

Abstract
The immune system is the key target for vaccines and immunotherapeutic approaches aimed at blunting infectious diseases, cancer, autoimmunity, and implant rejection. However, systemwide immunomodulation is undesirable due to the severe side effects that typically accompany such strategies. In order to circumvent these undesired, harmful effects, scientists have turned to tailorable biomaterials that can achieve localized, potent release of immune‐modulating agents. Specifically, “stimuli‐responsive” biomaterials hold a strong promise for delivery of immunotherapeutic agents to the disease site or disease‐relevant tissues with high spatial and temporal accuracy. This review provides an overview of stimuli‐responsive biomaterials used for targeted immunomodulation. Stimuli‐responsive or “environmentally responsive” materials are customized to specifically react to changes in pH, temperature, enzymes, redox environment, photo‐stimulation, molecule‐binding, magnetic fields, ultrasound‐stimulation, and electric fields. Moreover, the latest generation of this class of materials incorporates elements that allow for response to multiple stimuli. These developments, and other stimuli‐responsive materials that are on the horizon, are discussed in the context of controlling immune responses.
Keywords: biomaterials, environmentally‐responsive materials, immunotherapies, stimuli‐responsive materials, vaccines
“Stimuli‐responsive” biomaterials hold strong promise for delivery of immunotherapeutic agents to disease‐relevant sites with high spatial and temporal accuracy. This review provides an overview of stimuli‐responsive biomaterials that can modulate the immune system in response to environmental changes in pH, temperature, enzymes, redox environment, photo‐stimulation, molecule‐binding, magnetic fields, ultrasound‐stimulation, electric fields, and multiple stimuli.
1. Introduction
Vaccines are the most successful immunotherapeutic intervention to date, virtually eliminating many life‐threatening, infectious diseases such as polio and measles. Conventionally, vaccines are composed of weakened or killed microbial agents that stimulate the host's immunological memory and result in long‐term immunity. Current vaccination strategies exploit heat‐killed microorganisms, live‐attenuated viral agents, small fragments of disease‐causing organisms, or antigen‐encoding nucleic acids to induce immunological memory.[ 1 ] Although vaccines have been generated to treat a broad swath of infectious diseases, their adoption to other fatal infections such as the Ebola virus, as well as other conditions (e.g., breast cancer), has proven ineffective to date.[ 2 , 3 ] The complexity, evasion, and evolving nature of these conditions complicate the development of efficacious prophylactics and therapies. Moreover, current vaccine delivery routes, including intramuscular or subcutaneous administration, rely on immune cell recognition of vaccine‐associated antigens, which are always not readily accessible and detectable to the host's immune system while in soluble form.
Given these limitations, enterprising scientists are now seeking to innovate vaccines that incorporate stimuli‐responsiveness with a goal to efficiently target and deliver vaccine agents to immune compartments that induce potent responses and long‐term immunological memory. Often referred to as “smart” materials, stimuli‐responsive materials can respond to physicochemical triggers, such as pH, temperature, ionic state, or magnetism, for controlled and on‐demand release of therapeutic agents. For instance, the acidic nature of tumors has been exploited to facilitate pH‐reactive biomaterial degradation and targeted therapeutic delivery to tumor‐specific sites.[ 3 ] Stimuli‐responsive materials can range from self‐assembled, polymeric constructs to biomolecules, contributing to a novel class of biomaterials used in controlled release drug delivery applications.[ 4 ] Moreover, recent research has explored the use of stimuli‐responsive materials as novel vaccination and immunotherapy systems to induce robust, targeted, and safer immune responses. This review focuses on innovative strategies currently under development that employ environment‐responsive biomaterials for vaccine and immunomodulatory applications. All of the stimuli‐responsive materials covered in this review are summarized in Table 1 .
Table 1.
A cumulative list describing the stimulus and induced effect of each stimuli‐responsive biomaterial covered in this review article
| Stimulus | Material | Induced effect | Source |
|---|---|---|---|
| pH‐Responsive | |||
| Acidic pH | Protein‐based monomeric microparticles | Degradation for antigen release in endosome/lysosome | Kwon et al.[ 5 ] |
| Acidic pH | Microgels of copolymerized acrylamine with bisacrylamine acetal links | Degradation for antigen release and membrane disruption of endosome/lysosome | Murthy et al.[ 6 ,7] |
| Acidic pH | Poly(propylacrylic acid)/PLGA blend microparticles | Degradation for antigen release and membrane disruption of endosome/lysosome | Yang et al.,[ 8 ] Fernando et al.[ 9 ] |
| Acidic pH | Micelle of DC membrane, histidine‐modified stearic acid‐grafted chitosan, and OVA antigen | Degradation for antigen release and membrane disruption of endosome/lysosome | Yang et al.[ 10 ] |
| Acidic pH | Nanogel composed of methoxy triethylene glycol methacrylate and PFPMA polymer blocks | Degradation for IMDQ‐based TLR7/8 agonist and antigen release in endosome/lysosome | Nuhn et al.[ 11 ] |
| Acidic pH | Poly(amidoamine) polymer containing acetal or ketal linkages | Degradation for cargo release in endosome/lysosome | Jain et al.[ 12 ] |
| Acidic pH | Microparticles composed of pH‐sensitive crosslinkers and poly(amidoamine) backbones and functionalized with anti‐DEC‐205 monoclonal Abs | Degradation for vaccine release in endosome/lysosome | Kwon et al.[ 13 ] |
| Acidic pH | NPs mainly composed of dimethylaminoethyl methacrylate, propylacrylic acid, and butyl methacrylate | Membrane disruption of endosome/lysosome for antigen and CpG adjuvant release | Wilson et al.[ 14 ] |
| Acidic pH | NPs mainly composed of dimethylaminoethyl methacrylate, propylacrylic acid, and butyl methacrylate | Membrane disruption of endosome/lysosome for siRNA release | Convertine et al.[ 15 ] |
| Acidic pH | NPs composed of 2‐(N,N‐diethylamino)ethyl methacrylate, butyl methacrylate, N,N‐dimethylacrylamide, and pyridyl disulfide groups | Membrane disruption of endosome/lysosome for antigen release | Wilson et al.[ 16 ] |
| Acidic pH | Micelles composed of N‐(2‐hydroxypropyl) methacrylamide, pyridyl disulfide groups, propylacrylic acid, dimethylaminoethyl methacrylate, and butyl methacrylate | Membrane disruption of endosome/lysosome for antigen release | Keller et al.[ 17 ] |
| Acidic pH | Virosomes composed of viral fusion membrane | Membrane fusion within endosome/lysosome for antigen release | Bungener et al.[ 18 ] |
| Acidic pH | Liposomes modified with succinylated poly(glycidol) and 3‐methylglutarylated poly(glycidol) | Membrane fusion within endosome/lysosome for antigen release | Yuba et al.[ 19 ] |
| Acidic pH | Liposomes modified with succinylated poly(glycidol) | Membrane fusion within endosome/lysosome for antigen release | Watarai et al.[ 20 ] |
| Acidic pH | Liposome modified with MGlu‐HPG | Membrane fusion within endosome/lysosome for antigen release | Yoshizaki et al.[ 21 ] |
| Neutral pH | Hollow macroporous microparticles made with Eudragit S100, a copolymer composed of methacrylic acid‐methyl methacrylate copolymer | Release of cargo through pore opening in neutral intestinal pH | Kumar et al.[ 22 ] |
| Thermo‐responsive | |||
| Heat | PNIPA hydrogel | Release of loaded antigen while acting as an adjuvant due to inherent properties of hydrogel material | Shakya et al.[ 23 ] |
| Heat | Poly(vinylcaprolactam) hydrogel | Gel phase change above 33 °C | Makhaeva et al.[ 24 ] |
| Heat | Poly(vinyl methyl ether) hydrogel | Gel phase change above 36 °C | Moerkerke et al.[ 25 ] |
| Heat | Poloxamer 407 (PEO and PPO) or chitosan‐MC | Gel phase change at 37 °C | Kojarunchitt et al.[ 26 ] |
| Heat | PNIPA hydrogel coated with a dialysis membrane | Increased drug release at 37 °C compared to 10 °C | Zhang et al.[ 27 ] |
| Heat | PEG‐PCL‐PLA‐PCL‐PEG hydrogel containing vaccine‐encapsulated PLGA NPs | Gelation at 37 °C for sustained vaccine release | Bobbala et al.[ 28 ] |
| Heat | Thermoresponsive gel containing pluronic surfactant, carbopol, and hydroxypropyl methylcellulose | Gelation at 37 °C sublingually for sustained release or antigen and bacterial heat‐labile toxin adjuvant | White et al.[ 29 ] |
| Heat | Lysolipid thermally sensitive liposome | Release of doxorubicin drug intratumorally when externally heated to 40–45 °C | “Study of ThermoDox with standardized radiofrequency ablation (RFA) for treatment of hepatocellular carcinoma (HCC)”[ 30 ] |
| Heat | Electronically powered device that regulates temperature of thin poly(N‐isopropylacrylamide‐co‐acrylamidobenzophenone) hydrogel layer | Drug release upon allowing gel to reach 37 °C body temperature | Yang et al.[ 31 ] |
| Enzyme‐responsive | |||
| MMP2‐responsive | Liposomes modified with MMP‐2 cleavable long PEG strands with tumor targeting mAb 2C5 and shorter PEG strands with cell‐penetrating TATp peptides | Cleavage of long PEG strands to reveal cell‐penetrating TATp at tumor sites and deliver anti‐cancer drugs | Zhu et al.[ 32 ] |
| MMP‐responsive | Mesoporous silica NPs sealed with bovine serum albumin conjugated via PLGLAR MMP substrate | Release of DOX anticancer drug from particles at tumor site | Liu et al.[ 33 ] |
| Glycosyl hydrolase‐responsive | Drug‐loaded HNTs with dextrin stoppers | Intracellular anticancer drug delivery | Dzamukova et al.[ 34 ] |
| Cathepsin B‐responsive | MCM‐41 silica mesoporous NPs with cathepsin B peptide substrate caps | Release of drug cargo to intracellular tumor cell sites | De la Torre et al.[ 35 ] |
| Redox‐responsive | |||
| GSH | Micelle of single disulfide bond‐bridged block polymer of poly(ε‐caprolactone) and poly(ethyl ethylene phosphate) | Intracellular anticancer drug delivery | Wang et al.[ 36 ] |
| GSH | PSSN10 micelles consisting of POEG hydrophilic block and PNLG hydrophobic block with NLG919 motifs attached via redox‐sensitive linkages | Intracellular tumor co‐delivery of the IDO inhibitor NLG919 and loaded DOX | Sun et al.[ 37 ] |
| GSH | IL‐2/Fc PEG disulfide‐containing nanogel backpack conjugated to T cells | Induction of T cell expansion at tumor site | Xie et al.[ 38 ] |
| GSH | HA‐deoxycholic acid micelles containing disulfide bonds | Intracellular release of paclitaxel to tumor cells | Li et al.[ 39 ] |
| GSH | O,N‐Hydroxyethyl chitosan‐octylamine micelles containing disulfide bonds | Intracellular release of paclitaxel to tumor cells | Huo et al.[ 40 ] |
| GSH | Micelles consisting of PEG‐PCL disulfide bonded to docetaxel | Intracellular release of docetaxel to tumor cells | Zhang et al.[ 41 ] |
| ROS | Micelle of TPGS, HA, and arylboronic ester | Intracellular release of DOX to tumor cells | Su et al.[ 42 ] |
| Hypoxia | Hydrophobically modified 2‐nitroimidazole derivative conjugated to carboxymethyl dextran | Intracellular release of DOX to tumor cells | Thambi et al.[ 43 ] |
| Hypoxia | 4‐Nitrobenzyl (3‐azidopropyl) carbamate and mPEG‐poly(γ‐propargyl‐L‐glutamate) copolymer | Intracellular release of DOX to tumor cells | Zhang et al.[ 44 ] |
| Photo‐responsive | |||
| Visible light (>410 nm) | Acridin‐9‐methanol fluorescent organic NPS | Delivery of chlorambucil to cancer cell nuclei | Jana et al.[ 45 ] |
| Red light (670 nm) | Pheophorbide A grafted with polyethylenimine | Release of antigen into cytosol of APCs | Zhang et al.[ 46 ] |
| UV light (365 nm) | Hydrogel composed of an 8‐arm PEG alkyne with an azide‐functionalized photodegradable crosslinker | Hydrogel degradation and modulation of VICs cell phenotype | Kirschner et al.[ 47 ] |
| Blue light (405 nm) or UV light (365 nm) | NPs made up of poly(ethyleneimine) functionalized with 4,5‐dimethoxy‐2‐nitrobenzyl chloroformate | Release of retinoic acid to differentiate leukemia cells | Boto et al.[ 48 ] |
| Red light (630 nm) | Porfimer sodium (Photofrin) | Release of ROS in stimulated cancer tissues | Dos Santos et al.[ 49 ] |
| Red light (630 nm) | 5‐Aminolevulinic acid (Levulan) | Release of ROS in stimulated cancer tissues | Dos Santos et al.[ 49 ] |
| Red light (665 nm) | Silica NPs with covalently incorporated iodobenzylpyropheophorbide | Release of ROS in stimulated cancer tissues | Ohulchanskyy et al.[ 50 ] |
| Green light (550 nm) | Solid lipid NPs loaded with hypericin | Release of ROS in stimulated cancer tissues | Youssef et al.[ 51 ] |
| Molecule‐responsive | |||
| Tumor‐specific marker glycoprotein α‐fetoprotein | Imprinted gel formed by lectin–glycoprotein–antibody complexes | Shrinkage of gel in simulated tumor environment | Miyata et al.[ 52 ] |
| Adenosine or thrombin | Hydrogel made by crosslinking DNA aptamers with linear polyacrylamide chains | Dissolving of hydrogel in the presence of stimulus | Yang et al.[ 53 ] |
| ATP | Nanocarrier consisting of an ATP‐responsive DNA motif with DOX, protamine, and a HA crosslinked shell | Controlled release of DOX at ATP‐rich cancer tissue | Mo et al.[ 54 ] |
| Fluorescein | Hydrogel made up of an 8‐arm PEG functionalized with single chain antibody fragments and an 8‐arm PEG‐fluorescein | Dissociation of hydrogel to release human papilloma virus type 16 vaccine booster | Gübeli et al.[ 55 ] |
