediatrics. Blood samples of 11 relatives from two generations of the three families diagnosed with MSUD were analyzed using exome and Sanger sequencing analyses.
Results
Novel pathogenic variants in BCKDHB (c.1103C>T, c.989A>G, and c.704G>A), and DBT (c.263_265delAAG) genes were identified in three pediatric patients with MSUD.
Conclusions
We have identified novel pathogenic variants in the MSUD‐related genes in the pedigree of the three patient's families. Our findings expand the mutational spectrum of MSUD and provide the scientific basis for genetic counseling for the patient's families.
Keywords: BCKD, BCKDHB, DBT, exome sequencing, maple sirup urine disease
Novel mutations from patients with MUSD.
1. INTRODUCTION
Maple sirup urine disease (MSUD, OMIM# 248600) is a biochemical genetic disorder which is inherited in an autosomal recessive way. MSUD is derived from the pathogenic mutations in the genes encoding three subunits of the complex branched‐chain α‐keto acid dehydrogenase (BCKAD) (Chuang & Shih, 2001). The BCKAD enzyme complex is composed of three catalytic constituents, including branched chain α‐ketoacid decarboxylase (El), dihydrolipoyl transacylase (E2), and dihydrolipoamide dehydrogenase (E3). The thiamine‐dependent enzyme E1 consists of two α (E1α, encoded by the BCKDHA gene (OMIM 608348)) and two β (E1β, encoded by the BCKDHB gene (OMIM 248611)) subunits, while E2 and E3 are encoded by the DBT (OMIM 248610) and DLD (OMIM 246900) genes, respectively (Matsuda et al., 1990). BCKAD is located in the outer mitochondrial membrane and plays a key role in the catabolism of the essential branched‐chained amino acids (BCAAs), such as leucine, isoleucine, and valine (Chuang et al., 1991). The failure of BCKAD activity has been demonstrated as the main cause of MSUD (Baird, Wojcinski, Wise, & Godkin, 1987). Mutations in the catalytic components have been shown to be associated closely with impairment of the BCKAD activity (Sofia et al., 2008). Based on the clinical symptoms and severity, the clinical phenotype of MSUD has been classified into classical, intermediate, intermittent, and thiamine‐responsive forms (Chuang et al., 2001). Recent survey data showed that the worldwide incidence of MSUD is estimated approximately 1:185,000. The incidence in certain ethnic groups has been found to be higher, for example, in Philippines (2015), newborn screening is 1:82,354 (Mary et al., 2016); Mennonites population in Pennsylvania, at 1 in 176 births (Chuang et al., 2001); in Malaysia: detected 25 patients were MSUD and most of the patients occurred in Malay ethnic (Yunus, Kamaludin, Mamat, Choy, & Ngu, 2012). The clinical characteristics of this disorder include the following symptoms: maple sirup odor of urine and cerumen 12–24 hr after birth, poor breastfeeding by 2–3 days of age, high levels of BCAAs in plasma, ketonuria, irritability, and lethargy. The severe symptoms, such as encephalopathy and intermittent apnea may lead to failure of the respiratory tracts, coma, and death in the 1st weeks of life if left untreated (Kerstin et al., 2009).
Recent genetic studies have revealed the occurrence of mutations in BCKDHB gene to be higher in frequency compared to that in DBT gene in the patients with MSUD (Ali & Ngu, 2018). In the present study, we describe identification of novel mutations in BCKDHB and DBT genes in three Vietnamese patients with MSUD using the exome sequencing analysis (WES).
2. MATERIALS AND METHODS
2.1. Ethics approval and consent to participate
All the followed procedures were in accordance with the ethical standards of the responsible committee on human experimentation (Ethical Committee at Institute of Genome Research, No. 18/QD‐NCHG) and with the Helsinki Declaration of 1975, as revised in 2013 (World Medical Association, 2013).
2.2. Patient clinical information
Three newborn patients (including one girl and two boys, between 2 and 13 days of age) from three unrelated Vietnamese families were diagnosed with MSUD at the Metabolic Clinic, National Hospital of Pediatrics. Diagnosis was based on the clinical presentations and biochemical tests using quantitative plasma amino acid analysis (Yunus et al., 2012).
2.3. Exome and Sanger sequencing and protein modeling
Exome and Sanger sequencing and protein modeling were performed as described in Supporting Information 1. The GenBank reference sequences used for Sanger validations are: BCKDHB, NG_009775.2 and DBT, NG_011852.2.
The genomic DNA of the individual blood samples were extracted using the standard method (QIAamp DNA Blood Mini Kit, Germany) and subsequently subjected to the exome sequencing (ES) using HiSeq2500 sequencer. Library preparation for sequencing was carried out using the SureSelect V5 exome capture kit. All data were aligned to the GRCh37/hg19 reference human genome using Burrows–Wheeler Alignerv 0.7.10. Variants were qualitatively trimmed using Genome Analysis Toolkit v3.4 which was annotated for functional effect by SnpEff v4.1. Variations were screened by filtering out with indicators Sift_Pred = Damaging (D) or NA (normal) and followed by analysis of the variants occurred in four genes linked to MSUD. To confirm variants identified by ES, DNA of the family members were amplified for exons 1, 6, 9, and 10 of the BCKDHB (ENSG00000083123) and exon 4 of the DBT (ENSG00000137992) genes and sequenced by the Sanger method.
