

Abstract
Background
GLE1 (GLE1, RNA Export Mediator, OMIM#603371) variants are associated with severe autosomal recessive motor neuron diseases, that are lethal congenital contracture syndrome 1 (LCCS1, OMIM#253310) and congenital arthrogryposis with anterior horn cell disease (CAAHD, OMIM#611890).
The clinical spectrum of GLE1‐related disorders has been expanding these past years, including with adult‐onset amyotrophic lateral sclerosis (ALS) GLE1‐related forms, especially through the new molecular diagnosis strategies associated with the emergence of next‐generation sequencing (NGS) technologies. However, despite this phenotypic variability, reported congenital or ALS adult‐onset forms remain severe, leading to premature death.
Methods
Through multidisciplinary interactions between our Neuropediatric and Medical Genetics departments, we were able to diagnose two siblings presenting with congenital disorder, using an NGS approach accordingly to the novel French national recommendations.
Results
Two siblings with very similar clinical features, meaning neuromuscular disorder of neonatal onset with progressive improvement, were examined in our Neuropediatrics department. The clinical presentation evoked initially congenital myopathy with autosomal recessive inheritance. However, additional symptoms such as mild dysmorphic features including high anterior hairline, downslanted palpebral fissures, anteverted nares, smooth philtrum with thin upper‐lip, narrow mouth and microretrognathia or delayed expressive language and postnatal growth retardation were suggestive of a more complex clinical presentation and molecular diagnosis. Our NGS approach revealed an unexpected molecular diagnosis for these two siblings, meaning the presence of the homozygous c.1808G>T GLE1 variant.
Conclusions
We here report the mildest phenotype ever described, in two siblings carrying the homozygous c.1808G>T GLE1 variant, further widening the clinical spectrum of GLE1‐related diseases. Moreover, by reflecting current medical practice, this case report confirms the importance of establishing regular multidisciplinary meetings, essential for discussing such difficult clinical presentations to finally enable molecular diagnosis, especially when NGS technologies are used.
Keywords: congenital, GLE1, mild, mutation, myopathy, NGS
GLE1 variants have initially been associated to lethal congenital autosomal recessive motor neuron diseases and later‐onset ALS forms, and recently to less severe clinical presentations. We report the mildest phenotype ever described, in two siblings carrying a homozygous c.1808G>T GLE1 variant, further widening the clinical spectrum of GLE1‐related diseases.

Abbreviations
- ALS
Amyotrophic Lateral Sclerosis
- AMC
Arthrogryposis Multiplex Congenita
- CAAHD
Congenital Arthrogryposis with Anterior Horn Cell Disease
- HGVS
Human Genome Variation Society
- IP6
Inositol hexakisPhosphate
- LAAHD
Lethal Arthrogryposis with Anterior Horn Cell Disease
- LCCS1
Lethal Congenital Contracture Syndrome 1
- mRNP
messenger RiboNucleoProtein
- NGS
Next‐Generation Sequencing
1. INTRODUCTION
GLE1 (GLE1, RNA Export Mediator, OMIM#603371) variants are associated with severe autosomal recessive motor neuron diseases, that are lethal congenital contracture syndrome 1 (LCCS1, OMIM#253310) and lethal arthrogryposis with anterior horn cell disease (LAAHD) (Mäkelä‐Bengs et al., 1998; Nousiainen et al., 2008).
The clinical spectrum of GLE1‐related disorders has been expanding these past years, considering new molecular diagnosis strategies related to the emergence of next‐generation sequencing (NGS) technologies.
Indeed, description of cases with survival beyond the perinatal period broaden the clinical spectrum and led to the nomenclature modification of LAAHD to congenital arthrogryposis with anterior horn cell disease (CAAHD, OMIM#611890) (Paakkola et al., 2018; Said et al., 2017; Smith et al., 2017; Tan et al., 2017). Therefore, it appears more accurate to define GLE1‐related disorders under a larger designation of arthrogryposis multiplex congenita (AMC) (Smith et al., 2017).
