

Abstract
The pandemic COVID‐19 outbreak has been caused due to SARS‐CoV‐2 pathogen, resulting in millions of infections and deaths worldwide, the United States being on top at the present moment. The long, complex orf1ab polyproteins of SARS‐CoV‐2 play an important role in viral RNA synthesis. To assess the impact of mutations in this important domain, we analyzed 1134 complete protein sequences of the orf1ab polyprotein from the NCBI virus database from affected patients across various states of the United States from December 2019 to 25 April 2020. Multiple sequence alignment using Clustal Omega followed by statistical significance was calculated. Four significant mutations T265I (nsp 2), P4715L (nsp 12), and P5828L and Y5865C (both at nsp 13) were identified in important nonstructural proteins, which function either as replicase or helicase. A comparative analysis shows 265 T→I, 5828 P→L, and 5865Y→C are unique to the United States and not reported from Europe or Asia; while one, 4715 P→L is predominant in both Europe and the United States. Mutational changes in amino acids are predicted to alter the structure and function of the corresponding proteins, thereby, it is imperative to consider the mutational spectra while designing new antiviral therapeutics targeting viral orf1ab.
Keywords: COVID‐19, mutational spectra, ORF1ab polyprotein, pandemic, SARS‐CoV‐2, United States of America
Highlights
Four significant mutations T265I (nsp 2), P4715L (nsp 12), P5828L and Y5865C (both at nsp 13) were identified in USA.
265 T→I, 5828 P→L, and 5865Y→C are unique mutations for the United States.
Mutational changes in amino acids are predicted to alter the structure and function of the corresponding proteins.
Mutational spectra should be considered while designing new antiviral therapeutics targeting viral orf1ab.
1. INTRODUCTION
SARS‐CoV‐2 is the responsible pathogen for pandemic COVID‐19. Positive‐stranded, RNA genomes of coronaviruses are around 27 to 32‐kb in length, of which about 2/3rd encompasses the viral Orf1ab gene and expresses the largest and most complex polyproteins of any RNA viruses. The open reading frame 1 (ORF1), functions as replicase, replicase/transcriptase, or polymerase polyproteins, and is translated into ORF1a (~486 kDa, major product) and ORF1b (~306KDa) polyproteins in the host cell. Virus‐encoded proteinases including Papain‐like protease (PLPs) and 3 C like protease (3CL Pro) cleaves ORF1 into 16 nonstructural proteins (nsps). 1 , 2 ORF1a comprising nsps (nsp 1 to nsp 10) play an important role in coping with cellular stresses and maintaining the functional integrity of the cellular components along with the pivotal roles in viral replication. On the contrary, ORF1b encodes viral RNA‐dependent RNA polymerase (nsp 12), helicase (nsp 13), exonuclease (nsp14), a polyU (Uridylate) specific endonuclease (nsp15), and methyltransferase (nsp16). Hence, the majority of these nsps play an important role in viral pathogenesis and promising target for antiviral drug targeting and vaccine synthesis. 3
At present, the United States is one of the worst affected countries globally in terms of affected individuals and the number of deaths. Until 25 April, the United States was reported to have 860 772 positive cases and 44 053 deaths. 4 This adverse condition led us to investigate the sequence of the viral whole‐genome reported to the NCBI virus database. As on 25 April 2020 (till 12 noon, IST), around 1134 Orf1ab polyprotein sequences have been submitted from the United States alone. Different states of the United States like Washington DC, New York, Connecticut, Idaho, Georgia, etc have uploaded sequences of the Orf1ab polyprotein into the database. As COVID‐19 originated in Wuhan and was found to extend to different parts of the globe with variations in its virulence, it is imperative to identify the mutations that occurred in the Orf1ab polyprotein and consequent impact in protein structure and interaction with the host body. Hence, the present study aims (a) to identify the mutations observed in the orf1ab polyprotein, (b) to predict the conformational changes of SARS‐CoV‐2 polyprotein due to the mutations, and (c) to identify the signature pattern, if any for the United States.
