

Abstract
The recently emerged 2019 Novel Coronavirus (SARS‐CoV‐2) and associated COVID‐19 disease cause serious or even fatal respiratory tract infection. Observing the spread, illness and death caused by COVID‐19, the World Health Organization (WHO) declared COVID‐19 a pandemic. To date, there is no approved therapeutics or effective treatment available to combat the outbreak. This urgent situation is pressing the world to respond with development of novel vaccine or a small molecule therapeutics for SARS‐CoV‐2. In line with these efforts, the structure of several proteins of SARS‐CoV‐2 has been rapidly resolved and made publicly available to facilitate global efforts to develop novel drug candidates. In this paper, we aim to find out the small molecule inhibitors for ADP‐ribose phosphatase of SARS‐CoV‐2. In order to identify potential inhibitors, we applied sequential E‐pharmacophore and structure‐based virtual screening (VS) of MolPort database containing 113687 number of commercially available natural compounds using Glide module. Six potential inhibitors having admirable XP glide score range from −11.009 to −14.684 kcal/mol and good binding affinity towards active sites were identified. All the molecules are commercially available for further characterization and development by scientific community. The in vitro activity of selected inhibitors can be done easily which will provide useful information for clinical treatment of novel coronavirus pneumonia.
Keywords: Drug design, Molecular docking, MolPort database, SARS-CoV-2, Virtual screening
The sequential E‐pharmacophore and structure based virtual screening of MolPort database containing 113687 nos of natural compounds has been performed using Glide. Six potential inhibitors of ADP‐ribose‐1′′‐phosphate of SARS‐CoV‐2 nsP3 having admirable XP glide score range from −11.009 to −14.684 and good binding affinity towards active site of protein were identified. The inhibitors reported here are commercially available for further characterization and development by scientific community.
1. Introduction
Coronaviruses (CoVs) are enveloped, positive‐sense, single‐stranded RNA viruses, belonging to the Coronavirinae sub‐family and Coronaviridae family of the order Nidovirales, which are divided into four genera (α, β, γ and δ).1, 2 In the past two decades, two highly pathogenic human CoVs, including Severe Acute Respiratory Syndrome (SARS)3 in the year 2002 and Middle East Respiratory Syndrome (MERS)4 in 2012, emerging from animal reservoirs, have led to global epidemics with morbidity and mortality. Since late December, 2019, an outbreak of respiratory disease caused by a novel coronavirus (SARS‐CoV‐2) that was first detected in Wuhan City, Hubei Province, China has rapidly spread to worldwide.5, 6, 7 The SARS‐CoV‐2 belongs to the β genus like MERS‐CoV and SARS‐CoV.8 The type of pneumonia caused by the SARS‐CoV‐2 is a highly infectious disease.9 Though most infected patients only suffer from mild symptoms such as fever and cough associated with a good prognosis, the disease can progress into fatal cases of pneumonia and acute respiratory failure, especially in aged peoples.10 Person to person high dissemination capacity of SARS‐CoV‐2 made it pandemic to public health throughout the world and prompted the WHO to declare the SARS‐CoV‐2 outbreak as Global Public Health Emergency of International Concern.11, 12 According to WHO, up to this date, about 7553182 peoples are infected with 423349 death cases (as of June 13, 2020).13 Several groups of researchers are working on the development of vaccines and drug molecules to prevent and treat the disease caused by SARS‐CoV‐2,14, 15, 16, 17, 18 but no successful treatment has yet been developed. While drug repurposing19, 20, 21, 22, 23 may be a short‐term and non‐specific solution to treat COVID‐19 patients, development of more targeted inhibitors is highly desirable.
Potential anti‐coronavirus therapies can be divided into two categories depending on the target, one is acting on the human immune system or human cells, and the other is on coronavirus itself. The therapies acting on the coronavirus itself include preventing the synthesis of viral RNA through acting on the genetic material of the virus, inhibiting virus replication through acting on critical enzymes of virus, and blocking the virus binding to human cell receptors or inhibiting the virus's self‐assembly process through acting on some structural proteins. Human CoVs genome has several conserved structural proteins such as‐ Spike (S) glycoprotein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein and at least four non‐structural proteins such as‐ 3‐chymotrypsin‐like protease (3CLpro), papain‐like protease (PLpro), helicase, and RNA‐dependent RNA polymerase (RdRp). The spike is a transmembrane glycoprotein that plays a pivotal role in mediating viral infection through binding the host receptor.24 It uses angiotensin‐converting enzyme‐2 (ACE2) as a receptor and thereby promotes the virus‐cell membrane fusion during virus infection.25 The non‐structural proteins, 3CLpro and PLpro are two important protease, play a crucial role in viral replication and transcription process through the extensive proteolysis of two replicase polyproteins, pp1a and pp1ab.26 The proteolytic cleavage of pp1a and pp1ab produces 16 non‐structural proteins nsP1 to nsP16. These nonstructural proteins (nsP1–nsP16) are assembled and form the replication‐transcription complex which regulate the numerous functions of virus replication viz. replication of viral genome, sub‐genomic RNA processing and packaging of new virion.27 Interrupting any replication process would become a potential molecular target to develop therapeutics against coronavirus. Amongst the nsPs, the