3. RESULTS
3.1. Clinical findings
Three patients were all diagnosed with MSUD by doctors of the National Hospital of Pediatrics. The patient's parents and siblings of are asymptomatic. Two of the three patients showed the classic phenotype at 2–13 days of age. The diagnosis was performed based on their clinical features and biochemical tests. Clinical details of the patients are presented as follows:
Patient 01: The patient is the second child. His sister has normal development. After the birth, the patient presented respiratory failure due to congenital pleural effusion and transferred to Neonatal Intensive Care Unit (NICU) at 2 days of age. Evaluations showed bilateral pleural effusion and ketonuria. The patient was then treated with glucose infusion, breastfeeding pause, mechanical ventilation. After 5 days of treatment, his condition became better: spontaneous breathing without respiratory failure. However, after 2 days of fresh milk feeding, he presented lethargy, coma, and respiratory failure. The tests presented metabolic acidosis, hyponatremia, hyperleucinemia (2,887 μmol/L), hyperalle‐isoleucinemia (56 μmol/L). He was further treated with hemofiltration, breastfeeding pause, glucose infusion (10 mg kg−1 min−1). Consequently, the symptoms were treated (the level of plasma leucine was 502 μmol/L). He was discharged after 2 months of treatment. Now, he is 12 months old with DQ 60% and two recurrences of the symptoms (Table S2).
Patient 02: The patient is the third child. His two siblings (18 years elder sister, 14 years elder brother) develop normally. When the 13‐day‐old boy was admitted in the NICU, he presented lethargy, intermittent cyanosis, and mild hypertonia. The biomedical test results showed normal cerebral fluid, elevated CRP, and ketonuria. He was diagnosed with pneumonia/septicemia and treated with antibiotics, nutrition, and oxygen by nasal prongs. After 19 days of treatment, he presented coma, convulsion, apnea, and hyperleucinemia (3,450 μmol/L). The treatment was then changed to hemofiltration, glucose infusion (10 mg kg min−1), vitamin B1 (20 mg/day). After 48 hr of hemofiltration, he was alert (the plasma leucine level was 425 μmol/L). Now, he is 18 months old with DQ 75% and no recurrence (Table S2).
Patient 03: The patient is the third child. Her two siblings (9 years elder sister, 5 years elder brother) have normal growth. She was hospitalized in NICU with clinical signs as poor breastfeeding and lethargy at 6 days old. Evaluations revealed normal cerebral fluid, abnormal brain MRI, ketonuria, and hyperleucinemia (3,569 μmol/L). She was treated with the following manner: breastfeeding halt, glucose infusion (10 mg kg min−1), thiamine supplement, and hemofiltration. After 48 hr of hemofiltration, the patient's condition was better: alert with the plasma leucine level of 532 μmol/L. She was discharged after 3 weeks of treatment. Now she is 18 months and normally developed (Table S2).
3.2. ES and Sanger analysis
ES analysis of the three patients revealed novel pathogenic variants in the BCKDHB gene, of those two new heterozygous missense mutations were recorded (NM_000056.3, c.989A>G, E330G and c.1103C>T, P368L on chromosome 6) in the patient 01 and two heterozygous missense mutations were found in the patient 02 (NM_000056.3, c.1A>T, M1L and c.704G>A, C235Y on chromosome 6). In addition, a homozygous disruptive_inframe_deletion mutation was also identified in the DBT gene of the patient 03 (NM_0001918.3, c.263_265delAAG, E88del on chromosome 1) (Table 1). In which, E330G has been reported in gnomAD at an allele frequency of 3.98e‐6, and is not found in ClinVar; P368L and C235Y is not found in ExAC, gnomAD, and ClinVar; M1L has been reported in gnomAD at an allele frequency of 1.29e‐5, and is not found in ExAC or ClinVar; E88del has been reported in ClinVar as VUS, and is not found in ExAC or gnomAD.
TABLE 1.
Novel mutations in BCKDHB and DBT genes in the patients with MSUD
| Patient | Sex | Age of onset (day) | Gen | Chrom | Exon | Type of mutation | Coding DNA number | Protein number |
|---|---|---|---|---|---|---|---|---|
| 01 | Male | 2 | BCKDHB | 6 | 9/11 | Missense | c.989A>G | p.E330G |
| 10/11 | Missense | c.1103C>T | p.P368L | |||||
| 02 | Male | 13 | BCKDHB | 6 | 1/11 | Missense | c.1A>T | p.M1L |
| 6/11 | Missense | c.704G>A | p.C235Y | |||||
| 03 | Female | 6 | DBT | 1 | 4/11 | Small deletion | c.263_265delAAG | p.E88del |
Abbreviation: MSUD, maple sirup urine disease.
In the patient 01, the two mutations that occurred in exon 9 and 10 of BCKDHB gene are predicted to cause the replacement of Glu with Gly at the 330 position and Pro with Leu at the 368 position, respectively (Table 1 and Figure 1a). The segregation of disease‐causing variants in the patient and patient's family was analyzed using Sanger sequencing. The result showed that the heterozygous mutation occurred in exon 9 (E330G) is inherited from the patient's mother while the variant observed in exon 10 (P368L) is originated from the patient's father. This was consistent with the fact that the patient's sister is normal person and she does not carry any of these mutations (Figure 1b,c).