1.1. Methodology
1.1.1. ORF1ab protein sequence retrieval from the database
The protein sequences were retrieved from the “NCBI Virus” database, the specific input was “SARS‐CoV‐2”. Then, the output was refined with a sequence length 7050 to 7100, as the length of the target orf1ab polyprotein is 7096. A total of 1307 sequences were retrieved, among them, 125 sequences were from Asia, 42 from Europe, and 1134 solely from the United States.
1.1.2. Screening of submitted sequences and selection of study sample
Incomplete sequences or sequences with undetermined residues (mentioned as X) were eliminated. A total of 867 orf1ab polyprotein complete sequences deposited from 31 different states of the United States were considered for the present study (Group A). All the above‐mentioned sequences from Asia (Group B) and Europe (Group C) were also selected.
1.1.3. Multiple sequence alignment (MSA) and analysis of mutational spectra
Clustal Omega 5 was employed to align multiple sequences of each of the above‐mentioned groups. Then, sequences of Group A were subdivided into 31 subgroups depending on the regional source from which the sequence was originated, and MSA was conducted encompassing all subgroups using the ancestral orf1ab polyprotein sequence of Wuhan (YP_009742608, comprising 7096 residues) as the reference sequence. 6 , 7 Gap opening penalty and gap extension penalty was set at 12 and 2, respectively, to ensure that unnecessary gaps are not created during alignment and alterations are visualized easily. Alignment results were thoroughly screened to find out the exact location of the mutations and alteration linked to that position.
1.1.4. Calculation of statistical significance to detect signature mutations of the United States
The number of occurrences of each mutated variant in Groups A, B, and C were calculated (suppose a, b, and c) and then divided by the total number of sequences submitted under that group (suppose x for Group A, y for Group B, and z for Group C). The proportion of each variant in Group A would be a/x and likewise for the other Groups. Then, the P value was calculated through the Z‐score using the proportion of the mutant and sample size to establish whether the occurrence of that mutant variant in the United States is significantly higher in comparison to Asia and Europe. Two‐tailed P values were calculated using 0.05 as the significance level. 8 Thus, an attempt has been made to identify the signature mutations in the region of orf1ab polyprotein.
1.1.5. Homology modeling and simulation of protein structure
Structures of the associated nonstructural proteins for the wild type were reported at the I‐Tasser server, however, the structures were not available for the varied amino acid (AA) alteration. To identify the alteration, the Homology Modeling method by I‐Tasser was used to generate the secondary structure, which was then superimposed with the wild type using UCSF Chimera and PyMOL for easy visualization and comparison.
2. RESULTS AND DISCUSSION
To assess the structural variation and identify the signature mutations, if any, among the viral strain(s) identified from the United States, all 1134 sequences, submitted to the NCBI Virus database (December 2019 to 25 April 2020) were retrieved. Following the exclusion criteria mentioned above, 867 complete sequences from 31 different states of the United States were obtained. For each subgroup of Group A, mutations observed in more than 5% studied population were taken into considerations and are shown in Table 1.
Table 1.