SARS‐CoV nsP3 multi‐domain protein is the largest replicase subunit and plays an essential role during the formation of virus replication complex.28 Recently, the structure of several proteins of SARS‐CoV‐2 has been resolved and made publicly available to facilitate global efforts to develop novel drug candidates.29, 30, 31, 32, 33 Along these efforts, the nonstructural protein (PDB ID: 6 W02, Resolution: 1.5 Å) ADP‐ribose phosphatase of nsP3 of SARS‐CoV‐2 has been deposited in the RCSB protein databank.34 Saikatendu et al. observed that ADP‐ribose‐1′′‐phosphatase (ADRP) domain of SARS nsP3 is a phosphatase that removes the terminal 1′′‐phosphate group of ADP‐ribose‐1′′‐phosphate (Appr 1′′‐p) in vitro in SARS‐CoV.35 Thus, inhibition of ADP‐ribose‐1′′‐phosphatase function may block the functional nsP‐complex formation and may reduce the rapid multiplication of this virus, and can be potentially effective target to combat COVID‐19. So, the molecule has to be identified to target the active site of nsP3 to stop the effective multiplication of this virus. Most of the earlier efforts to target SARS‐CoV‐2 resulted in identification of several main protease (Mpro) inhibitors targeting the catalytic dyad of the protein defined by HIS‐41 and CYS‐145 residues.36 To the best of our knowledge, there is no report in the development of potential inhibitors targeting the nsP3 of SARS‐CoV‐2. Recently, Fischer and co‐workers reported 11 drug‐like compounds as inhibitors for coronavirus protease.37 In another study, Zheng et al. reported 13 compounds from different Chinese herb with the potential to directly inhibit SARS‐CoV‐2.38 Wu et al. screened ZINC drug database, Chinese database of natural products, and 78 anti‐viral drugs against human ACE2 targets and reported 135 inhibitors for SARS‐CoV‐2.39 In this study, we aim to find out whether any small molecule is available in the natural product databank that exhibits the inhibitory properties against the SARS‐CoV‐2 nsPs. So, this work has been designed to address the problem. In order to identify potential inhibitors, sequential E‐pharmacophore and structure based virtual screening (VS) of MolPort database (https://www.molport.com) containing 113687 numbers of natural products and modified natural compounds were performed using Glide.40, 41, 42 Six potential inhibitors having admirable XP glide score ranges from −11.009 to −14.684 and good binding affinity towards active site were identified. This study predicts a variety of commercially available natural compounds that may inhibit novel coronaviruses and provides researchers with information on compounds that may be effective. The inhibitors reported here are commercially available for further characterization and development by scientific community. Therefore, the in vitro activity can be done easily which will provide useful information for clinical treatment of novel coronavirus pneumonia.
2. Results and Discussion
2.1. Protein selection and docking validation
The X‐ray crystal structure (Figure S1) of viral protein (PDB ID: 6 W02, Resolution: 1.5 Å)34 was retrieved from RCSB protein databank and protein was prepared using ‘Protein Preparation Wizard’ workflow in Maestro 12.2.43 The Root Mean Square Deviation (RMSD) is used to measure the quality of reproduction of a crystallographic binding pose using docking method. The lower the value of RMSD the better is the reproduction by docking with respect to the original crystallographic bound conformation. The maximum acceptable range of RMSD value is <3.0 Å. The RMSD was calculated by superimposing the docked co‐ligand on its originally bound X‐ray crystallographic conformation (Figure 1). The result of docking validation showed that the RMSD value of co‐ligands of 6 W02 was found to be 1.946 Å which lies within the acceptable range. This lower RMSD value (1.946 Å) of the SARS‐CoV‐2 protein (6 W02) indicates that the docking protocol is able to reproduce similar docking pose of the ligand with respect to its original crystallographic bound conformation. Therefore, the docking protocol is validated.
Figure 1.
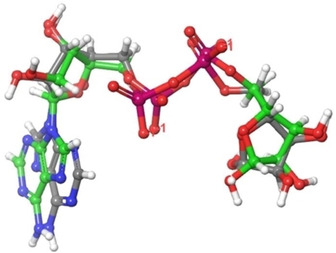
Superposition of docked co‐ligand (ash) on its originally bound X‐ray crystallographic ligand (green)–enzyme complex.
2.2. E‐Pharmacophoe based VS of MolPort database
Both, the Ligand‐based and structure‐based approaches are recognized as integral parts of drug discovery, and each method offers particular strengths. There is no specific wet lab for SARS‐CoV‐2 target protein inhibitors to generate the common pharmacophores of known inhibitors. Although structure‐based approach may be useful but this approach requires database searching which is a time‐consuming process. The E‐Pharmacophore approach has enjoy the convenience of both ligand‐ and structure‐based approach.44 The prepared protein‐coligand complex is used for building of E‐pharmacophoric hypothesis. The possible E‐pharmacophore sites‐ A1, A12, N22, N21, R23, D20, A10 were generated from the protein‐ligand (N3) complex by using default options. The pharmacophores were superimposed on the co‐ligand (N3) of 6 W02 as shown in Figure 2.
Figure 2.
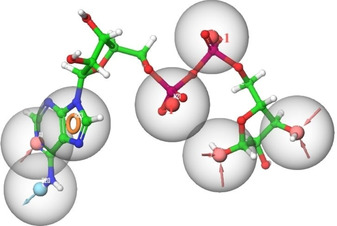
The possible number of Phase generated E‐pharmacophore hypotheses superimposed on co‐ligands A1, A10, A12 (pink, hydrogen bond acceptors), D20 (sky, hydrogen bond donor), R23 (orange, aromatic ring), N21, N22 (red, negative group).
The E‐pharmacophore based virtual screening of 113687 number of natural products or natural product derivatives in MolPort database resulted in 6025 hits. The resulted hits match with minimum 04 (four) pharmacophores out of seven. The E‐pharmacophore based and structure based virtual screening workflow of MolPort database is shown in Figure 3.
Figure 3.