Interestingly, the clinical spectrum of GLE1‐related variants is not only congenital but also associated with adult‐onset amyotrophic lateral sclerosis (ALS) (Aditi, Glass, Dawson, & Wente, 2016; Kaneb et al., 2015). This is not surprising as the GLE1 codes for two isoforms (hGLE1A and hGLE1B) with multiple independent roles, from nuclear export mRNA regulation to initiation and termination of translation (Alcázar‐Román, Bolger, & Wente, 2010; Bolger, Folkmann, Tran, & Wente, 2008; Bolger & Wente, 2011; Folkmann et al., 2013; Kaneb et al., 2015; Murphy & Wente, 1996).
Indeed, GLE1 plays an essential role in RNA‐dependent DEAD‐box ATPases modulations implicated in messenger ribonucleoprotein (mRNP) complexes regulation, thus having a critical effect in mRNA’s processing (Jarmoskaite & Russell, 2014). Over the past years, numerous genes encoding mRNP components and regulators were associated with ALS such as TAR DNA‐binding protein (TARDBP) (Sreedharan et al., 2008), Fused in Sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009), hnRNPA1 and hnRNPA2 (Kim et al., 2013).
Therefore, it is comprehensible that variants of such genes, by playing a direct or indirect upstream role in mRNA expression, may have an impact on several underlying pathophysiological mechanisms logically displaying phenotypic heterogeneity.
GLE1 variants seem to make no exception to this rule with variable effects on motor neurons resulting in a large phenotypical spectrum.
Nevertheless, despite this phenotypic variability, reported congenital or ALS adult‐onset forms remain severe, leading to premature death. Here, we describe two siblings with a much milder and atypical GLE1‐associated phenotype, combining moderate cognitive impairment and neuromuscular impairment (initially considered as a congenital myopathy).
2. METHODS
Through multidisciplinary interactions between the Neuropediatric and Genetics departments we included DNA of two affected siblings for genetic analysis, using NGS approach accordingly to the novel French national recommendations (Krahn et al., 2019).
This family gave informed consent, according to the Declaration of Helsinki, for molecular diagnosis of all four individuals explored (the two asymptomatic parents and two affected siblings). We also obtained consent for medical publication (including pictures of the siblings).
Genetic analysis consisted in clinical exome sequencing using the ClearSeq Inherited Disease Panel (Agilent technologies, CA, USA) on an Ion Proton platform (ThermoFisher Scientific, CA, USA). Sequencing data interpretation was initially focused on 44 genes associated with neuromuscular disorders (Supporting Information) (Kaplan & Hamroun, 2013), before extending analysis to other lists of genes following the novel French national recommendations (Krahn et al., 2019).
NGS findings were systematically confirmed, as well as variant familial segregation analysis performed by targeted Sanger sequencing on a 3500XL Genetic Analyzer® (ThermoFisher Scientific).
For the sequence variant nomenclature, we followed the Human Genome Variation Society (HGVS) recommendations and used the GLE1 transcript reference NM_001003722.1.
3. RESULTS
The two affected siblings are born from healthy Caucasian (with Flemish origin) consanguineous parents (second cousins). Patient II.1 (Figure 1a,b) was born at 41 week of gestation with normal birth parameters: weight: 3,590 g (50th centile), length: 54 cm (95th centile) and head circumference: 36.5cm (81st centile). Neonatal adaptation was normal (APGAR score: 10–10). He had clenched fists with adductus thumbs and hypomobility and slow spontaneous movement. Within the first month of life, he presented feeding difficulties that improved with thickened food. In the first year of life, flexion contractures of his upper and lower limbs were noticed. He could stand at 12 months and was able to walk at 3 years old. Expressive language was also delayed as he said only few words at 3 years of age.
Figure 1.