| Novobiocin | Hydrogel of PEG, gyrase B, and coumermycin | Dissociation of hydrogel to release hepatitis B vaccine booster | Gübeli et al.[ 56 ] |
| Magnetic field‐responsive | |||
| Magnetic field | Iron oxide core NPs coated with β‐cyclodextrin and pluronic polymer (F127) | Release of anticancer drug curcumin to tumor cells | Yallapu et al.[ 57 ] |
| Magnetic field | Iron‐oxide NPs with an aminosilane coating | Heating of particles at tumor site cause damage | Maier‐Hauff et al.[ 58 ] |
| Magnetic field | Iron‐oxide MNCs loaded with CpG‐ODN and coated with cancer cell membrane and anti‐CD205 | Guided delivery and retention of therapy at LN following injection | Li et al.[ 59 ] |
| Magnetic field | Iron‐oxide NPs with a polyethyleneimine coating | Enhanced transfection of malaria DNA vaccine | Al‐Deen et al.[ 60 ] |
| Magnetic field | Iron‐oxide NPs | Enhanced adenovirus delivery to target cells | Sapet et al.[ 61 ] |
| Ultrasound‐responsive | |||
| Ultrasound | Liposomes co‐modified with single stranded DNA aptamers and poly(NIPMAM‐co‐NIPAM) | Release of calcein or DOX to cancer tissues | Ninomiya et al.[ 62 ] |
| Ultrasound | PS‐based liposome nanobubble conjugates | Release of paclitaxel to cancer tissues | Chandan et al.[ 63 ] |
| Ultrasound | PEG bubble liposomes containing perfluoropropane as a contrast agent | Delivery of plasmid DNA for tissue‐specific gene delivery | Suzuki et al.[ 64 ] |
| Ultrasound | Mixture of mannosylated lipoplexes and bubble liposomes | DC and macrophage‐specific localized gene delivery | Un et al.[ 65 ] |
| Electric field‐responsive | |||
| Electric field | Vinyl monomer and HA crosslinked into a hydrogel | Controlled release of model drug | Sutani et al.[ 66 ] |
| Electric field | Polymethacrylic acid and sodium alginate hydrogel | Conformational change of gel shape | Kim et al.[ 67 ] |
| Electric field | Chondroitin 4‐sulphate crosslinked with ethylene glycol diglycidyl ether to form a hydrogel | Controlled release of various peptides and proteins | Jensen et al.[ 68 ] |
| Electric field | Polymethacrylic acid and poly(vinyl alcohol) hydrogel | Conformational change of gel shape | Kim et al.[ 69 ] |
| Electric field | Chitosan and polyacrylonitrile hydrogel | Conformational change of gel shape | Kim et al.[ 70 ] |
| Electric field | Sodium alginate and polyacrylic acid hydrogel | Release of hydrocortisone | Yuk et al.[ 71 ] |
| Electric field | Agarose–carbomer 934P gel | Release of hydrocortisone | Hsu et al.[ 72 ] |
| Electric field | PPy‐coated electrode | Release of dexamethasone | Wadhwa et al.[ 73 ] |
| Electric field | Biotin‐doped PPy film | Release of streptavidin and any attached biotinylated drugs | George et al.[ 74 ] |
| Electric field | PPy NPs with a PLGA‐PEG‐PLGA hydrogel | Release of fluorescein or daunorubicin | Ge et al.[ 75 ] |
| Electric field | PEDOT nanotubes on loaded electrospun PLGA nanoscale fibers | Release of dexamethasone | Abidian et al.[ 76 ] |
| Electric field | Drug‐loaded carbon nanotubes sealed with PPy | Release of dexamethasone | Luo et al.[ 77 ] |
| Multi‐stimuli‐responsive materials | |||
| Visible light (>410 nm), acidic pH | Spiropyran–coumarin | Delivery of chlorambucil to cancer cells | Barman et al.[ 78 ] |
| NIR light (808 nm), acidic pH | Self‐assembled micelles pluronic copolymer P123‐conjugated DOX prodrug and cypate‐conjugated PEG‐block‐poly(diisopropanolamino ethyl methacrylate) | DOX delivery to drug resistant tumor cells | Yu et al.[ 79 ] |
| GSH, acidic pH | NP composed of chitooligosaccharide‐disulfide‐PCL | Targeted DOX delivery to cancer cells | Xu et al.[ 80 ] |
| Heat, acidic pH | NPs formed from PLGA, pluronic F127, chitosan, and HA | Delivery to DOX and irinotecan to cancer stem‐like cells | Wang et al.[ 81 ] |
| NIR light (650–900 nm), acidic pH, GSH | NPs made from a 3‐arm PEG‐a‐PCL‐SS‐P(NIPAM‐co‐DMA) star quaterpolymer loaded with cypate | Targeted release of paclitaxel | An et al.[ 82 ] |
| GSH, acidic pH, trypsin | Keratin/DOX complex NPs formed via ionic gelation | DOX delivery to cancer cells | Li et al.[ 83 ] |
| Red light (660 nm) and hypoxia | Chlorin e6‐doped‐azobenzene‐glycol chitosan‐PEG mesoporous silica nanocarrier | Delivery of CpG ODN adjuvant to DCs and PDT‐assisted disruption of tumor cells to release antigen | Im et al.[ 84 ] |
| Prospective responsive materials | |||
| Complimentary DNA | DNA‐crosslinked polyacrylamide hydrogel | Gel to solution transition | Yurke et al.[ 85 ] |
| Complimentary DNA | DNA‐crosslinked polyacrylamide hydrogel | Swelling or shrinking | Murakami et al.[ 86 ] |
| Complimentary aptamer sequence | Aptamer‐functionalized hydrogel | Release of contained protein, like VEGF or platelet‐derived growth factor BB | Battig et al.[ 87 ] |
| K+ ion | Hydrogel of the copolymer poly(N‐isopropylacrylamide‐co‐benzo‐15‐crown‐5‐acrylamide) | Release of model drug VB12 | Mi et al.[ 88 ] |
| Ba2+ or Na+ ion | A porous microcapsule with linear grafted PNIPAM‐co‐PBCAm chains in the pores | Pore opening for the release of model drug VB12 | Chu et al.[ 89 ] |
2. Vaccines and Immunity—An Overview
Vaccines function by triggering an immune response, usually within hours by engaging the innate immune system and, subsequently adaptive immunity. Innate immunity consists of multiple stages of defense: 1) physical barriers (e.g., skin and mucosal layers) protecting against outside invaders; 2) engagement of the complement system enhancing the ability of immune system cells to clear the infection; and 3) recruitment of first responders (e.g., macrophages and neutrophils) and release of special chemical mediators (e.g., cytokines).[ 90 ] Once an infection occurs (stage 3), nonspecific innate immune cells trigger local inflammation and recruitment of phagocytic immune system cells. These early responder cells are primarily neutrophils, as well as macrophages, and dendritic cells (DCs), which engulf invading pathogens and degrade them internally.[ 91 ] Activated macrophages secrete cytokines and chemokines that further recruit and activate more phagocytic cells to fight the infection. The innate immune response is specialized to respond quickly and in a potent manner to an invading pathogen. This first‐line innate immune response may be sufficient to clear invading pathogen, or the adaptive immune response may be recruited to attack the pathogen in an antigen‐specific manner.[ 92 ] With respect to vaccines, this lack of this adaptive response could greatly hinder the generation of long‐lasting immunological memory that is essential to vaccine function.
Conventional vaccines aim to induce a long‐lasting response capability in the host, which provides protective immunity from subsequent challenge by the same pathogen, as outlined in Figure 1 . Enabled by professional antigen‐presenting cells (APCs) (e.g., DCs, macrophages, and B cells), lymphocytes (e.g., T cells, B cells, and natural killer cells) are the primary effectors of adaptive immunity. These adaptive responders can recognize and eliminate infection through a number of mechanisms including the production of antigen‐specific antibodies (Abs) and release of cytotoxic granules. Additionally, a small fraction of lymphocytes generated during the adaptive immune response can survive far beyond the end of the pathogenic insult, often for the lifetime of the host. This dormant lifelong protective immunity can be quickly and robustly reactivated in the event of a subsequent insult from the same pathogen, the desired hallmark of vaccination.
Figure 1.
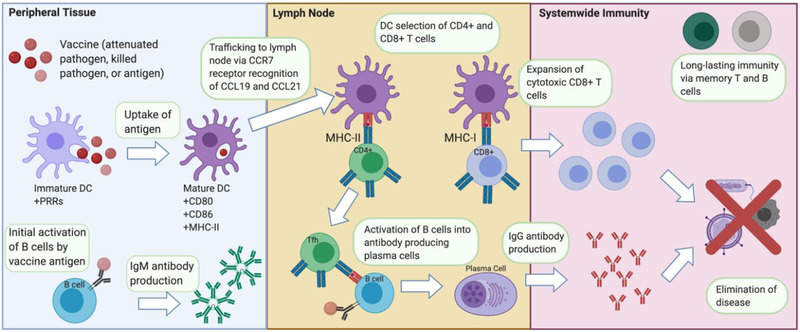
Immunological cascade following vaccine injection. In the peripheral tissue, the vaccine gets taken up by resident immature DCs, inducing maturation. B cells with affinity toward the vaccine antigen will begin producing IgM Abs for a temporary initial adaptive response. Mature DCs traffic to the LN where they present the vaccine antigens to select CD4+ and CD8+ T cells. Clonally selected T and B cells undergo rapid proliferation and differentiate into Tfh cells and antigen‐specific plasma cells that form the core of cellular and humoral immunity, respectively. Following this immune response, long‐lasting memory T and B cells remain in the body to provide adaptive immunity against “secondary” exposure from the real pathogen threat.
The activation of adaptive immune cells depends on a subset of innate cells called professional APCs, chiefly DCs. Immature DCs form in the bone marrow, reside in the blood stream, and surveil for pathogenic agents in peripheral tissues, such as the skin and mucosa. Immature DCs possess a multitude of receptors on their surface, including pathogen recognition receptors (PRRs), which can recognize common features of many pathogens, referred to as pathogen‐associated molecular patterns (PAMPs). Dendritic cells “mature” when they interact with pathogens through these recognition molecules, and this maturation is denoted by the expression of CD80, CD86, and major histocompatibility complex (MHC) cell surface proteins. Additionally, upregulation of CCR7 on maturing DCs allows for migration to the draining lymph nodes (LNs) following secretion gradients of CC‐chemokine ligand 19 (CCL19) and CCL21 from T cells in these organs. At the LNs, DCs present specific pathogenic peptides on their surface through MHC‐I and MHC‐II in mice (or human leukocyte antigen (HLA) in humans). Additionally, DCs express co‐stimulatory molecules to interact with and activate T cells.[ 93 ] CD4+ T follicular helper cells (Tfh) activate B cells through T cell receptor recognition of MHC‐II presented antigenic peptide. Upon activation, B cells mature into plasma cells that produce Abs specific for the pathogenic threat. This production of pathogen‐specific Abs is often referred to as the humoral response. On the other hand, the cell‐mediated response is where naïve T cells are activated to become CD8+ cytotoxic T lymphocytes (CTLs) or CD4+ T helper (TH) cells subsequent to synapsing with MHC I or MHC II, respectively, on the surface of APCs. The CTLs are responsible for killing infected cells (as well as tumor cells), while Tfh cells assist in maturation of B cells into plasma cells, aid the development of memory B cells, and help with activation of CTLs and macrophages.
The long‐term specific adaptive immune response involves highly specialized “memory” T and B cells, which are quickly activated in the event of a subsequent infection by the same pathogen. Memory B and TH cells mount immediate protection in peripheral tissues and re‐challenge responses in secondary lymphoid organs (LNs, spleen, etc.) upon re‐encounter with the same pathogen. Memory B cells differentiate into short‐lived plasma cells to produce antibody at a much faster pace than naïve B cells.[ 94 ] Furthermore, in subsequent re‐infections, memory TH cells play an important role in selecting B cells to undergo affinity maturation for a stronger selectivity toward the invading pathogen. Memory TH also secretes specific cytokines to quickly induce the appropriate isotype class switching of these TH‐activated B cells for humoral immune response. CD8+ CTLs are also important in immunizations, particularly in the context of cancer immunotherapies. Elimination and prevention of tumors rely on potent and sustained CD8+ CTL responses that can overcome established tolerance to self‐antigens. This can be achieved through multiple methods such as cytokine stimulation (IL‐2, IL‐15, and IL‐21), chimeric antigen receptors, or DC presentation of antigen.[ 95 ]
The primary goal of vaccine immunity is to drive plasma cell antibody production and the development of memory B cells. A major requirement for effective vaccination is the activation of DCs, propelling them to traverse to the LNs and activate T and B cells. Recently, PAMPS have been investigated as stimulatory adjuvants (immunomodulatory molecules) in vaccines for orchestrating APCs toward a more robust immune response. Historically, two adjuvants—aluminum salts and monophosphoryl lipid A that are approved by the Food and Drug Administration (FDA)—have been used in vaccines. More recently, the FDA also approved a novel flu vaccine named FLUAD, which contains MF59 adjuvant, an oil‐in‐water emulsion of squalene oil.[ 96 ] FLUAD is the first adjuvanted seasonal flu vaccine marketed in the United States. The MF59 adjuvant, resembling in size to a large virion, stimulates an influx of immune inflammatory cells, including monocytes and macrophages. These cells release cytokines, including CCL2 and interleukin‐18 (IL‐18), to induce a greater inflammatory response. The co‐administered vaccine antigens are taken up by monocytes, while the MF59 adjuvant upregulates CCR7 and induces monocyte differentiation into DCs and their homing to the LNs. Another newly FDA‐approved adjuvant is cytosine phosphoguanine 1018 (CpG 1018), included in the formulation of the hepatitis B vaccine HEPLISAV‐B.[ 97 ] CpG oligodeoxynucleotides (ODNs) are synthetic agonists of Toll‐like receptor 9 (TLR9), inducing an enhanced, virus‐specific Th1 immune response. Despite the efficacy of vaccines at eradicating several deadly diseases such as polio and smallpox, there are limitations to these prophylactics as evidenced by recent outbreaks (e.g., Ebola, Zika, and COVID‐19). Soluble vaccines, even with the addition of an adjuvant, often fail to generate a strong, long‐lasting immune response against the disease antigen. There is promising potential to overcome these limitations with the current development of therapies utilizing specialized stimuli‐responsive materials for sustained release of vaccine components to immune‐relevant tissues like the LN.