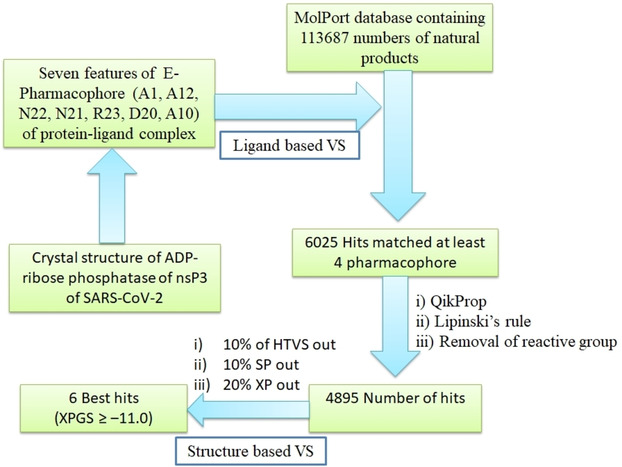
E‐Pharmacophore based and structure based virtual screening workflow for identification of inhibitors for ADP‐ribose phosphatase of nsP3 of SARS‐CoV‐2.
2.3. Structure based VS and molecular docking analysis
The 6025 hits resulted after E‐pharmacophore based VS, are subjected to structure based Virtual Screening (VS) using Glide. In the structure based VS workflow, output pharmacophore matched hits were subjected to filtration through QikProp,45 followed by Lipinski rule and removal of active functional group. After filtration, the number of hits has been reduced to 4895. In each stage of VS (HTVS to SP to XP) lesser number of molecules is forwarded to the next level with higher binding accuracy. Finally, six best hits having XP glide scores >−11.0 kcal/mol were selected. The ranges of fitness score of six best hits were 0.865 to 1.242. Out of six hits, fitness score of five are >1.00 and remaining one (MolPort‐035‐700‐887) has fitness score of 0.865. These hits with their superimposed pharmacophore are shown in Figure 4.
Figure 4.
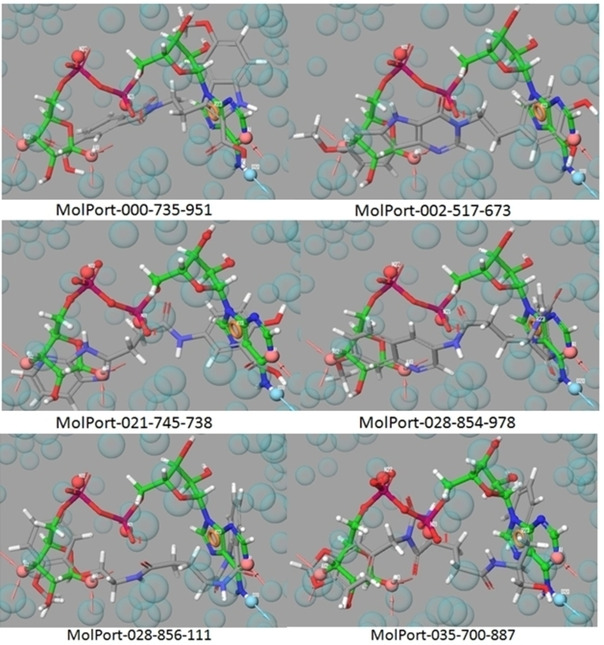
The E‐pharmacophore matched hits MolPort‐000‐735‐951, MolPort‐002‐517‐673, MolPort‐021‐745‐738, MolPort‐028‐854‐978, MolPort‐028‐856‐111, and MolPort‐035‐700‐887 along with co‐ligand.
The range of XP glide score of selected hits are −11.009 to −14.684 kcal/mol. The 2D and 3D interactions of the six hits with different active site amino acid residues of 6 W02 are shown in Figure 5 and Figure 6, respectively. The result showed that all selected hits have excellent glide score as well as good binding affinity towards active site of protein. The XP glide score of co‐ligand APR‐201 of nsP3 (PDB ID: 6 W02) is −8.2 kcal/mol and the active site interacting amino acid residues are ASP‐22 (2.07 Å), ILE‐23 (2.50 Å), ALA‐38 (1.98 Å), ASN‐40 (2.63 Å), VAL‐49 (2.51 Å), ALA‐50 (2.32 Å), ILE‐131 (2.77 Å), PHE‐132 (2.15 Å). The 2D, 3D interactions and binding pose of co‐ligand (APR‐201) with the active site of amino acid residues of 6 W02D is shown in Figure 7. The fitness score of MolPort‐000‐735‐951 with E‐pharmacophores is 1.002 and XP glide score is −14.684 kcal/mol. The interacting active site amino acid residues was ALA‐38 (1.67 Å), VAL‐49 (1.70 Å), LEU‐126 (1.80 Å), GLN‐130(2.17 Å), PHE‐132 (π‐π) and ALA‐154 (2.68 Å). This hit exhibits three common amino acid interactions (ALA‐38, VAL‐49 and PHE‐132) with co‐ligand APR‐201 in active site and all the interactions occur at a shorter distance as compared to co‐ligand. The fitness score of MolPort‐002‐517‐673 is 1.088 and XP glide score is −11.192 kcal/mol. This molecule interacts with nsP3 using VAL‐49 (2.20 Å), GLY‐130(2.00 Å), PHE‐132(two π‐π), ALA‐154 (1.85 Å), and PHE‐156 (2.30 Å) active site amino acid residues. This hit has two common interacting amino acid residues (VAL‐49 and PHE‐132) with co‐ligand. These two interactions also take place from a shorter distance as compared to APR‐201. The fitness score of MolPort‐021‐745‐738 is 1.242 and XP glide score is −11.386 kcal/mol. The interacting active site amino acid residues are ILE‐23 (2.67 Å), LEU‐126 (1.08 Å), SER‐128 (2.24 Å), PHE‐132 (2.42 Å) and ALA‐154 (1.95 Å). The common interacting active site amino acid residues of this hit with co‐ligand are ILE‐23 and PHE‐132. The XP glide score of MolPort‐028‐854‐978 is −11.356 kcal/mol and fitness score is 1.057. The interacting amino acid residues with active site of nsP3 are VAL‐49 (2.50 Å), LEU‐126 (1.67 Å), PHE‐132 (two π‐π), ALA‐154 (2.06 Å), and ASP‐157 (2.02 Å). This hit interacts with two common amino acids namely VAL‐49 and PHE‐132 as well as with co‐ligand. And the interactions are comparable with co‐ligand. The XP glide score of MolPort‐028‐856‐111 is −11.009 kcal/mol and fitness score with E‐pharmacophores is 1.051. Its active site interacting amino acid residues are ILE‐23 (1.98 Å), LEU‐126 (1.93 Å), SER‐128 (2.17 Å), ALA‐154 (2.06 Å) and PHE‐156 (π‐π). The compound MolPort‐035‐700‐887 has a fitness score of 0.865 with E‐pharmacophores and XP glide score is −13.313 kcal/mol. The interacting amino acid residues are ILE‐23 (2.16 Å), ALA‐38 (2.43 Å), LEU‐126 (1.62, 2.52 Å) and PHE‐152 (2.23 Å, π‐π). This hit has two common interacting amino acid residue with co‐ligand (ILE‐23 and ALA‐38). The interacting distance between all the selected hits and active site amino acid residues is shown in the parenthesis.