FIGURE 1.
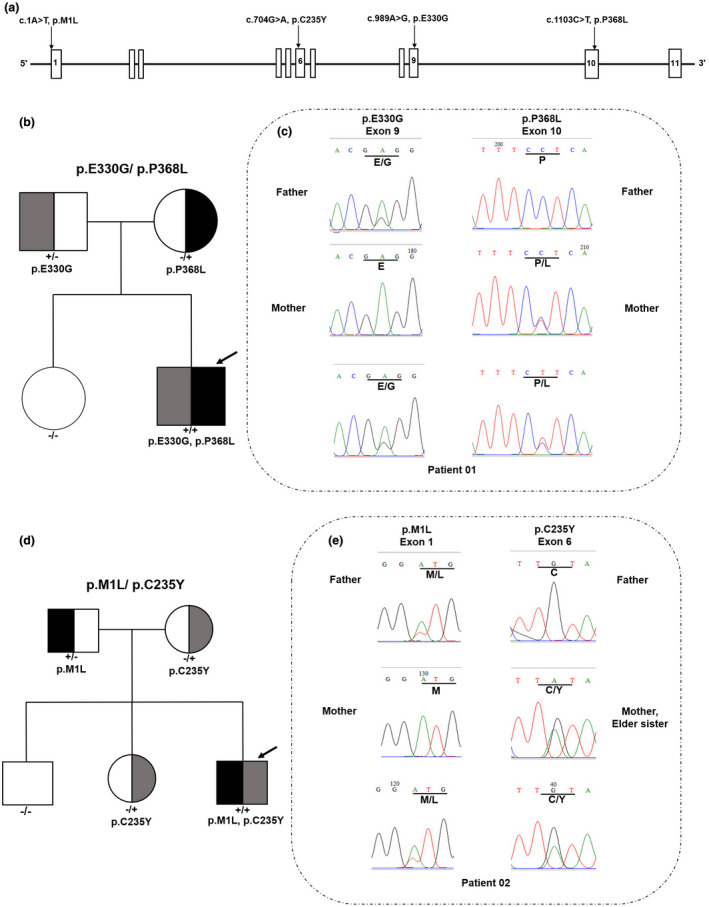
Pedigrees and Sanger sequences of mutations are detected in the families of patient 01 and 02. (a) Location of the mutation in the BCKDHB gene. (b) Pedigree presentation of the family of patient 01. Heterozygous individuals, half black (+/−: p.P368L mutation) or gray (−/+: p.E330G mutation). The compound heterozygous proband: both halves filled (+/+: both of the mutations). The patient's sister had only wild‐type alleles (−/−: wild type [WT], no half black or gray area). (c) Sanger sequence diagram: two heterozygous mutations in exon 9 and 10 of BCKDHB gene (c.989A>G and c.1103C>T) caused replacement of Glu with Gly at the 330 position and Pro with Leu at the 368 position, respectively. (d) Pedigree presentation of the family of patient 02. Heterozygous individuals: half black (+/−: p.M1L mutation) or gray (−/+: p.C235Y mutation). The compound heterozygous proband: both halves filled (+/+: both of the mutations). The patient's brother had only wild‐type alleles (−/−: wild type [WT], no half black or gray area). (e) Sanger sequence diagram: two heterozygous mutations in exon 1 and 6 of BCKDHB gene (c.1A>T and c.704G>A) caused replacement of Met with Leu at the 1 position and Cys with Tyr at the 235 position, respectively. “Male” and “Female” are represented by squares and circles, respectively. The proband is indicated by an arrow
In the BCKDHB gene of the patient 02, the initiator codon and missense pathogenic variant were detected in exon 1 and 6 (c.1A>T, M1L; c.704G>A, C235Y), respectively. In exon 6, a novel missense mutation at c.704G>A is predicted to cause replacement of Cys by Tyr at 235 position and this mutation was found to be inherited from the patient's mother (Figure 1a). The variant in exon 1 at the ATG initiating codon, is predicted to lead to a change in the protein translation process. This mutation was shown to be derived from the patient's father (Figure 1d,e). Furthermore, Sanger sequencing analysis indicated that, the mutation occurred at c.704G>A is passed on to the patient and the patient's sister from their mother (Figure 1d,e). The initialization code mutation that occurred at 1A>T was inherited from the patient's father. However, this genetic mutation was not found in both of the patient's unaffected brother and sister (Figure 1d,e).
In addition, an in‐frame deletion mutation detected in patient 03 was found in exon 4 of the DBT gene. This mutation was inherited from the patient's parents, and is predicted to cause deletion of the trio of bases coding for the amino acid Glu at the 88 position (NM_001918.3: c.263_265delAAG, E88del) (Figure 2a). The Sanger sequencing analysis of the family members also indicated that the patients' parents, brother, and sister are not affected by the disease because they all carried only one copy of the pathogenic variant (Figure 2b,c).
FIGURE 2.