2.1. Vaccine Administration Site Considerations
Vaccine efficacy hinges upon successful delivery of antigen and adjuvant to the correct cell types with high spatial and temporal accuracy.[ 98 , 99 ] As such, the route of injection is a critical factor affecting the success of a vaccine. Typical vaccine injection routes are intramuscular or subcutaneous due to ease of administration. However, these sites contain relatively low numbers of DCs,[ 100 ] which are critical to vaccine function.[ 101 ] Alternatively, peripheral tissues, e.g., skin and mucosa, contain a high abundance of DCs that make up 1–3% of the cells in these tissues,[ 102 ] making intradermal injection seem much more attractive as a vaccination route.[ 100 ] However, ultimately the efficacy of vaccines relies upon successful delivery to the LNs, the immune command centers of the body. While moderate amounts of antigen can be transported to the LNs via lymph drainage, generation of a robust immune response via these delivery routes hinges upon antigen‐specific DC migration to draining LNs, and activation of LN‐resident T and B cells to produce adaptive and memory immune response for long‐term immunity. Since the delivery of vaccine components to the LNs is critical to successful immunization, a more ideal vaccine route may be direct administration to these immune command centers. Andorko et al. speculated that this method would circumvent vaccine accumulation and degradation at the site of injection, while delivering more therapeutic cargo to LNs.[ 103 ] This group has recently investigated intra‐LN delivery of vaccine agents by directly injecting vaccine components into the LNs.[ 104 ] This approach yielded promising results with potent, long‐lasting humoral and cellular immunity. However, the size and anatomical positions of the LNs make translation of this vaccination method difficult.
Mucosal epithelia are also sites that are under constant immune surveillance, especially by DCs. Most immune challenges develop in the mucosal regions, including the digestive and respiratory tracts, which possess special lymphoid tissues referred to as the mucosal‐associated lymphoid tissues.[ 105 ] These tissues contain membranous microfold (M cells) responsible for transfer of antigenic material across the mucosal membrane to DCs and adaptive immune cells that generate humoral (antibody; IgA) responses, as well as helper T cell responses. Similarly, in the gut, immunosurveillance is performed by special immune organs called the gut‐associated lymphoid tissues (GALT). Although most vaccines are administered via parenteral routes, there is great potential in intranasal, intravaginal, or oral delivery methods. For instance, a recently investigated recombinant rhinovirus‐based vaccine delivered mucosally showed potent and protective HIV‐specific immune response.[ 106 ] Specifically, the vaccination elicited stronger multifunctional CD8+ T cell response and higher antibody titers. Mucosal delivery routes have long been avoided by scientists because of the many physical, chemical, and mechanical barriers that reduce the transfer of vaccine components into the blood. One such barrier is mucin, a protein that is secreted from mucin‐filled vesicles from specialized epithelial cells; this biomolecule forms a fibrous matrix that hinders the interaction and eventually translocation of vaccine agents and adjuvants across the mucosal epithelial layer. Mitigating the challenges of drug delivery is an active area of biomaterials research and some interesting strategies have been developed to promote mucosal delivery. For example, poly(ethylene glycol) (PEG)‐coated nanoparticles (NPs) have been developed to mitigate particle aggregation that typically prevents vaccine loaded particles from infiltrating the mucosal layer.[ 107 ] Additionally, PEG acts as a mucoadhesive “glue” that entangles with mucosa fibers and assists with penetration.[ 108 ] While the mucosal barrier may hinder delivery to the bloodstream, other therapies target the immune cells within the mucosal lining. For example, pulmonary vaccine delivery nanocarrier formulations that target pulmonary DCs to yield an enhanced immune response using minimal dosing.[ 109 ]
Uncovering the potential of biomaterials is crucial in developing long‐lasting effective vaccines. For example, thermo‐responsive hydrogels undergo gelation at physiological temperatures, making them immensely useful for creation of a sustained release depot of vaccine components from the injection site.[ 23 , 28 , 29 ] Moreover, spatial and temporal control over immunotherapeutic release from biomaterials is important in the context of other applications such as prevention of implant rejection, delivery of anti‐cancer drugs to tumors, and targeted drug release to inflamed or infected tissues.
3. Biomaterials for Vaccine and Immunotherapeutic Delivery
In the last few decades, novel biomaterials have been extensively investigated to produce more potent therapeutics for a plethora of diseases, including immune conditions. Biomaterials have the potential to assist vaccine delivery to various tissue targets, primarily due to the temporal and spatial control provided by these vehicles. Polymeric, lipid, and protein‐based particulate drug delivery vehicles provide a myriad of formulations to protect the vaccine components while accomplishing delivery to the target tissue or intracellular locations. Additionally, biomaterial‐based vaccine carriers can contribute to the immunogenicity of vaccine components by mimicking pathogenic features, thereby providing cues to immune cells for the generation of powerful and enduring immune responses.[ 110 ]
The development of stimuli‐responsive biomaterials has further buoyed the application of biomaterials in vaccine and immunotherapeutic applications. These special classes of biomaterials undergo specific physicochemical changes upon interaction with external or internal stimuli. Intracellular environments, such as the lysosome, have distinct properties including enzyme activity, redox gradients, and ionic charge differences that have been exploited by smart biomaterials. These localized conditions can be found not only in certain cell compartments, but also in different microenvironments (e.g., tumor microenvironments), tissues, and organs. On the other hand, exogenous stimuli consist of thermal, photo, magnetic, ultrasound, and electric field triggers. Figure 2 highlights some of the general, stimuli‐responsive biomaterial strategies under development, for both internal and external stimuli.
Figure 2.
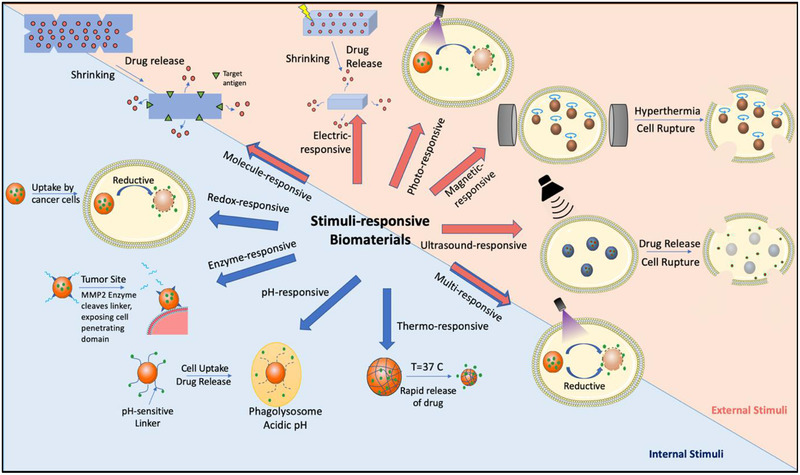
Overview of the different types of stimuli‐responsive biomaterials for vaccine and immunotherapeutic delivery. The stimulus can come from an external source including electric field, light, magnetic field, and ultrasound signal. Alternatively, intrinsic stimuli found within intracellular compartments or specific tissues can also be used to activate materials. These stimuli include reductive environments, changes in pH, enzymatic cleavage, and temperature change. Certain types of materials respond to stimuli that can be found intrinsically or externally, such as therapies sensitive to multiple stimuli and molecule‐responsive hydrogels that can react to intrinsic or exogenous analytes.
3.1. pH‐Responsive Materials
Polymeric materials responsive to pH change are among the most researched smart biomaterials for vaccine applications. The ability to activate vaccine agents intracellularly through lysosome cleavable chemistry is a powerful way to control immunogenic pathways. pH‐Cleavable microparticles have been developed to encapsulate vaccine agents with high efficiency and release their vaccine components at site‐specific targets.[ 5 ] Furthermore, pH‐sensitive biomaterials, such as aliphatic monomers with cleavable acetal linkages, have enabled controlled antigen presentation by APCs to induce desired immune responses. APCs scavenge potentially infectious materials and present the antigenic components on their surface to T and B cells. Endogenous cytosolic pathways typically lead to MHC‐I‐mediated responses and CD8+ CTL activation, while exogenous extracellular material pathways mostly result in MHC‐II‐mediated CD4+ T cell response. There is also the potential of cross‐presentation, where endogenous pathways can lead to MHC‐II‐mediated presentation and exogenous pathways to MHC‐I antigen presentation. It is highly desirable in vaccine applications to control which pathways are activated, as a CTL response may be more effective for cancer and viral vaccines, while a TH cell response is a better candidate for long‐term immunological memory against extracellular pathogens.[ 111 ] Most antigens delivered extracellularly lead to MHC‐II response after degradation in the lysosome. However, escape of antigenic material from the lysosome into the cytosol can enhance MHC‐I presentation.[ 5 ] Using pH‐degradable hydrogels or polymeric formulations that degrade in the endosome and result in its membrane disruption can lead to an MHC‐I meditated antigen presentation.[ 6 , 7 , 8 , 9 ] Recently, interesting work on antigen delivery has been done by Yang et al. on using biomimetic micelles composed of DC membrane, histidine‐modified stearic acid‐grafted chitosan, and OVA antigen.[ 10 ] These micelles use the pH‐responsive properties of histidine for endosomal escape and antigen delivery, while using DC membrane as a natural adjuvant.
Another advantage of pH‐responsive vaccines is their ability to passively target the LNs (through lymphatic drainage of biomaterial and/or immune cell trafficking to the LNs following phagocytic uptake) to enhance the immune response. Vaccine adjuvants, including agonists of TLRs, can boost the immune response. However, these adjuvants may result in systematic inflammation and toxicity when in soluble form. To address this issue, researchers have developed pH‐degradable micro‐ and nanomaterials, such as imidazoquinoline (IMDQ)‐based TLR7/8 agonist conjugated nanogels that passively diffuse to the draining LN to induce immune activation.[ 11 ] These pH‐degradable polymeric nanogels are composed of self‐assembled amphiphilic block copolymers of hydrophilic PEG‐like structures and hydrophobic pentafluorophenyl methacrylate (PFPMA). Upon uptake by DCs and macrophages in the LN, these pH‐sensitive biomaterials will degrade in the lysosome and showcase their IMDQ‐based TLR7/8 agonist as an adjuvant to induce a more potent and long‐term immune response. Passive delivery to the draining LN combined with pH responsive co‐delivery of antigen and TLR7/8 agonist adjuvant successfully induced a multifold increase in magnitude of T and B cell immune response.[ 11 ]
In addition to pH‐responsive hydrogel adjuvant vaccines, protein‐based vaccines also exploit the acidic pH of the phagosome for releasing antigen and boosting the immune response. For instance, acid degradable amino acid‐based microparticles encapsulating vaccine agents were surface‐functionalized with anti‐DEC‐205 monoclonal Abs targeting the DEC‐205 endocytosis receptors only expressed by epidermal Langerhans DCs and thymic endothelial cells. The microparticles composed of pH‐sensitive crosslinkers and poly(amidoamine) backbones are stable at physiological pH, but degrade and release vaccine components at the acidic pH of the DC lysosome.[ 12 ] These vaccines have presented great promise in delivering vaccine antigens and adjuvants to DCs to elicit a potent immune response.[ 13 ]
An even more magnified immunostimulatory effect can be achieved through multimodal therapies that combine co‐delivery of antigen and adjuvant with pH‐responsive endosomal membrane disruption. For example, Wilson et al. developed micellar endosomolytic NPs for delivery of immune‐stimulatory CpG ODNs and protein agents to augment immune responses.[ 14 ] This formulation has previously been used to release siRNA from the endosome (without use of an adjuvant).[ 15 ] These NPs are composed of an amphiphilic diblock copolymers of dimethylaminoethyl methacrylate and polyacrylic acid allowing for a shift to a more hydrophobic state at endosomal pH, thus resulting in membrane disruption and release of endosomal contents into the cytosol. Measured at 23 nm in hydrodynamic diameter, the NPs are surface‐functionalized with CpG ODN complexed via electrostatic interactions and bound to thiolated ovalbumin (OVA) via thiol‐reactive disulfide groups to enable dual delivery of vaccine antigens and immunostimulatory adjuvants. Immunization of mice with subcutaneous injections of the NP formulations significantly enhanced CD8+ T cell response, measured up to 18‐fold compared to administration of the free agents.[ 14 ] This research group also subsequently developed other endosomolytic delivery vehicles. Wilson et al. created a formulation of branched copolymers of 2‐(N,N‐diethylamino)ethyl methacrylate and butyl methacrylate, extended with N,N‐dimethylacrylamide copolymerized with thiol‐reactive pyridyl disulfide groups showed that hyperbranched architecture could result in higher MHC‐I antigen presentation compared to crosslinked, linear, and soluble formulations.[ 16 ] Furthermore, Keller et al. characterized a neutral polymeric micelle composed of N‐(2‐hydroxypropyl) methacrylamide with pyridyl disulfide groups, and a copolymer of propylacrylic acid, dimethylaminoethyl methacrylate, and butyl methacrylate; this nanocarrier displayed endosome‐releasing activity and an enhanced CD8+ T cell response.[ 17 ]
Outside of membrane disruption methods, membrane fusion can also be used to escape the endosome in a pH‐dependent manner for MHC‐I specific immune stimulation. For example, one approach uses a viral fusion protein‐incorporated liposomes have been produced to instigate the immune system, while risking unwanted immune responses.[ 18 ] Also, liposomes modified with pH‐responsive fusogenic polymers such as succinylated poly(glycidol) or 3‐methylglutarylated poly(glycidol) have the ability to fuse with endosomal membrane upon uptake by phagocytic cells and release their content intracellularly to elicit a stronger immune response.[ 19 ] Once inoculated in mice, fusogenic liposomes result in induction of TH1 and TH2 immune response mediating humoral and cell‐mediated immunity.[ 20 ] In a similar approach, Yoshizaki et al. used pH‐sensitive 3‐methyl‐glutarylated hyperbranched poly(glycidol) (MGlu‐HPG) polymer‐modified liposomes to deliver OVA antigen to DCs for an anti‐tumor immune response (Figure 3 ).[ 21 ]
Figure 3.