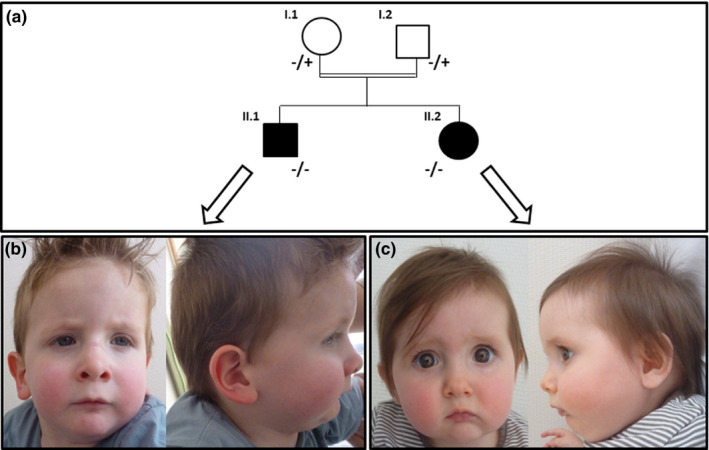
(a) Pedigree and familial segregation analysis of c.1808G>T [p.(Arg603Leu)] GLE1 variant. (+) indicates non mutated allele and (‐) indicates mutated allele for the c.1808G>T [p.(Arg603Leu)] GLE1 variant. (b) Pictures face and profile of patient II.1 at 30 months old. (c) Pictures face and profile of patient II.2 at 10 months old. The two siblings have mild shared dysmorphic features including high anterior hairline, downslanted palpebral fissures, anteverted nares, smooth philtrum with thin upper‐lip, narrow mouth and microretrognathia.
At the last examination, he was 6 years and 10 months old. He weighed 16.4 kg and was 102.7 cm tall (both < −2 standard deviation). He had postnatal growth retardation that has started since he was 15 months old. He had no microcephaly: his head circumference was 51 cm (−1 standard deviation). He had mild dysmorphic features including high anterior hairline, downslanted palpebral fissures, anteverted nares, smooth philtrum with thin upper‐lip, narrow mouth, and microretrognathia. He walked with triple lower limb joints contracture but sometimes fell. He had peroneal muscle atrophy, patellar hyperreflexia, and hyperlordosis. He was able to say short sentences. He wore glasses for hypermetropia.
Many complementary investigations were performed. He had normal brain MRI scan with spectroscopy at 2 years old. Electroneuromyography (EMG) at 2.5 years showed rich EMG signals reflecting a myogenic pattern but also associated with a neurogenic component (decreased nerve conduction velocity and amplitude, especially for peroneal nerves). Creatine kinase level was normal (79 UI/L). Specific assays for diagnosis of mucopolysaccharidoses were normal including skeletal X‐ray that has been done because of hepatomegaly (which disappeared spontaneously after 3 years old). Skeletal muscle biopsy at 3.5 years old evidenced no specific abnormalities: few fibers with vacuolation on electron microscopy and mild increase of PAS positive material. Other analyses were normal, in particular immunohistochemistry. Peripheral blood karyotyping was normal.
Patient II.2 (Figure 1a,c), the younger sister of the propositus, was born at 39 weeks of gestation, after uneventful pregnancy, with normal birth parameters: weight 2,730 g (11st centile), length 50 cm (68th centile) and head circumference 35cm (75th centile). Neonatal adaptation was normal (APGAR score: 10–10). During neonatal period, nystagmus and vertical talus feet have been observed with improvement after few weeks. At 13 months old, she had thoracolumbar kyphosis, preserved deep tendon reflexes and hips movement disability. Two short respiratory arrests have been reported at 6 months and 12 months old due to swallowing disorders. She quickly recovered and improved with swallow therapy.
At last examination, she was 4 years old. She weighed 12 kg (< −2 standard deviation) and was 92.6 cm tall (< −2 standard deviation). She had no microcephaly: her head circumference was 48.5 cm (normal). She was able to walk on all fours, to stand and to walk with support. She could say small sentences with good vocabulary. She also wore glasses for hypermetropia.
Her clinical presentation was similar to her brother regarding lower limb: triple lower limb joints contracture, patellar hyperreflexia but no Achilles tendon reflexes and kyphosis aspect. She also had dysmorphic features similar to her brother (Figure 1b,c). Cardiac ultrasound and examination were normal. Array‐CGH was performed and did not evidence any anomaly. Fewer investigations have been performed on patient II.2 as exhaustive analyses have been done for patient II.1.
In summary, the two siblings had very similar clinical features: neuromuscular disorder of neonatal onset with progressive improvement. This clinical presentation evoked congenital myopathy with autosomal recessive inheritance.
