

Abstract
Low-grade fibromyxoid sarcoma, also known as Evans tumor, is a low-grade sarcoma that most commonly arises in the deep soft tissue of the proximal extremities or trunk in young adults. It is very rare in the viscera as a primary site, with only a few cases reported in the literature. Here, we present a case of Evans tumor occurring in an unusual and rarely reported location; an intrathoracic mass arising from the diaphragmatic pleura.
Keywords: Low-grade fibromyxoid sarcoma, Evans tumor, hyalinized rosettes, giant rosettes, hyalinizing spindle cell tumor
Introduction
Low-grade fibromyxoid sarcomas (LGFMS), first described by Evans,1 were once considered rare but are now thought to have been historically underreported due to misclassification. They consist of deceptively bland appearing spindle cells in a combination of collagenous to myxoid stroma. These tumors tend to arise in young adults, with no specific gender predilection, as a slow-growing, painless mass in the deep soft tissue of the proximal extremities or trunk. Rarely, these tumors present in other locations such as viscera. These tumors are treated with surgical excision but may experience repeated recurrences and a high rate of metastasis, in spite of their relatively low-grade appearance, with the lungs as the most common metastatic site.2,3 These tumors are genetically characterized by translocations involving the FUS gene, most commonly fusions of FUS-CREB3L2 (in more than 90% of cases) and FUS-CREB3L1 (in less than 10% of cases).2,4,5
Case description
A 32-year-old man with a prior smoking history and no significant past medical history presented with a persistent cough. Subsequent imaging revealed a lobular heterogeneous enhancing mass containing calcifications arising in the left hemithorax with attachment to the left diaphragm and abutting the left epicardium (Figures 1 and 2). The mass was surgically removed. Grossly, the tumor was well circumscribed, measured up to 11 cm in greatest dimension, and arose from the parietal pleura with attachment to both lung and diaphragm and focally involving visceral pleura with no extension into the underlying lung parenchyma.
Figure 1.
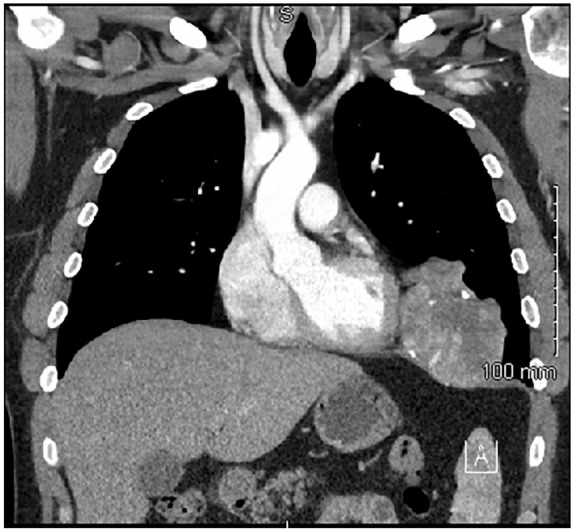
Coronal CT imaging of the thorax showing a tumor within the left hemithorax attached to the diaphragm and abutting the left epicardium.
Figure 2.
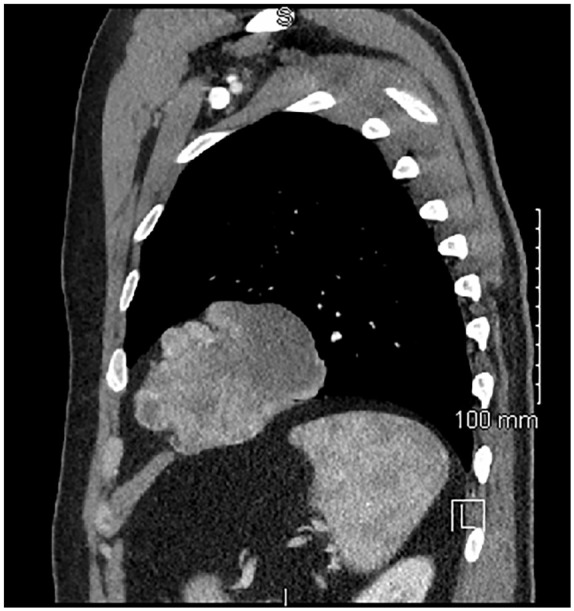
Sagittal CT imaging of the left thorax showing the tumor’s relationship to the diaphragm.
