

Abstract
Immunotherapy is a powerful treatment strategy being applied to cancer, autoimmune diseases, allergies, and transplantation. Although therapeutic monoclonal antibodies (mAbs) have demonstrated significant clinical efficacy, there is also the potential for severe adverse events, including cytokine release syndrome (CRS). CRS is characterized by the rapid production of inflammatory cytokines following delivery of therapy, with symptoms ranging from mild fever to life‐threating pathology and multi‐organ failure. Overall there is a paucity of models to reliably and accurately predict the induction of CRS by immune therapeutics. Here, we describe the development of a humanized mouse model based on the NOD‐scid IL2rgnull (NSG) mouse to study CRS in vivo. PBMC‐engrafted NSG, NSG‐MHC‐DKO, and NSG‐SGM3 mice were used to study cytokine release in response to treatment with mAb immunotherapies. Our data show that therapeutic‐stimulated cytokine release in these PBMC‐based NSG models captures the variation in cytokine release between individual donors, is drug dependent, occurs in the absence of acute xeno‐GVHD, highlighting the specificity of the assay, and shows a robust response following treatment with a TGN1412 analog, a CD28 superagonist. Overall our results demonstrate that PBMC‐engrafted NSG models are rapid, sensitive, and reproducible platforms to screen novel therapeutics for CRS.
Keywords: cytokine release syndrome, cytokine storm, humanized mouse, immune toxicity, therapeutic
Abbreviations
- ATG
anti‐thymocyte globulin
- BRG
BALB/c‐Rag2 null IL2r γ null
- CRS
cytokine release syndrome
- GVHD
graft‐versus‐host disease
- IL
interleukin
- IL2rg
IL‐2 receptor subunit γ
- IR
Irradiated
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- MHC‐DKO
NSG‐H2‐Ab1 em1mvw H‐2K tm1Bpe H‐2D tm1Bpe
- NOD
NOD/ShiLtJ
- NOG
NOD/Shi‐scid/IL‐2R γ null
- NRG
NOD‐Rag1 null IL2r γ null
- NSG
NOD‐scid IL2rγ null
- PBL
peripheral blood lymphocyte
- PBMC
peripheral blood mononuclear cell
- PD‐1
programmed death‐1
- SCID
severe combined immunodeficiency
- SGM3
Tg(CMV‐IL3,CSF2,KITLG)
- TNF
tumor necrosis factor
1. INTRODUCTION
Immunotherapeutics target the immune system to provide clinical benefits and are powerful treatment strategies being applied to cancer, autoimmune diseases, allergies, and transplantation. 1 , 2 , 3 , 4 Numerous approaches for immunotherapies, including cell‐based therapies such as CAR‐T, cytokine delivery, and monoclonal antibodies (mAbs) have demonstrated efficacy in the clinic. 5 Many mAb‐based therapies target antigens expressed on the surface of immune cells, including lymphocytes and innate immune cells, and can block ligand‐receptor interactions, potentiate ligand‐receptor interactions, and stimulate complement‐dependent cytotoxicity (CDC), antibody‐dependent cellular cytotoxicity (ADCC), and antibody‐dependent cellular phagocytosis (ADCP). 6 , 7 However, with the success of mAb‐based therapies there is also the potential for severe adverse events, including cytokine release syndrome (CRS). 8
CRS is characterized by the rapid production of cytokines following exposure to infectious agents, immune stimulating agents and therapeutics, and symptoms range from mild, including self‐limiting fever, to a systemic inflammatory event with severe pathology and multi‐organ failure. 9 , 10 In addition, the pathology associated with SARS‐CoV‐2 infection and COVID‐19 appears to be in part mediated by cytokine storm in patients. 11 The first reports of CRS stimulated by immunotherapies were in patients that were treated with anti‐CD3 mAb (muromonab or OKT3), and this response was characterized by the rapid and self‐limiting release of inflammatory cytokines and fever. 12 Since this initial observation CRS has been documented with several antibody‐based therapies, including anti‐thymocyte globulin (ATG), rituximab, obinutuzumab, alemtuzumab, brentuximab, dacetuzumab, and nivolumab. 13 One of the more severe cases of CRS was in patients treated with TGN1412 mAb, an anti‐CD28 superagonist developed by TeGenero AG. 14 A single infusion with TGN1412 stimulated a systemic inflammatory response characterized by cytokine release and the development of life‐threatening conditions including multi‐organ failure. 15 Surprisingly, preclinical studies done with TGN1412 showed no evidence of cytokine storm potential in both in vitro assays and in vivo animal models, including non‐human primates, 16 , 17 highlighting the need for novel screening platforms. 18 , 19 , 20 There remains a significant gap between pre‐clinical testing and clinical trials to efficiently predict CRS.
Humanized mouse models provide a potential translational bridge for the study and prediction of CRS in vivo. 21 Previous studies have suggested the potential of using immunodeficient mice engrafted with human immune systems to study CRS, but to date these have not emerged as a standard preclinical screening tool. 22 , 23 , 24 , 25 , 26 , 27 , 28 Here, we describe the development of a humanized mouse model based on the NOD‐scid IL2rgnull (NSG) mouse to study CRS in vivo. 29 , 30 We used PBMC engrafted NSG, NSG‐SGM3, and NSG‐MHC‐DKO mice to study cytokine release in response to several immunotherapeutics, including anti‐CD3, anti‐CD28, Keytruda, anti‐thymocyte globulin (ATG), and a TGN1412 analog. Our data show that PBMC‐NSG and PBMC‐SGM3 mice capture the variation in cytokine release between individual human PBMC donors. Moreover, cytokine release responses were demonstrated even in genetically modified NSG mice that do not express MHC class I or class II and do not develop acute GVHD following PBMC injection. Overall the PBMC engrafted NSG, NSG‐MHC‐DKO, and NSG‐SGM3 mouse models enabled the detection of cytokine release stimulated by several mAbs and are rapid, sensitive, and reproducible platforms to screen novel therapeutics for inflammatory events.
2. MATERIALS AND METHODS
2.1. Engraftment of human PBMC in NSG mice
Female NOD.Cg‐Prkdcscid Il2rgtm1Wjl /SzJ mice (NSG, stock number 005557), NOD.Cg‐Prkdcscid Il2rgtm1Wjl Tg(CMV‐IL3,CSF2,KITLG)1Eav/MloySzJ (NSG‐SGM3, stock number 013062, express human stem cell factor, GM‐CSF, and IL3 29 , 31 ), and NSG‐H2‐Ab1em1mvw H‐2K1tm1Bpe H‐2D1tm1Bpe mice (NSG‐MHC‐DKO, stock number 025216) have been described previously 30 , 32 and were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were housed in a specific pathogen free facility in microisolator cages, given autoclaved food and maintained on acidified autoclaved water at The Jackson Laboratory or alternated weekly between acidified autoclaved water and sulfamethoxazole‐trimethoprim medicated water (Goldline Laboratories, Ft. Lauderdale, FL) at the University of Massachusetts Medical School. All animal procedures were done in accordance with the guidelines of the Animal Care and Use Committee of The Jackson Laboratory and the University of Massachusetts Medical School and conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, Eighth Edition 2011).
