

Abstract
Maspin repression is frequently observed in prostate cancer; however, molecular mechanism(s) causing the loss is not completely understood. Here we demonstrate that inhibition of class I histone deacetylases (HDACs) mediate re-expression of maspin plays an essential role in suppressing proliferation and migration capability in prostate cancer cells. Human prostate cancer LNCaP and DU145 cells treated with HDAC inhibitors sodium butyrate and trichostatin A resulted in maspin re-expression. Interestingly, exploration into the molecular mechanisms demonstrate that maspin repression in prostate tumor and human prostate cancer cell lines occurs via epigenetic silencing through increase in HDAC activity/expression, independent of promoter DNA hypermethylation. Furthermore, transcriptional activation of maspin was accompanied with the suppression of HDAC1 and HDAC8 with significant p53 enrichment at the maspin promoter associated with an increase in Histone H3/H4 acetylation. Our results provide evidence of maspin induction as a critical epigenetic event altered by class I HDACs in the restoration of balance to delay proliferation and migration ability of prostate cancer cells.
Keywords: Prostate Cancer, Maspin, Metastasis, Epigenetics, Histone Deacetylases, Histone acetylation
Introduction
Acquisition of metastatic capability in advanced-stage prostate cancer leads to clinically incurable malignancy and accounts for the majority of deaths from this disease1–3. Metastasis involves multiple sequential steps, including invasion, intravasation, extravasation and tumor growth at the secondary site4,5. SERPINs (serine protease inhibitors) control matrix-degrading proteases and growth factors, which are secreted in high levels by prostate cancer cells6,7. Maspin (SERPINB5) is a unique member of the SERPIN family that regulates cell motility, invasion, and contributes to tumor metastasis7–9. Loss of maspin has been frequently identified in prostate cancer cell lines and tumor specimens that correlates with high-grade tumor stage10,11. Studies document that Maspin gene is differentially regulated at the transcriptional level, and its subsequent loss in prostate luminal cells could be due to negative regulation of androgen response elements inhibiting its expression11. Another major regulatory mechanism for maspin involves its interaction with p53 through direct binding to the p53 consensus-binding site(s) on maspin promoter12. Loss of p53 function affects several target genes, and influences p53-dependent transcriptional activity towards pro-apoptosis mediators including Bax and Waf1/p2113–15. Furthermore, maspin overlaps with p53 function as demonstrated by mutant p53 studies accelerating cancer development and subsequent progression16.
Maspin is widely considered as a class II tumor suppressor gene7,8. Evidence suggests that maspin activity is suppressed during cancer progression through epigenetic modifications, without mutations or deletions in the coding regions7,17. Some studies have demonstrated tissue and cell-specific maspin expression directly correlates with DNA methylation18,19. In fact, cytosine methylation, deacetylation of histone proteins, and chromatin accessibility occurs at the 5’ regulatory region of the Maspin gene20–22. Additionally, 5-aza-2’-deoxycytidine exposure to maspin-null malignant cells resulted in induction of maspin23. Maspin exert endogenous inhibitory effect on class I HDACs, especially HDAC1 in prostate epithelial cells24. Class I HDAC levels are increased in prostate cancer, and their aberrant expression correlates with decreased tumor suppressor activity, drug resistance, and poor prognosis25,26. However, the involvement of epigenetic process in maspin loss in prostate cancer remains unclear.
Here we demonstrate that loss of maspin expression in prostate cancer is not due to the epigenetic process of DNA methylation but aberrant expression of class I HDACs. Notably, exposure of prostate cancer cells with HDAC inhibitors resulted in increased maspin expression. Further studies into the molecular mechanism(s) identified that maspin levels were induced by inhibition of HDAC1 and HDAC8 and enhanced binding of p53 to the maspin promoter with concomitant increase in Histone H3/H4 acetylation.
Materials and Methods
Antibodies and reagents.
Anti-Maspin (SC-22762), anti-p53 (SC-126), anti-HDAC1 (SC-7872), anti-HDAC2 (SC-6296), anti-HDAC3 (SC-11417), anti-HDAC8 (SC-11405), anti-actin (SC-47778), anti-α-GAPDH (SC-47724), goat anti-mouse IgG-HRP (SC-2005), bovine anti-goat IgG-HRP (SC-2350) and goat anti-rabbit IgG-HRP (SC-2004) were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-acetyl histone H3 (07–593) that recognizes Histone H3 acetylated on lysine 9 and 18; anti-acetyl histone H4 (06–598) recognizes acetylated histone H4 on lysine 5, 8, 12 and 16 and total anti-histone H3 (05–928) and anti-histone H4 (07–108) antibodies purchased from Upstate-Millipore (Temecula, CA).
Human prostate tissue specimens.
Samples of post-surgical discarded human prostate tissue were procured from the Tissue Procurement Facility of the University Hospitals Cleveland Medical Center and the Midwestern Division of the Cooperative Human Tissue Network. Consent was not prerequisite for these discarded tissues per hospital policies and the Institutional Review Board. Approval for these studies was confirmed by the Institutional Review Board at the University Hospitals Cleveland Medical Center. Tissue specimens were obtained from patients having undergone surgical procedures for prostatic disease without having received any adjuvant therapy. The Gleason grading was performed by a surgical pathologist with specific genitourinary experience. Following procurement, the tissue samples were snap-frozen in liquid nitrogen and stored at −80°C until further use.
Immunohistochemistry.
Benign (n=8) and cancer tissues (=24) of various Gleason grades were utilized for immunohistochemistry using standard protocol. Images were acquired with Olympus MicroSuite™ Five Software (Soft Imaging System, Lakewood, CO) and the staining intensity was graded semi-quantitatively. Each specimen was assigned a score on a scale from 0 to 3 designated as 0 (negative), 1+ when 10–20% of cells stain (weak), 2+ when 20–50% of cell stain (moderate) and 3+ when >50% of cell stain (strong) intensity. A scale of 0 to 4 was assigned for the percentage of positive cells where 0 (no positive cells), 1= <10% positive cells, 2= 10–50% positive cells, 3= 51–80% positive cells and 4= >80% positive cells, reported as the immunoreactive score (IRS) range between 0–12, as previously described27.
Cell culture and treatment.
Human prostate cancer LNCaP, 22Rv1, DU145, PC-3 cells, and virally transformed prostate epithelial PZ-HPV-7 cells were obtained from American Type Culture Collection (Manassas, VA) and were grown in appropriate culture medium as per vendor’s recommendation. Cells 60–70% confluence were treated with 10 μM 5-Aza-2′-deoxycytidine (Sigma Aldrich, St. Louis, MO), 1–25 nM trichostatin A (Sigma Aldrich), 0.1–1 mM sodium butyrate (Sigma Aldrich) for 48 h. The cells were harvested and lysed using whole-cell lysis buffer or were processed for cytosolic and nuclear lysate.
Cytosolic and nuclear lysate preparation.
The cytosolic and nuclear fractions were prepared as previously reported27. All the fractions were stored at −80°C and were used for Western blotting.
Western blotting.
Immunoblotting was performed on benign or cancer tissues by resolving on polyacrylamide gels, transferring and incubation with primary antibody, IgG secondary antibody and detection as previously reported27.
Bisulfite treatment and methyl‐specific PCR.
Genomic DNA was used from human prostate cancer tissue for bisulfite treatment applying EZ DNA Methylation–Gold Kit (Cat# D5005, Zymo, Orange County, CA) as per previous publication28. The computational approach was used to identify the CpG island in Maspin genome (Locus name: NC_000018.10:63476958–63505085 Homo sapiens chromosome 18, GRCh38.p13 Primary Assembly). EMBOSS CgPLOT (EMBL) program was searched with the parameters: length 200 %GC>50.0 P(CpG)/exp ≥0.60 (Supplemental figure 1). Primers to perform MS‐PCR on the maspin promoter were designed using the Methyl Primer Express® Software v1.0 (Applied Biosystems, Foster City, CA). Maspin–Methylated Forward: 5’-GAGTAGGAGAGGAGTGTCGTC-3’, Methylated Reverse: 5’-CGACCGTATCTAAACAAAAACA-3’; Unmethylated Forward 5’-TTTGAGTAGGAGAGGAGTGTTGTT-3’ and Unmethyated Reverse 5’-CAACAATATCTAAACAAAAACAACA-3’ were used. PCR products were resolved on acrylamide-TBE gels. Images were pictured and quantified using the Kodak Image Station 2000R.