Histologic findings
Histologically, the tumor was composed of uniform and bland appearing spindle cells interspersed among large areas of hyalinized rosettes, so-called “giant rosettes” (Figures 3 and 4). By immunohistochemistry, the tumor was positive for EMA, bcl-2, TLE-1 (Figure 5), and MUC4 (Figure 6) and negative for STAT-6; ruling out solitary fibrous tumor (SFT). FISH for SYT gene rearrangement was performed and was negative ruling out synovial sarcoma. Given the overall morphology and the phenotype, the final diagnosis was low-grade fibromyxoid sarcoma with giant collagen rosettes (previously described as hyalinizing spindle cell tumor with giant rosettes).
Figure 3.

Low power view demonstrating hyalinized spindle cell rosettes and cellular spindle cell component along with arcades of thin-walled vessels (H&E, 4×).
Figure 4.

High power view showing the loose fascicular arrangement of the spindled tumor cells in a myxocollagenous stroma (H&E, 10×).
Figure 5.

Weak to moderate nuclear positivity with TLE1 (4×).
Figure 6.

Cytoplasmic staining of tumor cells with MUC4 (4×).
Discussion
Intrathoracic LGFMS are very rare6–16 as a primary site and metastasis from an extrathoracic site needs to be excluded. These tumors usually arise in young adults, but age extremes can also be seen. The clinical presentation of an intrathoracic mass can be varied, depending on exact location and size, but common symptoms include persistent cough, chest pain, dyspnea on exertion, and pleural effusion. It is not unusual for cases to be asymptomatic and found coincidentally on imaging as a slow-growing mass. In spite of its deceptively bland appearance, LGFMS has reportedly shown a progressive course with high rates of recurrence and metastasis. These rates are somewhat controversial when compared to some studies, such as one by Folpe et al.17 where a recurrence rate of 9%, a rate of metastasis of 6%, and a rate of death of 2% was reported in 54 of the 73 patients who were followed up. However, only 16 of these 54 patients had follow-up data extending over 4 years. Studies such as this one argue that the aggressive potential of LGFMS has been overestimated because many of the cases originally studied were not initially diagnosed or treated as sarcomas and were only studied once patients presented with metastasis. In a more recent study by Evans, 33 patients were followed up for a minimum of 5 years, revealing a recurrence rate of 64%, a metastasis rate of 45%, and a death rate of 42%. The study also concluded that LGFMS has a low rate of recurrence and metastasis within the first 5 years, with data showing that local recurrence can occur up to 15 years after excision, while metastasis can occur up to 45 years after excision, although the median interval to metastasis was 5 years and the median interval to local recurrence was 3.5 years.3 These studies all agree that the morphology of the tumor does not seem to indicate a worse prognosis; however, dedifferentiation can lead to shorter survival.3
Most intrathoracic cases of LGFMS in our research have had little to no, or short-term follow-up. Jakowski et al.6 thoroughly reported a review of six intrathoracic LGFMS cases. To summarize their findings, all cases were in adults, two arose in the anterior mediastinum,10,11 one from the pleura,7 two from the lung,8,9 and their own case report arising from the epicardium.6 Of these, six cases, only one presented with local recurrence after 9 years follow-up.11 However, two cases had no follow-up data in this review. Liang and Xu12 presented a case of a right hemithorax LGFMS arising from the pleura but presented no follow-up data. Maeda et al.13 presented two cases: one in the anterior mediastinum, which presented with local recurrence after 5 years, and one in the superior mediastinum involving the chest wall in stable condition after 5 years. Steiner et al.14 published a case report of a 12-year-old girl with an LGFMS occupying the left hemithorax that was stable after 1-year follow-up, marking our only known pediatric intrathoracic case. Higuchi et al.15 described an LGFMS arising from the right posterior chest wall that was stable after 1.5-year follow-up. Finally, Tominaga et al.16 described a case of LGFMS within the right hemithorax arising from the posterior chest wall with no complications after 2.5 years of follow-up. Ultimately, it was concluded that our case, to the best of our knowledge, represents the first LGFMS case to arise from the diaphragm and present as an intrathoracic mass. A summary of the findings of our literature search can be found in Table 1.