After the first genetic testings (karyotype and array‐CGH), we performed targeted sequencing of 44 genes associated with neuromuscular disorders (Kaplan & Hamroun, 2013) in patient II.1 (propositus), following our diagnosis strategy (Supporting information). This analysis did not reveal any convincing molecular diagnosis.
However, considering the atypical clinical presentation, we decided in agreement with the clinicians to broaden the molecular analysis. This strategy follows the recent and novel NGS strategy recommendations (Krahn et al., 2019). A multidisciplinary team meeting allowed discussing the clinical presentation and regular follow‐up data of the two siblings: diagnosis of arthrogryposis was evoked. Indeed, patients were both born with some arthrogryposis features as clenched fists with adductus thumbs, hypomobility and/or vertical talus feet. Therefore, by focusing on the appropriate genes list, that is the "Fœtal and Neonatal Arthrogryposes ‐ Unique exhaustive genes list" (Krahn et al., 2019), we identified an homozygous GLE1 variant, c.1808G>T [p.(Arg603Leu)], located in exon 13, for patient II.1. Segregation analysis showed that it is inherited from healthy heterozygous parents and that patient II.2 also carries this homozygous variant (Figure 1a). To our knowledge, this variant has never been reported at homozygous status in individuals. It has a very low frequency in the general population (allele frequency = 3.98e‐6; GnomAD, http://gnomad.broadinstitute.org/, 30 June 2019) and it is predicted to be damaging by several bioinformatics tools [PolyPhen2 (Adzhubei et al., 2010), Mutation Taster (Schwarz, Cooper, Schuelke, & Seelow, 2014) and UMD‐predictor (Salgado et al., 2016)].
According to the ACMG classification, it is a likely pathogenic (class 4) variant (Richards et al., 2015). Interestingly, this specific c.1808G>T GLE1 variant has been reported as pathogenic once in patients database [Clinvar (Landrum et al., 2018)] and recently in the literature, associated with another likely pathogenic GLE1 variant on the second allele (compound heterozygous status) (Tan et al., 2017). The phenotype associated with this initial description is also a moderate clinical presentation but not as mild as the one we report for these two siblings (Table 1).
Table 1.
Clinical features for GLE1 variants recently associated with congenital phenotypes
| Clinical features | Smith et al., 2017 | Said et al., 2017 | Paakkola et al. 2018 | Tan et al., 2017 | Our patients | ||||
|---|---|---|---|---|---|---|---|---|---|
| Case 1 | Case 2 | A | B | Patient 1 | Patient 2 | One patient | Patient II.1 | Patient II.2 | |
| Age onset | NR | Prenatal | Birth | Birth | Birth | Birth | Prenatal | Birth | Birth |
| Birth weight (g) | NR | 2,955 | 2,480 | 2,930 | 3,210 | 3,210 | 2,675 (36gw) | 3,590 | 2,730 |
| Birth head circumference (cm) | NR | NR | 35 | 34.5 | 36 | 36.5 | 33.5 | 36.5 | 35 |
| Neonatal respiratory distress | + | + | + | NR | + | + | + | − | − |
| Gastrostomy tube feeding | − | + | − | − | + | + | + | − | − |
| Dysmorphic features | + | + | + | + | + | + | + | + | + |
| Head circumference outcome | NR | Microcephaly | NR | Normal | NR | NR | Normal | Normal | Normal |
| Walk | NR | − |
+ (3y8m) |
− | NR | NR | − | abnormal | abnormal |
| Language | NR | Sign language | + | + | NR | NR | Sign language | Language delay | Language delay |
| Intercurrent disease | NR | NR | NR | Frequent pneumonia | Pneumonia | Pneumonia, progressive neurological symptoms | NR | − | − |
| Outcome Age at last examination | Died at 2 w |
Improvement 12 y |
Improvement 4 y 8 m |
Died 4 y |
Died 6 m |
Died 6 m |
Improvement 26 m |
Improvement 5 y |
Improvement 3 y |
| GLE1 variant (NM_001003722.1) |
Compound heterozygous c.100−7_100−3delTCTCT c.1882−2A>G |
Homozygous c.2078C>T |
Compound heterozygous c.1706G>A c.1750C>T |
Homozygous c.2015T>C |
Compound heterozygous c.1808G>T c.1997G>T |
Homozygous c.1808G>T |
|||
| Muscle biopsy | − | No specific abnormalities | NR | NR |
Neurogenic atrophy Atrophic and hypertrophic muscle fibers |
Neurogenic atrophy Atrophic and hypertrophic muscle fibers |
No specific abnormalities | NR | |
Abbreviations: gw, gestational week; m, months; NR, not reported; w, weeks; y, years.