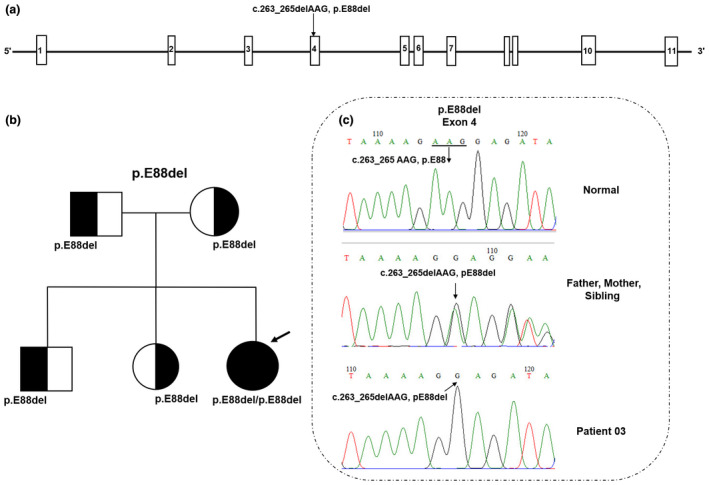
Pedigrees and Sanger sequences of mutations detected in the family of patient 03. (a) Location of the mutation (E88del) in the DBT gene. (b) Pedigree chart of the family showing inheritance of homozygous chromosomal segments. The patient's parents, elder brother, and elder sister are all heterozygous. The patient is homozygous and carries two mutant alleles. (c) Sanger sequence diagram: the homozygous mutation in exon 4 of DBT gene (c.263_265delAAG) caused the loss of Glutamic acid at the 88 position. “Male” and “Female” are indicated by squares and circles, respectively. The proband is shown by an arrow. DNA sequencing of the healthy control shows the wild‐type (WT) sequence
The 3‐dimensional (3D) structural protein modeling was performed using PDB file to predict mutations in BCKDHB and DBT protein structures. The Human X‐ray crystal structure for BCKDHB and DBT proteins were obtained from the protein data bank (RCSB‐PDB). 1X7Y and 1K8O (RCSB PDB code) were used as templates for BCKDHB and DBT, respectively. The 3D structures of normal and mutant BCKDHB and DBT proteins are shown in Figure 3. As a result, 3D modeling prediction indicated that three out of five pathogenic variants are presented with strong modifications in the structures of the mutant proteins caused by the alterations in their amino acid sequence (Figure 3). Three missense variants (E330, P368, and C235) in BCKDHB gene were mapped on E1β subunit (Figure 3d). The novel mutations were confirmed by an extensive search on the Human Gene Mutation Database and Clinvar‐NCBI.
FIGURE 3.
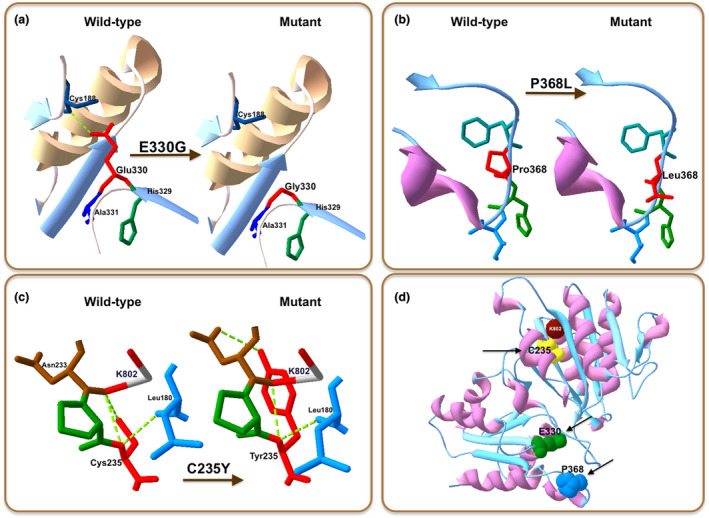
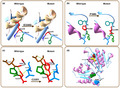
Structural illustration of the new missense mutations in BCKDHB heterotetramer. (a) p.E330G mutation. (b) p.P368L mutation. (c) p.C235Ymutation. (d) Three missense mutations (C235, E330, and P368) identified in E1β subunit are represented by yellow, green, and blue spheres with arrows, respectively. K802 reaction center is presented as a red sphere. Wild‐type and mutated residues are located on the right and left hand, respectively (in accordance with the symbol shown in the image)
4. DISCUSSION
Maple sirup urine disease is an inherited autosomal recessive disease. Patients with MSUD are not able to use branched chain amino acid as BCAAs, leading accumulation of BCAAs products in the blood, urine, and CSF. This has been implicated with severe clinical consequences, such as neurotoxicity for brain tissue and progressive neurodegeneration (Ali & Ngu, 2018; Deepti et al., 2015). Early and accurate diagnosis by means of genetic tests is vital, and interventional medical care can be effective in the curative path for many cases. To date, more than 470 mutations associated with MSUD have been reported in Clinvar. There are approximately 38% of mutations occurring in the BCKDHB gene (Wenjie et al., 2018; Nellis & Danner, 2001; Theodoros et al., 2009) which has been described as the major gene causing MSUD, followed by BCKDHA gene and DBT gene (Peter et al., 2017). Although inheritance of MSUD adheres to a simple autosomal recessive pattern, mutations in each of the three BCKDH specific have been shown to cause MSUD (Nellis & Danner, 2001).