Table 1.
Reported cases of LGFMS in literature.
| Reference | Age/gender | Origin | Size/focality | Histology | Local recurrence/follow-up (Mos) |
|---|---|---|---|---|---|
| Galetta et al.10 | 41M | Anterior mediastinum | 7 cm, single mass | LGFMS with rosettes | Unknown |
| Higuchi et al.15 | 20F | Right posterior chest wall | 6 cm, single mass | LGFMS | No/18 |
| Jakowski and Wakely6 | 44F | Right heart/epicardium | 12 cm, single mass | LGFMS with rosettes | No/7 |
| Kim et al.9 | 50F | Lung | 7.5 cm, single mass with metastatic pleural nodules | LGFMS with rosettes | Unknown |
| Kim et al.7 | 37M | Pleura | Unknown size, single mass | LGFMS | Unknown |
| Liang and Xu12 | 42F | Right hemithorax, pleura | 11 cm, single mass | LGFMS | Unknown |
| Maeda et al.13 | 19F | Anterior mediastinum | 23 cm, single mass | LGFMS with rosettes | Yes/60 |
| Maeda et al.13 | 50M | Superior mediastinum | 13cm, single mass | LGFMS | No/60 |
| Magro et al.8 | 20F | Lung | 2 cm, multiple masses | LGFMS with rosettes | No/12 |
| Steiner et al.14 | 12F | Left hemithorax | 23 cm, single mass | LGFMS | No/12 |
| Takanami et al.11 | 35M | Anterior mediastinum | 9 cm, single mass | LGFMS | Yes/108 |
| Tominaga et al.16 | 70M | Right posterior chest wall | 18 cm, single mass | LGFMS | No/30 |
| Current case | 32M | Left diaphragmatic pleura | 11 cm, single mass with multiple chest wall implants | LGFMS with rosettes | No/29 |
LGFMS: low-grade fibromyxoid sarcomas.
Although no long-term follow-up data about the prognosis of intrathoracic LGFMS could be found in our research, it is safe to assume that the recurrence and metastasis rate of intrathoracic LGFMS is similar to LGFMS arising in classic locations, and these patients are recommended to be closely followed up after diagnosis and resection, especially long-term. As for our patient, he has been receiving frequent imaging surveillance and has had no instances of local recurrence or metastasis over the last 29 months since surgical resection.
Histologically, LGFMS are characterized by cytologically bland spindle cells with indistinct cytoplasm and rare to absent mitotic figures. Fibrous and myxoid zones are often seen. The fibrous areas tend to be paucicellular with abundant feathery stromal collagen with short fascicles or whorled patterns of growth, and occasionally collagen rosettes can be seen. The myxoid zones are more cellular than the fibrous zones and have prominent vasculature. The separate entity previously described as a hyalinizing spindle cell tumor with giant rosettes (HSCT) is now considered a variant of LGFMS and is characterized by prominent paucicellular hyalinized nodules bordered by plump tumor cells with a background morphology identical to classic LGFMS. This entity was ultimately considered as part of the same spectrum of LGFMS due to its morphological similarity, clinical presentation, prognosis, and the presence of the same genetic translocation.3,6,17,18 A small proportion (approximately 10%) of LGFMS cases have areas with atypia, increased cellularity or epithelioid morphology. In most cases, recurrences and metastases resemble the primary neoplasm; however, recurrences may show hypercellular nodular areas with increased mitotic activity.3 Progression to undifferentiated pleomorphic, round cell, or primitive round cell sarcomas has also been reported.1
Some LGFMS cases have foci that are indistinguishable from sclerosing epithelioid fibrosarcoma (SEF) and are thus called hybrid SEF-LGFMS. These tumors are also MUC4 positive and a good proportion of them harbor FUS-CREB3L2 fusion that is characteristic of LGFMS. SEF is a rare deeply located slow-growing malignant tumor with distinctive features characterized by neoplastic epithelioid cells arranged in nests, cords, and strands within a densely hyalinized stroma that sets it apart morphologically from LGFMS. In 2007, Guillou et al.4 suggested a potential relationship between LGFMS and SEF. This observation is reinforced both immunophenotypically and at the molecular level. Recent studies have found FUS-CREB3L2 gene fusion in 10% of hybrid SEF/LGFMS cases, recurrent EWSR1-CREB3L1 fusion transcripts by RT-PCR in 30% of pure SEF cases, and splits and deletions of the EWSR1 and/or CREB3L1 genes by FISH in 60% of pure SEF cases.19 In addition, MUC4 gene on chr 3q29 is upregulated in SEF just like LGFMS and is associated with corresponding MUC4 overexpression at the protein level in up to 78% of cases. In fact, MUC4 immunostain is a specific and highly sensitive marker for LGFMS that was described by Doyle et al.20 in 2011. The authors at the time proved its potential use as a diagnostic marker for LGFMS after global gene expression profiling performed at their laboratory had identified differential upregulation of the mucin 4 (MUC4) gene in LGFMS compared with histologically similar tumor mimics.