2.2. Flow cytometry
Human immune cell populations were monitored in PBMC‐engrafted mice using mAbs specific for the following human antigens; CD45‐PerCPCy5.5 (clone HI30), CD3‐FITC (clone UCHT1), CD19‐APC (clone HIB19), CD33‐PE/Cy7 (clone P67.6), CD14 APC‐Cy7 (clone HIB19), and CD56‐PE (clone HCD56). All antibodies were purchased from BioLegend (San Diego, CA). Whole blood was collected in heparin, and then 100 μL of blood was washed with FACS buffer (PBS supplemented with 2% of fetal bovine serum (FBS) and 0.02% of sodium azide) and pre‐incubated with rat anti‐mouse FcR mAb (clone 2.4G2, BD Biosciences) to block binding to mouse Fc receptors. Specific mAbs were then added to the samples and incubated for 30 minutes at 4°C. Stained samples were washed and treated with BD FACS lysing solution. At least 50 000 events were acquired on LSR II or FACSCalibur instruments (BD Biosciences). Data analysis was performed with FlowJo (Tree Star, Inc, Ashland, OR) software.
2.3. Induction of cytokine release in PBMC‐NSG, PBMC‐NSG‐MHC‐DKO, and PBMC‐NSG‐SGM3 mice and quantification of human cytokines
Mice were either preconditioned with irradiation (100 cGy, IR) at least 4 hours before human PBMC injection or left non‐irradiated (non‐IR). Cryopreserved human PBMC were purchased commercially from Astarte Biologics (Bothell, WA), AllCell Technologies (Chicago, IL), Lonza (Walkersville, NC), and STEMCELL Technologies (Vancouver, Canada), see Table S1. PBMCs were washed twice with PBS after thawing, then, injected intravenously (IV) into NSG, NSG‐MHC‐DKO, or NSG‐SGM3 mice (strains described in Table S2) at the indicated cell numbers. Following PBMC injection, mice were observed daily for overall health including general appearance of the fur, mobility, and body weights.
To stimulate cytokine release, PBMC‐NSG, PBMC‐NSG‐MHC‐DKO, and PBMC‐SGM3 mice were injected intravenously (IV) with human‐specific antibodies including, OKT3 (anti‐CD3, 0.5 mg/kg, BioLegend, San Diego CA, ANC28 (anti‐CD28, 1 mg/kg, Millipore‐Sigma, St. Louis, MO), KEYTRUDA (anti‐PD‐1, 5 mg/kg, Merck Oncology, Kenilworth, NJ), ATG (anti‐thymocyte globulin, 1 mg/kg, Sanofi Genzyme, Cambridge, MA), or a TGN1412 analog (anti‐CD28, 0.5, 1 and 2 mg/kg, Creative Biolabs, Shirley, NY). PBS injection was used as a negative control in all experiments. Mice were bled at the indicated time points and serum was collected and analyzed for human cytokines (IFN‐gamma, IL10, IL6, IL2, IL4, and TNF) using a BD Cytometric Bead Array (CBA) Human Th1/Th2 Cytokine kit II (BD‐Biosciences, San Jose, CA).
2.4. Monitoring of body temperatures of NSG mice
Rectal temperatures of PBMC‐NSG and PBMC‐SGM3 mice were measured before treatment and again immediately before each time‐point bleed. Temperature was measured by the insertion of a rectal thermocouple probe and waiting until a stable reading was obtained.
2.5. In vitro assay for evaluating cytokine release by human PBMC
To evaluate in vitro cytokine release, PBMC were incubated in 96 well tissue culture plates at 37°C/5% CO2 for 48 hours in the presence of OKT3, anti‐CD28, or PBS. One day prior to addition of PBMC, plates were coated overnight with OKT3 (0.1 mg/mL) and anti‐CD28 (0.2 mg/mL) in PBS at 4°C. Cryopreserved PBMC were thawed, resuspended in RPMI supplemented with 10% FBS, 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, 2 mM L‐glutamine, 1% (v/v) nonessential amino acids solution, 50 μM 2‐mercaptoethanol, and 1 mM sodium pyruvate, and washed once. Viability counts were performed, and 1 × 105 viable PBMC were added to each well. Supernatants were harvested 48 hours later, and cytokine levels were measured by a BD CBA Th1/Th2 II kit.
2.6. Statistical analysis
Statistical analyses were performed using GraphPad PRISM 8.0 software. Data significance (P values) was calculated using one‐way and two‐way ANOVAs and Bonferroni's multiple comparisons test. All error bars represent the SEM. For all statistical analyses, significance is defined as P < .05 or indicated as NS (none significant).
3. RESULTS
3.1. Cytokine profiles in the PBMC‐NSG model captures donor variability in response to in vivo stimulation with therapeutic antibodies
To evaluate the in vivo production of human cytokines following treatment with monoclonal antibodies, we used PBMC‐injected NSG (PBMC‐NSG) mice that were preconditioned with irradiation (100 cGy) and injected IV with 20 million PBMC. NSG mice support rapid and efficient engraftment with human PBMC that results in survival and activation of human T cells and the development of acute‐xenogeneic (xeno) GVHD. We evaluated the conditions for PBMC engraftment regarding preconditioning with irradiation, animal health, cell number, and engrafting immune cell subsets as described in Figures [Link], [Link], [Link]. We next evaluated 10 donors for therapeutic‐induced cytokine release in NSG‐PBMC mice. NSG mice were irradiated and injected with 20 million PBMC from 10 different donors (4692, 4625, 4668, 362, 366, 345, 213, 364, 353, 309). Six days later, PBMC‐NSG mice were treated with PBS, OKT3, or anti‐CD28 mAb (ANC28), and sera was collected 6 hours later for human cytokine analysis (Figure 1). Treatment with OKT3 stimulated significant amounts of IFN‐gamma for 8 out of 10 donors (greater than 2000 pg/mL for donors 4692, 4625, 4668, 362, 366, 345, 213, 309, Figure 1A). Moreover, anti‐CD28 treatment also induced human IFN‐gamma for three donors (4692, 4668, 362). Treatment with OKT3 stimulated significant amounts of human IL10 for 8 out of 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 309), with donor 4692 also showing an IL10 response after treatment with anti‐CD28 (Figure 1B). Treatment with OKT3 stimulated significant amounts of human IL6 for 9 out of 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 353, 309), with donors 4692, 362, and 345 also showing an IL6 response after treatment with anti‐CD28 (Figure 1C). Treatment with OKT3 stimulated significant release of human IL2 for all 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 364, 353, 309), but no IL2 responses were observed after anti‐CD28 treatment (Figure 1D). Treatment with OKT3 stimulated significant amounts of human IL4 for 9 out of 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 364, 309), with donors 4692, 4668, 362, and 366 also showing an IL4 response after treatment with anti‐CD28 (Figure 1E). Treatment with OKT3 stimulated significant amounts of human TNF for all 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 364, 353, 309), but minimal TNF responses were observed after anti‐CD28 treatment (Figure 1F). In addition, we observed that by 6 hours after treatment, levels of human T cells were significantly decreased in the blood of NSG‐PBMC mice injected with OKT3 or anti‐CD28, as compared to PBS‐treated mice (Figure S2D). Together these data highlight the ability of the PBMC‐NSG model to capture the donor variability in cytokine release following treatment with OKT3 and anti‐CD28.