Figure 5.
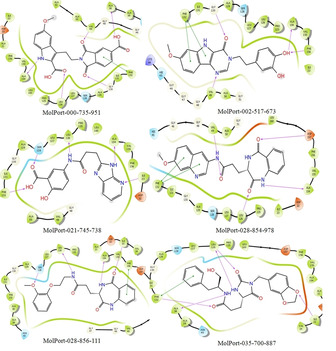
The 2D docking poses of compounds MolPort‐000‐735‐951, MolPort‐002‐517‐673, MolPort‐021‐745‐738, MolPort‐028‐854‐978, MolPort‐028‐856‐111, and MolPort‐035‐700‐887. The hydrogen bond (donor and acceptor) interactions are depicted by purple colors line, and π‐π stacking are presented by sky colors line.
Figure 6.
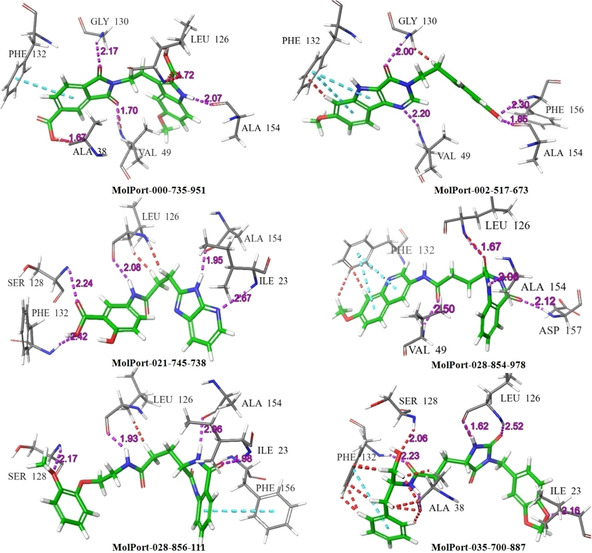
The 3D docking poses of compound MolPort‐000‐735‐951, MolPort‐002‐517‐673, MolPort‐021‐745‐738, MolPort‐028‐854‐978, MolPort‐028‐856‐111, and MolPort‐035‐700‐887. The hydrogen bond (donor and acceptor) interactions are shown by doted purple line, and π‐π stacking are presented by doted sky color line.
Figure 7.
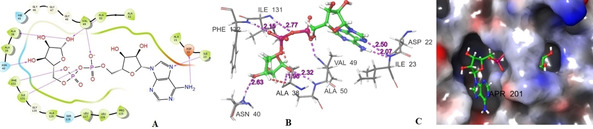
The 2D (Fig. A), 3D (Fig. B) interactions and binding pose (Fig. C) of co‐ligand (APR‐201) with active site amino acid residues of 6 W02.
From the above findings it is observed that the XP glide score of all the selected hits is much higher than that of the co‐ligand APR‐201. This indicates that the selected hits have more binding affinity than its original X‐ray crystallographic co‐ligand. Except MolPort‐035‐700‐887 (fitness score 0.865), the fitness score of all hits is greater than 1.0 and matches with at least four E‐pharmacophores out of seven. Among the six hits, the interacting distance of common amino acid residues of MolPort‐000‐735‐951 and MolPort‐002‐517‐673 is shorter than compared to co‐ligand APR‐201. Further, the best docked pose of the selected hits shows that all the hits are deeply inserted into the active site cavities and bonded pose is similar to co‐ligand (Figure 8). The type of interactions of these inhibitors in the active sites are H‐bond donor, H‐bond acceptor and π‐π staking. The minimum interacting amino acid residues of the selected inhibitors are five and maximum six, and interact from very close distance. It indicates that there is good binding affinity of these ligands towards the active site. The details of selected hits and co‐ligand such as‐ fitness score, XP Glide score and interacting amino acid residues along with distance are shown in Table 1.
Figure 8.