4. DISCUSSION
We here describe a relatively moderate congenital phenotype associated with the homozygous c.1808G>T [p.(Arg603Leu)] GLE1 genotype, for two siblings. To date, only nine patients have been reported in the literature for GLE1‐associated congenital disorders.
The phenotype associated with this c.1808G>T homozygous GLE1 genotype, could even be considered as the mildest described to date, based on literature reports (Table 1) (Paakkola et al., 2018; Said et al., 2017; Smith et al., 2017).
Indeed, GLE1 variants were initially associated with severe autosomal recessive motor neuron diseases either for lethal congenital forms (Mäkelä‐Bengs et al., 1998; Nousiainen et al., 2008) or later‐onset ALS forms (Aditi, Glass, Dawson, & Wente, 2016; Kaneb et al., 2015), leading in both cases to premature death. Even if the recently expanding GLE1 phenotype spectrum includes less severe clinical presentations, the case we report here, associated with a c.1808G>T [p.(Arg603Leu)] GLE1 homozygous genotype, constitutes the mildest phenotype reported to date, in comparison with previously reported congenital moderate phenotypes (Table 1) (Paakkola et al., 2018; Said et al., 2017; Smith et al., 2017; Tan et al., 2017).
Interestingly, this c.1808G>T GLE1 variant has already been described in a compound heterozygous association with the c.1997G>T [p.(Gly666Val)] variant with a milder phenotype as usually observed for GLE1 variants (Tan et al., 2017).
Tan and colleagues reasonably hypothesized that the c.1997G>T [p.(Gly666Val)] GLE1 variant could be responsible for this milder phenotype as the p.Gly666 residue is present only in the hGLE1B isoform. However, we report here a milder phenotype than the one described by Tan and colleagues for two siblings with the homozygous c.1808G>T GLE1 variant. Our report confirms that the c.1808G>T [p.(Arg603Leu)] GLE1 variant seems also associated with a moderate pathogenic effect leading to a milder phenotype. This p.(Arg603Leu) variant is located near the carboxy‐terminus end of the protein, in the region of the GLE1 protein implicated in inositol hexakisphosphate (IP6) binding domain. Therefore, future research should focus on the possible impact of this specific GLE1 variant on the interaction with IP6 and the potential consequences on mRNA export and translation termination (Alcázar‐Román, Bolger, & Wente, 2010; Alcázar‐Román, Tran, Guo, & Wente, 2006).
Furthermore, this case report also emphasizes the utility of NGS technologies to elucidate atypical and/or overlapping phenotypes.
Indeed, as this clinical case demonstrates, a well‐established gene panel NGS approach associated with a sequential gene filtering clinically oriented strategy can be sufficient to resolve these types of complex diagnosis (Krahn et al., 2019).
Moreover, by reflecting current medical practice, this case report confirms the importance of establishing regular multidisciplinary meetings, essential for discussing such difficult clinical presentations to finally enable molecular diagnosis. Thus, leading sometimes to the reassessment of the initial clinical indication, as in our context the widening of GLE1‐associated phenotypical indication to congenital slowly progressing muscle disease, as previously suggested by Tan and colleagues (Tan et al., 2017).
This description also reveals the importance of taking into account the mutational GLE1 combination to better comprehend clinical presentation as well as the severity and evolution of the disease. In the present case, the homozygous c.1808G>C GLE1 genotype seems to be associated with a milder phenotype, thus further expanding the GLE1 clinical spectrum.