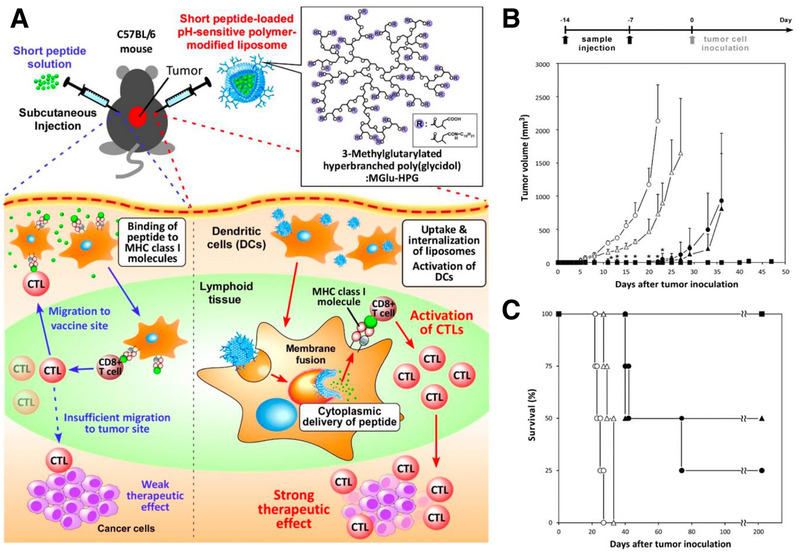
A) Schematic of a pH‐responsive anti‐cancer immunotherapy. Tumor‐specific peptides are loaded into MGlu‐HPG polymer‐modified liposomes, which are taken up by DCs in lymphoid tissues. In response to endosomal/lysosomal acidic pH levels, these particles release the peptide into the cytosol of the DCs via membrane fusion, allowing for MHC class I loading of the peptide and a stronger immune response than if the peptide was administered alone. B,C) Mice were immunized through treatment of OVA‐I solution (open circles), OVA‐I‐loaded liposomes (closed circles), OVA‐II solution (open triangles), OVA‐II‐loaded liposomes (closed triangles), and OVA‐loaded liposomes (closed squares) 14 and 7 days before tumor cell inoculation against OVA antigen. pH‐responsive liposome formulations showed higher resistance to tumor growth and increased survival. Adapted with permission.[ 21 ] Copyright 2016, Multidisciplinary Digital Publishing Institute.
pH‐Responsive materials can also be designed to release therapeutic cargo at neutral pH. For example, pH‐responsive macroporous microparticles have been developed for controlled release of drugs via oral delivery.[ 22 ] In these formulations, freeze‐drying of MPs induces pore closure. Upon subsequent exposure to neutral intestinal pH, the pores open and release the contained drug. The presence of macroporous surface enables facile and highly efficient loading of active drugs, such as peptide antigens. These encapsulated drugs are protected from the acidic stomach pH (2.0) and are released rapidly in the desired intestinal pH (7.1). In vitro experiments observed pores remained closed at acidic pH for 2 h and released drug at pH 7.1 for 4 h.[ 22 ] Potentially, this approach can be exploited for vaccine delivery at specific mucosal surfaces.
3.2. Thermo‐Responsive Materials
Another intrinsic stimulus that researchers have utilized for responsive material‐based therapy is temperature change. Thermo‐responsive biomaterials possess the unique property of changing solvation state based on temperature. These biomaterials take advantage of physiological temperature differences as cues for biomaterial conformational changes. The most researched group of thermo‐responsive biomaterials for drug delivery applications are hydrogel‐based polymers, such as poly(N‐isopropylacrylamide) (PNIPAM), poly(N‐vinyl caprolactam) (PVC), poly(methyl vinyl ether) (PMVE), poly(2‐oxazoline) (POx), and naturally derived cellulose polymers.[ 112 ]
Thermo‐responsive hydrogels receive temperature cues and transition from a liquid in ambient temperature to a gel formation in physiological temperature without the need for chemical or other environmental stimulations. This conversion from solution to gel formation is called the “sol–gel transition”. Below a critical temperature, known as the lower critical solution temperature (LCST), the hydrogel is in liquid soluble state. Upon an increase in temperature above the LCST, hydrogels become insoluble and gel‐out of the solution due to the hydrophobicity of the polymer chains induced by the increased temperature. On the other hand, the upper critical solution temperature (UCST) is the critical temperature at which some hydrogels become immiscible and result in gel formation at temperatures lower than the UCST. These changes are explained by thermodynamics of the system involving a balance between entropy, enthalpy, and free energy of the hydrogel and the surrounding water molecules. The unique properties of thermo‐responsive hydrogels and cross‐linkage capability with various moieties, such as dextran, enable controlled release of drugs or vaccine agents that can be loaded into hydrogels by simple mixing at room temperature. Hydrogels can be tuned to take hours or weeks to disintegrate in the body and release the drug components.[ 113 ]
For instance, PNIPAM becomes insoluble with an increase in temperature to around 32–33 °C and is a major candidate for controlled release applications at physiological conditions.[ 112 ] Furthermore, PNIPAM has inherent adjuvant properties that have been applied to the immune setting. For instance, Shakya et al. demonstrated this effect of this thermo‐responsive polymer by inducing a significant IgG response in conjunction with collagen type II (CII) in a rheumatoid arthritis (RA) mouse model.[ 23 ] Other thermo‐responsive biomaterials, such as PVC, are soluble in water and organic solvents with a transition temperature of 33 °C.[ 24 ] Further, PMVE has phase‐change temperature of ≈36 °C, making it very remarkably useful for physiological activation of drugs.[ 25 ] Other modified thermo‐responsive polymers include poloxamer 407, consisting of polyethylene oxide (PEO) and polypropylene oxide (PPO) as well as chitosan‐methyl cellulose (MC) formulations. These polymers are free‐flowing liquids at room temperature and transform to stable gels at physiological conditions, with the ability to create an immobilized biomaterial depot for sustained antigen release and induced immunological response. For instance, one thermo‐responsive therapy involved subcutaneous injections of MC formulations containing soluble antigens and adjuvants in the dorsal skinfold of C57BL/6 mice demonstrated antigen release of up to 14 days after administration.[ 26 ] Alternatively, Zhang et al. also developed a novel drug‐loaded PNIPAM hydrogels coated in a dialysis membrane that demonstrates rapid release of the drug components at increased temperatures to account for inflammation and disease conditions that require rapid responsive release of drugs.[ 27 ] This specialized hydrogel released the model drug 5‐fluorouracil at a high rate under increased temperature conditions of 37 °C compared to 10 °C. More recently, researchers have developed pentablock copolymers made of PEG, poly‐caprolactone (PCL) and poly‐lactic acid (PLA) for sustained release of vaccines.[ 28 ] These copolymers exhibited increased stability, sustained release of immunomodulatory agents and elicitation of a more potent and prolonged immune response compared to an earlier generation of triblock copolymers.[ 26 ] In vivo studies with pentablocker polymers demonstrated sustained release and significantly higher CD8+ T cell expression compared to other formulations of triblock hydrogels or poly(lactic‐co‐glycolic acid) (PLGA) NPs.[ 28 ] Additionally, thermo‐responsive gel formulations have been developed for delivery of adjuvants sublingually. The vaccine formulation is delivered as a liquid and upon administration at physiological temperatures, gel formation enables vaccine antigens to adhere to mucosal surfaces and reach targeted cells. Furthermore, the gel form protects vaccine components from degradation by salivary enzymes and allows for slow controlled release without rapid clearance.[ 29 , 114 ]
Future smart vaccination strategies could further take advantage of immunomodulatory and thermo‐responsive properties of hydrogels to allow prolonged controlled release of vaccine adjuvants. Hypothetically, sustained vaccine efflux could prime the innate and adaptive immune response for long‐term and more potent immunological memory. A novel vaccine formulation can exploit the liquid to gel formation of hydrogels to carry vaccine agents via oral administration both avoiding needle injection discomfort and greatly enhancing the immune response. The vaccine formulation would form a gel upon entering the body serving as a depot for gradual vaccine release rather than multiple vaccination strategies in conventional vaccination series.
Beyond hydrogels, ThermoDox, a lysolipid thermal‐responsive liposome, is currently in phase III clinical trials for efficient delivery of chemotherapeutic agents in primary liver cancer. The most recent data demonstrated increased patient survival by 2.1 years over the placebo treatment and is a promising alternative to current treatments.[ 30 ] Also, implantable thermo‐regulated drug delivery systems have been developed to activate drug‐release via pulsating Peltier device, an element creating heat flow between two surfaces.[ 31 ] Exploiting thermo‐regulation of the body at 37 °C, PNIPAM with LCST at 32 °C can be utilized for rapid release of drug by cooling the implantable device and slow release at physiological temperature. Although the test was executed in vitro, further in vivo testing may demonstrate a novel way to control drug release via external temperature stimuli.[ 31 ] Perhaps, a skin‐patch like Peltier device would allow for site‐specific controlled drug release of intradermal drug injections allowing to target specific cancer lesions under the skin and tissues.
3.3. Enzyme‐Responsive Materials
Site‐specific enzymatic activity enables a powerful class of enzyme‐responsive drug delivery materials for activation of drug load at pathologically active sites, such as inflamed or cancerous tissues. Enzymes play a major role in cellular and metabolic processes and their dysregulation underscores the pathology of many diseases, rendering enzymatic expression as potential targets for bio‐catalytically activated drug delivery modalities. For instance, malignant tumors possess genes encoding specific enzymes, which can be exploited to unmask anticancer drugs at the site of the tumor. Overexpression of various enzymes (e.g., cathepsin B or matrix metalloprotease 2 (MMP2)) at tumor sites compared to their relatively low expression in healthy tissues empowers formulation of anticancer vaccines for enzyme‐dependent targeted and controlled release of drug agents. As such, novel nanomaterials, including polymeric, phospholipids, and inorganic materials, have been utilized with integration of enzyme‐cleavable moieties in their side groups or main chain.[ 115 ]
A commonly used enzyme‐responsive material is liposome formulations. For instance, liposome nanocarriers have been synthesized to be enzymatically activated at high concentrations of MMP2 protease, a hallmark of solid tumors, and release anticancer drugs. These liposomes are decorated with a longer length PEG (3400 MW) containing an MMP2‐cleavable linker that function to prevent non‐specific interactions and also hide a second layer of shorter PEG strands (2000 MW) which display cell‐penetrating peptides. At tumor sites, MMP2 substrates are cleaved and protective long‐chained PEGs are detached, thus allowing for exposure of cell‐penetrating components on liposomes and their entry into tumor cells to release their drug payload intracellularly.[ 32 ] Similarly, MMP2‐responsive mesoporous silica NPs have been created for release of antitumor drugs, such as doxorubicin hydrochloride (DOX), at the microenvironment of tumors. In this vein, Liu et al. subcutaneously injected HepG2 cells in nude mice to induce tumor formation. After 4 days, 20 nude mice with similar tumor sizes were treated with different formulations of DOX NPs. A significant reduction in tumor weight was observed for the NP treated group compared to the control saline group with free administration of DOX.[ 33 ]
While extracellular enzymes have been targeted in enzyme‐responsive drug delivery systems, intracellular lysosomal enzymes have also shown great promise for controlled release of drug agents. Halloysite nanotubes (HNTs) coated with dextrin and loaded with drugs are used as an efficient carrier for intracellular controlled release of drug through glycosyl hydrolase cleavage of dextrin.[ 34 ] These HNTs are tubule aluminosilicate clays with an external diameter of 50–60 nm and internal diameter of 12–15 nm with a length of about 1 × 10−6 m.[ 116 ] The surface dextrin coating serves as a cap on the end of the hollow tubes to prevent drug leakage until it is cleaved at the targeted site. The HNTs are readily taken up by cells and hydrolysis of the dextran cap by cellular glycosyl hydrolases releases the drug payload intracellularly. Other enzyme‐induced drug delivery systems exploit the high cathepsin B expression in lysosomes of tumor cells for targeted anticancer drug release. Mesoporous silica NPs surface‐functionalized with a cathepsin B cleavage sequence, Gly‐Phe‐Leu‐Gly (GFLG), and tumor‐targeting sequence, Arg‐Gly‐Asp‐Ser (RGDS), function to release loaded drug upon cleavage of the capping peptide, therefore enhancing antitumor activity.[ 35 ]
3.4. Redox‐Responsive Materials
Responsive biomaterials have also been developed to respond to key discrepancies in the reduction and oxidation (redox) environment in the body. Redox reactions involve transfer of electrons between chemical reagents, breaking old covalent bonds and forming new ones; in tissues, the redox state is determined by the balance between various oxidative species and antioxidants. Redox‐responsive drug delivery platforms exploit reducible chemistries at the target sites to break bonds anchoring drug molecules and release drug payload. This strategy enables drugs to remain inactive until delivered to the targeted site and reduce undesirable off‐target effects. In this realm, disulfide bonds are of great interest as they are prone to cleavage by glutathione (GSH), found intracellularly and also in excess concentration at cancerous tissues. Micelles have been developed from self‐assembled amphiphilic copolymers that contain GSH‐cleavable crosslinking reagents within the core or outer layer of the structure. This design allows for rapid disassembly of the micelle in GSH‐containing target tissues and cell compartments to release drug payload in a controlled manner.[ 117 ] For instance, a targeted intracellular drug delivery method utilizing disulfide bond‐bridged nanocarriers loaded with DOX has shown greater efficacy and less cytotoxicity when DOX is released at the tumor site due to the reductive intracellular environment.[ 36 ] A similar approach has also be used to co‐deliver DOX and the indoleamine 2,3‐dioxygenase (IDO) inhibitor NLG919 through disulfide containing polymeric micelles to tumor cells with high efficacy (Figure 4 ).[ 37 ] Redox‐responsive materials targeting tumor sites have also been combined with adoptive cell cancer immunotherapy. Xie et al. developed redox‐responsive nanogels that release IL‐2, a potent cytokine for expanding T cells during cancer therapies.[ 38 ] These nanogels can backpack onto T cells prior to adoptive T‐cell transfer cancer therapy and induce T cell expansion at the tumor site via IL‐2 release. Additionally, Li et al. demonstrated that hyaluronic acid (HA)‐deoxycholic acid micelles modified with disulfide bioreducible linkages mitigate cytotoxicity and enable intracellular delivery of paclitaxel‐loaded micelles. In vivo investigation of paclitaxel micelles in tumor‐bearing mice exhibited more than 2.5 fold tumor targeting capability relative to free drug control.[ 39 ] These nanocarriers are among a few of the many different modified biomaterials, including chitosan[ 40 ] and PEG‐PCL micelles,[ 41 ] which have been developed to respond to intracellular redox cues to release drug molecules.