In this study, we describe the clinical, biochemical, and molecular characteristics of the first cases of MSUD in Vietnam. All the patients in the study were diagnosed by the specific clinical and biochemical characteristics of MSUD at 2–13 days after birth while their siblings are the normal and healthy peoples. Sanger sequencing analyses resulted in identification of five pathogenic variants occurred in two genes (BCKDHB and DBT) of the three patients with MSUD. Four pathogenic variants in BCKDHB gene were identified in patients 01 (E330G and P368L) and 02 (M1L and C235Y), and not in their respective unaffected family members. Apathogenic variant (E88del) in DBT gene was detected in patient 03 who was homozygous and in the parents who were heterozygous carriers for the mutation.
The mutations occurred in BCKDHB (1X7Y‐RCSB PDB code) was visualized by the 3D protein modeling prediction. As shown in Figure 3, three out of four pathogenic variants were shown to change remarkably the structure of BCKDHB protein. Inside, the pathogenic variant E330G in exon 9 caused the change of Glu to Gly, subsequently leading to the loss of hydrogen bonding with the Cys188 (Figure 3a). The change of the negatively charged Glu with hydrophobic nonpolar Gly not only affected the local electrostatic interactions, but also the hydrogen bonds. The pathogenic variant also caused the removal of the hydrogen bonding of the Glu330‐β‐Cys188‐β link (Figure 3a), leading to the possible change of the E1β stability (Abiri et al., 2016). Accordingly, Glu330Lys variant is located near the end of the alpha helix and beta sheets of the β subunit secondary structures of the BCKDC, and, therefore, involved in the redistribution of adjacent amino acids near the β‐β’ interfaces (Abiri et al., 2016).
The variant P368L detected in exon 10 of the BCKDHB gene was predicted to cause the replacement of Pro by Leu, leading to the loss of an α‐amino group (Figure 3b). In addition, the residue P368 is located on the C‐terminal domain of the interfaces of the β and α’ subunit (Arnthor et al., 2000). Therefore, when the mutation is occurred, the E1β stability might be changed, leading to the MSUD. This was in the good agreement with the previous studies that the mutations occurred in exon 10 of the BCKDHB gene are associated closely with the pathogenesis of MSUD, for example,: mutation C382 (Imtiaz et al., 2017), mutation Y383 (Henneke et al., 2003), mutation R359 (Lui et al., 2019). Based on the phenotype presented here, we propose the P368 variant also to be contributing to the cause of MSUD.
The C235Y variant is predicted to cause a single nucleotide substitution on exon 6 (c.704G>A). As the result, the 3D protein modeling pattern showed that in the normal case, the Cys235 site has a hydrogen bond with Leu180 and two hydrogen bonds with the K802 reaction center (Figure 3c). However, when a mutation occurred, Cys was replaced by Tyr at position 235, leading to changes in the BCKDHB protein structure, including the loss of the hydrogen bond with the K802 reaction center and the production of an aromatic ring. K802 is one of the two essential positions of K+ ion channels located in the β subunit near the C‐terminal domain of the α subunit (Shimomura, Kuntz, Suzuki, Ozawa, & Harris, 1988). The known MSUD mutations were reported to affect the functions of E1β by interfering with the cofactor and K+ sites, the packing of hydrophobic cores, and the precise arrangement of residues at or near several subunit interfaces (Arnthor et al., 2000). Therefore, the K+ position in the E1β structure of humans is very important for protein stability and function (Arnthor et al., 2000; Chuang, Wynn, Song, & Chuang, 1999). As reported by Lui et al. (2019), the mutation that caused the deletion of a thymine at position 705 (c.705delT) in exon 6 was classified as a likely pathogenic agent (Lui et al., 2019).
The fourth variant at c.1A>T was predicted to cause the change of Met to Leu in codon 1 in exon 1 (M1L). This mutation has been reported previously in the prenatal tests for a Saudi Arabian woman (Imtiaz et al., 2017). So far, mutations of the translation‐initiator ATG (Met1) have been rarely found in the genetic diseases (Huang, Cheng, Reid, & Chen, 1999). In case of the patient, the mutation took place at the opening code of exon 1, resulting in the lack of the first 70 amino acid residues corresponding to the N‐terminus domain of the β subunit in the protein. In addition, the N‐terminus truncation would affect the insertion of the protein into the lipid bilayer (Singer, 1990). This mutation type has also been demonstrated as the cause of several disorders, such as Rhmod syndrome (Huang et al., 1999), Idiopathic infantile nystagmus of Korean patient (Choi, Shin, Seo, Jung, & Choi, 2015).
The fifth variant was identified in exon 4 of the DBT gene (c.263_265delAAG, p.E88del). The patient carried two identical alleles, each one was inherited from the father or mother. In the normal case, Glu88 has a hydrogen bond with Leu121. When the mutation occurred, three AAG (3‐bp) nucleotides were taken away, resulting in the loss of Glutamic acid. As shown by 3D modeling, the mutation not only caused a loss of an amino acid but also changed the entire order of the chain and made the chain ends shorter than the origin.