Promoter fragmentation analysis.
Promoter fragment DNA analysis for maspin using combined bisulfite restriction analysis (COBRA) was performed to study the locus-specific CpG methylation status of maspin gene promoter as per the manufacturer’s protocol. Maspin promoter was amplified using the primer Forward 5’-ACGTATGCATGTAACTCACAGCCC-3’; Reverse 5’-ACGTATGCATGTAACTCACAGCCC-3’ designed using the EMBOSS Cpgplot software to detect putative CpG islands on the maspin gene promoter. Gels were stained with ethidium bromide and quantified on the Kodak Image Station 2000R.
Maspin DNA methylation analysis on TCGA dataset.
The Cancer Genome Atlas (TCGA), patient data of prostate adenocarcinoma (PARD) was used and compared with normal (n=49) and prostate cancer patients (n=340) with reference to maspin including Cg20490031, Cg11862144, Cg26753302, Cg20837735, Cg08411049, and Cg1542214 datasets. The data was analyzed statistically at the adjusted p threshold value of 0.05.
HDAC activity assay.
HDAC activity was measured using HDAC Fluorometric Cellular Activity Assay Kit (Biomole, Plymouth, PA) in a 96-well format according to the manufacturer’s protocol.
Acid extraction of histone proteins.
Histone extraction was performed using the protocol from Upstate-Millipore (Temecula, CA) as described previously28. Samples were resolved on an SDS page gel and transferred to a nitrocellulose membrane that was probed for total Histone H3 and H4 and acetylated Histone H3/H4 (Lys 9/18) and H4 proteins.
Transfection of cell packaging.
293T cells were used for packaging PLK0.1 (control vector-only), shHDAC‐1 (4814), shHDAC‐2 (4818), shHDAC‐3 (4826), shHDAC‐8 (4849) together with second-generation packaging construct (pCMV‐dR8.74), and pMD2G transfected using lipid transfection (Lipofectamine/PLUS reagent; Invitrogen Corporation, Grand Island, NY) as per vendor’s protocol. Subsequently, the collected media was layered onto prostate cancer LNCaP cells to infect them with the virus as previously described29.
Immunoprecipitation.
LNCaP cells silenced for HDAC 1, 2, 3, and 8, along with those harboring the empty vector were either treated with vehicle or TSA. Cells were lysed using total lysis buffer, and protein was immunoprecipitated with p53 antibody (SC-126) or maspin (SC-22762) followed by the addition of Protein A/G agarose beads (SC-2003). Following day the immune-precipitated proteins were washed with lysis buffer electrophoresed by SDS-PAGE and analyzed by Western blotting.
Biological network analysis of maspin.
The biological network analysis of maspin (SERPINB5) was performed using https://www.pathwaycommons.org. The interaction showed biological binding and expression networks associated with SERPINB5.
Chromatin Immunoprecipitation (ChIP) assay.
ChIP assay was performed on Class I HDAC silenced LNCaP cells and the cells with empty vector as previously described29 following PCR amplification for 30 cycles with the conditions: stage 1, 95 °C for 5 min (1 cycle); stage 2, 95 °C for 30 s and 62 °C for 30 s and 72 °C for 1 min (30 cycles); and stage 3, 72 °C for 7 min (1 cycle). The primers used to assess the binding of p53 to the maspin promoter are Maspin-ChIP-p53 Forward: 5’-ATATTTCACCTTCCGGTCCTG-3’, Maspin-ChIP-p53 Reverse: 5’-CCTACTCAAGCCTCTTGGCA-3’. PCR products were subjected to electrophoresis on a 2% ethidium bromide–agarose gel and visualized under UV light.
Wound healing assay.
LNCaP cells harboring PLK0.1 and Sh-HDAC 1, 2, 3, and 8 were plated into six-well culture dishes to 60–70% density. A minimum of three scratch wounds were made per plate using a sterile pipette tip. The cells were washed with 1x PBS solution to remove floating cells. Cells were then incubated 37°C in a 5% CO2 humidified incubator and photographed. Cell migration areas were calculated using Image J software [29].
Cell doubling time.
LNCaP cells silenced for HDAC 1, 2, 3, and 8, and transfected with empty vector PLK0.1 were trypsinized and counted using a new Bauer chamber at a time interval of 24, 48, and 72 h, and the numbers were recorded.
Statistical analysis.
Digital images of Western Blotting and the scratch analysis were quantified using Image J software (NIH), and statistical analysis was performed using a two‐tailed Student’s t‐test. The level of significance designated statistically significant are as follows: *P ≤ 0.05 and **P ≤ 0.001.
Results
Maspin and HDAC1 expression in benign and malignant prostate cancer specimens.
Maspin and HDAC1 expression was determined in surgically removed clinical specimens. Immunoblotting was performed for maspin and HDAC1, and GAPDH as a loading control in 16 paired matched benign and prostate cancer specimens (Supplemental figure 2A). Boxplot analysis exhibits a marked difference (P<0.05) in the maspin and HDAC1 expression between benign and cancer specimens (Figure 1 A&B). Compared to benign tissue, maspin expression was markedly decreased in the majority of cancer specimens. In contrast, HDAC1 expression was higher in the majority of cancer specimens compared to benign tissue.
Figure 1:
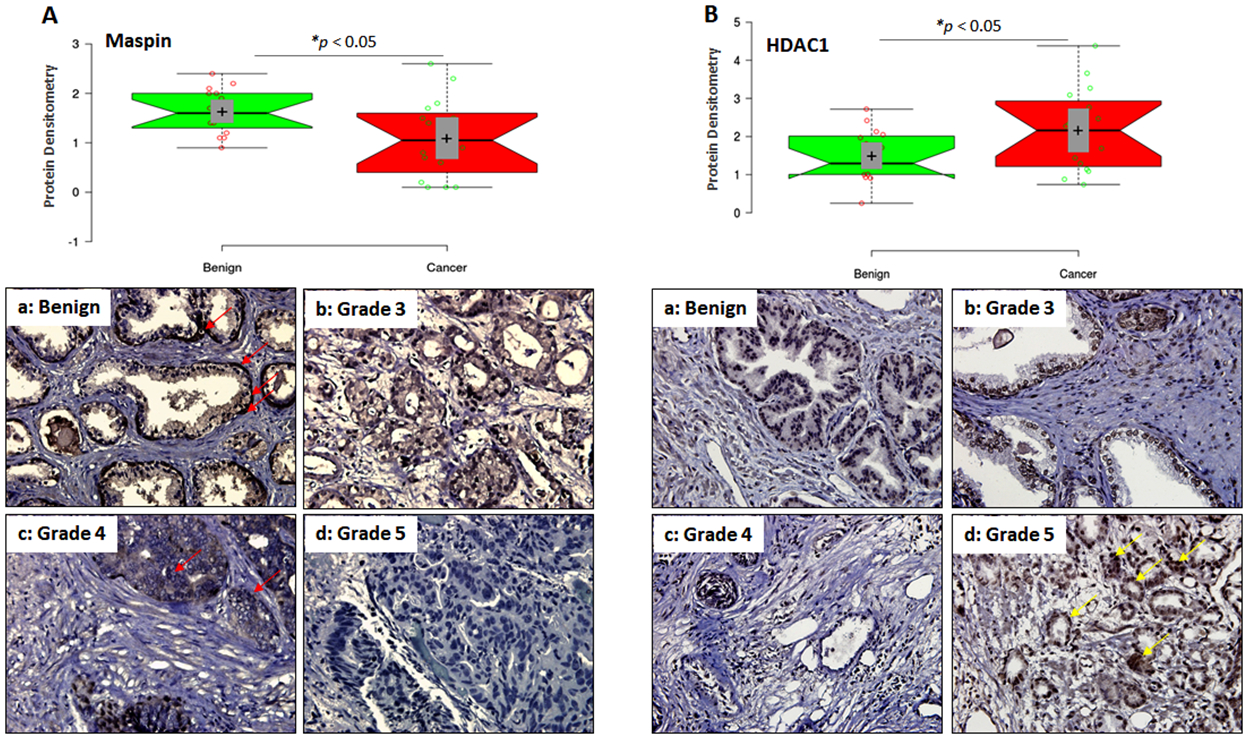
Maspin and HDAC1 expression in benign and prostate cancer specimens. (A) Densitometic analysis of maspin normalized to GAPDH from 16 paired specimens shown in the boxplot. Bars ± SD, *p < 0.05, compared to benign tissue. Immunostaining for maspin in representative samples of benign prostate tissue and cancer specimens of various Gleason grades. Magnification ×400. Red arrow represents maspin expressing cells. (B) Densitometic analysis of HDAC1 normalized to GAPDH from 16 paired specimens shown in the boxplot. Bars ± SD, *p < 0.05, compared to benign tissue. Immunostaining for HDAC1 in representative samples of benign prostate tissue and cancer specimens of various Gleason grades. Magnification ×400. Yellow arrow represents HDAC1 expressing cells.