Region‐wise list of mutations at orf1ab polyprotein in the United States
| Name of the state | Sample size | Mutation Position | Location in orf1ab | Amino acid alteration | Individual with mutated variant | % Carrying mutated variant |
|---|---|---|---|---|---|---|
| Washington | 500 | 265 | nsp 2 | T→I | 206 | 41.2 |
| 4715 | nsp 12 | P→L | 226 | 45.2 | ||
| 5828 | nsp 13 | P→L | 255 | 51.0 | ||
| 5865 | nsp 13 | Y→C | 255 | 51.0 | ||
| New York | 150 | 265 | nsp 2 | T→I | 114 | 76.0 |
| 3884 | nsp 7 | S→L | 28 | 18.7 | ||
| 4715 | nsp 12 | P→L | 135 | 90.0 | ||
| 6245 | nsp 14 | A→V | 24 | 16 | ||
| Virginia | 52 | 265 | nsp 2 | T→I | 17 | 32.7 |
| 3483 | nsp 5 | L→F | 5 | 9.6 | ||
| 4715 | nsp 12 | P→L | 38 | 73.1 | ||
| 5828 | nsp 13 | P→L | 10 | 19.2 | ||
| 5865 | nsp 13 | Y→C | 10 | 19.2 | ||
| Idaho | 21 | 265 | nsp 2 | T→I | 18 | 85.7 |
| 4715 | nsp 12 | P→L | 19 | 90.5 | ||
| Connecticut | 17 | 265 | nsp 2 | T→I | 11 | 64.7 |
| 4715 | nsp 12 | P→L | 14 | 82.4 | ||
| 5828 | nsp 13 | P→L | 1 | 5.9 | ||
| 5865 | nsp 13 | Y→C | 1 | 5.9 | ||
| California | 16 | 265 | nsp 2 | T→I | 4 | 25.0 |
| 4715 | nsp 12 | P→L | 5 | 31.3 | ||
| Massachusetts | 16 | 4715 | nsp 12 | P→L | 15 | 93.8 |
| Georgia | 13 | 971 | nsp 3 | P→L | 8 | 61.5 |
| 3606 | nsp 6 | L→F | 2 | 15.3 | ||
| 4715 | nsp 12 | P→L | 3 | 23.1 | ||
| 6158 | nsp 14 | F→L | 8 | 61.5 | ||
| Utah | 10 | 265 | nsp 2 | T→I | 4 | 40 |
| 4715 | nsp 12 | P→L | 5 | 50 | ||
| 5828 | nsp 13 | P→L | 4 | 40 | ||
| 5865 | nsp 13 | Y→C | 4 | 40 | ||
| Minnesota | 8 | 265 | nsp 2 | T→I | 3 | 37.5 |
| 4715 | nsp 12 | P→L | 3 | 37.5 | ||
| 5828 | nsp 13 | P→L | 3 | 37.5 | ||
| 5865 | nsp 14 | Y→C | 3 | 37.5 | ||
| Florida | 8 | 3606 | nsp 6 | L→F | 1 | 12.5 |
| 4715 | nsp 12 | P→L | 5 | 62.5 | ||
| 5828 | nsp 13 | P→L | 1 | 12.5 | ||
| 5865 | nsp 13 | Y→C | 1 | 12.5 | ||
| Illionis | 7 | 265 | nsp 2 | T→I | 1 | 14.3 |
| 4715 | nsp 12 | P→L | 3 | 42.8 | ||
| 5828 | nsp 13 | P→L | 1 | 14.3 | ||
| 5865 | nsp 13 | Y→C | 1 | 14.3 | ||
| Iowa | 7 | 4715 | nsp 12 | P→L | 7 | 100 |
| Pennsylvania | 7 | 4715 | nsp 12 | P→L | 7 | 100 |
| New Hampshire | 4 | 265 | nsp 2 | T→I | 3 | 75 |
| 4715 | nsp 12 | P→L | 4 | 100 | ||
| 4764 | nsp 12 | L→F | 1 | 25 | ||
| N. Carolina | 4 | 265 | nsp 2 | T→I | 1 | 25 |
| 4715 | nsp 12 | P→L | 1 | 25 | ||
| 5828 | nsp 13 | P→L | 1 | 25 | ||
| 5865 | nsp 13 | Y→C | 1 | 25 | ||
| Texas | 3 | 2124 | nsp 3 | T→I | 2 | 66.7 |
| 3606 | nsp 6 | L→F | 2 | 66.7 | ||
| 4715 | nsp 12 | P→L | 2 | 66.7 | ||
| Arizona | 3 | 265 | nsp 2 | T→I | 1 | 33.3 |
| 4715 | nsp 12 | P→L | 1 | 33.3 | ||
| 5828 | nsp 13 | P→L | 2 | 66.7 | ||
| 5865 | nsp 13 | Y→C | 2 | 66.7 | ||
| Ohio | 3 | 3606 | nsp 6 | L→F | 1 | 33.3 |
| 4715 | nsp 12 | P→L | 2 | 66.7 | ||
| Rhode Island | 3 | 265 | nsp 2 | T→I | 1 | 33.3 |
| 4715 | nsp 12 | P→L | 3 | 100 | ||
| Nebraska | 2 | 392 | nsp 2 | G→D | 2 | 100 |
| 876 | nsp 3 | A→T | 2 | 100 | ||
| 4715 | nsp 12 | P→L | 2 | 100 | ||
| Nevada | 2 | 3606 | nsp 6 | L→F | 1 | 50 |
| 6669 | nsp 15 | A→V | 1 | 50 | ||
| S. Carolina | 2 | 971 | nsp 3 | P→L | 2 | 100 |
| 5401 | nsp 13 | P→L | 1 | 50 | ||
| 6158 | nsp 14 | F→L | 2 | 100 | ||
| New Orlando | 2 | 265 | nsp 2 | T→I | 2 | 100 |
| 4715 | nsp 12 | P→L | 2 | 100 | ||
| Kansas | 1 | 4715 | nsp 12 | P→L | 1 | 100 |
| Louisiana | 1 | 265 | nsp 2 | T→I | 1 | 100 |
| 3352 | nsp 5 | L→F | 1 | 100 | ||
| 4715 | nsp 12 | P→L | 1 | 100 | ||
| Maryland | 1 | 4715 | nsp 12 | P→L | 1 | 100 |
| Missouri | 1 | 4715 | nsp 12 | P→L | 1 | 100 |
| New Jersey | 1 | 4715 | nsp 12 | P→L | 1 | 100 |
| 6528 | nsp 15 | G→D | 1 | 100 | ||
| Hawaii | 1 | 4715 | nsp 12 | P→L | 1 | 100 |
| Wisconsin | 1 | 6679 | nsp 15 | L→del | 1 | 100 |
| Total | 867 |
A mutation at AA location 265 (Thr→Ile) of nsp2 is observed among ~50% states (subgroups) and 44.2% sequences in the United States (Table 1). Nsp2 is an important domain that takes care of the functional integrity of mitochondria and copes with cellular stress. 9 In addition to this, nsp2 co‐operates with nsp4 in viral replication. Nsp12 is important for its RNA‐dependent RNA polymerase (RdRp) activity and Pro→Leu in 4715 at this domain is a significant alteration, reported in 28 out of 31 states. Nsp 13 functions as a helicase protein, which is a key enzyme in viral replication, and therefore, two mutations, P5828L and Y5865C observed in this domain, are expected to alter in structure‐function and interaction with the host's target site. These two mutations, in particular, are observed and presumed to be linked and found in equal proportions (0.321), among affected individuals from 9 out of 31 states.
All these four mutations are widely observed throughout the country, but some mutations are consistently observed in specific regions (Figure 1). Georgia and South Carolina are two neighboring states sharing common borders in the South East region of the United States and both carry the mutant variant Leucine at the position of 971 (P→L) at nsp 3 and 6158 (F→L) at nsp14 in more than 60% cases. Another mutation, 3606 L→F is found in nsp6, and individuals from Florida, Texas, Ohio, Nevada, and Georgia had this mutation with a frequency of more than 10%. Nsp 6 plays a role in the initial induction of autophagosomes from the host endoplasmic reticulum, but gradually limits the expansion of these phagosomes which are unable to deliver viral components to lysosomes. It is important to note here that 3606 L→F mutations were found in Italy while analyzing Group C (present study), which further strengthens our previous reporting. 10
Figure 1.