Due to the late interval to recurrence or metastasis and morphological pitfalls of LGFMS, one should carefully deliberate on a differential diagnosis and work-up of tumors with overlapping morphology. Bearing this in mind, the differential diagnosis in our case was wide and included SFT, synovial sarcoma, perineurioma, mesothelioma, spindle cell melanoma, low-grade dedifferentiated liposarcoma, myxoma, and extraintestinal gastrointestinal stromal tumor (GIST), neuroblastoma-like schwannoma, and myxofirosarcoma. Due to the tumor’s association with the pleura and its spindled and collagenous morphology, an SFT was considered. SFTs are usually cytologically bland, and negativity for STAT6 immunohistochemical stain is the most sensitive method to rule it out. Both TLE1 and bcl2 immunostains were positive in our case; hence, synovial sarcoma entered the differential diagnosis. Monophasic synovial sarcoma is characterized by monotonous spindle cells in varied patterns within a variably hyalinized to myxoid stroma thus overlapping with the tumor morphology in our case. Molecular testing for the SYT gene break apart by FISH, a highly specific test that is positive in about 95% of synovial sarcomas, was performed and was negative thus ruling it out. Based on the tumor positivity for EMA and the similar morphological features, perineurioma was a consideration. Perineuriomas consist of bland spindle cells in loose fascicular, trabecular, or reticular architecture that can also have collagenous to myxoid stroma similar to LGFMS; however, they lack giant collagen rosettes unlike our case. Due to the tumor’s peripheral thoracic location and its relationship with the pleura, mesothelioma was considered. Sarcomatoid mesothelioma mimics many mesenchymal neoplasms but does not stain for any mesenchymal markers other than vimentin and is usually strongly positive for the epithelial marker keratin, effectively ruling it out from our differential diagnosis. Spindle cell melanoma is also in the differential diagnosis given its notorious nature of being a universal mimicker but S100 immunostain was negative in the tumor cells, which mitigated this diagnosis. Dedifferentiated liposarcoma is characterized by its transition from an atypical lipomatous tumor component to a nonlipomatous, cellular sarcomatous component. The amount of the lipomatous component may vary and the morphology of the sarcomatous component can be high or low grade. A prominent myxomatous sarcomatous component can appear cytologically low grade thus mimicking LGFMS. Identification of lipomatous components and more conventional high-grade areas is useful in distinguishing the two. In our case, a lipomatous component was not identified thus ruling it out. Myxomas tend to be hypocellular haphazard proliferations of small spindled or stellate cells with bland cytologic features in a diffusely myxoid stroma; however, unlike our case, the lack of transition between the cellular areas and the fibrous and myxoid stroma makes myxoma less likely. The morphology of extraintestinal GIST can be quite variable, and although DOG1, an immunostain that is highly specific for GIST, has been reported in LGFMS,21 the tumor in our case was negative. The presence of collagen rosettes in our case may be confused with the characteristic rosettes seen in neuroblastoma-like schwannoma. The rosettes in the latter are comprised of a collagen core and surrounded by small S100-positive Schwann cells. The neoplastic cells in our case were negative for S100, which ruled it out. Last, myxofibrosarcoma is commonly confused with LGFMS; however, unlike our case, myxofibrosarcoma is uniformly myxoid, has a greater degree of nuclear pleomorphism and is MUC4 negative.