Next we performed immunohistochemistry in benign and cancer specimens of various Gleason grades. Maspin is uniformly expressed in the basal cells in benign specimens (red arrow). In comparison, maspin expression was markedly decreased in cancer specimens and diminished in neoplastic cells. A progressive decrease in maspin expression was noted that corresponded to increase in the Gleason grade (Grade 3 and 4) with complete loss noted in high-grade (Grade 5) tumors (Figure 1A). In contrast, HDAC expression in the benign tissue exhibited a characteristic pattern with minimal variation in the nuclei of stromal cells. Luminal cells showed a homogenous moderate HDAC1 positivity and the basal cells were mostly negative. However, in tumor specimen high abundance of HDAC1 was noted with progressive increase in grade (Grade 3 and 4); strong nuclear positivity in high-grade tumor (Grade 5; yellow arrow) and lower expression in stromal cells (Figure 1B). In summary, an inverse association was noted between maspin and HDAC1 expression during prostate tumor progression (Supplemental figure 2B).
Promoter fragmentation and methylation of maspin promoter in benign and malignant prostate cancer specimens.
Sequence analysis of cDNA and genes from benign and tumor tissue demonstrate that the sequence and structure of maspin gene were not altered in the malignant cells. This indicates that maspin may possibly be downregulated due to epigenetic modification(s) rather than a mutation in tumor tissue. In few human cancers methylation of maspin promoter has been reported to be the cause of its repression leading to loss of expression and function30–32. Hence we investigated whether loss of maspin in prostate cancer is due to promoter fragmentation or changes in methylation status in prostate specimens. We employed methyl-specific PCR in a subset of benign and cancer samples from the same individuals to identify whether methylation and promoter fragmentation were observed in the maspin promoter. As shown in figure 2A, no changes were found in the promoter fragmentation or the methylation of CpG islands on maspin gene, whereas 3 out of 10 samples exhibited promoter fragmentation in high-grade tumor specimens and were negative for methylation. Our data suggest that CpG island methylation or promoter fragmentation did not have a role in the loss of expression of maspin, at least, in prostate cancer.
Figure 2:
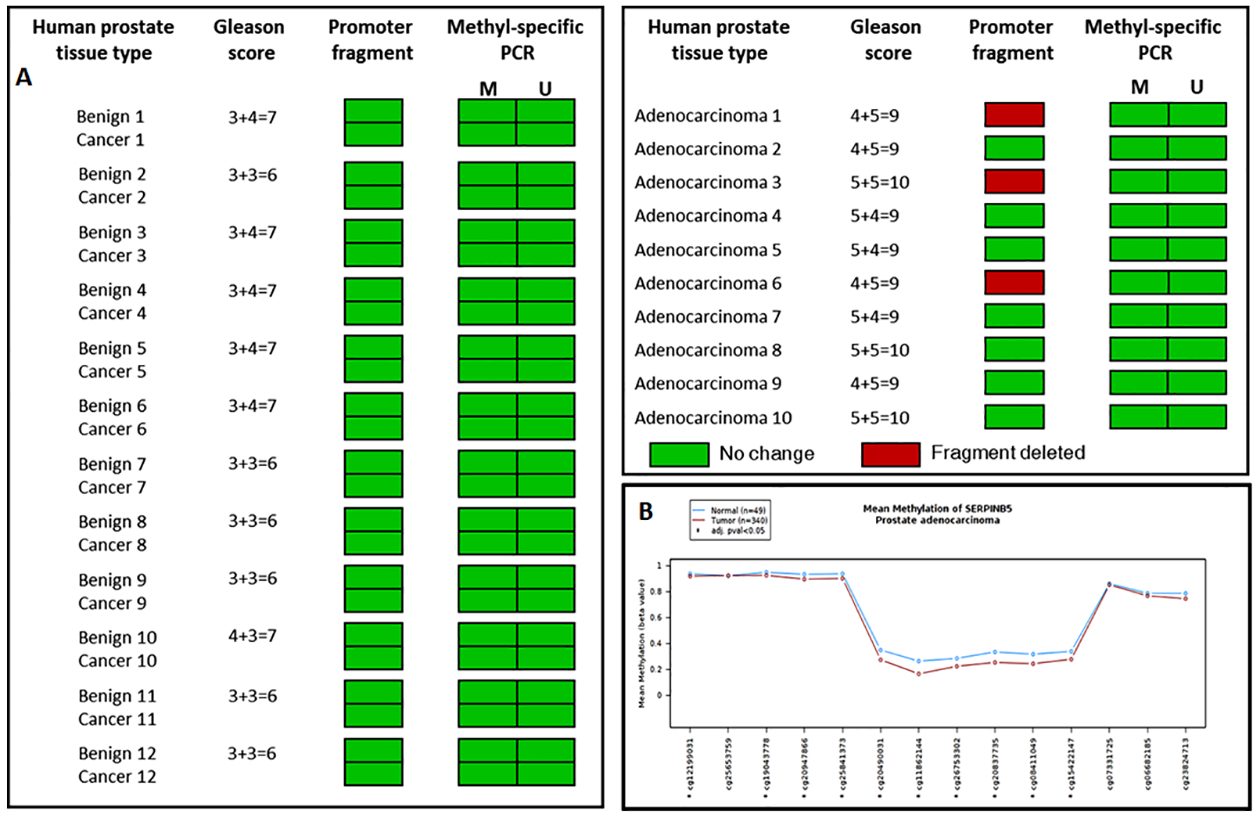
Epigenetic alterations in maspin. (A) Maspin promoter fragment analysis and methylation status by methyl-specific-PCR performed in human benign prostate tissue and cancer specimens of various Gleason score. Green box indicate, no fragment deletion and methylation, whereas red box indicate promoter fragment deletion. (B) Mean methylation difference of maspin in the Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) data between normal and prostate tumor samples. No statistical difference was noted between the two groups.
Visualization of maspin DNA methylation in TCGA dataset.
Next, we used the Wanderer (public domain web portal) to determine the interactive DNA methylation from 450k methylation array pattern of maspin (SERPINB5) in clinical specimens. As shown in figure 2B, Cg probes identified mean methylation difference of maspin in the Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) data between normal and prostate tumor samples. The difference between the normal and tumor specimens were statistically insignificant.
Class I HDAC inhibition increases maspin expression in human prostate cancer cells.
Our previous observation pointed out decreased maspin expression and higher HDAC1 expression in clinical specimens, which exhibited minimal involvement of DNA methylation or promoter fragmentation (Figure 1&2). To further confirm, we treated a panel of prostate cancer cells (22Rv1, LNCaP, DU145 and PC-3) with 10 μM DNA methyltransferase inhibitor 5-Aza-2′-deoxycytidine (5-AZA-dC). We used PZ-HPV-7 as the prostate epithelial cell line possessing high levels of maspin, compared to other human prostate cancer cell lines. Treatment of cells with 5-AZA-dC did not increase the expression of maspin in any cell line, confirming that DNA methylation does not play a role in altering maspin expression in prostate cancer (Supplemental figure 3).