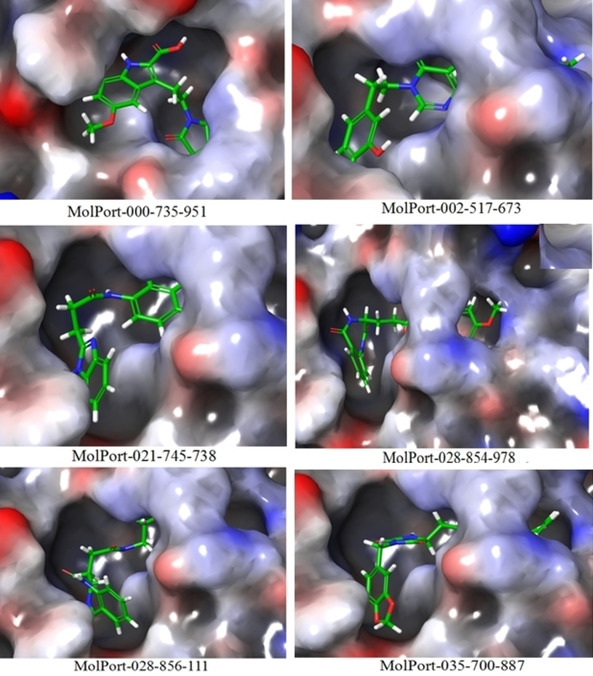
Individual binding pose of six selected hits in the active site of 6 W02.
Table 1.
The fitness score, XP Glide score and interacting amino acid residues along with distance (Å) of six hits in the active site.
|
Data Source Registration ID |
Fitness score |
XPGS (kcal/mol) |
Interacting active site amino acid residues and their interacting distance (Å) |
|---|---|---|---|
|
MolPort‐000‐735‐951 |
1.002 |
−14.684 |
ALA‐38 (1.67 Å), VAL‐49 (1.70 Å), LEU‐126 (1.80 Å), GLN‐130(2.17 Å), PHE‐132 (π‐π), ALA‐154 (2.68 Å) |
|
MolPort‐002‐517‐673 |
1.088 |
−11.192 |
VAL‐49 (2.20 Å), GLY‐130(2.00 Å), PHE‐132 (two π‐π), ALA‐154 (1.85 Å), PHE‐156 (2.30 Å) |
|
MolPort‐021‐745‐738 |
1.242 |
−11.386 |
ILE‐23 (2.67 Å), LEU‐126 (1.08 Å), SER‐128 (2.24 Å), PHE‐132 (2.42 Å), ALA‐154 (1.95 Å) |
|
MolPort‐028‐854‐978 |
1.057 |
−11.356 |
VAL‐49 (2.50 Å), LEU‐126 (1.67 Å), PHE‐132 (two π‐π), ALA‐154 (2.06 Å), ASP‐157 (2.02 Å) |
|
MolPort‐028‐856‐111 |
1.051 |
−11.009 |
ILE‐23 (1.98 Å), LEU‐126 (1.93 Å), SER‐128 (2.17 Å), ALA‐154 (2.06 Å), PHE‐156(π‐π) |
|
MolPort‐035‐700‐887 |
0.865 |
−13.313 |
ILE‐23 (2.16 Å), ALA‐38 (2.43 Å), LEU‐126 (1.62, 2.52 Å), PHE‐152 (2.23 Å, π‐π) |
|
APR‐201 |
– |
−8.2 |
ASP‐22 (2.07 Å), ILE‐23 (2.50 Å), ALA‐38 (1.98 Å), ASN‐40 (2.63 Å), VAL‐49 (2.51 Å), ALA‐50 (2.32 Å), ILE‐131 (2.77 Å), PHE‐132 (2.15 Å). |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.4. ADME analysis of selected hits
Almost 40 % of drug candidates have been found to be unsuccessful in clinical trials due to poor ADME properties. QikProp detects problematic drug candidates in the early stage minimizing the wastage of money, time and resources. The QikProp predicts the properties and descriptors of organic molecules by comparing with 95 % of known drugs. According to the Lipinski's rule, a molecule will be a drug like when molecular weight is <500, octanol‐water partition coefficient (QPlogPo/w) should be <5.0, hydrogen bond donor groups (donorHB) should be <5.0, and hydrogen bond acceptor groups should be ≤10. For six selected hits, total 48 numbers of predicted QikProp properties and descriptors are presented in Table 2 and Table 3. Out of 48 predicted properties most of the properties lie within the acceptable range. Only few properties like Caco‐2 cell permeability in nm/sec (QPPCaco) and apparent MDCK cell permeability in nm/sec (QPPMDCK) for molecules MolPort‐000‐735‐951 and MolPort‐021‐745‐738 defy the acceptable range. The predicted IC50 value for blockage of HERG K+ channels (QPlogHERG) for two molecules MolPort‐000‐735‐951 and MolPort‐035‐700‐887 lie slightly above the acceptable range. The data source registration ID, structures, IUPAC name and commercial sources of the six hits are shown in Table 4.
Table 2.
QikProp properties and descriptors of six selected inhibitors.