FIGURE 1.
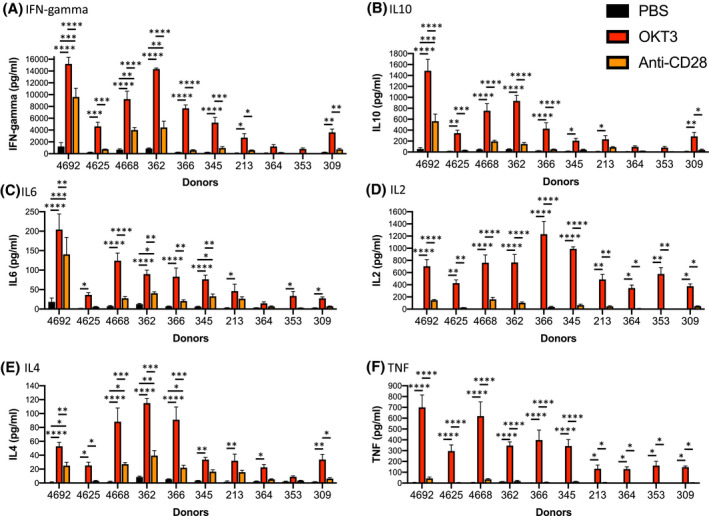
Variation in donor‐specific cytokine release in PBMC‐NSG mice. Irradiated (100 cGy) NSG mice were injected IV with PBMC (20 × 106 cells) from 10 donors (4692, 4625, 4668, 362, 366, 345, 213, 364, 353, 309). On day 6 post‐engraftment, PBMC‐NSG mice were treated IV with PBS, OKT3 (0.5 mg/kg), or anti‐CD28 (1 mg/kg) as described in the Materials and Methods. Six hours after treatment sera were collected for human cytokine analysis including, (A) IFN‐gamma, (B) IL10, (C) IL6, (D) IL2, (E) IL4, and (F) TNF. Cytokine levels (pg/mL) ± SEM are shown. Each experimental group represents three to five mice. *P < .05, **P < .01, ***P < .001, ****P < .0001
3.2. Evaluation of cytokine release in NSG‐MHC‐DKO mice that do not develop acute‐GVHD
As described above, injection of PBMC into irradiated NSG mice resulted in the development of acute xeno‐GHVD as the engrafting human T cells respond to mouse MHC. 33 It is possible that this acute xeno‐GVHD in PBMC‐NSG mice will alter the magnitude and the specific human cytokines released during the response to therapeutic antibodies. To address the potential contribution of the acute GHVD, we used a newly developed NSG strain (NSG‐MHC‐DKO) that lacks the expression of mouse MHC. 32 This mouse strain supports the engraftment of functional human T cells but does not develop the acute GVHD due to the lack of murine MHC xenogeneic antigens, the primary targets of the xeno‐GVHD response. NSG and NSG‐MHC‐DKO mice were irradiated (100 cGy), and then, injected with 20 million PBMC from donor 4536 (Figure 2). Percentages of human CD45+ cells were evaluated in the peripheral blood at day 5 and day 10 (Figure 2A), and human immune cell subsets were identified (Figure 2B). Percentages of human CD45+ cells were similar between NSG and NSG‐MHC‐DKO mice on day 5 and slightly higher in NSG mice on day 10 (Figure 2A). CD3+ T cells were the dominant immune cell population at day 5 and day 10, and NK cells were detectable at day 5 in both mouse strains (Figure 2B). Next, we evaluated the cytokine release response in PBMC‐NSG and PBMC‐NSG‐MHC‐DKO mice. Six days post‐PBMC injection, mice were treated with PBS, OKT3 or Keytruda, and sera were collected 6 hours later for human cytokine analysis (Figure 2C‐H). The levels of human IFN‐gamma, IL10, IL6, IL2, and IL4 were similar between the two mouse strains (Figure 2C‐G), while levels of TNF were slightly higher in NSG mice (Figure 2H). Together these data indicate that the cytokine release detected in PBMC‐NSG mice treated with antibody therapeutics is induced by the drug and not dependent on the development of acute xeno‐GVHD.
FIGURE 2.
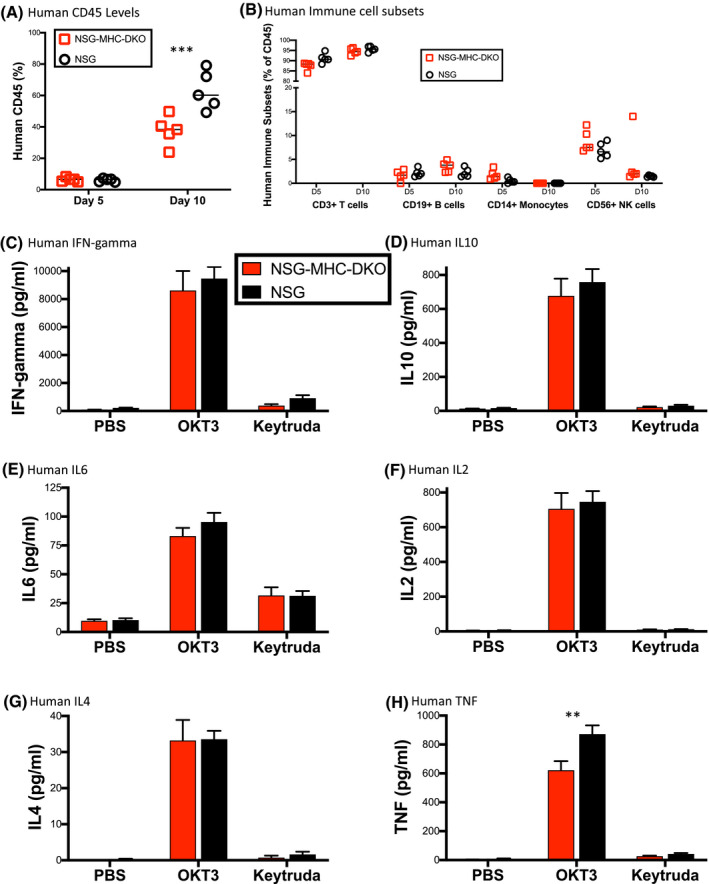
Cytokine release in NSG‐MHC‐DKO mice that do not develop acute xeno‐GVHD. NSG and NSG‐MHC‐DKO mice were irradiated and then injected with 20 million PBMC from donor 4536. A, levels of human CD45+ and (B) human immune cell subsets (CD3+ T cells, CD19+ B cells, CD14+ monocytes, and CD56+ NK cells) were evaluated by flow cytometry in the peripheral blood. Each symbol represents an individual mouse. C‐H, Irradiated NSG and NSG‐MHC‐DKO mice were injected IV with PBMC (20 × 106 cells) from donor 4536. Day 6 post‐PBMC engraftment, mice were treated IV with PBS, OKT3 (0.5 mg/kg), and Keytruda (5 mg/kg) as described in the Materials and Methods. Six hours after treatment serum was collected for human cytokine analysis including, (C) IFN‐gamma, (D) IL10 and (E) IL6, (F) IL2, (G) IL4, and (H) TNF. The results are representative of three independent experiments. Each experimental group represents five mice. Cytokine levels (pg/mL) ± SEM are shown. **P < .01, ***P < .001
3.3. Comparison of cytokine release using the in vivo PBMC‐NSG model and an in vitro PBMC assay