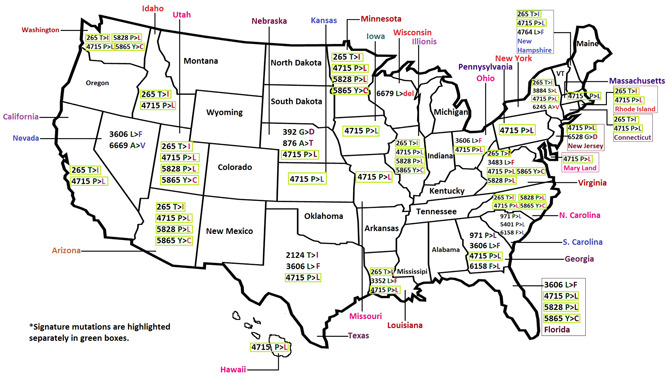
Mutational hotspots and the corresponding variants found in different geographical regions of the United States
To study the implication of these mutations, we have compared the prevalence of T265I in the European and Asian populations with that of the United States population. Strikingly, Threonine at 265 is the wild type AA found in all sequences from Europe and 119 out of 125 sequences from Asia. Therefore, the mutated AA Isoleucine is present in only 4.8% patient population in Asia, null in the European population so far, but observed in 44.2% patient population (383 out of 867) in the United States, establishing it to be a signature mutation of the country (P ≤ .0001).
Let us consider the mutation 4715 P→L, which is quite common in Europe and present in countries like Spain, France, Greece, etc, and the AA variant Leucine is found in 51.6% (16 out of 31 sequences deposited by European countries). In the United States, the frequency of Leucine is 58.1%, not significantly higher in comparison to Europe (P = .47) but clearly so compared with Asia (<0.0001). However, the mutant Leucine at 4715 is consistently observed in America. Two other most frequent mutations in the United States are 5828 P→L and 5865 Y→C, found in equal proportion (both observed 278 out of 867), presumed to be linked, and exclusively found in the United States, so far (Table 2).
Table 2.
The signature mutations of the United States and proportional presence of those mutations in the Asian and European continent
| Mutational hotspot in orf1ab pp | Location in orf1ab pp | United States (Grp 1) | Asia (Grp 2) | P value (Grp 1 and Grp 2) | Europe (Grp 3) | P value (Grp 1 and Grp 3) |
|---|---|---|---|---|---|---|
| T265I | nsp 2 | 383/867 = 0.442 | 6/125 = 0.048 | <.00001 | 0/31 = 0.0 | <.00001 |
| P4715L | nsp 12 or RdRp | 504/867 = 0.581 | 14/125 = 0.112 | <.00001 | 16/31 = 0.516 | .47152 |
| P5828L | nsp 13 or Helicase | 278/867 = 0.321 | 0/125 = 0 | <.00001 | 0/31 = 0.0 | .00014 |
| Y5865C | nsp 13 or Helicase | 278/867 = 0.321 | 0/125 = 0 | <.00001 | 0/31 = 0.0 | .00014 |
The four signature mutations are located in nsp 2, nsp 12, and nsp 13, respectively (Figure 2). Nonstructural protein 2 is assumed to be responsible in the modulation of the host cell survival signaling pathway by interacting with PHB and PHB2 in the host body. 9 , 11 , 12 Change of a polar AA (threonine) to a nonpolar one (isoleucine) makes it hydrophobic and induces structural alteration in that domain, which is observed by simulating the structure of the nsp2 protein harboring mutated allele through homology modeling using I‐Tasser. 13 , 14 , 15
Figure 2.