Next, we treated LNCaP and DU145 cells with class I HDAC inhibitors such as sodium butyrate (NaB) and trichostatin A (TSA). Treatment of LNCaP and DU145 cells with NaB and TSA exhibited a dose-dependent increase in maspin expression in these cell lines (Figure 3A&B). Clinical data suggests that translocation of maspin into the nucleus is associated with improved survival of several human cancers33. Following NaB and TSA treatment maspin expression was upregulated in both cytosolic and nuclear fractions; however its distribution in the nuclear fraction increased in a dose-dependent manner in both cell lines (Figure 3A&B). These results correlate with a decrease in HDAC activity in LNCaP and DU145 cells post-NaB and TSA treatment (Figure 3A&B).
Figure 3:
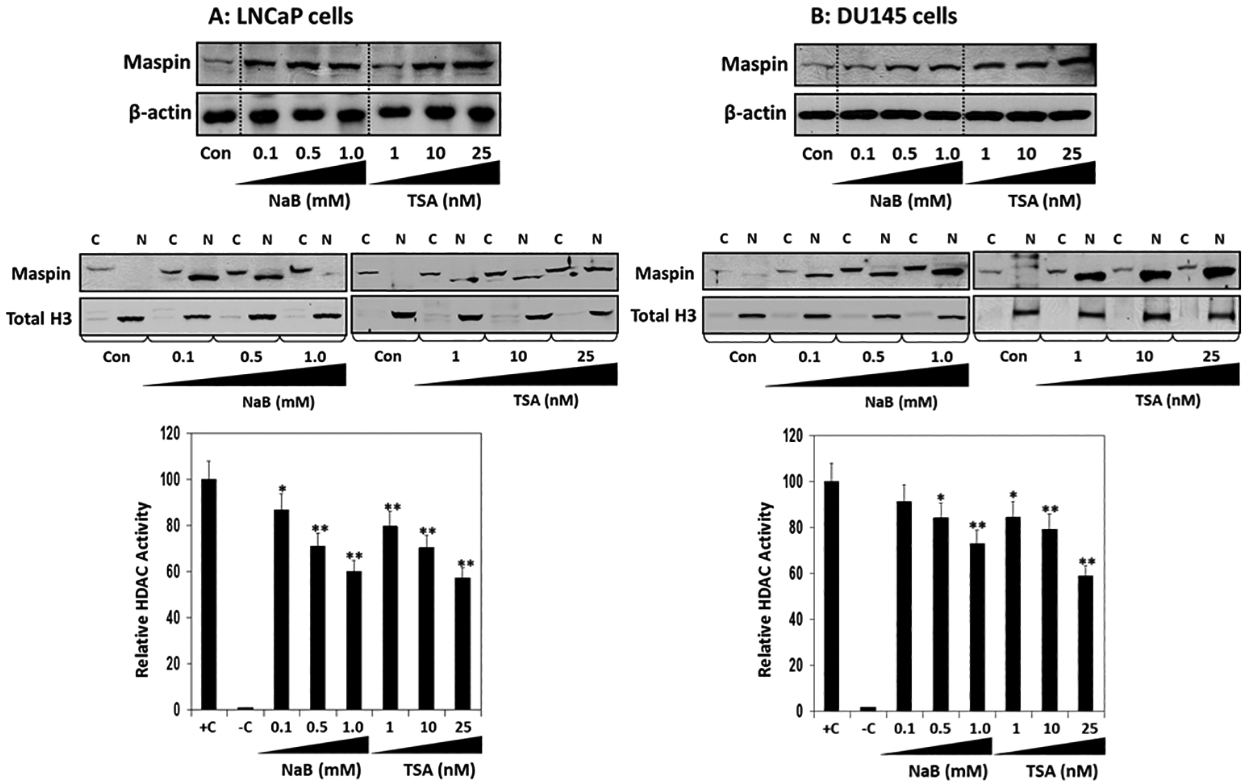
Effect of histone deacetylase inhibitors on maspin expression in prostate cancer cells. (A) Human prostate cancer LNCaP and (B) DU145 cells treated with indicated concentrations of NaB and TSA for 48 h. Total cell lysates, cytosolic and nuclear fractions were prepared, electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-maspin and anti-β-actin and Total histone H3 antibodies. β-actin and histone H3 serves as loading controls. HDAC activity was measured in these cells. Bars ± SD of three independent assays. *p < 0.05, **p < 0.001, compared to control.
Class I HDAC inhibition increases histone acetylation in human prostate cancer cells.
Next, we elucidated whether downregulation of HDAC activity post-treated with NaB and TSA in prostate cancer cells was associated with decrease in class I HDACs. Western blotting was performed on LNCaP and DU145 cell lysates treated with and without NaB and TSA. Exposure of cells to NaB markedly decreased the levels of HDAC 1, 2, 3 and 8 in LNCaP cells, whereas a modest decrease in HDAC 1, 2, and 3 was noted in DU145 cells. Treatment of cells with TSA showed a marked decrease in HDAC 1, 2, 3 and 8 in LNCaP cells and in HDAC 1, 2 and 3 in DU145 cells but not in HDAC8 (Figure 4 A&B). Our next objective was to determine whether decreased levels of HDACs following NaB and TSA treatment of prostate cancer cells led to an increase in acetylation of histone proteins. Yan and Boyd (2006) have previously identified a chromatin region able to permit or restrict gene expression based on Histone H3K4me/H3ac levels34. Immunoblotting of acid extracted protein obtained from prostate cancer cells treated with TSA exhibited significantly increased acetylation of Histone H3/H4, compared to untreated controls in both cell lines. Although the increase in acetylation noted after TSA treatment for Histone H3/H4 in both cell lines was higher than the increase observed after treatment of cells with NaB (Figure 4 A&B).
Figure 4:
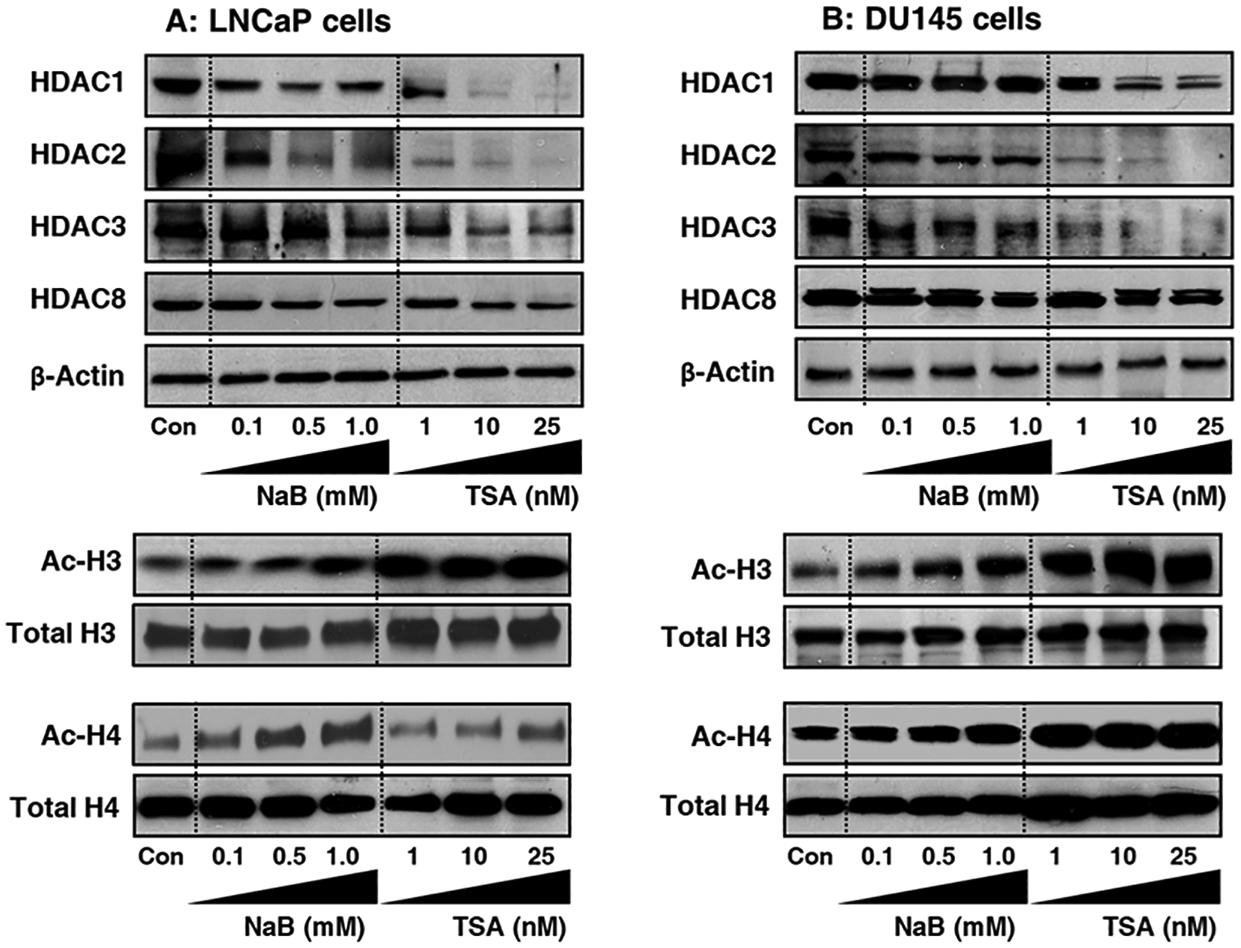
Effect of HDAC inhibitors on class I HDAC expression and histone H3/H4 acetylation in prostate cancer cells. (A) Human prostate cancer LNCaP and (B) DU145 cells treated with indicated concentrations of NaB and TSA for 48 h. The lysates were prepared, electrophoresed by SDS‐PAGE, followed by immunoblotting with HDAC 1, 2, 3 and 8 antibodies. β-actin serves as loading control. Histone extraction was performed on cells treated with HDAC inhibitors for 48 h. The immunoblots were probed for acetylated anti-Histone H3/H4 antibodies. Total histone H3/H4 serves as loading control.