In vitro whole‐blood or PBMC assays are currently the primary tools used to determine effects of new biologics on cytokine release during screening. 8 Here, we have compared the in vivo and in vitro assays for stimulation of cytokine release following stimulation with OKT3 and anti‐CD28 using five PBMC donors (213, 309, 345, 362, and 366, (Figure 3). IR (100 cGy) PBMC‐NSG mice were treated with PBS, OKT3, or anti‐CD28 6 days after PBMC injection, and 6 hours after treatment sera were recovered for cytokine analysis. For the in vitro assay, PBMC were incubated with plate‐bound OKT3 or anti‐CD28 for 2 days, and then, culture supernatants were collected for cytokine analysis. While the human cytokine profiles stimulated by OKT3 for the in vivo and in vitro assays were similar with regard to the donor production of cytokines, the in vivo PBMC‐NSG model was more sensitive than the in vitro assay for detection of cytokine release responses from specific donors, including IFN‐gamma production by donor 213 (Figure 3A,B), IL10 production by donors 213, 309, 345, and 362 (Figure 3C,D), IL6 production for donors 213 and 309 (Figure 3E,F), IL2 production for donors 213 and 345 (Figure 3G,H), and TNF production for donors 213 and 309 (Figure 3I,J). Cytokine release stimulated by anti‐CD28 was also similar between the in vivo and in vitro models, with the one exception of donor 345, which showed an IFN‐gamma response with the in vitro assay but not the in vivo model (Figure 3A,B). Overall levels of human cytokines detected are higher for the in vitro assay, but the extremely high amounts of cytokines produced by specific donors (eg, donor 366 for IL10 and donor 345 for IL6), complicates the interpretation of the data sets and statistical analyses. These data suggest that the in vivo PBMC‐NSG model is a sensitive platform for evaluation of CRS and may enable the identification of responders that would not be detected by standard approaches.
FIGURE 3.
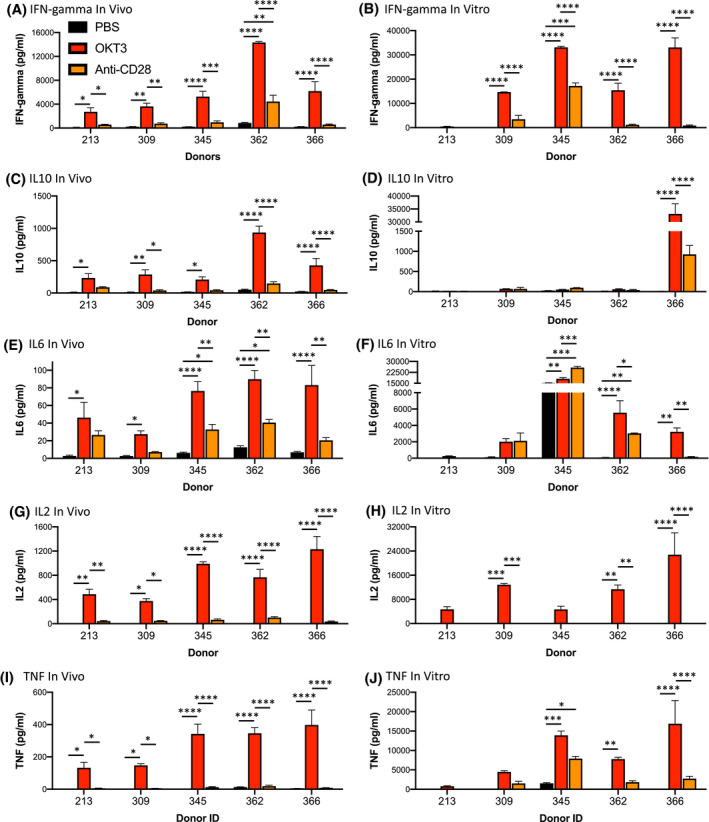
Comparison of cytokine release using the in vivo PBMC‐NSG model and an in vitro PBMC assay. PBMC from five donors were evaluated for in vivo (A, C, E, G, and I) and in vitro (B, D, F, H, and J) cytokine release. For in vivo cytokine release, irradiated (100 cGy) NSG mice were injected IV with PBMC (20 × 106 cells) and 6 days post‐injection, mice were treated IV with PBS, OKT3 (0.5 mg/mL), or anti‐CD28 (1 mg/mL) as described in the Materials and Methods. Six hours after treatment sera were collected for human cytokine analysis. For in vitro cytokine release, PBMC were added to plates that were coated overnight with PBS, OKT3, or anti‐CD28 as described in the Materials and Methods. After 48 hours supernatants were collected for human cytokine analysis. Cytokine quantification included: (A, B) IFN‐gamma; (C, D) IL10; (E, F) IL6; (G, H) IL2; (I, J) TNF. Cytokine levels (pg/mL) ± SEM are shown. Each in vivo experimental group represents three to five mice, and in vitro results were performed in duplicate for each donor. *P < .05, **P < .01, ***P < .001, ****P < .0001
3.4. Cytokine release is reproducible between experiments with PBMC using the same donor
We next tested the reproducibility of the cytokine release response for an individual donor. For this experiment, PBMC from donor 213 were used to setup five independent experiments in NSG mice (Figure 4). Six days after injection, PBMC‐NSG mice were treated with the indicated therapeutics or control PBS, and levels of human cytokines were evaluated 6 hours later. OKT3 and anti‐CD28 were used as treatments in two experiments, and OKT3 and Keytruda were used in three experiments. Following treatment with OKT3, PBMC‐NSG mice engrafted with cells from donor 213 showed consistent release of human IFN‐gamma (Figure 4A), IL10 (Figure 4B), IL6 (Figure 4C), and TNF (Figure 4D) as compared to the control PBS. In addition, donor 213 did not show significant cytokine responses to anti‐CD28 or Keytruda. Together these data demonstrate the reproducibility of donor‐specific cytokine release in PBMC‐NSG mice.
FIGURE 4.