Figure 4.
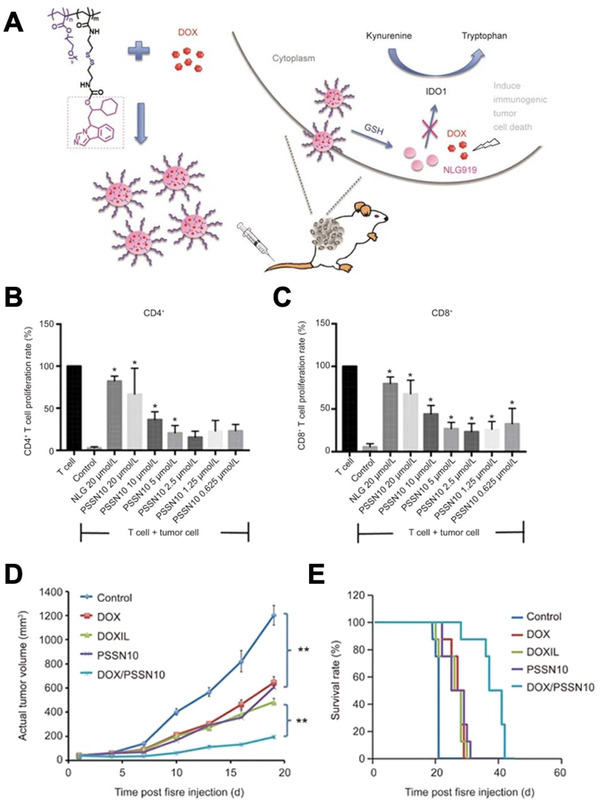
A) Diagram showing a redox‐responsive anti‐cancer immunotherapy consisting of PSSN10 micelles for co‐delivery of the IDO inhibitor NLG919 (NLG) and loaded DOX. Introduction of the micelles into the cytoplasm of tumor cells blocks immune suppressive pathways through NLG and induces cytotoxicity through DOX. B,C) PSSN10 micelles induce an increase in CD4+ and CD8+ T cell proliferation in the presence of tumor cells comparable to free NLG and positive control conditions. D,E) PSSN10 micelles loaded with DOX prevent tumor growth and increase survival rate in a mouse model when compared to free DOX, DOXIL (clinical liposomal form of DOX), unloaded PSSN10, and untreated conditions. Adapted with permission.[ 37 ] Copyright 2017, Nature.
Redox‐responsive materials may also take advantage of the accumulation of reactive oxygen species (ROS) at various target sites, including inflammation and cancer. ROS include hydrogen peroxide (H2O2), superoxides and hydroxyl radicals, which are generated excessively by mitochondria under environmental stress and increased metabolic activity. Similar to drug delivery platforms responsive to GSH, ROS‐responsive nanocarriers stay dormant until at the target site, where they change conformation to release drug components. ROS‐reactive drug carriers may be assembled through numerous chemical linkages exploiting diselenides, arylboronic esters, thiolethers, aryloxalates, or ferrocene, which react with H2O2 to unload drug cargo.[ 118 ] For instance, Ma et al. developed an amphiphilic block copolymer, PEG‐PUSeSe‐PEG, which self‐assembles in aqueous conditions to form micelles that can disassemble by breakage of Se‐Se bonds in presence of H2O2. Additionally, a ROS‐reactive NP system was developed linking HA and D‐α‐tocopherol polyethylene glycol 1000 succinate (TPGS) using arylboronic ester, which readily degrades in redox environment. The release of TPGS would induce ROS regeneration and further disassembly of the NPs. The antitumor efficacy and cancer targeting was demonstrated in tumor‐bearing mice by significant reduction of tumor size and increase in fluorescent intensity in tumors 24 h post‐injection.[ 42 ]
Another class of redox responsive materials is those responding to hypoxia, a condition of low oxygen pressure within the tissue. These materials can target solid tumor tissue, known to contain hypoxic microenvironments. For example, Thambi et al. developed a hypoxia‐responsive polymeric NP composed of a hydrophobically modified 2‐nitroimidazole derivative conjugated to carboxymethyl dextran.[ 43 ] These NPs successfully released the anticancer drug DOX at a higher rate with higher toxicity to hypoxic cancer cells compared to control normoxic cells. Also, Zhang et al. created another hypoxia responsive NP made up of the hydrophobic small molecule, 4‐nitrobenzyl (3‐azidopropyl) carbamate and mPEG‐poly(γ‐propargyl‐L‐glutamate) copolymer chains, with promising results successfully delivering DOX to cancer cells.[ 44 ]
Intrinsic stimuli‐responsive materials can be a powerful tool to release immunotherapeutics in a conditional manner. As outlined in this portion of the review, there are many promising materials that take advantage of changes in pH, temperature, enzymatic activity, and redox environment. However, with these parameters, the exact levels of the intrinsic properties vary depending on the individual and other variables, such as physiological condition. This may result in unwanted material accumulation or activation in off‐target sites. Furthermore, these many of these technologies have yet to be clinically tested, so full understanding of their behavior in the body and toxicity is unknown. Nevertheless, with future improvements and testing of these materials have strong potential to rapidly advance future vaccines and immunotherapies.
3.5. Photo‐Responsive Materials
While internal stimuli, such as pH, temperature, enzyme, and redox are crucial to stimuli responsive drug delivery systems, external stimuli also play a major role in release of drug cargo at the target site. Specifically, peripheral interventions such as light, lead to high spatiotemporal control of drug release. Photo‐controllable drug delivery systems offer many advantages, including noninvasive stimulation, ease of administration, and most notably high precision. The ability to focus light with tunable irradiation conditions on specific tissues empowers precise wavelength‐specific activation and drug release, thus significantly reducing off‐target cytotoxicity. For instance, organic NPs composed of acridin‐9‐methanol achieve both nuclear targeting and photo‐controlled release of drug agents.[ 45 ] Acridin‐based compounds have unique features that allow for tight binding to DNA and disassociation upon light stimulation, enabling cell nucleus targeting and temporal control over drug release. Additionally, Barman et al. developed a multi‐responsive, anti‐cancer drug delivery system for sequential pH‐induced activation and optically activated drug release. For this, they caged drugs in acid‐activated spiropyran and photo‐responsive shells composed of coumarin moieties enabling site‐specific and dose‐dependent release of drugs at the tumor site through an external light source.[ 78 ]
A myriad of liposomes, polymeric micelles, polymerosomes, nanogels, and mesoporous silica NPs with crosslinked photo‐reactive groups have been developed for localized and controllable drug release.[ 119 ] For example, DC immunotherapy vaccines composed of light‐responsive, pheophorbide‐A grafted polyethylenimine OVA NPs have been developed to enable rupture of the phagolysosome upon light exposure and release of immunomodulatory agents in the cytosol to induce desired immune activation. The release of these agents in the cytosol facilitates a shift from MHC II to MHC I presentation and therefore enhanced CD8+ T cell response resulting in a more potent anticancer immune response.[ 46 ] Additionally, photodegradable hydrogels have been developed to spatiotemporally control stiffness and topography of fibroblast to myofibroblast transition in presence of valvular interstitial cells (VICs) in real‐time. Through mass‐loss upon illumination with light, the hydrogels can modulate VIC phenotype towards myofibroblast phenotype. Photodegradability is achieved by integration of a photolabile nitrobenzyl ether moiety into a PEG‐azide.[ 47 ] Moreover, Boto et al. recently demonstrated remote activation of light‐inducible retinoic acid (RA)‐encapsulated NPs to accumulate in leukemia cells and activate by exposure to UV light. RA enables differentiation of leukemia cells and is a therapeutic strategy for blood cancers. Light‐degrading NPs disassemble in response to light and can be triggered in the cytoplasm of leukemia cells to prevent off‐target cytotoxicity (Figure 5 ).[ 48 ]
Figure 5.
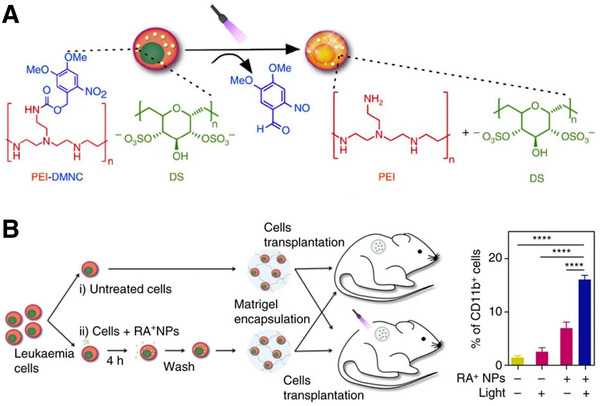
A) Design of a photo‐responsive NP therapy (RA+NPs) that allows for triggered release of RA within leukemia cells. B) Leukemia cells were either treated or untreated with RA+NPs, then encapsulated in Matrigel and implanted in mice. Light therapy successfully induced differentiation of leukemia cells in the RA+NP‐treated group shown by CD11b‐positive populations. Adapted with permission.[ 48 ] Copyright 2017, Nature.
Another fascinating subarea of light‐responsive therapeutics is photodynamic therapies (PDTs). These therapies consist of a photosensitizer, light, and oxygen. The photosensitizer is a photosensitive molecule that can be administered intravenously or topically. Upon stimulation by light, the photosensitizer transfers the light energy to tissue oxygen, locally generating ROS. This class of therapy has been extensively studied for use as an anti‐tumor treatment. These photosensitizers can be activated by light while within the tumor, releasing ROS. There are three mechanisms by which PDTs are effective against tumors.[ 120 ] First, the direct damage via photosensitizer‐induced oxidative stress directly kills many tumor cells within the stimulated area. Second, PDT damages the tumor vasculature, thereby reducing nutrient availability to cancer cells. Lastly, PDT simulates an immune response at the tumor site. The phototoxic damage causes inflammation and increases immune cell infiltration. This allows for DC uptake of tumor‐associated antigens from tumor cells that have undergone necrosis or apoptosis, increasing the probability of anti‐tumoral immunity.[ 121 ] Many PDTs have been through clinical trials and approved.[ 49 ] For example, porfimer sodium (Photofrin) has been clinically approved for bladder cancer and 5‐aminolevulinic acid (Levulan) has been approved for treating skin cancer. Researchers have also investigated combining these photo‐responsive drugs with biomaterial nanocarriers to achieve controlled delivery while protecting the photosensitizer from enzymatic degradation. For example, Ohulchanskyy et al. developed a silica NP covalently modified with the photosensitizer iodobenzylpyropheophorbide that had high tumor cell targeting properties with retained phototoxic effects.[ 50 ] Other formulations have been developed such as solid lipid NPs loaded with hypericin, a natural photosensitizer.[ 51 ]
3.6. Molecular Recognition‐Responsive Materials
A relatively newer development in the smart materials realm is the design of biomaterials that can release therapeutic cargo in response to the binding of a definite molecule(s). These molecular recognition‐responsive materials all share a similar method of action involving competitive binding to a target, allowing for conditional release of cargo. A major advantage of these materials is that they have high specificity, allowing for controlled, targeted delivery only to sites where large enough amounts of analyte are located. Target locations for drug release are usually characterized by higher levels of signature surface receptors or signaling proteins. Researchers are using various techniques to create materials that can release therapeutics in response to binding to location‐specific, disease‐associated molecules. Molecularly imprinted polymer (MIP) formulations can be tailored to possess a high affinity for an antigen or ligand template molecule. These materials can be created by mixing the template molecule with free monomers and crosslinking agents. After polymerization, the template molecules can be washed out, leaving behind a recognition scaffold with specific affinity for the molecules of interest. These MIP hydrogels can undergo physical changes in response to binding to their specific template molecule. For example, a molecularly imprinted hydrogel was created using lectin and antibody molecules to shrink in response to a target tumor‐specific biomarker glycoprotein α‐fetoprotein (AFP).[ 52 ]
Another method for recognizing target ligands is use of aptamer‐crosslinked hydrogels. Aptamers are structures of folded nucleic acids that can recognize proteins and other molecules with high specificity and sensitivity. These aptamer sequences can be duplexed with complementary sequence of DNA in a hydrogel. In the presence of an increased concentration of aptamer targets, the aptamer will disassociate with its complementary strand to competitively bind to its target, thereby degrading the hydrogel and releasing any contained drugs. For example, Yang et al. developed different aptamer‐based hydrogels that can specifically respond to adenosine or thrombin.[ 53 ] Mo et al. created a similar nanogel as a nanocarrier that can respond to excess levels of ATP associated with cancer metabolism.[ 54 ] These nanocarriers are composed of the anticancer drug DOX, a duplexed ATP‐binding aptamer, crosslinked HA and protamine. At the tumor site, HA is degraded by tumor hyaluronidases and DNA aptamers bind to ATP, causing the structure to release DOX.
The previously discussed molecular‐responsive materials are responsive to intrinsic stimuli. However, materials sensitive to extrinsic molecular stimuli have also been investigated. Gübeli et al. created an implantable drug depot made of a PEG hydrogel with anti‐fluorescein single‐chain antibody fragments (scFvs).[ 55 ] This formulation can be injected intramuscularly to create a depot of vaccine cargo that can be released upon remote‐controlled activation via an oral dose of fluorescein. A variation of this approach in a vaccine platform as a booster dose was shown to be effective in immunization against hepatitis B. This vaccine hydrogel depot was designed using the antibiotic molecule novobiocin as the stimulus for timed release.[ 56 ] The externally molecularly triggered group had a higher hepatitis B surface antigen‐specific antibody titer than the non‐stimulated group and similar level to the booster dose treated group, demonstrating this method's efficacy (Figure 6 ). By circumventing the need for multiple vaccine booster injections, these therapies could be a valuable tool for increasing patient compliance.
Figure 6.