Our results showed that the isolated mutation is the homozygous mutation occurred at position 88 of the DBT protein. This mutation was observed in the mature peptide chain (62‐482), exactly in the lipoyl‐binding domain extending from position 65 to position 138. Glutamic acid at position 88 would be lost after the mutation occurred, leading to the disconnection between lipoyl Co‐factor and the mutated protein. This may induce a defect in the protein lipoyl‐binding function of the E2 ("Homo sapiens dihydrolipoamide branched chain transacylase E2 (DBT), nuclear gene encoding mitochondrial protein, mRNA"). In addition, location of this mutation in the protein domains, such as (Biotin/lipoyl: 66‐138) and (Single hybrid motif: 64‐146) was also observed. This caused the loss of an amino acid in the functional region of the protein, leading to the dysfunction of the DBT protein (Chang, Chou, Chuang, Chuang, & Huang, 2002).
So far, several deletion mutations occurred in the DBT gene of the MSUD type 2 have been identified. Imtiaz et al. (2017) reported two mutations that caused loss of the 1 bp and 2 bp segments in the Saudi Arabian patient (Imtiaz et al., 2017). The 2 bp (AT) fragment loss on exon 2 of the DBT gene was also reported by Fisher et al. (1993) (Fisher et al., 1993). Chi et al. (2003) identified a large deletion mutation (4.7 kb) occurred in the DBT gene when studying MSUD patients in the Austronesian aboriginal tribe Paiwan of Taiwan (Chi et al., 2003). In a case of classic MSUD, Herring, Litwer, Weber, and Danner (1991) identified a 124 bp deletion in the DBT gene encoding the E2 component of the BCKDH complex (Herring et al., 1991).
In addition, genetic analyzes of BCKDHB gene revealed that the E330G, P368L, and C235Y mutations occurred in the E1 subunit are most likely responsible for the enzymatic dysfunction of the BCKDH complex. This was deduced by their occurrence at the amino acid residues which are highly conserved across species (including Pan paniscus, Nomascus leucogenys, Pongo abelii, Delphinaterus leucas, Monodon monoceros, Marmota marmota marmota, Castor fiber, Oryctolagus cuniculus, and Castor canadensis) (Figure 4a). Similarly, the E88del mutation occurred in the E2 subunit of DBT protein was also found to be highly conserved among species (including Bos indicus, Capra hircus, Lepisosteus oculatus, Pongo abelii, Pan troglodytes, Lipotes vexillifer, Oryctolagus cuniculus, and Anolis carolinensis) (Figure 4b).
FIGURE 4.

Evolutionary conservation of three novel mutations among different species. (a) Three mutations (C235Y, E330G, and P368L) in BCKDHB gene. (b) The mutation E88del in DBT gene
Mutations in the human E1β subunit might be able to induce MSUD via defects in protein structure, because loss of enzyme's stability and function has been shown to have correlation with disease severity (Park et al., 2011). This primary study was conducted on only three patients, and all the mutations were found to be occurred on BCKDHB and DBT genes but not in BCKDHA gene. Therefore, our results provide the scientific basis for further studies to determine the existence and origin of the mutation as well as the genotype‐phenotype correlation of MSUD in the Vietnamese community.
In conclusion, five mutations in the BCKDHB and DBT genes were identified in the three Vietnamese newborns with MSUD. This is the first report on genetic analysis of the Vietnamese MSUD patients using WES method. All the mutations were predicted to potentially destabilize formation of the E1 and E2 complexes. Therefore, this study contributes to more insightful understanding of the MSUD‐related mutations. In addition, the discovery of mutations in the patient's family is extremely important for disease management and genetic counseling for the patient's families themselves.
With the initial results of genetics of metabolic disorders in Vietnamese newborns that we report in the present study, the patients' families have got genetic understandings for themselves and their children after becoming mature. Hopefully, in the future, in combination of the results achieved from screening statistic projects of Vietnamese newborns and our research, we will have a database of mutant genes in Vietnamese people with MSUD disease.
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AUTHOR CONTRIBUTIONS
All authors have substantively contributed to this work. HHN is the principal investigator who designed the study and gave ideas to preparation of the manuscript. TTNN and CDV: performed data analysis and wrote the manuscript; NKN, NLN, and THNT: conducted the experiments related to the study. All authors read and approved the final version of the manuscript.
CONSENT FOR PUBLICATION
The patient's parents totally agreed with publication of this work.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant from Vietnam Academy of Science and Technology (KHCBSS.02/18‐20). We sincerely thank all the patients and their families for participating in this study.
Nguyen TTN, Vu CD, Nguyen NL, Nguyen TTH, Nguyen NK, Nguyen HH. Identification of novel mutations in BCKDHB and DBT genes in Vietnamese patients with maple sirup urine disease. Mol Genet Genomic Med. 2020;8:e1337 10.1002/mgg3.1337
Thi T. N. Nguyen and Chi D. Vu have contributed equally to this work.