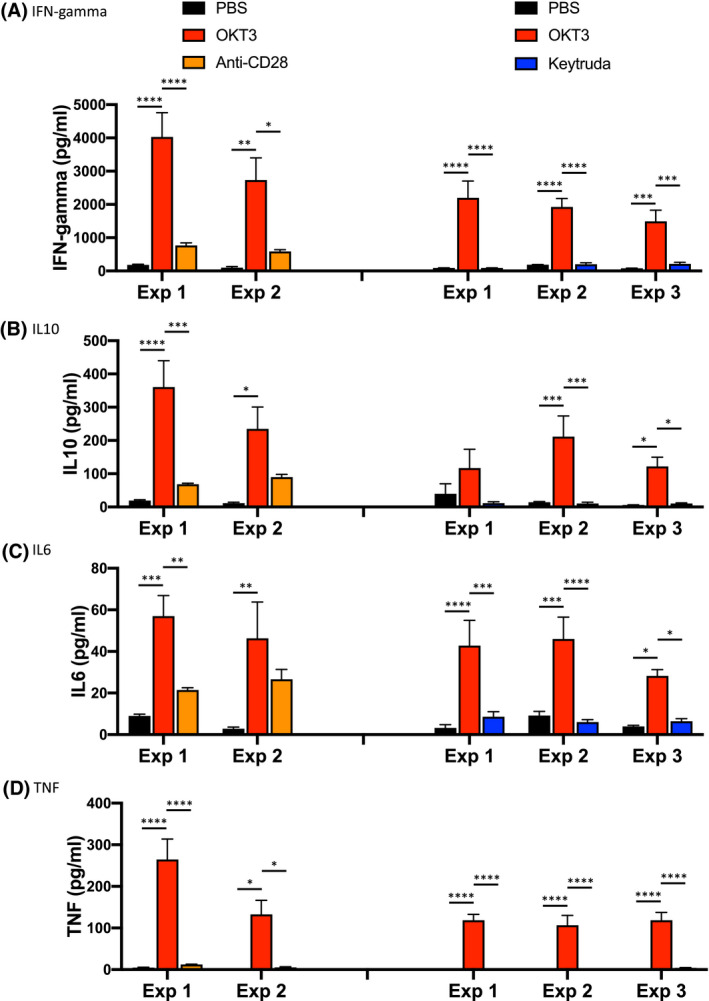
Donor‐specific cytokine release in PBMC‐NSG mice is reproducible between experiments. PBMC from donor 213 were used to setup five independent experiments on different days. Irradiated NSG mice were injected IV with PBMC (20 × 106 to 30 × 106 cells) from donors 213. Day 6 post engraftment mice were treated IV with either PBS, OKT3 (0.5 mg/mL), anti‐CD28 (0.2 mg/mL) in one experiment, or in a second experiment, with PBS, OKT3 (0.1 mg/mL), or Keytruda (5 to 10 mg/kg) as described in the Materials and Methods. Six hours after treatment serum was collected for human cytokine analysis including, (A) IFN‐gamma, (B) IL10, (C) IL6, and (D) TNF. Cytokine levels (pg/mL) ± SEM are shown. Each experimental group represents five to five mice. *P < .05, **P < .01, ***P < .001, ****P < .0001
3.5. Cytokine release in PBMC‐engrafted NSG‐SGM3 (PBMC‐SGM3) mice
We next compared the induction of cytokine release between NSG and NSG‐SGM3 (SGM3) mice that were engrafted with PBMC. The SGM3 strain expresses human stem cell factor, GM‐CSF, and IL3. 29 , 31 NSG and NSG‐SGM3 mice were irradiated, and then, injected with PBMC from donor 3251. Both PBMC‐NSG and PBMC‐SGM3 mice supported human PBMC engraftment, and no significant differences were observed for the percentages of human CD45+ cells and the engrafting human immune cell subsets in the peripheral blood at day 5 (data not shown). Six days after injection the PBMC‐NSG and PBMC‐NSG‐SGM3 mice were treated with IV with PBS, anti‐CD28, Keytruda, or ATG and levels of human cytokines were evaluated 6 hours later (Figure S4). For NSG mice engrafted with PBMC from donor 3251, therapeutic treatment‐induced minimal IFN‐gamma and IL10 responses. In contrast, treatment of PBMC‐SGM3 mice with anti‐CD28 or ATG stimulated the significant release of human IFN‐gamma and IL10 and Keytruda stimulated increased IFN‐gamma but not IL10 (Figure S4). These data suggest that the PBMC‐SGM3 model is a more sensitive platform for testing cytokine release stimulated by antibody‐based therapeutics.
Given the sensitivity of the PBMC‐engrafted SGM3 model, we next evaluated the response of PBMC‐SGM3 mice to a TGN1412 analog, a CD28 superagonist antibody that stimulated a severe cytokine storm and systemic inflammation in healthy volunteers. 14 NSG‐SGM3 mice were irradiated and then, injected with 1.5 × 107 PBMC from donor 3251. Six days after injection PBMC‐SGM3 mice were treated with PBS, anti‐CD28 (ANC28, 1 mg/kg), or the TGN1412 analog (0.5, 1.0, or 2.0 mg/kg) (Figure 5). Sera samples were collected for cytokine analysis at 1, 2, 4, and 6 hours posttreatment. PBMC‐SGM3 mice treated with 1.0 and 2.0 mg/kg of the TGN1412 analog showed rapid cytokine release for all cytokines measured as compared to PBS‐treated control mice. Human IFN‐gamma peaked at 4 hours posttreatment with 1.0 mg/kg of the TGN1412 analog (6674.1 ± 1024 pg/mL) and 2.0 mg/kg (7532.2 ± 1080 pg/mL) (Figure 5A). Anti‐CD28 (ANC28) also induced IFN‐gamma in PBMC‐SGM3 mice, reaching peaked by 4 hours (5225.9 ± 928 pg/mL). Human IL10 was released in PBMC‐SGM3 mice treated with 1.0 and 2.0 mg/kg of the TGN1412 analog and anti‐CD28, with peak levels at 2 hours for anti‐CD28 (348.3 ± 40.6 pg/mL) and 2.0 mg/kg of the TGN1412 analog (309.2 ± 74.0 pg/mL) and at 4 hours for 1.0 mg/kg of the TGN1412 analog (275.3 ± 51.3 pg/mL) (Figure 5B). IL10 levels were sustained with anti‐CD28 treatment at the 6‐hour time point. Human IL6 was detectable in PBMC‐SGM3 mice treated with 1.0 and 2.0 mg/kg of the TGN1412 analog and anti‐CD28, with peak levels at 4 hours for 1.0 mg/kg (89.1 ± 17 pg/mL) or 2.0 mg/kg (131.5 ± 14.8 pg/mL) of the TGN1412 analog and at 6 hours for anti‐CD28 (108.1 ± 8.0 pg/mL) (Figure 5C). Human IL2 was detectable at 2‐, 4‐, and 6‐hours posttreatment with anti‐CD28, and the levels were significantly higher that any TGN1412 analog‐induced responses (Figure 5D). IL2 levels peaked with anti‐CD28 treatment at 6 hours (258.5.4 ± 34.4 pg/mL). TGN1412 analog‐stimulated IL2 release peaked at 4 hours for 0.5 (48.3 ± 9.8 pg/mL), 1.0 (70.8 ± 9.4 pg/mL), and 2.0 mg/kg (91.1 ± 3.1 pg/mL), and IL2 was still detectable at 6 hours for the 2.0 mg/kg dose. Human IL4 was detectable in PBMC‐SGM3 mice treated with 1.0 and 2.0 mg/kg of the TGN1412 analog and anti‐CD28, with peak levels at 4 hours for 1.0 mg/kg (51.1 ± 10 pg/mL) and 2.0 mg/kg (64.1 ± 10 pg/mL) of the TGN1412 analog and at 6 hours for anti‐CD28 (62.4 ± 8.8 pg/mL) (Figure 5E). Human TNF was detectable at 1‐ and 2‐hours posttreatment for 1.0 and 2.0 mg/kg of the TGN1412 analog and at 1 hour posttreatment for anti‐CD28 (Figure 5F). Levels peaked at the 1‐hour time point for 1.0 mg/kg (286.1 ± 117 pg/mL) and 2.0 mg/kg (601.6 ± 91.8 pg/mL) of the TGN1412 analog, and anti‐CD28 (294.8 ± 76.2 pg/mL). Overall these results show that the PBMC‐SGM3 model reproduced the rapid production of multiple cytokines observed from patients treated with TGN1412, 14 and suggest that this model may be an effective tool for predicting patient cytokine release syndrome variable responses to immunotherapy.