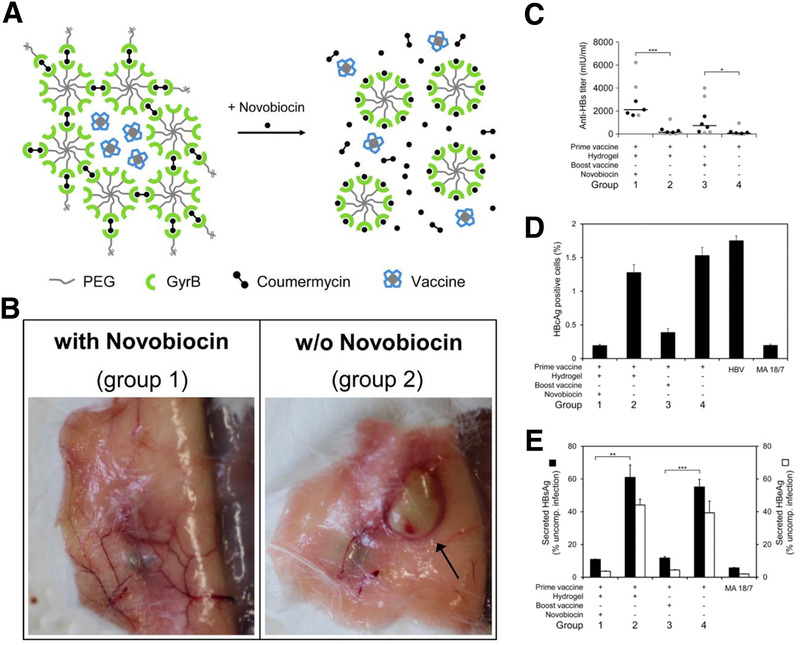
Example of a molecule‐responsive immunotherapy. A) This treatment consists of a vaccine‐loaded PEG hydrogel crosslinked using the protein gyrase B (GyrB) and coumermycin. Upon addition of the antibiotic novobiocin, the GyrB binds to novobiocin, undoing the crosslink and allowing for triggered release of the vaccine. B) Visual confirmation of hydrogel degradation upon novobiocin treatment. C–E) Mice given a primary vaccine then booster vaccine via hydrogel activated by novobiocin showed increased anti‐hepatitis B (HB) Abs, lower percentages of HB‐positive cell, and lower amounts of secreted HB antigen. Adapted with permission.[ 56 ] Copyright 2013, Nature.
3.7. Magnetic‐Responsive Materials
Drug delivery approaches have exploited other external triggering and targeting stimuli, including magnetism. In this realm, controllable magnetic NP systems rely on external magnetic guidance, local drug activation by induction heating with the alternating magnetic field, or a combination of both for therapeutic applications. Additionally, magnetite NPs (MNPs) can be detected noninvasively through magnetic resonance imaging (MRI), a widely used medical imaging technique that utilizes a magnetic field for imaging anatomical or physiological processes in the body. As such, there is an incredible opportunity for multifunctional MNP formulations which enable simultaneous magnetic drug delivery, MRI diagnostic and thermo‐sensitive chemotherapy for enhanced cancer therapeutics.[ 57 ]
One approach that has exhibited great promise in targeted drug delivery for cancer therapeutics is magnetic NP‐based hyperthermia. Magnetic hyperthermia is the result of remote heating of magnetic NPs by inducing oscillating current resulting in magnetic energy loss in the form of heat.[ 122 ] In 2013, European Medicines Agency approved NanoTherm for treatment of primary and recurrent glioblastoma.[ 22 ] In this therapy, aminosilane‐coated MNPs are injected intratumorally and an alternate magnetic field is applied for local heating of MNPs, thus destroying the tumor or sensitizing it for other cancer therapies, including immunotherapy.[ 58 ] Clinical trials for this therapy have demonstrated significant tumor size reduction and increase in patient survival rates when used in combination with radiotherapy.
Materials have also been developed to use magnetic field‐responsivity to travel to and remain localized at a specific site within the body following injection. Li et al. created a novel cancer vaccine composed of Fe3O4 magnetic nanoclusters (MNCs) that are coated with cancer cell membranes, decorated with CD8+ DC‐targeting anti‐CD205, and loaded with CpG‐ODN adjuvant.[ 59 ] Following a subcutaneous injection of MNCs, an applied magnetic field was used to target of the LNs using MRI as a guide. This therapy successfully guided and retained the nanotherapy to the LN, where cancer antigens and CpG‐ODN adjuvant were delivered to CD8+ DCs for a potent cellular immune response.
Another application of magnetic‐responsive NPs is in gene transfection and gene vaccine delivery. Magnetofection, magnetic‐assisted transduction using MNPs, has empowered highly efficient and effective transfection method without physical damage to the cell, unlike electroporation and other invasive methods.[ 123 ] In this transfection method, a magnetic field is applied at the desired site of transfection to localize the injected DNA‐coated MNPs and induce transduction via the close association of cells and MNPs. Al‐Deen et al. demonstrated effective delivery of a Malaria DNA vaccine in vitro with significantly greater transfection efficiency using magnetofection than previously achieved using other transfection methods.[ 60 ] Similarly, MNPs enhanced adenovirus transduction efficiency in vivo and in vitro thus minimizing off‐target adverse effects.[ 61 ]
3.8. Ultrasound‐Responsive Materials
Microbubbles, commonly used in ultrasound imaging, are increasingly prevalent in targeted drug delivery systems. Ultrasound‐reactive nanocarriers hold great promise as multifunctional platforms for therapeutic and diagnostic applications. The effect of ultrasound on micron‐ or nano‐sized particles and their target owe to mechanical and thermal processes triggering drug release by disruption of carrier vesicles through cavitation.[ 124 ] Similar to aforementioned MNPs, researchers have shown that particles at tumor sites can resonate via irradiation with high intensity ultrasound to produce heat for successive release of drug payload in the presence of thermo‐responsive polymers. The benign nature and controllable intensity of ultrasound enables a powerful class of targeted delivery vehicles, especially in destroying tumors surrounded by sensitive tissue, such as in glioblastoma. Drug‐encapsulated liposomes can release cargo by local irradiation of low intensity ultrasound without damage to surrounding tissue, thereby enabling highly specific and targeted drug delivery at the tumor site.
Thermo‐sensitive polymers work in conjunction with ultrasound‐induced localized hyperthermia to achieve rapid tumor destruction. Ninomiya et al. utilized calcein‐loaded liposomes modified with the thermo‐sensitive polymer, poly(NIPMAM‐co‐NIPAM), and crosslinked with single stranded DNA aptamers for targeting cancer cells. Upon exposure to 1 MHz ultrasound irradiation, liposomes collapsed due to local heating and released calcein.[ 62 ] Additionally, another efficacious ultrasound‐responsive formulation of phosphatidylserine (PS)‐based paclitaxel‐liposomes‐nanobubble conjugates (PSPLBC) have been investigated as an anticancer therapy for ultrasound‐sensitive image‐guided delivery of paclitaxel to tumor sites with promising results (Figure 7 ).[ 63 ] Moreover, FDA‐approved perfluorocarbon (PFC) ultrasound contrast agents currently used in ultrasound imaging can transform into a drug delivery system activated by exposure to external low frequency ultrasound. These droplets can transform into microbubbles and undergo cavitation with ultrasound irradiation, thus facilitating drug release.[ 117 ]
Figure 7.
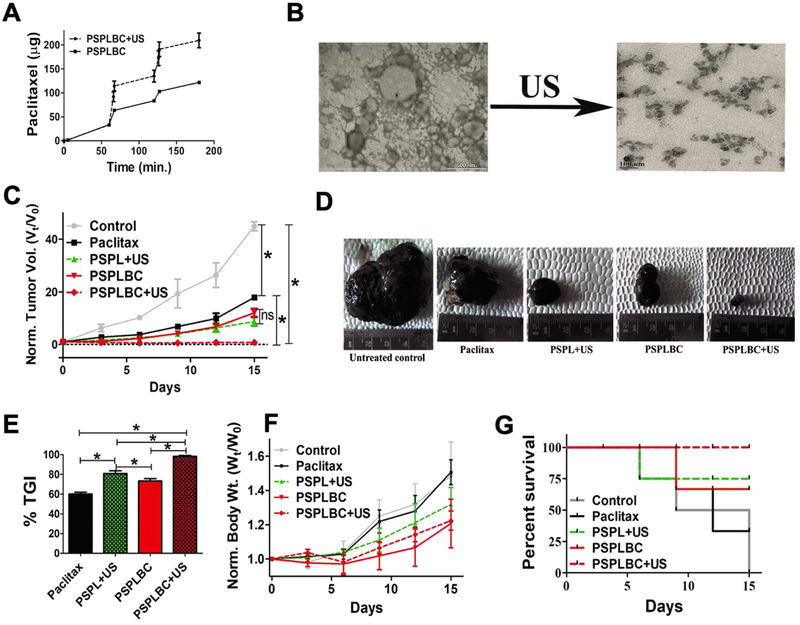
Ultrasound‐responsive drug delivery through PSPLBC. A) PSPLBCs release paclitaxel at a faster rate while exposed to ultrasound. B) Disruption of PSPLBCs confirmed through microscopy. C–G) PSPLBs displayed high antitumor efficacy shown through reduced tumor volume, higher % tumor growth inhibition (TGI), lower normalized body weight, and increased percent survival. Adapted with permission.[ 63 ] Copyright 2018, Nature.
It has also been demonstrated that gene therapy efficiency is enhanced using bubble liposomes which get activated with ultrasound exposure.[ 64 ] Un et al. developed an even more precisely targeted version of this gene delivery ultrasound system using a mixture of plasmid DNA encapsulated mannosylated lipoplexes, selective for macrophages and DCs, and bubble liposomes to enhance transfection with ultrasound. Applying ultrasound externally at the treatment site induces collapse of the liposomes and release of the plasmid DNA for more efficient transfection.[ 65 ] This APC‐targeting gene delivery technology could be used for new vaccines and other immunotherapies in the future.
3.9. Electric‐Field‐Responsive Materials
Applied electric fields are another external stimulus currently under investigation for delivery of therapeutic molecules. An advantage of using electric signals is that they do not require the large or specialized equipment needed for ultrasound, light, or magnetic signals. Additionally, these stimuli are easy to generate and control, allowing for facile, triggered release from electrically responsive materials.
Most polymers that exhibit electric‐responsive characteristics are polyelectrolyte hydrogels. These gels are composed of ionized (typically cationic) monomer units; the resulting gels have high water content, allowing for ion transport when electrically stimulated. Many of these materials have the unique property to shrink in response to an electric field, resulting in a forced convection of the entrapped drug cargo out of the gel matrix. For this reason, several polyelectrolyte gels have been investigated for use in electric‐stimulated delivery of therapeutic molecules. Natural polyelectrolyte hydrogels characterized include HA,[ 66 ] alginate,[ 67 ] chondroitin sulfate,[ 68 ] and chitosan.[ 125 ] Also synthetic polymer electric‐responsive gels have been developed from several materials including poly(vinyl alcohol)[ 69 ] and acrylonitrile.[ 70 ] Precisely controlled delivery of the anti‐inflammatory drug hydrocortisone has been achieved using chitosan,[ 125 ] sodium alginate/polyacrylic acid composites,[ 71 ] and agarose[ 72 ] polyelectrolyte gel formulations.
Another type of electric field responsive material is conductive polymers. Electrical stimulation of these materials results in a change in redox state that affects the release rate of the loaded therapeutics. Also, modification of the type and concentration of dopants (counter‐balancing ions used for formation of the polymer) during synthesis can affect its properties like stability and solubility. A widely studied conductive polymer is polypyrrole (PPy). By applying current through a PPy‐coated electrode, therapeutic drugs can be released in a controlled manner.[ 73 ] However, these delivery systems are limited in the molecular weight of the drug and choice of dopant. To address these issues, George et al. developed a platform system for attaching a range of molecules to a biotin dopant through biotin‐streptavidin coupling.[ 74 ] Electrical stimulation results in reduction of the PPy backbone, releasing the biotin and drug cargo.
For the above‐mentioned conductive materials, drug release and responsiveness depend on the available surface area. To circumvent this limitation, micro‐ and nano‐structured formulations have been developed for highly controllable delivery. For example, PPy can be formed into a conductive NP which can subsequently be embedded into a PLGA‐PEG‐PLGA temperature‐sensitive hydrogel that is liquid at room temperature but gels after injection into the body.[ 75 ] The resulting electric field responsive drug delivery system has high spatial, temporal, and dosage control. Another conductive polymer, poly(3,4‐ethylenedioxythiophene) (PEDOT), can be deposited as nanotubes on a drug‐loaded layer of electrospun PLGA nanoscale fibers.[ 76 ] When these nanotubes are actuated with an electric pulse, the pressure created releases the anti‐inflammatory drug dexamethasone from the PLGA matrix. This coating could potentially prevent unwanted immune responses on implants. Another electric field responsive material used to release dexamethasone is carbon nanotubes sealed with PPy films.[ 77 ] The drug is loaded into the inner cavity of the nanotubes and, upon electrical stimulation of the PPy layer, the anti‐inflammatory can be released in a controlled manner.
Extrinsically activated materials are a very exciting branch of stimuli‐responsive immunotherapy with many activation source options including light exposure, molecule binding, magnetic fields, ultrasound, and electric fields. However, a significant drawback of these stimuli is the requirement of specialized, expensive instrumentation. Molecule‐binding materials do not require equipment but are extremely difficult to design and fabricate. Furthermore, electrically stimulated materials have limitations in tissue penetration depth, so drug delivery is limited to topical or subdermal localization. Despite these disadvantages, many externally activated materials have been studied with positive results with an optimistic future considering that multiple of these therapies including PDT and magnetic hyperthermia have been clinically approved.
3.10. Multi‐Stimuli‐Responsive Materials
So far, we have discussed the application of single‐stimuli materials in the context of vaccine and immunotherapeutic drug delivery. More recently, scientists have started exploring the development of combinatorially stimulated materials for even greater control and precision of delivery and action of immunotherapeutic agents. Figure 8 outlines a hypothetical design of a therapy that combines multiple internal and external stimuli for high spatiotemporal control of drug release. One example of a multi‐stimuli‐responsive therapy is pH‐ and light‐responsive micelles that allow for triggered release of therapeutics at tumor sites as an effective cancer therapy.[ 78 , 79 ] The spatiotemporal fine‐tuning of stimuli prevents off‐target cytotoxicity associated with many drugs, including chemotherapy agents and enhances drug‐release for a more potent response. Another example of multi‐stimuli‐responsive materials is where dual‐responsive pH and redox saccharide‐based NPs were designed to induce maximal drug release specifically at the tumor site, a low pH and reductive GSH microenvironment.[ 80 ] Moreover, Wang et al. developed NPs made of a thermo‐responsive Pluronic F127 co‐polymer, a HA cancer homing moiety and a pH‐reactive chitosan/HA combination encapsulating DOX and hydrophobic irinotecan (CPT) anticancer drugs for an exceptionally enhanced anti‐cancer response.[ 81 ] Further, improved tumor ablation without regrowth was observed in mice treated with light, pH, and reduction responsive NPs encapsulating chemotherapeutic agents.[ 82 ] Li et al. developed keratin NPs that can respond to pH, reduction (GSH), and enzymes (trypsin) to achieve triple‐stimuli responsive co‐delivery of nitric oxide and DOX at cancerous tissues.[ 83 ] These particles demonstrated promising in vivo anti‐tumor efficacy similar to free DOX but with lowered side effects. Another interesting multi‐stimuli‐responsive therapy developed by Im et al. uses silica nanocarriers that respond to the hypoxic tumor microenvironment. The nanocarrier is a chlorin e6‐doped‐azobenzene‐glycol chitosan‐PEG mesoporous silica NP. In these conditions, the particles will detach the PEG portion and release the adjuvant CpG ODN to stimulate DCs and, upon combinatorial light activation, the photosensitizer chlorin e6 will disrupt the tumor tissue releasing cancer‐specific antigen for immunization.[ 84 ]
Figure 8.