Funding information
Vietnam Academy of Science and Technology, Grant/Award Numbers: KHCBSS.02/18‐20.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Abiri, M. , Karamzadeh, R. , Mojbafan, M. , Alaei, M. R. , Jodaki, A. , Safi, M. , … Zeinali, S. (2016). In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Metabolic Brain Disease, 32(1), 105–113. 10.1007/s11011-016-9867-1 [DOI] [PubMed] [Google Scholar]
- Ali, E. Z. , & Ngu, L. H. (2018). Fourteen new mutations of BCKDHA, BCKDHB and DBT genes associated with maple syrup urine disease (MSUD) in Malaysian population. Molecular Genetics and Metabolism Reports, 17, 22–30. 10.1016/j.ymgmr.2018.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnthor, Æ. , Jacinta, L. C. , Wynn, R. M. , Stewart, T. , Chuang, D. T. , & Hol, W. G. J. (2000). Crystal structure of human branched‐chain a‐ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure, 8(3), 277–291. 10.1016/S0969-2126(00)00105-2 [DOI] [PubMed] [Google Scholar]
- Baird, J. D. , Wojcinski, Z. W. , Wise, A. P. , & Godkin, M. A. (1987). Maple syrup urine disease in five hereford calves in Ontario. Canadian Veterinary Journal, 28(8), 501–511. [PMC free article] [PubMed] [Google Scholar]
- Chang, C. F. , Chou, H. T. , Chuang, J. L. , Chuang, D. T. , & Huang, T. (2002). Solution structure and dynamics of the lipoic acid‐bearing domain of human mitochondrial branched‐chain alpha‐keto acid dehydrogenase complex. The Journal of Biological Chemistry, 277(18), 16866–16873. 10.1074/jbc.M110952200 [DOI] [PubMed] [Google Scholar]
- Chi, C. S. , Tsai, C. R. , Chen, L. H. , Lee, H. F. , Mak, B. S. C. , & Yang, S. H. (2003). Maple syrup urine disease in the Austronesian aboriginal tribe Paiwan of Taiwan: A novel DBT (E2) gene 4.7 kb founder deletion caused by a nonhomologous recombination between LINE‐1 and Alu and the carrier‐frequency determination. European Journal of Human Genetics, 11, 931–936. 10.1038/sj.ejhg.5201069 [DOI] [PubMed] [Google Scholar]
- Choi, J. H. , Shin, J. H. , Seo, J. H. , Jung, J. H. , & Choi, K. D. (2015). A start codon mutation of the FRMD7 gene in two Korean families with idiopathic infantile nystagmus. Scientific Report, 5(13003), 1–6. 10.1038/srep13003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, D. , Fisher, C. , Lau, K. , Griffin, T. , Wynn, R. , & Cox, R. (1991). Maple syrup urine disease: Domain structure, mutations and exon skipping in the dihydrolipoyl transacylase (E2) component of the branched‐chain alpha‐keto acid dehydrogenase complex. Molecular Biology & Medicine, 8(1), 49–63. [PubMed] [Google Scholar]
- Chuang, D. T. , & Shih, V. E. (2001). Maple syrup urine disease (branched‐chain ketoaciduria) In Scriver C. R., Beaudet A. L., Sly W. S. & Valle D. (Eds.), The metabolic and molecular bases of inherited disease (Vol. II, 8th ed., pp. 1971–2005). McGraw‐Hill: New York. [Google Scholar]
- Chuang, J. L. , Wynn, R. M. , Song, J. L. , & Chuang, D. T. (1999). GroEL/GroES‐dependent reconsititution of a2b2, tetramers of human mitochondrial branched chain a‐ketoacid decarboxylase‐obligatory interaction of the chaperonins with an alpha beta dimeric intermediate. Journal of Biological Chemistry, 274, 10395–10404. 10.1074/jbc.274.15.10395 [DOI] [PubMed] [Google Scholar]
- Deepti, G. , Sunita, B. M. , Renu, S. , Sudha, K. , Ratna, D. P. , Jyotsna, V. , Ishwar, C. V. (2015). Identification of mutations, genotype‐phenotype correlation and prenatal diagnosis of maple syrup urine disease in Indian patients. European Journal of Medical Genetics, 58, 471–478. 10.1016/j.ejmg.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Fisher, C. W. , Fisher, C. R. , Chuang, J. L. , Lau, K. S. , Chuang, D. , & Cox, R. P. (1993). Occurrence of a 2‐bp (AT) Deletion Allele and a Nonsense (G‐to‐T) Mutant Allele at the E2 (DBT) Locus of Six Patients with Maple Syrup Urine Disease: Multiple‐Exon Skipping as a Secondary Effect of the Mutations. American Journal of Human Genetics, 52, 414–424. [PMC free article] [PubMed] [Google Scholar]
- Henneke, M. , Flaschker, N. , Helbling, C. , Müller, M. , Schadewaldt, P. , Gärtner, J. , & Wendel, U. (2003). Identification of twelve novel mutations in patients with classic and variant forms of maple syrup urine disease. Human Mutation, 22(5), 417–417. 10.1002/humu.9187 [DOI] [PubMed] [Google Scholar]
- Herring, W. J. , Litwer, S. , Weber, J. L. , & Danner, D. J. (1991). Molecular genetic basis of maple syrup urine disease in a family with two defective alleles for branched chain acyltransferase and localization of the gene to human chromosome I. American Journal of Human Genetics, 48, 342–350. [PMC free article] [PubMed] [Google Scholar]