FIGURE 5.
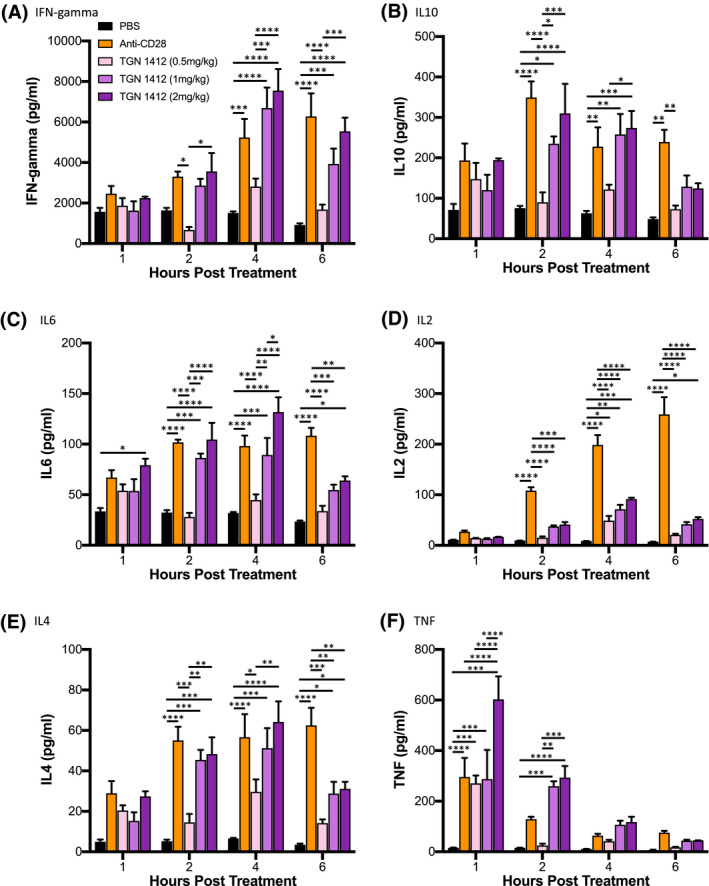
Stimulation of cytokine release by TGN1412 in PBMC‐SGM3 mice. NSG‐SGM3 mice were irradiated and then injected with 15 × 106 PBMC from donor 3251. Six days after injection PBMC‐NSG mice were treated IV with PBS, anti‐CD28 (1 mg/kg), or TGN1412 analog (0.5, 1.0, or 2.0 mg/kg). Serum samples were collected for human cytokine analysis at 1, 2, 4, and 6 hours posttreatment. Cytokine quantification included; (A) IFN‐gamma, (B) IL10 and (C) IL6, (D) IL2, (E) IL4, and (F) TNF. The results are representative of two independent experiments. Cytokine levels (pg/mL) ± SEM are shown. Each experimental group represents four mice. *P < .05, **P < .01, ***P < .001, ****P < .0001
4. DISCUSSION
The advent of antibody‐based immune therapies has revolutionized the treatment of several human disease states. 34 , 35 Over the past decade, a primary application of these immunotherapies has been the treatment of malignancies by targeting immune checkpoint molecules. 36 While antibody‐based therapeutics are enjoying an unprecedented level of success in the clinic by targeting cells of the immune system, a number of clinical trials have reported adverse reactions, including CRS. 13 Current platforms to screen biologic agents for induction of CRS include in vitro studies with human PBMC and traditional in vivo animal models such as non‐human primates. 16 However, there is still an urgent need for human‐based in vivo CRS screening platforms that are accurate, translatable, can easily be scaled and will ideally enable the study of patient‐specific responses. 8 Here, we describe a humanized mouse model that enables the rapid, sensitive, and reproducible screening of human‐specific therapeutics for induction of inflammatory events. This humanized platform is based on engraftment of PBMC into the NSG and NSG‐SGM3 strains. PBMC‐NSG and PBMC‐SGM3 mice demonstrated consistent cytokine release following treatment with antibody‐based therapeutics. Moreover, our results demonstrate that cytokine release was also observed in a novel NSG‐MHC‐DKO mouse strain that does not develop acute xeno‐GVHD following injection of human PBMC. Together our data demonstrate the utility of PBMC engrafted NSG, NSG‐MHC‐DKO, and NSG‐SGM3 mice as an easily scalable approach for screening immunotherapies for CRS.