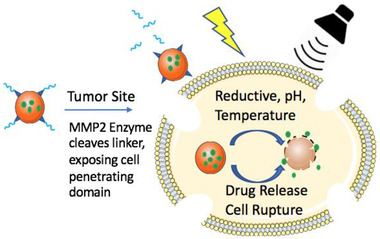
Hypothetical multi‐responsive drug delivery system. The proposed particle would respond to MMP2 enzyme cleavage to activate a cell penetrating domain, allowing for entry into the intracellular space of the tumor cell. The drug load would be delivered upon further activation via intracellular reductive activity, low pH, and external electric or ultrasound signals.
The disadvantage of such combinatory‐responsive systems is the inherent complexity when using multiple stimuli. Timely and localized release of a drug at the right dosage may be complicated by multi‐stimuli‐responsive particle systems. Further “proof‐of‐concept” studies will be necessary prior to translation of such drug delivery systems to clinical trials.
3.11. Prospective Stimuli‐Responsive Materials
Our discussion has centered on materials that respond to somewhat conventional stimuli. There are now several groups pursuing the design of materials that respond to previously untested stimuli. Some of these materials have not been extensively studied in vitro or in vivo for drug delivery or immunomodulation but hold strong promise for the future.
For example, DNA‐responsive materials could release therapeutic cargo to specific cells. Nucleic acids are advantageous as a stimulus due to their predictable binding kinetics to specific complementary sequences of target cells. These DNA‐responsive materials function by using single‐stranded DNA as a crosslinker in hydrogels. Upon exposure to a complementary target sequence, the DNA crosslinker could competitively bind to the target and the gel would undergo a gel‐to‐solution transition [ 85 ] or volume change[ 86 ] to release therapeutic contents. These materials have been shown to respond to mRNA or DNA sequences with accuracy to a single base‐pair mismatch. While still in the early stages of development, these materials show promise for targeting diseases like cancer that involve dysregulated gene expression or mutations.
As previously mentioned, aptamers can be bound to drugs in hydrogels and respond to targets via competitive binding with complementary DNA strands. These complementary sequences can be externally administered as a biomolecular trigger for timed release. Battig et al. tested two aptamer models for this approach to release vascular endothelial growth factor (VEGF) and platelet‐derived growth factor BB (PDGF‐BB).[ 87 ] These aptamer‐functionalized hydrogels were shown to successfully function as a controlled release platform with adjustable release rates at specified time points.
Other materials have been developed to release drugs after exposure to a high concentration of a specific ion. Extracellular potassium ion concentration is known to be localized to pathological sites in the body. Therefore, Mi et al. developed a smart hydrogel that can regulate drug release via K+ concentrations.[ 88 ] This hydrogel is a 3D crosslinked structure of the copolymer poly(N‐isopropylacrylamide‐co‐benzo‐15‐crown‐5‐acrylamide). The 15‐crown‐5 portion serves as an ion‐signal sensing receptor and PNIPA acts as an actuator. Together, the polymeric gel shrinks in response to K+, thereby releasing the contained drugs at pathogenic sites. Ion‐responsive microcapsule designs have also been investigated. The microcapsule is designed as a porous membrane with linear grafted poly (N‐isopropylacrylamide‐co‐benzo‐18‐crown[6]‐acrylamide) (PNIPAM‐co‐PBCAm) chains in the pores.[ 89 ] Benzo‐18‐crown[6] acrylamide (BCAm) serves as the ionic molecular recognition portion and PNIPAM acts as the swelling/shrinking actuator. When the target ion—in this case Ba2+ or Na+—is present in the microcapsule's environment, the PNIPAM‐co‐PBCAm shrinks to open the pores and release the encapsulated therapeutic cargo.
4. Future Perspectives and Conclusion
Over the past few decades, stimuli‐responsive biomaterials have been extensively researched and developed due to the congruent advances in nanobiotechnology and materials science. Many of such materials are currently in clinical trials for use in diagnosis, imaging and therapy of diseases. Drug delivery systems have advanced leaps and bounds since doxil was approved by the FDA in 1996 as the first nanomaterial delivery system. This delivery system encapsulated the anti‐cancer drug DOX within PEGylated liposomes to evade the immune system and prolong effects.[ 126 ] Since then, many new liposome‐based nanocarriers and various other therapeutic technologies have been approved to fight different types of cancer and immune conditions.
Responsive‐biomaterials could eventually play a major role in immune‐involved disease diagnosis and prevention. Future stimuli‐responsive intradermal smart implants could detect minute fluctuations in tissue microenvironments and the immune system in real‐time to warn of disease progression and autonomously relay the information to physicians and nurses. Apps on mobile phones could continuously monitor the information to make controllable actions via AI‐based algorithms. Smart vaccines or dermal patches could deliver NP therapeutics to specific cells based on cellular microenvironment, body temperature, or blood composition.
Stimuli‐responsive biomaterials will certainly play a vital role in future of nanomedicine. However, there are major hurdles to overcome before successful commercialization of such products. This relies on demonstrating efficacy and safety to pass the FDA regulatory hurdles. Many systems are too complicated to move past preclinical animal models. Therefore, feasibility should be accounted for in the research design. Despite these hurdles, stimuli‐responsive biomaterials are undoubtedly an immensely powerful tool as future immunotherapeutics. The precise nature of these materials is especially important in the context of vaccines and immunotherapies where success hinges on time‐sensitive delivery of cargo to specific tissues and subcellular compartments, while avoiding off‐target toxicity. This review has covered several different types of stimuli‐responsive materials that researchers have investigated; these collectively form an arsenal of tools that can be used for tailored, condition‐specific immunotherapies. With the right research goals and continuous innovation, more stimuli‐responsive biomaterials can transition from the lab‐bench to the clinical trials with the aim of alleviating patient suffering.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
J.S.L. gratefully acknowledges the grants NIH R35GM125012, NIH R03AI138191, and NIH R01AI139399. N.P. acknowledges the grants NIH T32GM099608 and NSF GRFP grant 1650042. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
Biographies
Noah Pacifici received his B.S. in Biological Engineering from Cornell University in 2017. He is currently a Ph.D. candidate in the Biomedical Engineering Department at the University of California, Davis under the supervision of Dr. Jamal S. Lewis. His current research focuses on investigating the mechanisms of vomocytosis, a pathogen escape phenomenon utilized by Cryptococcus neoformans, for the development of immunomodulatory biomaterials.

Amir Bolandparvaz is a Biomedical Engineering Ph.D. candidate at UC Davis with an entrepreneurial drive in multidisciplinary and translational research to improve human health. Amir received his B.Sc. in Biomedical Engineering from UC San Diego. His current research focuses on designing an epitope‐functionalized iron oxide NP system as a prophylactic for autoimmune autism.

Jamal S. Lewis is an assistant professor in Biomedical Engineering at the University of California, Davis. Prior to his professorship, Dr. Lewis was a senior scientist at OneVax, LLC and a postdoctoral associate in the J. Crayton Pruitt Family Department of Biomedical Engineering at the University of Florida, where he also received a Ph.D. in Biomedical Engineering in 2012.

References
- 1. Plotkin S. A., Orenstein W. A., Offit P. A., Vaccines, Saunders, Philadelphia, PA: 2008. [Google Scholar]
- 2. Behravan J., Razazan A., Behravan G., Current Drug Disc. Tech. 2019, 16, 251. [DOI] [PubMed] [Google Scholar]
- 3. Bråve A., Ljungberg K., Wahren B., Liu M. A., Mol. Pharmaceutics 2007, 4, 18. [DOI] [PubMed] [Google Scholar]
- 4. Hanagata N., Int. J. Nanomedicine 2017, 12, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwon Y. J., Standley S. M., Goodwin A. P., Gillies E. R., Fréchet J. M. J., Mol. Pharmaceutics 2005, 2, 83. [DOI] [PubMed] [Google Scholar]
- 6. Murthy N., Thng Y. X., Schuck S., Xu M. C., Fréchet J. M. J., J. Am. Chem. Soc. 2002, 124, 12398. [DOI] [PubMed] [Google Scholar]
- 7. Murthy N., Xu M., Schuck S., Kunisawa J., Shastri N., Fréchet J. M. J., Proc. Natl. Acad. Sci. USA 2003, 100, 4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang L., Bracho‐Sanchez E., Fernando L. P., Lewis J. S., Carstens M. R., Duvall C. L., Keselowsky B. G., Bioeng. Transl. Med. 2017, 2, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fernando L. P., Lewis J. S., Evans B. C., Duvall C. L., Keselowsky B. G., J. Biomed. Mater. Res., Part A 2018, 106, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang X., Yu T., Zeng Y., Lian K., Zhou X., Ke J., Li Y., Yuan H., Hu F., Biomacromolecules 2020, 21, 2818. [DOI] [PubMed] [Google Scholar]
- 11. Nuhn L., Vanparijs N., De Beuckelaer A., Lybaert L., Verstraete G., Deswarte K., Lienenklaus S., Shukla N. M., Salyer A. C. D., Lambrecht B. N., Grooten J., David S. A., De Koker S., De Geest B. G., Proc. Natl. Acad. Sci. USA 2016, 113, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jain R., Standley S. M., Fréchet J. M. J., Macromolecules 2007, 40, 452. [Google Scholar]
- 13. Kwon Y. J., James E., Shastri N., Fréchet J. M. J., Proc. Natl. Acad. Sci. USA 2005, 102, 18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson J. T., Keller S., Manganiello M. J., Cheng C., Lee C.‐C., Opara C., Convertine A., Stayton P. S., ACS Nano 2013, 7, 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Convertine A. J., Benoit D. S. W., Duvall C. L., Hoffman A. S., Stayton P. S., J. Controlled Release 2009, 133, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson J. T., Postma A., Keller S., Convertine A. J., Moad G., Rizzardo E., Meagher L., Chiefari J., Stayton P. S., AAPS J. 2015, 17, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keller S., Wilson J. T., Patilea G. I., Kern H. B., Convertine A. J., Stayton P. S., J. Controlled Release 2014, 191, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bungener L., Serre K., Bijl L., Leserman L., Wilschut J., Daemen T., Machy P., Vaccine 2002, 20, 2287. [DOI] [PubMed] [Google Scholar]
- 19. Yuba E., Kojima C., Harada A., Tana, Watarai S., Kono K., Biomaterials 2010, 31, 943. [DOI] [PubMed] [Google Scholar]
- 20. Watarai S., Iwase T., Tajima T., Yuba E., Kono K., Sci. World J. 2013, 2013, 903234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshizaki Y., Yuba E., Komatsu T., Udaka K., Harada A., Kono K., Molecules 2016, 21, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar A., Montemagno C., Choi H.‐J., Sci. Rep. 2017, 7, 3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shakya A. K., Kumar A., Nandakumar K. S., J. R. Soc., Interface 2011, 8, 1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makhaeva E. E., Tenhu H., Khokhlov A. R., Macromolecules 1998, 31, 6112. [Google Scholar]
- 25. Moerkerke R., Meeussen F., Koningsveld R., Berghmans H., Mondelaers W., Schacht E., Dušek K., Šolc K., Macromolecules 1998, 31, 2223. [Google Scholar]
- 26. Kojarunchitt T., Baldursdottir S., Dong Y.‐D., Boyd B. J., Rades T., Hook S., Eur. J. Pharm. Biopharm. 2015, 89, 74. [DOI] [PubMed] [Google Scholar]
- 27. Zhang X.‐Z., Zhuo R.‐X., Cui J.‐Z., Zhang J.‐T., Int. J. Pharm. 2002, 235, 43. [DOI] [PubMed] [Google Scholar]
- 28. Bobbala S., Tamboli V., McDowell A., Mitra A. K., Hook S., AAPS J. 2016, 18, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White J. A., Blum J. S., Hosken N. A., Marshak J. O., Duncan L., Zhu C., Norton E. B., Clements J. D., Koelle D. M., Chen D., Weldon W. C., Oberste M. S., Lal M., Hum. Vaccines Immunother. 2014, 10, 3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Study of ThermoDox with standardized radiofrequency ablation (RFA) for treatment of hepatocellular carcinoma (HCC) , https://clinicaltrials.gov/ct2/show/NCT02112656 (accessed: June 2020).