- Huang, C. H. , Cheng, G. J. , Reid, M. E. , & Chen, Y. (1999). Rhmod syndrome: A family study of the translation‐initiator mutation in the Rh50 glycoprotein gene. American Journal of Human Genetics, 64, 108–117. 10.1086/302215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz, F. , Al‐Mostafa, A. , Allama, R. , Ramzan, K. , Nada, A. T. , & Asma, I. T. (2017). Twenty novelmutations in BCKDHA, BCKDHB and DBT genes in a cohort of 52 Saudi Arabian patients with maple syrup urine disease. Molecular Genetics and Metabolism Reports, 11, 17–23. 10.1016/j.ymgmr.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstin, G. , Ali, D. , Turgay, C. , Serap, H. K. S. , Gülden, F. G. , Mübeccel, D. , … Udo, W. (2009). Molecular genetics of maple syrup urine disease in the Turkish population. The Turkish Journal of Pediatrics, 51(2), 97–102. [PubMed] [Google Scholar]
- Li, W. , Meng, X. , Wang, W. , Lv, J. , Sun, Y. , Lv, Y. , … Song, D. (2018). Silico analysis of a novel mutation c.550delT in a Chinese patient with maple syrup urine disease. Clinical Case Reports, 6, 1989–1993. 10.1002/ccr3.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui, D. Y. , Chu, X. , Liu, R. H. , Sun, Y. , Kong, Q. X. , & Li, Q. B. (2019). Paroxysmal spasticity of lower extremities as the initial symptom in two siblings with maple syrup urine disease. Molecular Medicine Reports, 19, 4872–4880. 10.3892/mmr.2019.10133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary, A. D. C. , Marilyn, A. T. , Cynthia, P. C. , Esphie, G. D. F. , Judy, S. M. , Cristine, P. L. , & Leslie, M. M. D. (2016). Plasma amino acid and urine organic acid profiles of Filipino patients with maple syrup urine disease (MSUD) and correlation with their neurologic features. Molecular Genetics and Metabolism Reports, 9, 46–53. 10.1016/j.ymgmr.2016.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, I. , Nobukuni, Y. , Mitsubuchi, H. , Indo, Y. , Endo, F. , Asaka, J. , & Harada, A. (1990). A T‐to‐A substitution in the E1α subunit gene of the branched‐chain α‐ketoacid dehydrogenase complex in two cell lines derived from Menonite maple syrup urine disease patients. Biochemmical and Biophysical Research Communications, 172, 646–651. 10.1016/0006-291X(90)90723-Z [DOI] [PubMed] [Google Scholar]
- Nellis, M. M. , & Danner, D. J. (2001). Gene preference in maple syrup urine disease. American Journal of Human Genetics, 68(1), 232–237. 10.1086/316950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H. D. , Lee, D. H. , Hong, Y. H. , Kang, D. H. , Lee, Y. K. , Song, J. H. , … Lee, Y. W. (2011). Three Korean patients with maple syrup urine disease: Four novel mutations in the BCKDHA gene. Annals of Clinical & Laboratory Science, 41(2), 167–173. [PubMed] [Google Scholar]
- Quental, S. , Macedo‐Ribeiro, S. , Matos, R. , Vilarinho, L. , Martins, E. , Teles, E. L. , … Prata, M. J. (2008). Molecular and structural analyses of maple syrup urine disease and identification of a founder mutation in a Portuguese Gypsy community. Molecular Genetics and Metabolism, 94, 148–156. 10.1016/j.ymgme.2008.02.008 [DOI] [PubMed] [Google Scholar]
- Shimomura, Y. , Kuntz, M. J. , Suzuki, M. , Ozawa, T. , & Harris, R. A. (1988). Monovalent cations and inorganic phosphate alter branchedchain a‐ketoacid dehydrogenase‐kinase activity and inhibitor sensitivity. Archives of Biochemistry and Biophysics, 266, 210–218. [DOI] [PubMed] [Google Scholar]
- Singer, S. J. (1990). The structure and insertion of integral protein in membranes. Annual Review of Cell Biology, 6, 247–296. 10.1016/0003-9861(88)90252-4 [DOI] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , … Cooper, D. N. (2017). The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136(6), 665–677. 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoros, G. , Jacinta, L. C. , Max, W. R. , Goula, S. , Mark, K. , David, T. C. , & Anthi, D. (2009). Maple syrup urine disease in Cypriot families: Identification of three novel mutations and biochemical characterization of the p.Thr211Met mutation in the E1alpha subunit. Genetic Testing and Molecular Biomarkers, 13(5), 657–664. 10.1089/gtmb.2009.0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Medical Association . (2013). World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. Journal of the American Medical Association, 310, 2191–2194. [DOI] [PubMed] [Google Scholar]
- Yunus, Z. M. , Kamaludin, D. P. A. , Mamat, M. , Choy, Y. S. , & Ngu, L. H. (2012). Clinical and biochemical profiles of maple syrup urine disease in Malaysian children. JIMD Reports, 5, 99–107. 10.1007/8904_2011_105 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.