Immunodeficient mice that support engraftment of functional human immune systems are being widely applied to the study of human immunobiology, including immune system development, homeostasis, and function. 21 One of the original immunodeficient mouse strains used for humanization was the CB17‐SCID mouse. 37 , 38 , 39 PBMC‐engrafted CB17‐SCID mice were used to evaluate cytokine release following treatment with OKT3 and modified versions of OKT3. 28 The results of this study showed that OKT3 treatment of PBMC‐engrafted CB17‐SCID mice stimulated the release of human cytokines including IFN‐gamma, IL10, TNF, and IL2. However, the model requires the depletion of mouse NK cells, and human cell chimerism levels in CB17‐SCID mice are highly variable. 40 , 41 The use of humanized mice to study human immune systems was advanced significantly by the development of immunodeficient mice, including NSG, NRG, NOG, and BRG mice that bear mutations in the IL2r common gamma chain (CD132). 42 The common gamma chain deficiency blocks the development of murine NK cells and increases levels of human immune cell chimerism following injection of human hematopoietic stem cells or PBMC. 30 , 43 , 44 , 45 , 46 PBMC‐engrafted NSG mice were shown to release human cytokines following treatment with OKT3, but not following treatment with ATG or Campath‐1H (anti‐CD52). 27 In an effort to improve PBMC engraftment and sensitivity for CRS responses, an NRG mouse expressing HLA‐DQ8 and lacking the expression of murine MHC class II was used as a recipient of PBMC. The PBMC‐engrafted NRG HLA‐DQ8+ Ab1null mice displayed cytokine release following treatment with OKT3 and a TG1412, including IFN‐gamma, IL10, and TNF, which were also detected in our experiments. 26 However, PBMC‐NSG mice in our experiments also produced significant levels of human IL6, IL2, and IL4 following OKT3 treatment (Figure 1), and PBMC‐engrafted NSG‐SGM3 mice produced human IL6, IL2, and IL4 following treatment with a TGN1412 analog (Figure 5). In addition, PBMC‐engrafted NRG HLA‐DQ8+ Ab1null also displayed significant pathology following treatment with OKT3 and TGN1412. 26 Recent reports have also demonstrated the efficacy of using the NOG‐BLT (bone marrow/liver/thymus model) 47 to assess in vivo cytokine release stimulated by OKT3 25 and a TGN1412 analog. 22 NOG‐BLT mice and NOG‐EXL (expressing human GM‐SCF and IL3) BLT mice were also used to demonstrate the induction of adverse reactions to anti‐PD‐1 therapy (nivolumab), including pneumonitis and hepatitis. 23 However, the BLT model presents several challenges for high‐throughput screening, which include limited availability of human fetal tissues, ethical concerns with the procurement, the requirement for survival surgery and the lengthy time period required for immune system development (8 to 12 weeks). 48
The 2006 clinical trial using healthy volunteers with TGN1412 resulted in severe systemic inflammation that became life threatening in all recipients and correlated with a rapid cytokine release syndrome. 14 High levels of inflammatory cytokines, including TNF, IFN‐gamma, IL10, IL8, IL6, IL4, IL2, IL1‐beta, were detected systemically within hours of treatment. The severe adverse reactions were not predicted from preclinical studies done with the TGN1412 antibody in vitro and in non‐human primates. 49 The pathologic response to TGN1412 in individuals correlated with the release of IL2, IFN‐gamma, and TNF by CCR7‐/CD28+ effector memory CD4 T cells. 50 CD28 is not expressed by cynomolgus and rhesus CCR7‐ effector memory CD4 T‐cells, which is a possible explanation for the lack of cytokine response in non‐human primates. Our results show that PBMC‐SGM3 mice treated with a TGN1412 analog have a robust and rapid cytokine response (Figure 5), including release of human IFN‐gamma, IL10, IL6, IL2, IL4, and TNF. Furthermore, the PBMC‐SGM3 model shows a more robust and sensitive readout for human cytokine release stimulated by therapeutics as compared to the PBMC‐NSG mice (Figure S4). While the immune cell engraftment profiles are similar between the PBMC‐SGM3 and PBMC‐NSG mice, this may be related to a heightened effector activation profile for human T cells in NSG‐SGM3 mice, as demonstrated in NSG‐SGM3‐BLT mice. 51 Interestingly, the CRS induced by Keytruda in both PBMC‐NSG and PBMC‐SGM3 mice was minor for most donors, matching the clinical response observed in the majority of patients. 52
Overall these results indicate that PBMC‐engrafted NSG, NSG‐MHC‐DKO, and NSG‐SGM3 mice described here are rapid, sensitive, and reproducible models to screen of human‐specific novel therapeutics for induction of inflammatory events. Moreover, our data suggest that the use of PBMC for immune system engraftment enables the rapid evaluation of patient‐specific cytokine responses to therapeutics, which will facilitate the translation to clinical screening. The humanized CRS model established here will provide a potential platform to test donor‐specific responses to therapeutic options. Studies are currently being conducted with patient PBMC to directly compare and validate these humanized models to patient‐specific CRS stimulated by immunotherapies. In additional, these models, specifically the NSG‐SGM3 mouse, may prove useful to study cytokine release by cell populations isolated from the lungs of acute respiratory distress syndrome (ARDS) patients, including alveolar macrophages from COVID patients.
CONFLICT OF INTEREST
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. MAB and DLG are consultants for The Jackson Laboratory. The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
C. Ye performed experiments and wrote the paper, H. Yang performed experiments, M. Cheng performed experiments L.D. Shultz wrote the paper, D.L. Greiner wrote the paper, M.A. Brehm performed experiments and wrote the paper, and J.G. Keck designed the study and wrote the paper.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Table S1
Table S2
ACKNOWLEDGMENTS
This work was supported, in part, by US National Institutes of Health (NIH) Office of the Director Grant R24 OD026440‐01 and the NIH National Institute of Diabetes and Digestive and Kidney Diseases‐supported Human Islet Research Network (https://hirnetwork.org) Grants U01DK104218‐02 (to MAB, DLG, and LDS), R01 AI132963 (to MAB and LDS), R24OD0426640 (MAB, DLG, and LDS), and DP 3DK111898 (to MAB).