Finally, to complete Smith and colleagues’ hypothesis of a single entity for AMC (Smith et al., 2017), involving the GLE1 mutational combination evaluation, we believe that the influence of possible modifier genes should also be considered to explain the GLE1‐associated phenotypical heterogeneity, from extremely severe lethal forms to milder clinical presentations such as the one described here.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
AUTHOR CONTRIBUTION
CDM, FAu, FAl, CB, NP, and BC performed a history and physical exam on the patient. MC, CDM, FAu, FR, MB, NL, MK, and BC performed literature review and assisted in manuscript preparation. MC, MB, NL, and MK contributed to data acquisition and analysis. MC, MK, MB belong to the Translational Neuromyology team within MMG, where this study took place. All authors read and approved the final manuscript.
ETHICAL APPROVAL
Written informed consent obtained by parent/guardian of patients.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank Karine Bertaux, Cécile Mouradian, Pierre Cacciagli, Jean‐Pierre Desvignes, David Salgado, and Christophe Béroud for their contribution to this work. We would like to thank GIPTIS (Genetics Institute for Patients, Therapies Innovation and Science) for its generous support. We also wish to thank the patients, families and health professionals whose participation, through the FILNEMUS and Anddi‐Rares networks, made possible this research.
Cerino M, Di Meglio C, Albertini F, et al. Extension of the phenotypic spectrum of GLE1‐related disorders to a mild congenital form resembling congenital myopathy. Mol Genet Genomic Med. 2020;8:e1277 10.1002/mgg3.1277
Funding information
This study was supported by GIPTIS (Genetics Institute for Patients, Therapies Innovation & Science); National Research Agency, Investment for the Future (Grant No. ANR‐10‐INBS‐09); AFM‐Telethon (under grant agreement #19272); Fondation Maladies Rares; FHU A*MIDEX project MARCHE n.ANR‐11‐IDEX‐001‐02 funded by the “Investissement d’avenir” French government program, managed by the French National Research Agency (ANR); APHM; INSERM and the European Community Seventh Framework Programme Grant FP7/2007–2013 (NEUROMICS; Grant No. 2012–305121).
DATA AVAILABILITY STATEMENT
All relevant data are within the paper and its Supporting Information file. Due to ethical and legal restrictions from Assistance Publique – Hôpitaux de Marseille related to protecting participant privacy, all additional data are available upon request pending ethical approval. Please submit all requests to initiate the data access process to the corresponding author, Dr. Cerino (mathieu.cerino@univ‐amu.fr).
REFERENCES
- Aditi, Glass, L. , Dawson, T. R. , & Wente, S. R. (2016). An amyotrophic lateral sclerosis‐linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Advances in Biological Regulation, 62, 25–36. 10.1016/j.jbior.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcázar‐Román, A. R. , Bolger, T. A. , & Wente, S. R. (2010). Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. The Journal of Biological Chemistry, 285(22), 16683–16692. 10.1074/jbc.M109.082370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcázar‐Román, A. R. , Tran, E. J. , Guo, S. , & Wente, S. R. (2006). Inositol hexakisphosphate and Gle1 activate the DEAD‐box protein Dbp5 for nuclear mRNA export. Nature Cell Biology, 8(7), 711–716. 10.1038/ncb1427 [DOI] [PubMed] [Google Scholar]
- Bolger, T. A. , Folkmann, A. W. , Tran, E. J. , & Wente, S. R. (2008). The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell, 134(4), 624–633. 10.1016/j.cell.2008.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, T. A. , & Wente, S. R. (2011). Gle1 is a multifunctional DEAD‐box protein regulator that modulates Ded1 in translation initiation. The Journal of Biological Chemistry, 286(46), 39750–39759. 10.1074/jbc.M111.299321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann, A. W. , Collier, S. E. , Zhan, X. , Aditi, , Ohi, M. D. , & Wente, S. R. (2013). Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell, 155(3), 582–593. 10.1016/j.cell.2013.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite, I. , & Russell, R. (2014). RNA helicase proteins as chaperones and remodelers. Annual Review of Biochemistry, 83, 697–725. 10.1146/annurev-biochem-060713-035546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneb, H. M. , Folkmann, A. W. , Belzil, V. V. , Jao, L.‐E. , Leblond, C. S. , Girard, S. L. , … Dion, P. A. (2015). Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Human Molecular Genetics, 24(5), 1363–1373. 10.1093/hmg/ddu545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, J.‐C. , & Hamroun, D. (2013). The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscular Disorders: NMD, 23(12), 1081–1111. [DOI] [PubMed] [Google Scholar]