- 31. Yang R., Gorelov A. V., Aldabbagh F., Carroll W. M., Rochev Y., Int. J. Pharm. 2013, 447, 109. [DOI] [PubMed] [Google Scholar]
- 32. Zhu L., Kate P., Torchilin V. P., ACS Nano 2012, 6, 3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Y., Ding X., Li J., Luo Z., Hu Y., Liu J., Dai L., Zhou J., Hou C., Cai K., Nanotechnology 2015, 26, 145102. [DOI] [PubMed] [Google Scholar]
- 34. Dzamukova M. R., Naumenko E. A., Lvov Y. M., Fakhrullin R. F., Sci. Rep. 2015, 5, 10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de la Torre C., Mondragón L., Coll C., Sancenón F., Marcos M. D., Martínez‐Máñez R., Amorós P., Pérez‐Payá E., Orzáez M., Chem. ‐ Eur. J. 2014, 20, 15309. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y.‐C., Wang F., Sun T.‐M., Wang J., Bioconjugate Chem. 2011, 22, 1939. [DOI] [PubMed] [Google Scholar]
- 37. Sun J., Chen Y.‐C., Huang Y.‐X., Zhao W.‐C., Liu Y.‐H., Venkataramanan R., Lu B.‐F., Li S., Acta Pharmacol. Sin. 2017, 38, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xie Y.‐Q., Arik H., Wei L., Zheng Y., Suh H., Irvine D. J., Tang L., Biomater. Sci. 2019, 7, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li J., Huo M., Wang J., Zhou J., Mohammad J. M., Zhang Y., Zhu Q., Waddad A. Y., Zhang Q., Biomaterials 2012, 33, 2310. [DOI] [PubMed] [Google Scholar]
- 40. Huo M., Liu Y., Wang L., Yin T., Qin C., Xiao Y., Yin L., Liu J., Zhou J., Mol. Pharmaceutics 2016, 13, 1750. [DOI] [PubMed] [Google Scholar]
- 41. Zhang H., Wang K., Zhang P., He W., Song A., Luan Y., Colloids Surf., B 2016, 142, 89. [DOI] [PubMed] [Google Scholar]
- 42. Su Z., Chen M., Xiao Y., Sun M., Zong L., Asghar S., Dong M., Li H., Ping Q., Zhang C., J. Controlled Release 2014, 196, 370. [DOI] [PubMed] [Google Scholar]
- 43. Thambi T., Deepagan V. G., Yoon H. Y., Han H. S., Kim S.‐H., Son S., Jo D.‐G., Ahn C.‐H., Suh Y. D., Kim K., Kwon I. C., Lee D. S., Park J. H., Biomaterials 2014, 35, 1735. [DOI] [PubMed] [Google Scholar]
- 44. Zhang P., Yang H., Shen W., Liu W., Chen L., Xiao C., ACS Biomater. Sci. Eng. 2020, 6, 2167. [DOI] [PubMed] [Google Scholar]
- 45. Jana A., Saha B., Banerjee D. R., Ghosh S. K., Nguyen K. T., Ma X., Qu Q., Zhao Y., Singh N. D. P., Bioconjugate Chem. 2013, 24, 1828. [DOI] [PubMed] [Google Scholar]
- 46. Zhang C., Zhang J., Sh G., Song H., Shi S., Zhang X., Huang P., Wang Z., Wang W., Wang C., Kong D., Li C., Mol. Pharmaceutics 2017, 14, 1760. [DOI] [PubMed] [Google Scholar]
- 47. Kirschner C. M., Alge D. L., Gould S. T., Anseth K. S., Adv. Healthcare Mater. 2014, 3, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boto C., Quartin E., Cai Y., Martín‐Lorenzo A., Cenador M. B. G., Pinto S., Gupta R., Enver T., Sánchez‐García I., Hong D., das Neves R. P., Ferreira L., Nat. Commun. 2017, 8, 15204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dos Santos A. F., de Almeida D. R. Q., Terra L. F., Baptista M. S., Labriola L., J. Cancer Metastasis Treat. 2019, 5, 25. [Google Scholar]
- 50. Ohulchanskyy T. Y., Roy I., Goswami L. N., Chen Y., Bergey E. J., Pandey R. K., Oseroff A. R., Prasad P. N., Nano Lett. 2007, 7, 2835. [DOI] [PubMed] [Google Scholar]
- 51. Youssef T., Fadel M., Fahmy R., Kassab K., Pharm. Dev. Technol. 2012, 17, 177. [DOI] [PubMed] [Google Scholar]
- 52. Miyata T., Jige M., Nakaminami T., Uragami T., Proc. Natl. Acad. Sci. USA 2006, 103, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang H., Liu H., Kang H., Tan W., J. Am. Chem. Soc. 2008, 130, 6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mo R., Jiang T., DiSanto R., Tai W., Gu Z., Nat. Commun. 2014, 5, 3364. [DOI] [PubMed] [Google Scholar]
- 55. Gübeli R. J., Hövermann D., Seitz H., Rebmann B., Schoenmakers R. G., Ehrbar M., Hamri G. C.‐E., Baba M. D.‐E., Werner M., Müller M., Weber W., Adv. Funct. Mater. 2013, 23, 5355. [Google Scholar]
- 56. Gübeli R. J., Schöneweis K., Huzly D., Ehrbar M., Hamri G. C.‐E., El‐Baba M. D., Urban S., Weber W., Sci. Rep. 2013, 3, 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yallapu M. M., Othman S. F., Curtis E. T., Gupta B. K., Jaggi M., Chauhan S. C., Biomaterials 2011, 32, 1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maier‐Hauff K., Ulrich F., Nestler D., Niehoff H., Wust P., Thiesen B., Orawa H., Budach V., Jordan A., J. Neuro‐Oncol. 2011, 103, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li F., Nie W., Zhang F., Lu G., Lv C., Lv Y., Bao W., Zhang L., Wang S., Gao X., Wei W., Xie H.‐Y., ACS Cent. Sci. 2019, 5, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Al‐Deen F. N., Ho J., Selomulya C., Ma C., Coppel R., Langmuir 2011, 27, 3703. [DOI] [PubMed] [Google Scholar]
- 61. Sapet C., Pellegrino C., Laurent N., Sicard F., Zelphati O., Pharm. Res. 2012, 29, 1203. [DOI] [PubMed] [Google Scholar]
- 62. Ninomiya K., Yamashita T., Kawabata S., Shimizu N., Ultrason. Sonochem. 2014, 21, 1482. [DOI] [PubMed] [Google Scholar]
- 63. Chandan R., Banerjee R., Sci. Rep. 2018, 8, 2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Suzuki R., Takizawa T., Negishi Y., Utoguchi N., Maruyama K., J. Drug Targeting 2007, 15, 531. [DOI] [PubMed] [Google Scholar]
- 65. Un K., Kawakami S., Suzuki R., Maruyama K., Yamashita F., Hashida M., Hum. Gene Ther. 2010, 21, 65. [DOI] [PubMed] [Google Scholar]
- 66. Sutani K., Kaetsu I., Uchida K., Radiat. Phys. Chem. 2001, 61, 49. [Google Scholar]
- 67. Kim S. J., Yoon S. G., Lee Y. H., Kim S. I., Polym. Int. 2004, 53, 1456. [Google Scholar]
- 68. Jensen M., Birch Hansen P., Murdan S., Frokjaer S., Florence A. T., Eur. J. Pharm. Sci. 2002, 15, 139. [DOI] [PubMed] [Google Scholar]
- 69. Kim S. J., Yoon S. G., Lee S. M., Lee S. H., Kim S. I., J. Appl. Polym. Sci. 2004, 91, 3613. [Google Scholar]
- 70. Kim S. J., Shin S. R., Lee J. H., Lee S. H., Kim S. I., J. Appl. Polym. Sci. 2003, 90, 91. [Google Scholar]
- 71. Yuk S. H., Cho S. H., Lee H. B., Pharm. Res. 1992, 09, 955. [DOI] [PubMed] [Google Scholar]
- 72. Hsu C.‐S., Block L. H., Pharm. Res. 1996, 13, 1865. [DOI] [PubMed] [Google Scholar]
- 73. Wadhwa R., Lagenaur C. F., Cui X. T., J. Controlled Release 2006, 110, 531. [DOI] [PubMed] [Google Scholar]
- 74. George P. M., LaVan D. A., Burdick J.‐A., Chen C.‐Y., Liang E., Langer R., Adv. Mater. 2006, 18, 577. [Google Scholar]
- 75. Ge J., Neofytou E., Cahill T. J., Beygui R. E., Zare R. N., ACS Nano 2012, 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Abidian M. R., Kim D.‐H., Martin D. C., Adv. Mater. 2006, 18, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Luo X., Matranga C., Tan S., Alba N., Cui X. T., Biomaterials 2011, 32, 6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Barman S., Das J., Biswas S., Maiti T. K., Singh N. P., J. Mater. Chem. B 2017, 5, 3940. [DOI] [PubMed] [Google Scholar]
- 79. Yu H., Cui Z., Yu P., Guo C., Feng B., Jiang T., Wang S., Yin Q., Zhong D., Yang X., Zhang Z., Li Y., Adv. Funct. Mater. 2015, 25, 2489. [Google Scholar]
- 80. Xu Y., Wang L., Li Y.‐K., Wang C.‐Q., Colloids Surf. A 2016, 497, 8. [Google Scholar]
- 81. Wang H., Agarwal P., Zhao S., Xu R. X., Yu J., Lu X., He X., Biomaterials 2015, 72, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. An X., Zhu A., Luo H., Ke H., Chen H., Zhao Y., ACS Nano 2016, 10, 5947. [DOI] [PubMed] [Google Scholar]
- 83. Li Y., Linb J., Zhia X., Lia P., Jianga X., Yuana J., Mater. Sci. Eng., C 2018, 91, 606. [DOI] [PubMed] [Google Scholar]
- 84. Im S., Lee J., Park D., Park A., Kim Y.‐M., Kim W. J., ACS Nano 2019, 13, 476. [DOI] [PubMed] [Google Scholar]
- 85. Yurke B., J. Biomech. Eng. 2004, 126, 104. [DOI] [PubMed] [Google Scholar]
- 86. Murakami Y., Maeda M., Biomacromolecules 2005, 6, 2927. [DOI] [PubMed] [Google Scholar]
- 87. Battig M. R., Soontornworajit B., Wang Y., J. Am. Chem. Soc. 2012, 134, 12410. [DOI] [PubMed] [Google Scholar]
- 88. Mi P., Ju X.‐J., Xie R., Wu H.‐G., Ma J., Chu L.‐Y., Polymer 2010, 51, 1648. [Google Scholar]
- 89. Chu L.‐Y., Yamaguchi T., Nakao S., Adv. Mater. 2002, 14, 386. [Google Scholar]
- 90. Mogensen T. H., Clin. Microbiol. Rev. 2009, 22, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Iwasaki A., Medzhitov R., Nat. Immunol. 2004, 5, 987. [DOI] [PubMed] [Google Scholar]
- 92. Clem A. S., J. Global Infect. Dis. 2011, 3, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Steinman R. M., Annu. Rev. Immunol. 2012, 300, 1. [DOI] [PubMed] [Google Scholar]
- 94. Sarkander J., Hojyo S., Tokoyoda K., Clin. Transl. Immunol. 2016, 5, e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jackson S. R., Yuan J., Teague R. M., Immunotherapy 2014, 6, 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tsai T. F., Infect. Chemother. 2013, 45, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. HEPLISAV‐B Information, https://www.fda.gov/vaccines-blood-biologics/vaccines/heplisav-b (accessed: July 2020).
- 98. Lewis J. S., Allen R. P., Exp. Biol. Med. 2016, 241, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lewis J. S., Roy K., Keselowsky B. G., MRS Bull. 2014, 39, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Romani N., Flacher V., Tripp C. H., Sparber F., Ebner S., Stoitzner P., Curr. Top. Microbiol. Immunol. 2012, 351, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haniffa M., Gunawan M., Jardine L., J. Dermatol. Sci. 2015, 77, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Toebak M. J., Gibbs S., Bruynzeel D. P., Scheper R. J., Rustemeyer T., Contact Dermatitis 2009, 60, 2. [DOI] [PubMed] [Google Scholar]
- 103. Andorko J. I., Tostanoski L. H., Solano E., Mukhamedova M., Jewell C. M., J. Visualized Exp. 2014, 2, e50984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jewell C. M., López S. C. B., Irvine D. J., Proc. Natl. Acad. Sci. USA 2011, 108, 15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Harris T. J., von Maltzahn G., Lord M. E., Park J.‐H., Agrawal A., Min D.‐H., Sailor M. J., Bhatia S. N., Small 2008, 4, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tomusange K., Wijesundara D., Gummow J., Wesselingh S., Suhrbier A., Gowans E. J., Grubor‐Bauk B., Sci. Rep. 2016, 6, 36658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vila A., Sánchez A., Evora C., Soriano I., Jato J. L. V., Alonso M. J., J. Aerosol Med. 2004, 17, 174. [DOI] [PubMed] [Google Scholar]
- 108. Peppas N. A., J. Biomater. Sci., Polym. Ed. 1998, 9, 535. [DOI] [PubMed] [Google Scholar]
- 109. Kunda N. K., Somavarapu S., Gordon S. B., Hutcheon G. A., Saleem I. Y., Pharm. Res. 2013, 30, 325. [DOI] [PubMed] [Google Scholar]
- 110. Purwada A., Roy K., Singh A., Acta Biomater. 2014, 10, 1728. [DOI] [PubMed] [Google Scholar]
- 111. Travers P., Walport M. J., Janeway C., Murphy K. P., Janeway's Immunobiology, Garland Science, New York: 2008. [Google Scholar]
- 112. Gandhi A., Paul A., Sen S. O., Sen K. K., Asian J. Pharm. Sci. 2015, 10, 99. [Google Scholar]
- 113. Klouda L., Mikos A. G., Eur. J. Pharm. Biopharm. 2008, 68, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lal M., White J., Zhu C., in Vaccine Adjuvants (Ed: Fox C.), Humana Press, New York: 2017, pp. 153–163. [Google Scholar]
- 115. Hu Q., Katti P. S., Gu Z., Nanoscale 2014, 6, 12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Shutava T. G., Fakhrullin R. F., Lvov Y. M., Curr. Opin. Pharmacol. 2014, 18, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 118. Saravanakumar G., Kim J., Kim W. J., Adv. Sci. 2017, 4, 1600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Rwei A. Y., Wang W., Kohane D. S., Nano Today 2015, 10, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dolmans D. E. J. G. J., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380. [DOI] [PubMed] [Google Scholar]
- 121. Nowis D., Stokłosa T., Legat M., Issat T., Jakóbisiak M., Gołąb J., Photodiagn. Photodyn. Ther. 2005, 2, 283. [DOI] [PubMed] [Google Scholar]
- 122. Bañobre‐López M., Teijeiro A., Rivas J., Rep. Pract. Oncol. Radiother. 2013, 18, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Plank C., Zelphati O., Mykhaylyk O., Adv. Drug Delivery Rev. 2011, 63, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pitt W. G., Husseini G. A., Staples B. J., Expert Opin. Drug Delivery 2004, 1, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ramanathan S., Block L. H., J. Controlled Release 2001, 70, 109. [DOI] [PubMed] [Google Scholar]
- 126. Barenholz Y. C., J. Controlled Release 2012, 160, 117. [DOI] [PubMed] [Google Scholar]