Ye C, Yang H, Cheng M, et al. A rapid, sensitive, and reproducible in vivo PBMC humanized murine model for determining therapeutic-related cytokine release syndrome. The FASEB Journal. 2020;34:12963–12975. 10.1096/fj.202001203R
REFERENCES
- 1. McDonald‐Hyman C, Turka LA, Blazar BR. Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Sci Transl Med. 2015;7:280rv282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moote W, Kim H, Ellis AK. Allergen‐specific immunotherapy. Allergy Asthma Clin Immunol. 2018;14:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wraith DC. The future of immunotherapy: a 20‐year perspective. Front Immunol. 2017;8:1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dougan M, Dranoff G, Dougan SK. Cancer immunotherapy: beyond checkpoint blockade. Ann Rev Cancer Biol. 2019;3:55‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parakh S, King D, Gan HK, Scott AM. Current development of monoclonal antibodies in cancer therapy. Recent Results Cancer Res. 2020;214:1‐70. [DOI] [PubMed] [Google Scholar]
- 6. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK Cell‐mediated targeting of tumor cells. Methods Mol Biol. 2016;1441:333‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Redman JM, Hill EM, AlDeghaither D, Weiner LM. Mechanisms of action of therapeutic antibodies for cancer. Mol Immunol. 2015;67:28‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grimaldi C, Finco D, Fort MM, et al. Cytokine release: a workshop proceedings on the state‐of‐the‐science, current challenges and future directions. Cytokine. 2016;85:101‐108. [DOI] [PubMed] [Google Scholar]
- 9. Murthy H, Iqbal M, Chavez JC, Kharfan‐Dabaja MA. Cytokine release syndrome: current perspectives. Immunotargets Ther. 2019;8:43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76:16‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pedersen SF, Ho YC. SARS‐CoV‐2: a storm is raging. J Clin Invest. 2020;130:2202‐2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chatenoud L, Ferran C, Reuter A, et al. Systemic reaction to the anti‐T‐cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon‐gamma [corrected]. N Engl J Med. 1989;320:1420‐1421. [DOI] [PubMed] [Google Scholar]
- 13. Shimabukuro‐Vornhagen A, Godel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018‐1028. [DOI] [PubMed] [Google Scholar]
- 15. Attarwala H. TGN1412: from discovery to disaster. J Young Pharm. 2010;2:332‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vessillier S, Eastwood D, Fox B, et al. Cytokine release assays for the prediction of therapeutic mAb safety in first‐in man trials—whole blood cytokine release assays are poorly predictive for TGN1412 cytokine storm. J Immunol Methods. 2015;424:43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finco D, Grimaldi C, Fort M, et al. Cytokine release assays: current practices and future directions. Cytokine. 2014;66:143‐155. [DOI] [PubMed] [Google Scholar]
- 18. Stebbings R, Eastwood D, Poole S, Thorpe R. After TGN1412: recent developments in cytokine release assays. J Immunotoxicol. 2013;10:75‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507‐3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731‐2738. [DOI] [PubMed] [Google Scholar]
- 21. Shultz LD, Keck J, Burzenski L, et al. Humanized mouse models of immunological diseases and precision medicine. Mamm Genome. 2019;30:123‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan H, Bhagwat B, Sanden D, et al. Evaluation of a TGN1412 analogue using in vitro assays and two immune humanized mouse models. Toxicol Appl Pharmacol. 2019;372:57‐69. [DOI] [PubMed] [Google Scholar]
- 23. Weaver JL, Zadrozny LM, Gabrielson K, Semple KM, Shea KI, Howard KE. BLT‐immune humanized mice as a model for nivolumab‐induced immune‐mediated adverse events: comparison of the NOG and NOG‐EXL strains. Toxicol Sci. 2019;169:194‐208. [DOI] [PubMed] [Google Scholar]
- 24. Semple KM, Gonzalez CM, Zarr M, Austin JR, Patel V, Howard KE. Evaluation of the ability of immune humanized mice to demonstrate CD20‐specific cytotoxicity induced by ofatumumab. Clin Transl Sci. 2019;12:283‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan H, Semple KM, Gonzalez CM, Howard KE. Bone marrow‐liver‐thymus (BLT) immune humanized mice as a model to predict cytokine release syndrome. Transl Res. 2019;210:43‐56. [DOI] [PubMed] [Google Scholar]
- 26. Weissmuller S, Kronhart S, Kreuz D, et al. TGN1412 induces lymphopenia and human cytokine release in a humanized mouse model. PLoS One. 2016;11:e0149093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brady JL, Harrison LC, Goodman DJ, et al. Preclinical screening for acute toxicity of therapeutic monoclonal antibodies in a hu‐SCID model. Clin Transl Immunol. 2014;3:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malcolm SL, Smith EL, Bourne T, Shaw S. A humanised mouse model of cytokine release: comparison of CD3‐specific antibody fragments. J Immunol Methods. 2012;384:33‐42. [DOI] [PubMed] [Google Scholar]
- 29. Wunderlich M, Chou FS, Link KA, et al. AML xenograft efficiency is significantly improved in NOD/SCID‐IL2RG mice constitutively expressing human SCF, GM‐CSF and IL‐3. Leukemia. 2010;24:1785‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz‐scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477‐6489. [DOI] [PubMed] [Google Scholar]
- 31. Miller PH, Cheung AM, Beer PA, et al. Enhanced normal short‐term human myelopoiesis in mice engineered to express human‐specific myeloid growth factors. Blood. 2013;121:e1‐e4. [DOI] [PubMed] [Google Scholar]
- 32. Brehm MA, Kenney LL, Wiles MV, et al. Lack of acute xenogeneic graft‐ versus‐host disease, but retention of T‐cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019;33:3137‐3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. King MA, Covassin L, Brehm MA, et al. Human peripheral blood leucocyte non‐obese diabetic‐severe combined immunodeficiency interleukin‐2 receptor gamma chain gene mouse model of xenogeneic graft‐versus‐host‐like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157:104‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goulet DR, Atkins WM. Considerations for the design of antibody‐based therapeutics. J Pharm Sci. 2020;109:74‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lai Y, Dong C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol. 2016;28:181‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christofi T, Baritaki S, Falzone L, Libra M, Zaravinos A. Current perspectives in cancer immunotherapy. Cancers. 2019;11:1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527‐530. [DOI] [PubMed] [Google Scholar]
- 38. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256‐259. [DOI] [PubMed] [Google Scholar]
- 39. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID‐hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241:1632‐1639. [DOI] [PubMed] [Google Scholar]
- 40. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118‐130. [DOI] [PubMed] [Google Scholar]
- 41. Shultz LD, Schweitzer PA, Christianson SW, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz‐scid mice. J Immunol. 1995;154:180‐191. [PubMed] [Google Scholar]
- 42. Shultz LD, Brehm MA, Garcia‐Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Traggiai E, Chicha L, Mazzucchelli L, et al. Development of a human adaptive immune system in cord blood cell‐transplanted mice. Science. 2004;304:104‐107. [DOI] [PubMed] [Google Scholar]
- 44. Hiramatsu H, Nishikomori R, Heike T, et al. Complete reconstitution of human lymphocytes from cord blood CD34+ cells using the NOD/SCID/gammacnull mice model. Blood. 2003;102:873‐880. [DOI] [PubMed] [Google Scholar]
- 45. King M, Pearson T, Shultz LD, et al. A new Hu‐PBL model for the study of human islet alloreactivity based on NOD‐scid mice bearing a targeted mutation in the IL‐2 receptor gamma chain gene. Clin Immunol. 2008;126:303‐314. [DOI] [PubMed] [Google Scholar]
- 46. Pearson T, Shultz LD, Miller D, et al. Non‐obese diabetic‐recombination activating gene‐1 (NOD‐Rag1 null) interleukin (IL)‐2 receptor common gamma chain (IL2r gamma null) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol. 2008;154:270‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Melkus MW, Estes JD, Padgett‐Thomas A, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST‐1. Nat Med. 2006;12:1316‐1322. [DOI] [PubMed] [Google Scholar]
- 48. Aryee KE, Shultz LD, Brehm MA. Immunodeficient mouse model for human hematopoietic stem cell engraftment and immune system development. Methods Mol Biol. 2014;1185:267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stebbings R, Findlay L, Edwards C, et al. “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179:3325‐3331. [DOI] [PubMed] [Google Scholar]
- 50. Eastwood D, Findlay L, Poole S, et al. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T‐cells. Br J Pharmacol. 2010;161:512‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD‐scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte‐macrophage colony‐stimulating factor and interleukin‐3. Immun Inflamm Dis. 2016;4:427‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hall KH, Liu Y, Jiang C, Harvey RD. New and worsening long‐term immune‐related adverse events with PD‐1/PD‐L1 pathway agents in patients with cancer. Pharmacotherapy. 2020;40:133‐141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Table S1
Table S2