|
MolPort ID |
#stars |
#amine |
#amidine |
#acid |
#amide |
#rotor |
#rtvFG |
CNS |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
0 |
0 |
0 |
2 |
0 |
6 |
0 |
−2 |
|
002‐517‐673 |
1 |
0 |
0 |
0 |
0 |
6 |
0 |
−2 |
|
021‐745‐738 |
0 |
0 |
0 |
1 |
0 |
6 |
0 |
−2 |
|
028‐854‐978 |
0 |
0 |
0 |
0 |
0 |
5 |
0 |
−2 |
|
028‐856‐111 |
0 |
0 |
0 |
0 |
1 |
8 |
0 |
−2 |
|
035‐700‐887 |
0 |
0 |
0 |
0 |
1 |
10 |
1 |
−2 |
|
AR# |
0–5 |
0–1 |
0 |
0–1 |
0–1 |
0–15 |
0–2 |
−2/+2* |
|
MolPort ID |
mol_MW |
dipole |
SASA |
FOSA |
FISA |
PISA |
WPSA |
volume |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
408.367 |
4.421 |
664.745 |
140.552 |
300.196 |
223.997 |
0 |
1171.638 |
|
002‐517‐673 |
351.361 |
0.719 |
611.79 |
161.157 |
195.794 |
254.84 |
0 |
1065.31 |
|
021‐745‐738 |
326.311 |
5.874 |
606.995 |
90.541 |
252.485 |
263.969 |
0 |
1031.41 |
|
028‐854‐978 |
404.424 |
4.565 |
699.589 |
158.971 |
195.453 |
345.165 |
0 |
1234.105 |
|
028‐856‐111 |
397.43 |
5.272 |
707.675 |
246.849 |
151.513 |
309.313 |
0 |
1246.038 |
|
035‐700‐887 |
439.467 |
5.846 |
715.198 |
270.177 |
164.084 |
280.938 |
0 |
1324.539 |
|
AR# |
130–725 |
1–12.5 |
300–100 |
0–750 |
7‐330 |
0‐450 |
0‐175 |
500–2000 |
|
MolPort ID |
donorHB |
accptHB |
dip 2/V |
ACxDN.5/SA |
glob |
QPpolrz |
QPlogPC16 |
QPlogPoct |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
3 |
7.75 |
0.016679 |
0.020193 |
0.808548 |
39.022 |
13.483 |
22.305 |
|
002‐517‐673 |
3 |
6.25 |
0.000485 |
0.017695 |
0.824543 |
35.061 |
11.94 |
19.452 |
|
021‐745‐738 |
3 |
6.75 |
0.03345 |
0.019261 |
0.813331 |
33.791 |
12.057 |
19.976 |
|
028‐854‐978 |
3 |
9.25 |
0.016886 |
0.022901 |
0.795347 |
43.321 |
14.219 |
24.294 |
|
028‐856‐111 |
3 |
9 |
0.022308 |
0.022028 |
0.79132 |
41.53 |
13.781 |
23.215 |
|
035‐700‐887 |
3 |
9.2 |
0.025799 |
0.02228 |
0.815547 |
43.116 |
14.347 |
24.008 |
|
AR# |
0–6 |
2–20 |
0–0.13 |
0–0.05 |
0.75‐0.95 |
13‐70 |
4–18 |
8–35 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 3.
QikProp properties and descriptors of six selected inhibitors.
|
MolPort ID |
QPlogPw |
QPlogPo/w |
QPlogS |
CIQPlogS |
QPlogHERG |
QPPCaco |
QPlogBB |
QPPMDCK |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
15.588 |
2.377 |
−4.593 |
−5.418 |
−2.083 |
0.904 |
−2.86 |
0.411 |
|
002‐517‐673 |
13.483 |
2.03 |
−4.125 |
−4.912 |
−5.586 |
137.781 |
−1.718 |
58.065 |
|
021‐745‐738 |
14.412 |
1.769 |
−3.973 |
−4.032 |
−5.004 |
10.12 |
−2.343 |
4.392 |
|
028‐854‐978 |
17.482 |
2.009 |
−4.902 |
−4.619 |
−6.49 |
138.811 |
−1.798 |
58.534 |
|
028‐856‐111 |
17.398 |
1.807 |
−4.007 |
−3.696 |
−5.009 |
259.514 |
−1.532 |
165.117 |
|
035‐700‐887 |
17.882 |
2.112 |
−3.379 |
−4.838 |
−4.482 |
161.081 |
−1.684 |
122.727 |
|
AR# |
4–45 |
−2‐6.5 |
−6.5–0.5 |
−6.5–0.5 |
concern −5 |
<25 poor |
−3.0–1.2 |
<25 poor |
|
MolPort ID |
QPlogKp |
IP(eV) |
EA(eV) |
#metab |
QPlogKhsa |
HOA |
PHOA |
SAfluorine |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
−5.687 |
8.316 |
1.589 |
2 |
−0.309 |
1 |
40.08 |
0 |
|
002‐517‐673 |
−3.654 |
8.238 |
0.647 |
5 |
−0.003 |
3 |
77.119 |
0 |
|
021‐745‐738 |
−4.667 |
8.66 |
0.626 |
4 |
−0.328 |
2 |
55.293 |
0 |
|
028‐854‐978 |
−3.426 |
8.767 |
1.075 |
5 |
−0.047 |
3 |
77.056 |
0 |
|
028‐856‐111 |
−2.454 |
9.033 |
0.519 |
5 |
−0.433 |
3 |
80.733 |
0 |
|
035‐700‐887 |
−2.594 |
9.078 |
0.162 |
5 |
−0.332 |
3 |
78.816 |
0 |
|
AR# |
−0.8–1.0 |
7.9–10.5 |
−0.9–1.7 |
1–8 |
−1.5–1.5 |
1,2,3 |
<25 % poor |
0‐100 |
|
MolPort ID |
SAamideO |
PSA |
#NandO |
RuleOfFive |
ringatoms |
in56 |
nonHatm |
RuleOfThree |
|---|---|---|---|---|---|---|---|---|
|
000‐735‐951 |
0 |
179.9 |
9 |
0 |
18 |
18 |
30 |
1 |
|
002‐517‐673 |
0 |
109.941 |
7 |
0 |
19 |
19 |
26 |
0 |
|
021‐745‐738 |
0 |
138.185 |
8 |
0 |
15 |
15 |
24 |
1 |
|
028‐854‐978 |
0 |
134.341 |
8 |
0 |
21 |
16 |
30 |
0 |
|
028‐856‐111 |
18.209 |
127.15 |
8 |
0 |
17 |
12 |
29 |
0 |
|
035‐700‐887 |
29.252 |
142.989 |
9 |
0 |
20 |
20 |
32 |
0 |
|
AR# |
0–35 |
7–200 |
2–15 |
4 |
|
|
|
Max 3 |
AR#: Acceptable range for 95 % known drugs, *CNS‐2 (inactive), +2 (active), HOA: Human Oral Absorption, PHOA: Percentage of Human Oral Absorption, ring atoms: Number of atoms in ring, inn56: Number of atoms in 5‐ or 6 member rings, nonHatom: Number of heavy atoms (Non‐hydrogen atoms).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 4.