Class I HDACs mediate epigenetic silencing of maspin in human prostate cancer cells.
Previous experiments demonstrate that exposure of prostate cancer cells with NaB or TSA significantly induced maspin expression, suggesting a role through epigenetic silencing via aberrant histone deacetylation. To further elucidate the mechanism of maspin silencing, class I HDACs (HDAC 1, 2, 3, and 8) were knockdown in LNCaP cells using specific shRNAs for each target. As shown in figure 5A, HDAC protein expression was considerably lower in corresponding knockdown cells when compared to vector control PLK0.1 cells. HDAC 1, 2, and 3 were markedly reduced, and a modest decrease in HDAC8 expression was noted in these knockdown cells. The expression of maspin was highest in HDAC1 and HDAC8 knockout cells, whereas p53 expression increased in all class I HDAC knockout cells (Figure 5A). These results suggest that HDAC1 and HDAC8 might play an important role in maspin regulation in prostate cancer.
Figure 5:
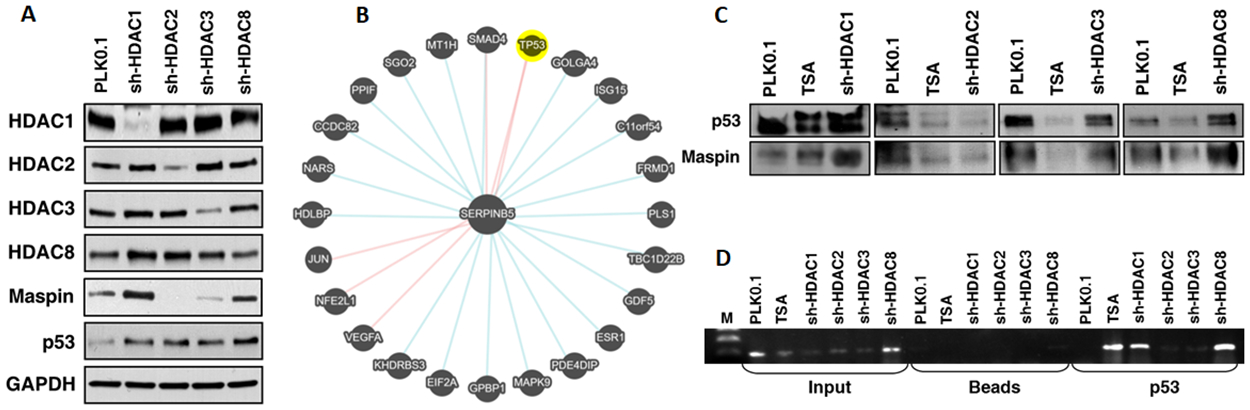
Effect of class I HDAC knockdown on maspin and p53 expression. (A) LNCaP cells were individually silenced for HDAC 1, 2, 3, and 8. The lysates were prepared, electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-HDAC 1, 2, 3 and 8, anti-maspin and anti-p53 antibodies. GAPDH serves as loading control. (B) Network analysis of maspin (SERPINB5) showing biological binding and expression associated with SERPINB5. (C) Class I HDAC silenced LNCaP cells were immunoprecipitated with maspin or p53. The association was ascertained by immunoblotting for p53 in maspin pulldown lysates and the vice versa for maspin expression. (D) Chromatin immunoprecipitation assay was performed to analyze the binding of p53 to the maspin promoter in the HDAC silenced LNCaP cells.
One of the transcriptional regulatory mechanisms for maspin is its association with the p53 signaling pathway12. Biological network analysis of maspin (SERPINB5) showed binding interaction with p53, JUN, NFE2L1, VEGFA and SMAD4, and it’s association with other transcription factors/genes include C11orf54, CCDC82, ESR1, EIF2A, FRMD1, GDF5, GOLGA4, HDLBP, ISG15, KHDRS3, MAPK9, MT1H, NARS, PDE4DIP, PLS1, PPIF, SGO2, SMAD4, and TBC1D22B (Figure 5B). Next, we performed immunoprecipitation by pulldown of p53 in HDAC silenced LNCaP cells. We observed that in the absence of HDAC1 and HDAC8, the binding/association between p53 and maspin significantly increased compared to the vector control PLK0.1 (Figure 5C). However, treatment with TSA resulted in increased p53 expression in only HDAC1 knockout cells. We did not observe this association in the LNCaP cells silenced for HDAC2 and HDAC3 (Figure 5C). Overall, upregulation of maspin expression upon pharmacological inhibition or genetic knockdown of class I HDACs strongly suggest that these histone-modifying enzymes mediate maspin repression in prostate cancer cells and decreases its association with p53.
Class I HDAC inhibition causes increased binding of p53 at the maspin promoter in human prostate cancer cells.
Maspin promoter possesses a p53 consensus site induced upon its binding [12]. Therefore, we next investigated if silencing of class I HDACs would facilitate increased binding of p53 to the maspin promoter. We performed ChIP assay in the LNCaP cells possessing the empty vector PLK0.1 and those silenced for class I HDACs. The HDAC inhibitor TSA was used as a positive control. Our results show that HDAC1 and HDAC8 knockout cells increased p53 binding and enrichment at the maspin promoter (Figure 5D). These results suggest that inhibition of class I HDACs could, in part, have ability to induce maspin expression and increases its association with p53.
Class I HDACs suppresses proliferation and migration ability in human prostate cancer cells.
Given the impact of maspin expression on tumor proliferation and metastasis, we determined the functional effect of class I HDACs inhibition on prostate cancer cell migration and proliferation activity. The wound healing assay was performed in LNCaP cells silenced for class I HDACs and those possessing the empty vector PLK0.1. Cell migration was monitored at different time points and compared to 0 h as the wounds significantly began to close by 24, 48, and completely by 72 h in LNCaP PLK0.1 cells. In shHDAC2 and shHDAC3 cells, wound closure was slow in comparison to vector control and was incomplete by the end of 72 h exhibiting approximately 40% closure. In shHDAC1 and shHDAC8 cells, the closure was almost similar to the 0 h exhibiting approximately 20% or less closure, suggesting these two class I HDACs might be responsible for regulating cell migration (Figure 6A). Next we investigated whether silencing of class I HDACs affected proliferation in prostate cancer cells. Compared to vector-transfected LNCaP cells, knockdown of individual class I HDACs significantly affected cell proliferation (P<0.0001) (Figure 6B). Collectively, our data showed that silencing of class I HDACs, especially HDAC1 and HDAC8 decreased migration and proliferation capability in human prostate cancer cells.