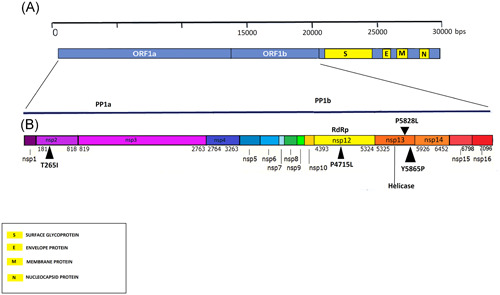
Mapping of orf1ab polyprotein and four signature mutations of the United States
Structure prediction for wild type nsps is already available in I‐Tasser; which gave us the opportunity to evaluate and superimpose three altered nsp structures (nsp 2, nsp 12, and nsp 13; against the said signature mutations), using UCSF Chimera 16 and PyMOL 17 for clear visualization of the alteration (Figure 3A). RdRp (nsp 12) is responsible for viral replication and transcription. Although SARS‐CoV‐2 and SARS‐CoV share about 79% genome similarity, 18 , 19 with regard to nsp12, the similarity increases as high as 98%, inferring the evolutionary significance of these conserved regions. 8 , 20 RdRp is a major antiviral drug target 21 , 22 and thus the structural alteration due to the mutant AA has important implications. Comparing the superimposed structure of RdRp, four small domains with the altered structure in the mutant sequence have been observed (Figure 3B) and these need to be investigated further. Helicase (nsp 13) is a multifunctional protein having a zinc‐binding domain in the N‐terminus exhibiting RNA and DNA duplex‐unwinding activities with 5′ to 3′ polarity. 11 , 12 Figure 3C exhibits some important structural alterations due to those two mutations present in close proximity near the N‐terminus; including a major structural alteration, where a loop or chain‐like structure is transformed into a helix, found near the N‐terminus of nsp 13. The length of nsp 13 or Helicase protein is 601 (5325‐5925 of orf1ab polyprotein) and the above‐mentioned alteration is found in the location 592 to 598, indicating a possible variation in functional outcome.
Figure 3.
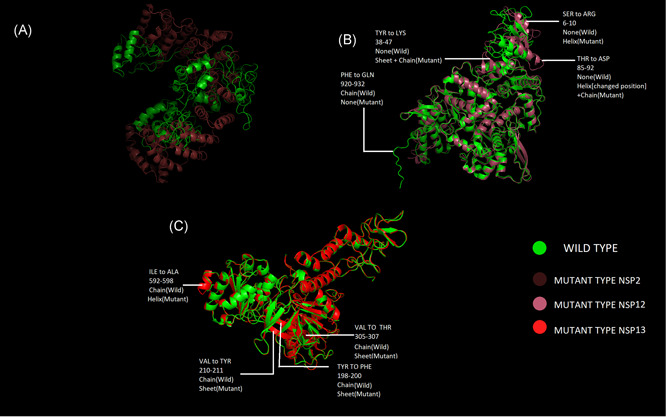
Structural comparison of nsp 2, nsp 12, and nsp 13 by superimposing the mutant structure over the wild type; (A) mutant nsp2 with corresponding wild type; (B) mutant nsp12 with corresponding wild type and (C) mutant nsp13 with corresponding wild type, compared through superimposition
3. CONCLUSIONS
The orf1ab polyprotein of SARS‐CoV‐2 encompasses mutational spectra. Compared with the European and Asian strain(s), four characteristic mutations, 265 T→I (nsp 2), 4715 P→L (nsp 12), 5828 P→L (nsp 13), and 5865Y→C (nsp 13) are observed, which can be considered as a signature pattern for the United States. It is noteworthy to mention here that 5828 P→L and 5865Y→C are exclusively found in the United States until now; whereas 265 T→I is found in very low abundance in Asia (4.42%) and not found at all in Europe. 4715 P→L is commonly found in both the United States (58.1%) and Europe (51.6%) but present in low abundance in Asia (11.2%). 971 P→L and 6158 F→L are frequently observed in Georgia and South Carolina, only representing the South‐East region of the country. All of the four signature mutations caused structural alteration in their respective nonstructural proteins (nsp 2, nsp 12, and nsp 13). Thereby, it is essential to consider the mutational spectra while designing new antiviral therapeutics targeting viral orf1ab.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
SB had the idea; SB, SS, RD, and KKM did the experiments; SB analyzed the data; SB and PB interpreted the data; SS, RD, KKM, and PB searched the literature; SS and RD did the referencing; SB and PB wrote the manuscript; SS and RD prepared figures; PB supervised the overall study.
ACKNOWLEDGMENTS
The authors acknowledge the UGC‐DAE‐CSR Kolkata Centre for providing a fellowship to Shuvam Banerjee.
Banerjee S, Seal S, Dey R, Mondal KK, Bhattacharjee P. Mutational spectra of SARS‐CoV‐2 orf1ab polyprotein and signature mutations in the United States of America. J Med Virol. 2021;93:1428–1435. 10.1002/jmv.26417
Sohan Seal and Riju Dey contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Harcourt BH, Jukneliene D, Kanjanahaluethai A, et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain‐like protease activity. J Virol. 2004;78(24):13600‐13612. 10.1128/JVI.78.24.13600-13612.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stobart CC, Sexton NR, Munjal H, et al. Chimeric exchange of coronavirus nsp5 proteases (3CLpro) identifies common and divergent regulatory determinants of protease activity. J Virol. 2013;87(23):12611‐12618. 10.1128/JVI.02050-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2‐human protein‐protein interaction map reveals drug targets and potential drug‐repurposing. Nature. 2020;583:459‐468. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. https://www.who.int/emergencies/diseases/novel-coronavirus-2019. Accessed 25 April 2020 (Time 12 noon, GMT +5:30).
- 5. Madeira F, Park YM, Lee J, et al. The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636‐W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:733‐745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flynn JA, Purushotham D, Choudhary MNK. Exploring the coronavirus epidemic using the new WashU virus genome browser. bioRxiv. 2020. 10.1101/2020.02.07.939124 [DOI] [Google Scholar]
- 8. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18:179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chowdhury I, Thompson WE, Thomas K. Prohibitins role in cellular survival through Ras‐Raf‐MEK‐ERK pathway. J Cell Physiol. 2014;229(8):998‐1004. 10.1002/jcp.24531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banerjee S, Dhar S, Bhattacharjee S, Bhattacharjee P. Decoding the lethal effect of SARS‐CoV‐2 (novel coronavirus) strains from global perspective: molecular pathogenesis and evolutionary divergence. bioRxiv. 2020. 10.1101/2020.04.06.027854 [DOI] [Google Scholar]
- 11. Zhang C, Zheng W, Huang X, Bell E, Zhou X, Zhang Y. Protein structure and sequence re‐analysis of 2019‐nCoV genome does not indicate snakes as its intermediate host or the unique similarity between its spike protein insertions and HIV‐1. J Proteome Res. 2020;19:1351‐1360. 10.1021/acs.jproteome.0c00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang X, Pearcs R, Zhang Y. Computational design of peptides to block binding of the SARS‐CoV‐2 spike protein to human ACE2. Aging. 2020;12:11263‐11276. 10.18632/aging.103416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7‐8. 10.1038/nmeth.3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roy A, Kucukural A, Zhang Y. I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725‐738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y. I‐TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pettersen EF, Goddard TD. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605‐1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 17. The PyMOL Molecular Graphics System , Version 1.2r3pre, Schrödinger, LLC.
- 18. Zheng J. SARS‐CoV‐2: an emerging coronavirus that causes a global threat. Int J Biol Sci. 2020;16(10):1678‐1685. 10.7150/ijbs.45053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graham RL, Sparks JS, Eckerle LD, Sims AC, Denison MR. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133(1):88‐100. 10.1016/j.virusres.2007.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, et al. Structure of RNA‐dependent RNA polymerase from 2019‐nCoV, a major antiviral drug target. bioRxiv. 2020. 10.1101/2020.03.16.993386 [DOI] [Google Scholar]
- 22. Velkov T, Carbone V, Akter J, et al. The RNA‐dependent‐RNA polymerase, an emerging antiviral drug target for the Hendra virus. Curr Drug Targets. 2014;15:103‐113. 10.2174/1389450114888131204163210 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.