The structure of the selected hits along with MolPort ID and Commercial sources.
|
Structure and MolPort ID of the compounds |
IUPAC name |
Commercial Sources |
|---|---|---|
|
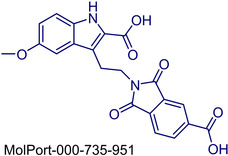
|
3‐(2‐(5‐Carboxy‐1,3‐dioxoisoindolin‐2‐yl)ethyl)‐5‐methoxy‐1H‐indole‐2‐carboxylic acid |
1. LabNetwork Compounds Country: United States CAS Number: 296265–99‐1 2. Aurora Building Blocks 6. Country: United States CAS Number: 296265‐99‐1 |
|
|
|
|
|
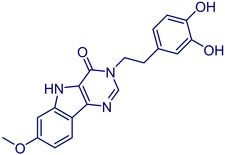
MolPort‐002‐517‐673 |
3‐(3,4‐Dihydroxy‐phenethyl)‐7‐methoxy‐3,5‐dihydro‐4H‐pyrimido[5,4‐b]indol‐4‐one |
1. Aurora Building Blocks 6. Country: United States CAS Number: 858747–55‐4. 2. LabNetwork Compounds Country: United States 3. Aurora Screening Compounds 1 Country: United States |
|
|
|
|
|
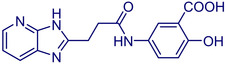
MolPort‐021‐745‐738 |
5‐(3‐(3H‐imidazo[4,5‐b]pyridin‐2‐yl)propanamido)‐2‐hydroxybenzoic acid |
1. LabNetwork Compounds Country: United States CAS Number: 1374523–48‐4 2. Chemieliva Pharmaceutical Product List. Country: China |
|
|
|
|
|
|
|
|
|
MolPort‐028‐854‐978 |
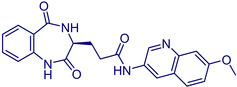
(S)‐3‐(2,5‐Dioxo‐2,3,4,5‐tetrahydro‐1H‐benzo[e][1,4]diazepin‐3‐yl)‐N‐(7‐methoxyquinolin‐3‐yl)propanamide |
1. Ambinter Screening Library Country: France CAS Number: 1574471–77‐4. 2. Princeton BioMolecular Research Screening Collection Country: United States |
|
|
|
|
|
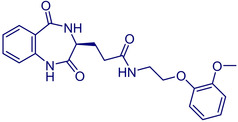
MolPort‐028‐856‐111 |
(S)‐3‐(2,5‐Dioxo‐2,3,4,5‐tetrahydro‐1H‐benzo[e][1,4]diazepin‐3‐yl)‐N‐(2‐(2‐methoxyphenoxy)‐ethyl)propanamide. |
1. Ambinter Screening Library Country: France CAS Number: 1574496–34‐6. 2. LabNetwork Compounds Country: United States 3. Aurora Screening Compounds 1 Country: United States |
|
|
|
|
|
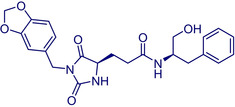
MolPort‐035‐700‐887 |
3‐(1‐(Benzo[d][1,3]dioxol‐5‐ylmethyl)‐2,5‐dioxoimidazolidin‐4‐yl)‐N‐(1‐hydroxy‐3‐phenylpropan‐2‐yl)propanamide |
1. Aurora Screening Compounds 1 Country: United States CAS Number: 2109196–54‐3. 2. Aurora Building Blocks 4 Country: United States CAS Number: 2109196–54‐3 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3. Conclusion
We have performed the virtual screenings of MolPort database containing 113687 number of natural and modified natural compounds using E‐pharmacophore as well as structure based virtual screening method. In this study, six potential inhibitors of SARS‐CoV‐2 having admirable XP glide score ranges from ‐11.009 to −14.684 kcal/mol and exhibit good binding affinity towards active site amino acid residues of nsP3. Out of seven number of E‐pharmacophores‐ A1, A10, A12, D20, R23, N21, and N22, six selected candidates match with at least four pharmacophores. In silico ADME properties also show that all the six inhibitors are within the acceptable range akin to 95 % known drugs. On the basis of XP glide score, binding affinity and ADME properties the compounds MolPort‐000‐735‐951 and MolPort‐035‐700‐887 are the best hits for potential inhibition of ADP‐ribose phosphatase of nsP3 of SARS‐CoV‐2. All the selected molecules are commercially available (Table 4). Therefore, in vitro experiments can be carried out which will facilitate global efforts in rapid development of suitable drug candidates against COVID‐19.
Supporting Information Summary
The supporting information contains methods and materials, structure of nsP3 protein and references.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are thankful to Schrodinger, Bangalore, India for providing Software support for this research work.
P. Debnath, B. Debnath, S. Bhaumik, S. Debnath, ChemistrySelect 2020, 5, 9388.
Contributor Information
Dr. Pradip Debnath, Email: pradipchem78@gmail.com.
Dr. Sudhan Debnath, Email: bcsdebnath@gmail.com.
References
- 1. Pillaiyar T., Meenakshisundaram S., Manickam M., Drug Discovery Today 2020, 25, 668–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen Y., Liu Q., Guo D., J. Med. Virol. 2020, 92, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CDC. 2018, https://www.cdc.gov/sars/about/fs-sars.html
- 4.
- 4a.WHO. 2018, http://www.who.int/emergencies/mers-cov/en/;
- 4b. Mohd H. A., Al-Tawfiq J. A., Memish Z. A., Virol. J. 2016, 13, 87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Israeli E., Harefuah 2020, 159, 70–71. [PubMed] [Google Scholar]
- 7.