- Kim, H. J. , Kim, N. C. , Wang, Y.‐D. , Scarborough, E. A. , Moore, J. , Diaz, Z. , … Taylor, J. P. (2013). Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature, 495(7442), 467–473. 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn, M. , Biancalana, V. , Cerino, M. , Perrin, A. , Michel‐Calemard, L. , Nectoux, J. , … Cossée, M. (2019). A National French consensus on gene lists for the diagnosis of myopathies using next‐generation sequencing. European Journal of Human Genetics: EJHG, 27(3), 349–352. 10.1038/s41431-018-0305-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski, T. J. , Bosco, D. A. , LeClerc, A. L. , Tamrazian, E. , Vanderburg, C. R. , Russ, C. , … Brown, R. H. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323(5918), 1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä‐Bengs, P. , Järvinen, N. , Vuopala, K. , Suomalainen, A. , Ignatius, J. , Sipilä, M. , … Peltonen, L. (1998). Assignment of the disease locus for lethal congenital contracture syndrome to a restricted region of chromosome 9q34, by genome scan using five affected individuals. American Journal of Human Genetics, 63(2), 506–516. 10.1086/301968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, R. , & Wente, S. R. (1996). An RNA‐export mediator with an essential nuclear export signal. Nature, 383(6598), 357–360. 10.1038/383357a0 [DOI] [PubMed] [Google Scholar]
- Nousiainen, H. O. , Kestilä, M. , Pakkasjärvi, N. , Honkala, H. , Kuure, S. , Tallila, J. , … Peltonen, L. (2008). Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nature Genetics, 40(2), 155–157. 10.1038/ng.2007.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakkola, T. , Vuopala, K. , Kokkonen, H. , Ignatius, J. , Valkama, M. , Moilanen, J. S. , … Uusimaa, J. (2018). A homozygous I684T in GLE1 as a novel cause of arthrogryposis and motor neuron loss. Clinical Genetics, 93(1), 173–177. 10.1111/cge.13086 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee , (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said, E. , Chong, J. X. , Hempel, M. , Denecke, J. , Soler, P. , Strom, T. , … Lessel, D. (2017). Survival beyond the perinatal period expands the phenotypes caused by mutations in GLE1. American Journal of Medical Genetics. Part A, 173(11), 3098–3103. 10.1002/ajmg.a.38406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado, D. , Desvignes, J.‐P. , Rai, G. , Blanchard, A. , Miltgen, M. , Pinard, A. , … Béroud, C. (2016). UMD‐predictor: A high‐throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Human Mutation, 37(5), 439–446. 10.1002/humu.22965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Smith, C. , Parboosingh, J. S. , Boycott, K. M. , Bönnemann, C. G. , & Mah, J. K. , Care4Rare Canada Consortium , … Bernier, F. P. (2017). Expansion of the GLE1‐associated arthrogryposis multiplex congenita clinical spectrum. Clinical Genetics, 91(3), 426–430. 10.1111/cge.12876 [DOI] [PubMed] [Google Scholar]
- Sreedharan, J. , Blair, I. P. , Tripathi, V. B. , Hu, X. , Vance, C. , Rogelj, B. , … Shaw, C. E. (2008). TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 319(5870), 1668–1672. 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, Q.‐K.‐G. , McConkie‐Rosell, A. , Juusola, J. , Gustafson, K. E. , Pizoli, C. E. , Buckley, A. F. , & Jiang, Y.‐H. (2017). The importance of managing the patient and not the gene: Expanded phenotype of GLE1‐associated arthrogryposis. Cold Spring Harbor Molecular Case Studies, 3(6), pii: a002063 10.1101/mcs.a002063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance, C. , Rogelj, B. , Hortobagyi, T. , De Vos, K. J. , Nishimura, A. L. , Sreedharan, J. , … Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323(5918), 1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
All relevant data are within the paper and its Supporting Information file. Due to ethical and legal restrictions from Assistance Publique – Hôpitaux de Marseille related to protecting participant privacy, all additional data are available upon request pending ethical approval. Please submit all requests to initiate the data access process to the corresponding author, Dr. Cerino (mathieu.cerino@univ‐amu.fr).