Figure 6:
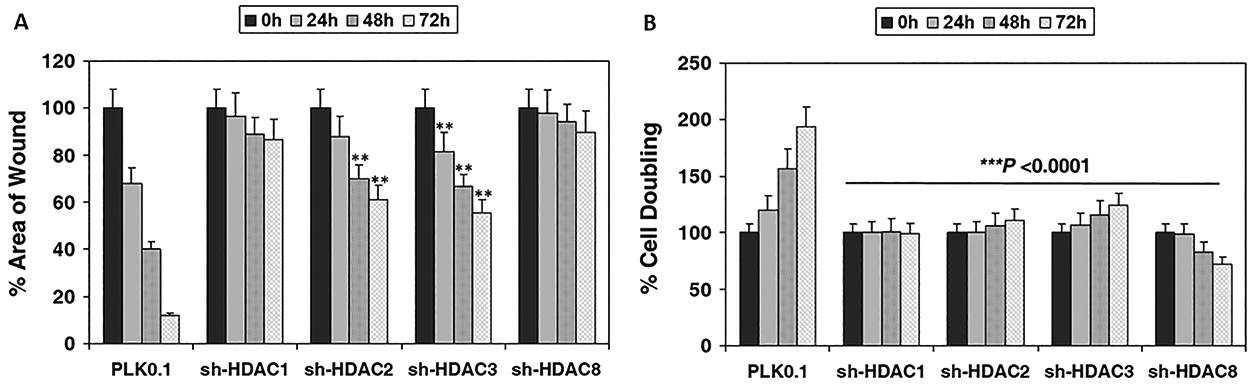
Effect of class I HDAC knockdown on migration and proliferation ability of prostate cancer cells. (A) Wound healing assay was performed in class I HDAC silenced LNCaP cells. Images were taken before (0 h) and after wound closure (24, 48, 72h). The results were expressed as the percentage of the remaining area determined by normalizing the area of wound after 24 h or 48 h (as indicated) to the initial wound area at 0 h (set to 100%). Each bar represents the mean of 5 fields measured ± SD. Two‐tailed Student’s t‐test was used to compare sh-HDAC2 and sh-HDAC3 groups and control. **P < 0.001 versus control. (B) Doubling time was assessed in LNCaP cells silenced for Class I HDACs at 24, 48 and 72h after plating 25,000 cells/well and allowed to attach overnight. The changes in doubling time were expressed as percentage. Two‐tailed Student’s t‐test was used to compare various sh-HDAC1–8 groups with control. ***P < 0.0001 versus control.
Discussion
This study determined that altered histone modification patterns at the maspin promotor lead to epigenetic silencing in prostate cancer cells, independent of promoter DNA hypermethylation. We used two human prostate cancer cells viz. LNCaP and DU145 as in vitro models representing different phenotypes. The major highlight of this work is that class I HDACs, especially HDAC1 and HDAC8, play a crucial role in regulating maspin expression. Importantly, inhibition of HDAC activity by NaB and TSA significantly inhibited cell proliferation and migration that correlated with maspin induction. Furthermore, inhibition of class I HDACs, either using pharmacological or genetic approach, resulted in an increase in Histone H3/H4 acetylation and enrichment of p53 at the maspin promoter.
Epigenetic studies suggest that DNA hypermethylation in the gene promoter region or chromatin modification play an important role in silencing of tumor suppressor genes leading to the initiation of prostate cancer21,26. We altered maspin expression with pharmacologic agents that modify epigenetic pathways such as DNA methyltransferase inhibitor 5‐Aza‐dC and HDAC inhibitors NaB and TSA. Previous studies have shown that maspin levels are controlled by the methylation status of CpG islands and treatment of maspin null cell lines with 5-AZA-dC resulted in maspin induction due to gene de-repression in few cancer types18–20. Our results demonstrate no maspin promoter hypermethylation in prostate cancer specimens, in accordance with a lack of maspin expression. Besides, human prostate cancer cells did not exhibit an increase in maspin expression after exposure to 5-AZA-dC. From the viewpoint of DNA methylation, maspin might be repressed in a cell/tissue type-specific manner and provides an insight that DNA methylation might not be the epigenetic process regulating maspin expression at least, in part, in prostate cancer.
Chromatin remodeling, like those of histone acetylation/deacetylation and chromatin‐binding proteins, influence local chromatin structure, in accordance with DNA methylation, regulating gene transcription21–26. HDACs are recruited to unique promoter regions responsible for silencing target genes by sequence-specific transcription factors26. Aberrant HDAC expression is observed in various human cancers, including prostate cancer, and their differential expression correlates with poor prognosis25,26. These epigenetic modifications play a vital role in the development and progression of cancer. Many of the tumor suppressor genes involved in the regulation of cell growth, differentiation, signal transduction, DNA repair, and others implicated in suppression of tumor metastasis and angiogenesis exhibit promoter hypermethylation, indicating that epigenetic inactivation of a gene is a common phenomenon in cancer development. Our findings provide evidence that treatment of prostate cancer cells with HDAC inhibitors viz. NaB and TSA could reverse maspin repression. Besides, genetic knockdown studies have confirmed that class I HDACs actively participate in maspin repression. Recent findings in prostate cancer demonstrate elevated HDAC activity results in deacetylation and repression of several essential genes35–37. Our results demonstrate decreased maspin expression in prostate cancer LNCaP and DU145 cells associate with aberrant expression of class I HDACs. Western blotting and ChIP studies showed an increase in maspin expression and p53 enrichment at the maspin promoter, with a corresponding increase in Histone H3/H4 acetylation in HDAC knockout prostate cancer cells.
Previous studies demonstrate that transcriptional repression of p53 results in the loss of maspin expression in prostate cancer cells with varying p53 status20. However, the mechanistic role of p53 and the involvement of molecular pathways in maspin re-repression is not addressed. To decipher the above, we employed two cells type’s viz. LNCaP cells which possess wild type p53 and DU145 cells which harbor mutant p53 with minimal expression of maspin transcript. Our results show that these two prostate cancer cells with a difference in p53 status demonstrate an induction in maspin expression after treatment with HDAC inhibitors. The present study indicates that suppression of class I HDACs regardless of p53 status, possibly results in the Histone H3/H4 acetylation, thereby enhancing the accessibility to the transcription machinery and its binding to the maspin promoter. Specific studies are needed to understand the role of class I HDACs in differential cellular response with varying p53 expression.
The stability and transcriptional activity of p53 is controlled by posttranslational modifications, which includes phosphorylation and acetylation38. We have previously demonstrated that p53 activation through acetylation can be achieved by inhibition of class I HDACs in prostate cancer LNCaP cells39. Increased acetylation of p53 enhances its binding on the promoters of downstream targets such as p21/Waf1, Bax, and maspin resulting in reduced proliferation and invasiveness in cancer cells39. Reports suggest that p300/CBP-mediated acetylation of p53 facilitates transcriptional activation of genes and stimulates sequence-specific DNA binding40. This synergistic enhancement of p53 transactivation activity may be the consequence of two pathways, including histone acetylation and p53 acetylation in prostate cancer cells accomplished by diminishing class I HDACs.
Aberrant expression of class I HDACs has been reported in human prostate cancer cell lines and clinical cancer specimens25,26. Studies document high HDAC1–3 levels in prostate cancer, and association of HDAC2 with shorter prostate-specific antigen relapse time41. Increased HDAC1 levels are linked with higher proliferation and invasiveness in prostate cancer cells and recognized as a potential therapeutic target25. HDACs function in association with multi-subunit transcriptional corepressor complexes, in sequence-specific manner deacetylating the catalytic core, resulting in transcriptional gene repression42–44. The presence of HDAC1 and HDAC2 are noted in the CoREST, mi2/NURD, and Sin3 complexes, and HDAC3 is associated with the catalytic activity of the N-CoR and SRMT corepressor complexes45. Inhibition of class I HDACs exhibit significant functional effects on malignant cells, however the molecular mechanisms underlying these effects requires further elucidation.
Reports suggest that class I HDACs mediate deacetylation of sequence-specific transcription factors leading to a reduction in DNA-binding activity and suppression of transcription46. HDAC-mediated covalent modifications were noted in E2F, sp1/sp3, p53, GATA1, and TFIIF transcription factors46,47. Reduction in class I HDACs alters the dynamic balance between HDAC and histone acetyltransferase activity towards increased acetylation of non-histone proteins, including p53. Reports suggest that p53 deacetylation by HDACs can be reversed by the use of HDAC inhibitors48. In our studies, inhibition of HDACs resulted in an increase in p53 enrichment and it’s binding to the maspin promoter along with acetylation of Histone H3/H4 in these cells (Figure 7). Additional studies are required to elucidate the effects of HDAC inhibition on p53 acetylation in human prostate cancer cells. It will also be interesting to investigate whether HDAC inhibition influences other molecular pathways in prostate cancer cells.
Figure 7:
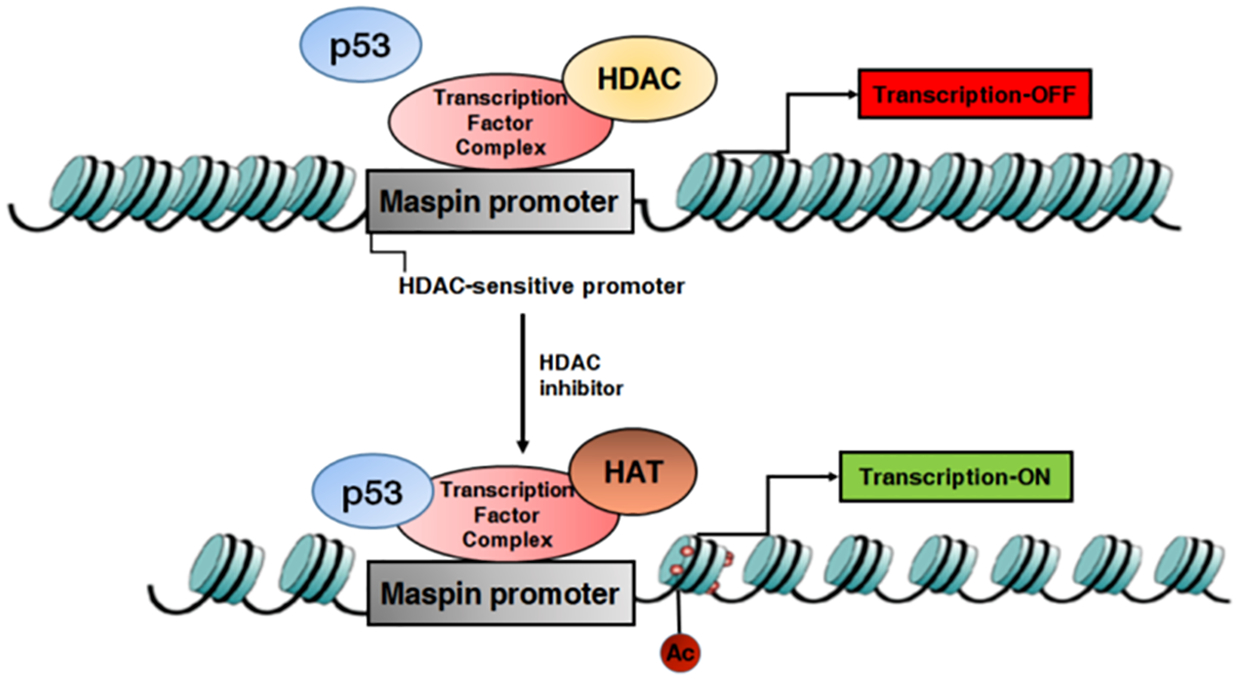
Schematic model demonstrating the role of class I HDACs on maspin inhibition. Class I HDACs binds to the maspin promoter along with the transcription factor complex. Silencing of class I HDACs lead to increase in p53 enrichment and its binding to the maspin promoter along with acetylation of Histone H3/H4 in prostate cancer LNCaP cells. HDAC, histone deacetylases; HAT, histone acetyltransferases.
In conclusion, our findings provide a detailed mechanistic insight into the epigenetic regulation of maspin and the role of class I HDACs in prostate cancer. Additional studies are required to fully elucidate the molecular mechanisms that lead to maspin repression, in particular, facilitating tumor invasion and metastasis.
Supplementary Material
Figure 1: CpG islands in maspin genome. Locus name: NC_000018.10:63476958–63505085 Homo sapiens chromosome 18, GRCh38.p13 Primary Assembly. EMBOSS CgPLOT (EMBL) program was used with search with the parameters: length 200 %GC>50.0 P(CpG)/exp >0.60. Various CpG islands are shown in the maspin gene.
Figure 2: (A) Western blot analysis of maspin and HDAC1 in benign and cancer specimens. Total cell extracts from tissues were prepared and electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-maspin, anti-HDAC1 and anti-GAPDH antibodies. GAPDH is used as a loading control. Benign and cancer tissue from same individuals are from a subset of 16 patients. (B) IRS score generated from immunostaining for maspin and HDAC1 tissues from benign prostate tissue and cancer specimens of various Gleason grades. An inverse association was noted between maspin and HDAC1 expression in grade-specific manner during tumor progression.
Figure 3: Effect of 5-AZA-dC on maspin expression in prostate cancer cells. Human prostate cancer 22Rv1, LNCaP, DU145 and PC-3 cells were treated with 10 μM concentration of 5-AZA-dC for 48 h. The lysates were prepared, electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-maspin and anti-β-actin antibodies. β-actin serves as a loading control. No changes in maspin expression were noted after 5-AZA-dC treatment.
Acknowledgements
This study was supported by funds from the United States Public Health Service Grants RO1CA115491, R21CA193080, R03CA186179, and VA Merit Award 1I01BX002494 to SG.
Abbreviations:
- 5-AZA-dC
5-aza-2’-deoxycytidine
- HDACs
histone deacetylases
- HATs
histone acetyltransferases
- NaB
sodium butyrate
- SERPINs
serine protease inhibitors
- TCGA
The Cancer Genome Atlas
- TSA
trichostatin A
Footnotes
Conflict of interest: The authors have no competing interest.
Data availability
The data that support the findings of this study which allows real-time access and visualization of gene expression and DNA methylation profiles from TCGA are openly available at the Wanderer web tool http://maplab.imppc.org/wanderer.
References
- 1.Kishan AU, Cook RR, Ciezki JP, et al. Radical Prostatectomy, External Beam Radiotherapy, or External Beam Radiotherapy With Brachytherapy Boost and Disease Progression and Mortality in Patients With Gleason Score 9–10 Prostate Cancer. Jama. 2018;319(9):896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frees S, Akamatsu S, Bidnur S, et al. The impact of time to metastasis on overall survival in patients with prostate cancer. World journal of urology. 2018;36(7):1039–1046. [DOI] [PubMed] [Google Scholar]
- 3.Kishan AU, Chu F-I, King CR, et al. Local Failure and Survival After Definitive Radiotherapy for Aggressive Prostate Cancer: An Individual Patient-level Meta-analysis of Six Randomized Trials. European Urology. 2020;77(2):201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suhail Y, Cain MP, Vanaja K, et al. Systems Biology of Cancer Metastasis. Cell systems. 2019;9(2):109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rycaj K, Li H, Zhou J, Chen X, Tang DG. Cellular determinants and microenvironmental regulation of prostate cancer metastasis. Seminars in cancer biology. 2017;44:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon BJ, Kwaan HC. Components of the Plasminogen-Plasmin System as Biologic Markers for Cancer. Advances in experimental medicine and biology. 2015;867:145–156. [DOI] [PubMed] [Google Scholar]
- 7.Sager R, Sheng S, Pemberton P, Hendrix MJ. Maspin: a tumor suppressing serpin. Current topics in microbiology and immunology. 1996;213 (Pt 1):51–64. [DOI] [PubMed] [Google Scholar]
- 8.Dzinic SH, Bernardo MM, Li X, et al. An Essential Role of Maspin in Embryogenesis and Tumor Suppression. Cancer research. 2017;77(4):886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teoh SS, Whisstock JC, Bird PI. Maspin (SERPINB5) is an obligate intracellular serpin. The Journal of biological chemistry. 2010;285(14):10862–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machtens S, Serth J, Bokemeyer C, et al. Expression of the p53 and Maspin protein in primary prostate cancer: correlation with clinical features. International journal of cancer. 2001;95(5):337–342. [DOI] [PubMed] [Google Scholar]
- 11.Berardi R, Morgese F, Onofri A, et al. Role of maspin in cancer. Clinical and translational medicine. 2013;2(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou Z, Gao C, Nagaich AK, et al. p53 regulates the expression of the tumor suppressor gene maspin. The Journal of biological chemistry. 2000;275(9):6051–6054. [DOI] [PubMed] [Google Scholar]
- 13.Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell death and differentiation. 2005;12(5):482–491. [DOI] [PubMed] [Google Scholar]
- 14.Machtens S, Kuczyk M, Serth J, Jonas U. P53 regulated maspin protein expression determines recurrence-free survival of patients with localised prostate cancer. Prostate cancer and prostatic diseases. 2000;3(S1):S27. [DOI] [PubMed] [Google Scholar]
- 15.Schumacher G, Bruckheimer EM, Beham AW, et al. Molecular determinants of cell death induction following adenovirus-mediated gene transfer of wild-type p53 in prostate cancer cells. International journal of cancer. 2001;91(2):159–166. [DOI] [PubMed] [Google Scholar]
- 16.Oshiro MM, Watts GS, Wozniak RJ, et al. Mutant p53 and aberrant cytosine methylation cooperate to silence gene expression. Oncogene. 2003;22(23):3624–3634. [DOI] [PubMed] [Google Scholar]
- 17.Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJ. Biological functions of maspin. Journal of cellular physiology. 2006;209(3):617–624. [DOI] [PubMed] [Google Scholar]
- 18.Costello JF, Vertino PM. Methylation matters: a new spin on maspin. Nature genetics. 2002;31(2):123–124. [DOI] [PubMed] [Google Scholar]
- 19.Futscher BW, Oshiro MM, Wozniak RJ, et al. Role for DNA methylation in the control of cell type specific maspin expression. Nature genetics. 2002;31(2):175–179. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama Y, Maesawa C, Ogasawara S, Terashima M, Masuda T. Cell-type-specific repression of the maspin gene is disrupted frequently by demethylation at the promoter region in gastric intestinal metaplasia and cancer cells. The American journal of pathology. 2003;163(5):1911–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maass N, Biallek M, Rosel F, et al. Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochemical and biophysical research communications. 2002;297(1):125–128. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Liu H, Yu J, et al. Methylation-induced silencing of maspin contributes to the proliferation of human glioma cells. Oncology reports. 2016;36(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugimoto S, Maass N, Takimoto Y, et al. Expression and regulation of tumor suppressor gene maspin in human bladder cancer. Cancer letters. 2004;203(2):209–215. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Yin S, Meng Y, Sakr W, Sheng S. Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer research. 2006;66(18):9323–9329. [DOI] [PubMed] [Google Scholar]
- 25.Burdelski C, Ruge OM, Melling N, et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Experimental and molecular pathology. 2015;98(3):419–426. [DOI] [PubMed] [Google Scholar]
- 26.Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3(6):300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla S, MacLennan GT, Fu P, et al. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia (New York, NY). 2004;6(4):390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. International journal of cancer. 2010;126(11):2520–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deb G, Shankar E, Thakur VS, et al. Green tea-induced epigenetic reactivation of tissue inhibitor of matrix metalloproteinase-3 suppresses prostate cancer progression through histone-modifying enzymes. Molecular carcinogenesis. 2019;58(7):1194–1207. [DOI] [PubMed] [Google Scholar]
- 30.Abd El-Maqsoud NM, Tawfiek ER. Loss of Maspin Expression in Bladder Cancer: Its Relationship with p53 and Clinicopathological Parameters. Journal of the Egyptian National Cancer Institute. 2010;22(1):1–12. [PubMed] [Google Scholar]
- 31.Denk AE, Bettstetter M, Wild PJ, et al. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment cell research. 2007;20(2):112–119. [DOI] [PubMed] [Google Scholar]
- 32.Sharma G, Mirza S, Parshad R, et al. Clinical significance of Maspin promoter methylation and loss of its protein expression in invasive ductal breast carcinoma: correlation with VEGF-A and MTA1 expression. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2011;32(1):23–32. [DOI] [PubMed] [Google Scholar]
- 33.Goulet B, Chan G, Chambers AF, Lewis JD. An emerging role for the nuclear localization of maspin in the suppression of tumor progression and metastasis. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2012;90(1):22–38. [DOI] [PubMed] [Google Scholar]
- 34.Yan C, Boyd DD. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Molecular and cellular biology. 2006;26(17):6357–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fialova B, Smesny Trtkova K, Paskova L, Langova K, Kolar Z. Effect of histone deacetylase and DNA methyltransferase inhibitors on the expression of the androgen receptor gene in androgen-independent prostate cancer cell lines. Oncology reports. 2013;29(5):2039–2045. [DOI] [PubMed] [Google Scholar]
- 36.Huang PH, Plass C, Chen CS. Effects of Histone Deacetylase Inhibitors on Modulating H3K4 Methylation Marks - A Novel Cross-Talk Mechanism between Histone-Modifying Enzymes. Molecular and cellular pharmacology. 2011;3(2):39–43. [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Kaplun A, Lonardo F, et al. HDAC1 inhibition by maspin abrogates epigenetic silencing of glutathione S-transferase pi in prostate carcinoma cells. Molecular cancer research : MCR. 2011;9(6):733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends in molecular medicine. 2010;16(11):528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thakur VS, Gupta K, Gupta S. Green tea polyphenols increase p53 transcriptional activity and acetylation by suppressing class I histone deacetylases. International journal of oncology. 2012;41(1):353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Chen Y, Chen Q, et al. The role of acetylation sites in the regulation of p53 activity. Molecular biology reports. 2020;47(1):381–391. [DOI] [PubMed] [Google Scholar]
- 41.Weichert W, Roske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. British journal of cancer. 2008;98(3):604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meier K, Brehm A. Chromatin regulation: how complex does it get? Epigenetics. 2014;9(11):1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Current opinion in genetics & development. 1999;9(2):140–147. [DOI] [PubMed] [Google Scholar]
- 44.Adams GE, Chandru A, Cowley SM. Co-repressor, co-activator and general transcription factor: the many faces of the Sin3 histone deacetylase (HDAC) complex. The Biochemical journal. 2018;475(24):3921–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochemical Society transactions. 2013;41(3):741–749. [DOI] [PubMed] [Google Scholar]
- 46.Olzscha H, Sheikh S, La Thangue NB. Deacetylation of chromatin and gene expression regulation: a new target for epigenetic therapy. Critical reviews in oncogenesis. 2015;20(1–2):1–17. [DOI] [PubMed] [Google Scholar]
- 47.Ng HH, Bird A. Histone deacetylases: silencers for hire. Trends in biochemical sciences. 2000;25(3):121–126. [DOI] [PubMed] [Google Scholar]
- 48.Juan LJ, Shia WJ, Chen MH, et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. The Journal of biological chemistry. 2000;275(27):20436–20443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1: CpG islands in maspin genome. Locus name: NC_000018.10:63476958–63505085 Homo sapiens chromosome 18, GRCh38.p13 Primary Assembly. EMBOSS CgPLOT (EMBL) program was used with search with the parameters: length 200 %GC>50.0 P(CpG)/exp >0.60. Various CpG islands are shown in the maspin gene.
Figure 2: (A) Western blot analysis of maspin and HDAC1 in benign and cancer specimens. Total cell extracts from tissues were prepared and electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-maspin, anti-HDAC1 and anti-GAPDH antibodies. GAPDH is used as a loading control. Benign and cancer tissue from same individuals are from a subset of 16 patients. (B) IRS score generated from immunostaining for maspin and HDAC1 tissues from benign prostate tissue and cancer specimens of various Gleason grades. An inverse association was noted between maspin and HDAC1 expression in grade-specific manner during tumor progression.
Figure 3: Effect of 5-AZA-dC on maspin expression in prostate cancer cells. Human prostate cancer 22Rv1, LNCaP, DU145 and PC-3 cells were treated with 10 μM concentration of 5-AZA-dC for 48 h. The lysates were prepared, electrophoresed by SDS‐PAGE, followed by immunoblotting with anti-maspin and anti-β-actin antibodies. β-actin serves as a loading control. No changes in maspin expression were noted after 5-AZA-dC treatment.