- 7a. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Lancet 2020, 395, 497–506; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Yang X., Yu Y., Xu J., Shu H., Xia J., Liu H., Wu Y., Zhang L., Yu Z., Fang M., Yu T., Wang Y., Pan S., Zou X., Yuan S., Shang Y., Lancet Respir. Med. 2020, 8, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020. , 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., Xia J., Yu T., Zhang X., Zhang L., Lancet 2020, 395, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Jiang F., Deng L., Zhang L., Cai Y., Cheung C. W., Xia Z., J. Gen. Intern. Med. 2020, 35, 1545–1549; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Wang C., Horby P. W., Hayden F. G., Gao G. F., Lancet 2020, 395, 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.World Health Organisation, Novel Coronavirus (2019-nCoV) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/.
- 13.WHO, International Health Regulations Emergency Committee on novel coronavirus in China.https://www.who.int/newsroom/events/detail/2020/01/30/defaultcalendar/international-health-regulations-emergency-committee-on-novel-coronavirus-in-china.
- 14. Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X., Zhu W., bioRxiv 2020. doi: 10.1101/2020.01.27.921627. [Google Scholar]
- 15. Ton A. T., Gentile F., Hsing M., Ban F., Cherkasov A., Mol. Inf. 2020. doi:10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang H., Saravanan K. M., Yang Y., Hossain M. T., Li J., Ren X., Pan Y., Wei Y., Interdiscip. Sci. 2020, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang D. H., Wu K. L., Zhang X., Deng S. Q., Peng B., J. Integr. Med. 2020, 18, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zumla A., Hui D. S., Azhar E. I., Memish Z. A., Maeurer M., Lancet 2020, 395, e35-e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guangdi L., Clercq E. D., Nat. Rev. Drug Discovery 2020, 19, 149–150. [DOI] [PubMed] [Google Scholar]
- 20. Sanders J. M., Monogue M. L., Jodlowski T. Z., JAMA, 2020. doi:10.1001/jama.2020.6019. [Google Scholar]
- 21. Liu W., Zhu H. L., Duan Y., Curr. Top. Med. Chem. 2020, 20, 603–605. [DOI] [PubMed] [Google Scholar]
- 22. Li Y., Zhang J., Wang N., Li H., Shi Y., Guo G., Liu K., Zeng H., Zou Q., bioRxiv. 2020, doi: https://doi.org/10.1101/2020.01.28.922922. [Google Scholar]
- 23. Zhou Y., Hu Y., Shen J., Huang Y., Martin W., Cheng F., Cell Discov. 2020, 6, 14. https://doi.org/10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bosch B. J., van der Zee R., de Haan C. A., Rottier P. J., J. Virol. 2003, 77, 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W., Moore M. J., Vasilieva N., Sui J., Wong S. K., Berne M. A., Somasundaran M., Sullivan J. L., Luzuriaga K., Greenough T. C., Choe H., Farzan M., Nature 2003, 426, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baez-Santos Y. M., John S. E. S., Mesecar A. D., Antiviral Res. 2014, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fehr A. R., Jankevicius G., Ahel I., Perlman S., Trends Microbiol. 2018, 26, 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imbert I., Snijder E. J., Dimitrova M., Guillemot J. C., Lecine P., Canard B., Virus Res. 2008, 133, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao, 2020, PDB id: 6LU7. doi: 10.2210/pdb6LU7/pd.
- 30.X. Wang, J. Lan, J. Ge, J. Yu, S. Shan, 2020, doi: 10.2210/pdb6 M0 J/pdb
- 31.J. Shang, G. Ye, K. Shi, Y. S. Wan, H. Aihara, F. Li, 2020, doi: 10.2210/pdb6VW1/pdb
- 32.Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Godzik, K. Michalska, A. Joachimiak. 2020, doi: 10.2210/pdb6VWW/pdb
- 33.L. Zhang, X. Sun, R. Hilgenfeld, 2020, doi: 10.2210/pdb6Y2E/pdb.
- 34.K. Michalska, Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Mececar, A. Joachimiak, 2020, doi: 10.2210/pdb6 W02/pdb
- 35. Saikatendu K. S., Joseph J. S., Subramanian V., Clayton T., Griffith M., Moy K., Velasquez J., Neuman B. W., Buchmeier M. J., Stevens R. C., Kuhn P., Structure 2005, 13, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409–412; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Khan S. A., Zia K., Ashraf S., Uddin R., Ul-Haq Z., J. Biomol. Struct. Dyn. 2020, 1–10; [DOI] [PubMed] [Google Scholar]
- 36c. Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M. M., J. Biomol. Struct. Dyn. 2020, 1–6; [DOI] [PubMed] [Google Scholar]
- 36d. Paasche A., Zipper A., Schäfer S., Ziebuhr J., Schirmeister T., Engels B., Biochemistry 2014, 53, 5930–5946. [DOI] [PubMed] [Google Scholar]
- 37. Fischer A., Sellner M., Neranjan S., Lill M. A., Smiesko M., ChemRxiv. 2020, Preprint. https://doi.org/10.26434/chemrxiv.11923239.v1. [Google Scholar]
- 38. Zhu Z., Wang X., Yang Y., Zhang Z., Mu K., Shi Y., Peng C., Xu Z., Zhu W., ChemRxiv 2020, https://doi.org/10.26434/chemrxiv.11959323.v1. [Google Scholar]
- 39. Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H., Acta Pharm. Sin. B 2020, 10, 766–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 41. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 42. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shaw D. E., Shelley M., Perry J. K., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 43.Schrödinger Release 2020-4: Maestro, Schrödinger, LLC, New York, NY, 2020
- 44. Salam N. K., Nuti R., Sherman W., J. Chem. Inf. Model. 2009, 49, 2356–2368. [DOI] [PubMed] [Google Scholar]
- 45.QikProp, Schrödinger, LLC, New York, NY, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary