

Abstract
Background
Cornelia de Lange syndrome (CdLS) comprises a recognizable pattern of multiple congenital anomalies caused by variants of the DNA cohesion complex. Affected individuals may display a wide range of phenotypic severity, even within the same family.
Methods
Exome sequencing and confirmatory Sanger sequencing showed the same previously described p.Arg629Ter NIPBL variant in two half‐brothers affected with CdLS. Clinical evaluations were obtained in a pro bono genetics clinic.
Results
One brother had relatively mild proportionate limb shortening; the other had complete bilateral hypogenesis of the upper arm with absence of lower arm structures, terminal transverse defects, and no digit remnants. His complex lower limb presentation included long bone deficiency and a deviated left foot. The mother had intellectual disability and microcephaly but lacked facial features diagnostic of the CdLS.
Conclusion
We describe a collaboration between a pediatrics team from a resource‐limited nation and USA‐based medical geneticists. Reports describing individuals of West Indian ancestry are rarely found in the medical literature. Here, we present a family of Afro‐Caribbean ancestry with CdLS presenting with phenotypic variability, including unusual lower limb abnormalities. The observation of this novel family adds to our knowledge of the phenotypic and molecular aspects of CdLS.
Keywords: Caribbean, Cornelia de Lange syndrome, exome sequencing
We found the same pathogenic NIPBL variant in two half‐brothers who are of Afro‐Caribbean ancestry. One of the brothers has a novel lower limb presentation.
1. INTRODUCTION
Cornelia de Lange syndrome (CDLS1, OMIM 122470) is a multisystem congenital, developmental disorder with an approximate incidence of 1 in 10,000–30,000 live births (Kline et al., 2007; Mannini, Cucco, Quarantotti, Krantz, & Musio, 2013). Dominant, typically de novo pathogenic variants in any of five different genes required for establishing the DNA cohesion complex during cell division may cause CdLS. The structural components of the cohesion complex are encoded by SMC3, SMC1A (Deardorff et al., 2007), and RAD21 (Deardorff, Wilde, et al., 2012). A histone deacetylase, HDAC8, regulates the release of the cohesion complex from the sister chromatids (Deardorff, Bando, et al., 2012). NIPBL (OMIM 608667) encodes the protein delangin, which regulates the interaction of the cohesion complex with the DNA that comprises the sister chromatids (Krantz et al., 2004).
Pathogenic variants in NIPBL typically cause the most severe presentations and contribute to about 60% of individuals with CdLS. Variants in HDAC8 cause the second most severe form of the syndrome, followed by RAD21, SMC1A, and SMC3. Various loss of function aberrations of these five genes have been found to underlie CdLS including nonsense, missense, insertions, deletions, rearrangements, and splice site mutations. A genotype‐phenotype correlation has been demonstrated for mutations of NIPBL. Gene translocations and gross deletions tend to cause the most severe phenotypic changes followed by nonsense and frameshift mutations. Gene regulatory and splicing errors are often less severe, and missense mutations are usually the least severe (Mannini et al., 2013).
Cornelia de Lange syndrome is a multisystem disorder with a variety of clinical features including developmental delay, intellectual disability, growth deficiency, microcephaly, limb defects, hypertrichosis, and characteristic facial dysmorphisms. Facial features typical of CdLS include long curly eyelashes, synophrys, depressed nasal bridge, small upturned nose, long philtrum, thin upper lip vermilion, and low‐set posteriorly angulated ears. Clinical features may vary widely in manifestation and severity among affected persons (Liu & Krantz, 2008). The range of limb defects in CdLS is wide and includes limb deficiency of a variable degree. The more severe end of the spectrum is characterized by defects involving primarily ulnar structures ranging from fifth finger hypoplasia to ulnar aplasia to absence of the forearm; structural malformations of the lower limbs are uncommon (Mehta et al., 2016).
Most individuals with CdLS inherit the disorder de novo likely because of the decreased reproductive fitness that is associated with the disorder. De novo pathogenic variants are usually seen in the NIPBL, HDAC8, and SMC1A, with affected parents reported in a small number of cases with a NIPBL mutation (Fitzpatrick & Kline, 2010). Mutations in NIPBL, RAD21, and SMC3 are autosomal dominant while HDAC8 and SMC1A are X‐linked dominant. Germline mosaicism in an unaffected parent with a mutation in NIPBL or SMC1A has been reported (Slavin et al., 2012). Additionally, somatic mosaicism has been observed in patients with variable presentations of typical CdLS (Huisman, Redeker, Maas, Mannens, & Hennekam, 2013; Nizon et al., 2016). Recurrence risk is low because of the high probability of new mutation in most reported individuals, but the possibility of germline and somatic mosaicism in a parent conveys increased risk in future pregnancies for the family (Deardorff, Noon, & Krantz, 2016; Slavin et al., 2012).
Genetic disorders are rarely diagnosed in the Caribbean because of a variety of factors including disparities of socioeconomic status, small populations among the many island nations, and geographical isolation (Roach, Warner, & Llanos, 2015). This report documents a successful collaboration between three different United States‐based clinical research groups and a team in the West Indies. Here, we present two half‐brothers with CdLS who have the same previously reported pathogenic variant in NIPBL. Their mother is presumed (but not confirmed) to have mosaicism for the variant. The observation of this family adds to our knowledge of the phenotypic and molecular aspects of CdLS.
2. CLINICAL REPORT
2.1. Individual 1
Individual 1 came to medical attention prenatally because of a discrepancy between gestational age and height of fundus in third trimester. He was born full term and was noted to be small for gestational age with a weight of 1.85 kg. He had grade I meconium at birth and his Apgar scores were 5/1, 7/5, and 8/10. His cry was absent on delivery, but he did not appear to be in distress. A day later, a cat‐like cry was noted. He did not show pallor, he was afebrile, acyanotic, and active. He had microcephaly (28.5 cm) with dysmorphic facial features including synophyrs, a depressed nasal bridge, long philtrum, low‐set ears with wide set eyes, and a wide mouth with macroglossia. He had normal breath sounds and widely separated breast buds. His reflexes and tone were normal, and the fontanelles were normotensive. The digits of his right upper limb were short, and a fully formed accessory digit was observed attached to the postaxial aspect of his right hand. The lower limbs were also short, and his left foot had a high plantar arch. His feet had a rocker bottom shape. After birth, he was feeding well, and he began to gain weight. Figure 1a,b shows the boy at an older age and the postaxial polydactyly as an infant. Over the next five years, he reportedly exhibited delay in developmental, language and motor functions. When he was five years old, a consultation with a pediatric geneticist was obtained, and the diagnosis of CdLS was suggested.
FIGURE 1.
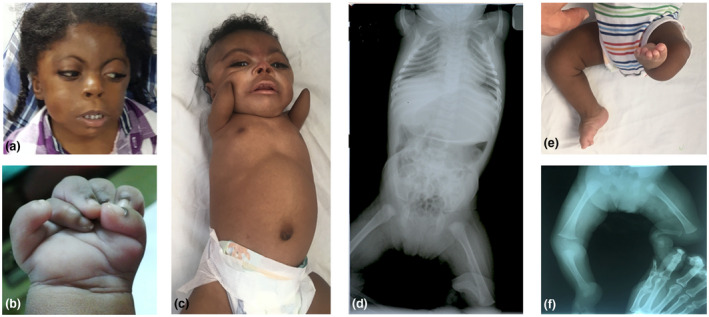
Photographs and radiographs of two individuals affected with CdLS. (a) The typical facial characteristics of CdLS of Individual 1 depicting arched eyebrows, anteverted nares, synophyrs, thin vermilion of the upper lip, and downturned corners of the mouth. (b) The postaxial polydactyly of Individual 1 from a photo taken in infancy. (c) Individual 2 with the typical facial features of CdLS and upper limb deficiency defects. (d) Plain radiograph of the whole body of Individual 2 as an infant showing the markedly shortened humeri and single bone in the lower left leg. (e) The left lower leg shortening with the deviation of the foot of Individual 2. (f) Plain radiograph of the left lower leg showing a single bone in Individual 2. CdLS, Cornelia de Lange syndrome
2.2. Individual 2
The younger half‐brother of Individual 1 was born with limb defects including symmetrical marked hypoplasia of both arms and malformations of the long bones of the legs. His birth weight, length and head circumference are not available. When he was 9 months old, developmental delay was evident and at this time an evaluation by a medical geneticist was obtained. He had the distinct facial features typical of CdLS including synophyrs. Limb deficiency defects included symmetrical bilateral upper tapered pointed upper arms with no visible elbow or forearm bones. (Figure 1c). The terminal transverse defects of both arms consisted of absence of digits, or digit remnants, and ended approximately two‐thirds of the way down the humeri (Figure 1d). His lower limb defects included a left‐sided single bone of the lower leg that was initially suggestive of a fibula (i.e., tibial agenesis), but the single bone is apparently thicker than a fibula, and it is likely a bowed and shortened tibia. In that case this long bone deficiency would represent fibular agenesis. His left foot deviated to the tibial‐preaxial side (Figure 1e,f).
2.3. Individual 3
The mother of the two affected brothers was a 35‐year‐old woman with moderate to severe intellectual disability. She had short stature, microcephaly (OFC 48 cm), and a distinctive facial appearance (Figure 2) including upturned nares. These features were not diagnostic for CdLS except in the context of her two sons. Extension and supination of both elbows showed limitation that were suggestive of a radial head dislocation or radioulnar synostosis, but these were not evident on plain radiography. She had a prominent elbow point and clinodactyly, but no brachydactyly. The mother's obstetric history was significant for a history of at least four miscarriages or stillbirths (Figure 3). Reportedly, she had multiple partners including different fathers for both affected individuals. We were told that she had two unaffected children, but they were not evaluated.
FIGURE 2.

The mother of Individuals 1 and 2. She has microcephaly and distinctive facial features but not diagnostic of CdLS. CdLS, Cornelia de Lange syndrome
FIGURE 3.
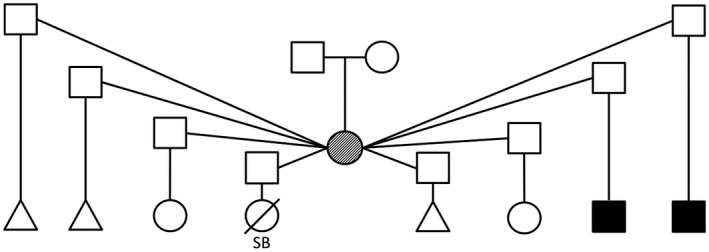
Family history is significant and shows four pregnancy losses, two unaffected children, and two children affected with Cornelia de Lange syndrome. Triangles describe spontaneous abortions, the partially filled symbol indicates intellectual disability and microcephaly, and filled symbols represent individuals affected with Cornelia de Lange syndrome
2.4. Genomic analysis
Cytogenomic microarray analysis (CMA) was offered to Individual 1 soon after he was born because of his presenting features. The mother was also tested because of her intellectual disability. No copy number variants (CNV) were found in the mother. A deletion of uncertain clinical significance of approximately 0.125 Mb was found in Individual 1 with 19 probes from this chip, hg19 arr 9p24.1p23 (8903382–9028184)x1. This deletion includes exon 7 in the 5′‐untranslated region of PTPRD and was not detected on the maternal array. However, this deletion is not known to be associated with disease and has been previously observed in healthy individuals (http://dgv.tcag.ca) in the Database of Genomic Variants and in an internal database.
Exome sequencing analysis of Individual 2 revealed a heterozygous variant in NIPBL (NM_015384.4) c.1885C>T; p.Arg629Ter. This variant was previously reported in a cohort of CdLS patients in Japan (Miyake et al., 2005), but photographic documentation of the facial phenotype was not provided. No other pathogenic, or likely pathogenic variants were found that might explain the relatively severe presentation in Individual 2.
To confirm the presence of the variant in Individual 2 and to determine if the sibling (Individual 1) carried the same variant, PCR using standard conditions was performed on genomic DNA with intronic primers from NIPBL, which flank the variant identified by exome sequencing: NIPBL‐F‐5′‐TGCACCTGTTTCTGTTCTTCA‐3′ and NIPBL‐R‐5′‐AGGCTCAACTATGGTGCTCTC‐3′. PCR products were treated with ExoSAP‐IT (USB, Cleveland, OH) to digest primers. Sanger sequencing (Genewiz.com) and analysis of electropherograms were done using standard protocols to confirm the variant in Individual 2 and demonstrated its presence in Individual 1.
The c.1885C>T; p.Arg629Ter variant was not found in Individual 3 by exome sequencing of DNA obtained from blood, urine, or saliva with a mean coverage depth of 70.20, 64.48, and 64.62, respectively. Manual review of the alignments failed to identify any reads harboring the mutant allele under the context of 43, 79, and 72 reference reads in the blood, urine, and saliva, respectively. The fathers of the two affected individuals were not available for testing, and we were unable to obtain exome sequencing from Individual 1.
3. DISCUSSION
Here, we describe two male half‐siblings with CdLS due to a previously reported pathogenic variant in NIPBL, p.Arg629Ter. The mother of these two affected individuals has intellectual disability and a complex obstetric history that includes multiple miscarriages and stillbirths. The variant was not detected in the mother in DNA obtained from blood, saliva, or urine. Although not proven, we strongly suspect that she has mosaicism for the NIPBL pathogenic variant because she had two children affected with the same variant who were reportedly fathered by different partners. Support for maternal mosaicism is also supplied by her overlapping phenotype (intellectual disability, distinct facial features, microcephaly, and short stature). Unfortunately, neither father is available for genetic testing so we cannot prove that they are unrelated. An additional weakness in our study is that since we cannot obtain either an exome analysis for Individual 1, or test the fathers, we are unable to confidently demonstrate that the two affected individuals are only half‐brothers.
Both germline and somatic mosaicism have been reported in families and persons with CdLS with variable phenotype (Huisman et al., 2013; Nizon et al., 2016; Slavin et al., 2012). Slavin et al., reported 12 families where there were 2 or more offspring with CdLS, a documented change in either NIPBL or SMC1A, and unaffected parents, strongly suggesting germline mosaicism. Huisman et al., (2013) and more recently Nizon et al., (2016) documented individuals with varying degrees of the clinical diagnosis of CdLS who had pathogenic variants of NIPBL only in buccal samples and not in lymphocytes. In the Huisman et al., series, the clinical picture of those with mosaicism and those without (variant present in blood and buccal cells) was considered not different. In the Nizon et al., study, two of the three individuals with somatic mosaicism of NIPBL did not have photographs of the face available, and therefore, cannot be fully evaluated for CdLS. The mother of the two individuals we report had some features of CdLS (short stature, microcephaly, and intellectual disability) but lacked facial features typical of CdLS (see Figure 2). We would posit that she has somatic mosaicism for the NIPBL variant, but we were not able to confirm this hypothesis with exome sequencing in the available tissues.
Individual 2 has a unique limb deficiency defect of the lower limbs. It is challenging to say with certainty what the single bone seen radiographically represents (Figure 1d,f). Since postaxial deficiency of the upper limbs (ulnar hypoplasia or aplasia) is relatively common in CdLS, one could conclude that it is likely that this limb defect is postaxial, that is, fibular agenesis. Additionally, unilateral fibular aplasia in general is more common than tibial agenesis (Lewin & Opitz, 1986). However, the foot deviates to the preaxial side suggesting that the absent bone may be the tibia. The severe limb deficiency defects in individuals with NIBPL‐related CdLS are reported to be upper limb malformations ranging from ulnar aplasia to absence of the forearms (like Individual 2) to split hand defects. To our knowledge, lower limb long bone deficiency such as the aplasia seen in Individual 2 is rare in CdLS (Pié et al., 2016); usually the CdLS lower limb abnormalities consist of small feet, 2–3 toe syndactyly, and occasionally club feet. Thus, the lower limb defect described in this report is a novel observation but likely still part of the variability of limb malformation in CdLS. The polydactyly in Individual 1 was observed in 6% of reported cases in one series of CdLS (Kline et al., 2007). Additionally, polydactyly was seen in a mouse CdLS model with Nipbl mutation (Lopez‐Burks, Santos, Kawauchi, Calof, & Lander, 2016). We did not observe any other pathogenic, or likely pathogenic variants from the exome analysis of Individual 2 that might explain his more severe limb defects. Since we were unable to obtain exome sequence from Individual 1, we do not know if there are any other variants that might explain his less severe phenotype.
Here, we describe two Afro‐Caribbean half‐brothers with CdLS due to mutation of NIPBL, one of whom has a unique limb defect presentation. A recent report described the facial dysmorphology of CdLS in diverse populations to aid in diagnosis in populations other than Caucasian; however, other aspects of the CdLS phenotype were not discussed (Dowsett et al., 2019). In the Caribbean, most people with genetic disorders remain undiagnosed (Roach et al., 2015). As a result of our efforts, we have helped the local medical establishment and the extended members of this family understand the disorder affecting these two individuals. Genetic counseling, including recurrence risk estimations to other family members, was provided.
4. MATERIALS AND METHODS
4.1. Editorial policies and ethical considerations
Verbal and written consent were obtained from the mother and grandmother of the affected individuals for genetic testing, release of clinical history, and publication of photos. Due to the small population size of the Caribbean island where our patients and family reside, our IRB has requested that we do not reveal their precise nationality or geographic locations. Clinical evaluations were performed at a pro bono pediatric genetics clinic at the request of locally registered consulting pediatricians.
4.2. Cytogenomic microarray analysis
DNA was isolated from whole blood by Qiagen Puregene (Venlo, Netherlands) for Molecular testing. Extracted DNA from the patient's specimen and a previously genotyped normal same sex control individual were differentially labeled and co‐hybridized to a 180K custom Agilent oligonucleotide array, which has genome wide backbone coverage with an average resolution of 30 kb (excluding LCRs and other repetitive sequences). It also includes targeted exonic coverage of approximately 1,700 genes with an average of 4.2 probes/exon. All microdeletion and microduplication syndromes known at the time of development as well as pericentromeric and subtelomeric regions are detected. Mitochondrial genome deletions greater than 2 kb are detected. Data were analyzed with an in‐house developed software program.
4.3. DNA sequencing
We conducted exome capture on DNA isolated from peripheral blood mononuclear cells of Individual 2 and DNA obtained from blood, urine, or saliva of the mother using the Twist Human Core Exome Kit (TWIST Bioscience), guided by the manufacturer's protocols. The libraries were subsequently sequenced for 101 cycles with a paired‐end mode on the Illumina NovaSeq 6000. Raw reads were aligned to the reference human genome using the Burrows‐Wheeler Aligner (BWA) (Li & Durbin, 2009). Single‐nucleotide variants and small insertions/deletions were captured using the genome analysis tool kit (GATK) (DePristo et al., 2011). Research exome sequencing was performed on individual 2 to a mean coverage depth of 82.50. ANNOVAR (Wang, Li, & Hakonarson, 2010) and SnpEff (Cingolani et al., 2012) were used to functionally annotate the variants. Variants generated were filtered against public variant databases (dbSNP, 1000 Genomes Project, NHLBI ESP6500SI, gnomAD, and Kaviar) and an additional >5,000 exome samples previously analyzed in our database hosted at Center for Applied Genomics (CAG) at The Children's Hospital of Philadelphia with a minor allele frequency of 1% (CAG database PMID accession numbers: 31263281 and 29905864). Subsequent gene prioritization was performed based on deleterious prediction and biological relevance by referring to the OMIM and HGMD. Sanger sequencing for the NIBPL variant was subsequently performed on both of the affected individuals and the mother.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
WT and PC wrote the first draft of the manuscript. TD, BN, JCC, and EJB provided patient care and clinical characterizations. Molecular investigations were carried out by MR, JLS, SHE, DL, HH, and EJB. All authors contributed to critical review and editing of the manuscript. AKS coordinated the project from its inception and supervised all writing. All authors approved the final manuscript. The relatively large number of authors for this piece is a product of the international collaborative aspect of the project.
ETHICS STATEMENT
This study was performed in accordance with the ethical standards detailed in the Declaration of Helsinki.
ACKNOWLEDGMENTS
We thank the family for participating in this study.
Thompson W, Carey PZ, Donald T, et al. Application of exome sequencing to diagnose a novel presentation of the Cornelia de Lange syndrome in an Afro‐Caribbean family. Mol Genet Genomic Med. 2020;8:e1318 10.1002/mgg3.1318
Wayne Thompson and Patrick Z. Carey have contributed equally to this work.
Funding information
St. George's University Small Research Grant Initiative 18017.
Contributor Information
Patrick Z. Carey, Email: John.Carey@hsc.utah.edu.
Andrew K. Sobering, Email: asobering@sgu.edu.
DATA AVAILABILITY STATEMENT
The NIPBL variant described in this report had been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) previously and can be found under accession numbers RCV000146527.3 and RCV000725320.1.
REFERENCES
- Cingolani, P. , Platts, A. , Wang, L. L. , Coon, M. , Nguyen, T. , Wang, L. , … Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6(2), 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff, M. A. , Bando, M. , Nakato, R. , Watrin, E. , Itoh, T. , Minamino, M. , … Shirahige, K. (2012). HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature, 489(7415), 313–317. 10.1038/nature11316.HDAC8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff, M. A. , Kaur, M. , Yaeger, D. , Rampuria, A. , Korolev, S. , Pie, J. , … Krantz, I. D. (2007). Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. American Journal of Human Genetics, 80(3), 485–494. 10.1086/511888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff, M. A. , Noon, S. E. , & Krantz, I. D. (2016). Cornelia de Lange syndrome Adam M., Ardinger H.& Pagon R. A. (Eds.), GeneReviews®. Seattle, WA: University of Washington; 1993–2019. 10.1016/j.jhsa.2015.07.023 [DOI] [Google Scholar]
- Deardorff, M. A. , Wilde, J. J. , Albrecht, M. , Dickinson, E. , Tennstedt, S. , Braunholz, D. , … Kaiser, F. J. (2012). RAD21 mutations cause a human cohesinopathy. American Journal of Human Genetics, 90(6), 1014–1027. 10.1016/j.ajhg.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , … Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806.A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsett, L. , Porras, A. R. , Kruszka, P. , Davis, B. , Hu, T. , Honey, E. , … Krantz, I. D. (2019). Cornelia de Lange syndrome in diverse populations. American Journal of Medical Genetics, Part A, 179A(2), 150–158. 10.1002/ajmg.a.61033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick, D. , & Kline, A. (2010). In Cassidy S., & Allanson J. (Eds.), Management of genetic syndromes (3rd ed.). Hoboken, NJ: John Wiley & Sons. [Google Scholar]
- Huisman, S. A. , Redeker, E. J. W. , Maas, S. M. , Mannens, M. M. , & Hennekam, R. C. M. (2013). High rate of mosaicism in individuals with Cornelia de Lange syndrome. Journal of Medical Genetics, 50(5), 339–344. 10.1136/jmedgenet-2012-101477 [DOI] [PubMed] [Google Scholar]
- Kline, A. D. , Krantz, I. D. , Sommer, A. , Kliewer, M. , Jackson, L. G. , FitzPatrick, D. R. , … Selicorni, A. (2007). Cornelia de Lange Syndrome: Clinical review, diagnostic and scoring systems, and anticipatory guidance. American Journal of Medical Genetics Part A, 143A(3), 1287–1296. 10.1002/ajmg.a.31757 [DOI] [PubMed] [Google Scholar]
- Krantz, I. D. , McCallum, J. , DeScipio, C. , Kaur, M. , Gillis, L. A. , Yaeger, D. , … Jackson, L. G. (2004). Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped‐B. Nature Genetics, 36(6), 631–635. 10.1038/ng1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, S. O. , Opitz, J. M. , & Reynolds, J. F. (1986). Fibular a/hypoplasia: Review and documentation of the fibular developmental field. American Journal of Medical Genetics Supplement, 2, 215–238. 10.1002/ajmg.1320250626 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , & Krantz, I. D. (2008). Cohesin and human disease. Annual Review of Genomics and Human Genetics, 9, 303–320. 10.1146/annurev.genom.9.081307.164211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Burks, M. E. , Santos, R. , Kawauchi, S. , Calof, A. L. , & Lander, A. D. (2016). Genetic enhancement of limb defects in a mouse model of Cornelia de Lange syndrome. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics, 172C(2), 146–154. 10.1002/ajmg.c.31491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini, L. , Cucco, F. , Quarantotti, V. , Krantz, I. , & Musio, A. (2013). Mutation spectrum and genotype‐phenotype correlation in Cornelia de Lange syndrome. Human Mutation, 34(12), 1589–1596. 10.1038/jid.2014.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, D. , Vergano, S. A. S. , Deardorff, M. , Aggarwal, S. , Barot, A. , Johnson, D. M. , … Krantz, I. D. (2016). Characterization of limb differences in children with Cornelia de Lange Syndrome. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics, 172, 155–162. 10.1002/ajmg.c.31498 [DOI] [PubMed] [Google Scholar]
- Miyake, N. , Visser, R. , Kinoshita, A. , Yoshiura, K.‐I. , Niikawa, N. , Kondoh, T. , … Kurosawa, K. (2005). Four novel NIPBL mutations in Japanese patients with Cornelia de Lange syndrome. American Journal of Medical Genetics, 135A(1), 103–105. 10.1002/ajmg.a.30637 [DOI] [PubMed] [Google Scholar]
- Nizon, M. , Henry, M. , Michot, C. , Baumann, C. , Bazin, A. , Bessières, B. , … Cormier‐Daire, V. (2016). A series of 38 novel germline and somatic mutations of NIPBL in Cornelia de Lange syndrome. Clinical Genetics, 89(5), 584–589. 10.1111/cge.12720 [DOI] [PubMed] [Google Scholar]
- Pié, J. , Puisac, B. , Hernández‐Marcos, M. , Teresa‐Rodrigo, M. E. , Gil‐Rodríguez, M. , Baquero‐Montoya, C. , … Ramos, F. J. (2016). Special cases in Cornelia de Lange syndrome: The Spanish experience. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics, 172C(2), 198–205. 10.1002/ajmg.c.31501 [DOI] [PubMed] [Google Scholar]
- Roach, A. , Warner, W. A. , & Llanos, A. A. M. (2015). Building capacity for human genetics and genomics research in Trinidad and Tobago. Revista Panamericana De Salud Publica/Pan American Journal of Public Health, 38(5), 425–430. [PMC free article] [PubMed] [Google Scholar]
- Slavin, T. P. , Lazebnik, N. , Clark, D. M. , Vengoechea, J. , Cohen, L. , Kaur, M. , … Krantz, I. D. (2012). Germline mosaicism in Cornelia de Lange syndrome. American Journal of Medical Genetics, Part A, 158A(6), 1481–1485. 10.1002/ajmg.a.35381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), 1–7. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The NIPBL variant described in this report had been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) previously and can be found under accession numbers RCV000146527.3 and RCV000725320.1.