The characteristic presence of giant rosettes in our case, along with the strong and diffuse tumor positivity for MUC4, was most consistent with low-grade fibromyxoid sarcoma with giant collagen rosettes (previously described as hyalinizing spindle cell tumor with giant rosettes).
Conclusion
Intrathoracic low-grade fibromyxoid sarcoma is a rare entity that should be considered in the differential diagnosis of visceral soft tissue tumors. Pathologic examination and a thorough immunohistochemical and molecular work-up play a vital role in establishing this rare and challenging diagnosis.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval: Our institution does not require ethical approval for reporting individual cases or case series.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent: Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
ORCID iD: Diandra Perez  https://orcid.org/0000-0001-9572-026X
https://orcid.org/0000-0001-9572-026X
References
- 1. Evans H. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance. Am J Clin Pathol 1987; 88: 615–619. [DOI] [PubMed] [Google Scholar]
- 2. Fletcher CD. WHO classification of tumors of soft tissue and bone: low-grade fibromyxoid sarcoma. Lyon: IARC Press, 2013. [Google Scholar]
- 3. Evans H. Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases eith long-term follow-up. Am J Surg Pathol 2011; 35(10): 1450–1462. [DOI] [PubMed] [Google Scholar]
- 4. Guillou L, Benhattar J, Gengler C, et al. Translocation-positive low-grade Fibromyxoid Sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma. Am J Surg Pathol 2007; 31(9): 1387–1402. [DOI] [PubMed] [Google Scholar]
- 5. Matsuyama A, Hisaoka M, Shimajiri S, et al. DNA-based polymerase chain reaction for detecting FUS-CREB3L2 in low-grade fibro-myxoid sarcoma using formalin-fixed, paraffin-embedded tissue specimens. Diagn Mol Pathol 2008; 17(4): 237–240. [DOI] [PubMed] [Google Scholar]
- 6. Jakowski JD, Wakely PE., Jr. Primary intrathoracic low-grade fibromyxoid sarcoma. Hum Pathol 2008; 39(4): 623–628. [DOI] [PubMed] [Google Scholar]
- 7. Kim SY, Kim MY, Hwang YJ, et al. Low-grade fibromyxoid sarcoma: CT, sonography, and MR findings in 3 cases. J Thorac Imaging 2005; 20: 294–297. [DOI] [PubMed] [Google Scholar]
- 8. Magro G, Fraggetta F, Manusia M, et al. Hyalinizing spindle cell tumor with giant rosettes: a previously undescribed lesion of the lung. Am J Surg Pathol 1998; 22: 1431–1433. [DOI] [PubMed] [Google Scholar]
- 9. Kim L, Yoon YH, Choi SJ, et al. Hyalinizing spindle cell tumor with giant rosettes arising in the lung: report of a case with FUS-CREB3L2 fusion transcripts. Pathol Int 2007; 57: 153–157. [DOI] [PubMed] [Google Scholar]
- 10. Galetta D, Cesario A, Margaritora S, et al. Primary mediastinal hyalinizing spindle cell tumor with giant rosettes. Ann Thorac Surg 2004; 77: 2206–2209. [DOI] [PubMed] [Google Scholar]
- 11. Takanami I, Takeuchi K, Naruke M. Low-grade fibromyxoid sarcoma arising in the mediastinum. J Thorac Cardiovasc Surg 1999; 118(5): 970–971. [DOI] [PubMed] [Google Scholar]
- 12. Liang W, Xu S. Imaging findings from a case of pleural low-grade fibromyxoid sarcoma similar to mesothelioma with pleural effusion. Clin Respir J 2014; 10: 120–124. [DOI] [PubMed] [Google Scholar]
- 13. Maeda E, Ohta S, Watadani T, et al. Imaging findings of thoracic low-grade fibromyxoid sarcoma: report of three cases. Jap J Radiol 2009; 27(9); 375–380. [DOI] [PubMed] [Google Scholar]
- 14. Steiner MA, Giles HW, Daley WP. Massive low-grade fibromyxoid sarcoma presenting as acute respiratory distress in a 12-year-old girl. Pediatr Radiol 2009; 39(4): 396–399. [DOI] [PubMed] [Google Scholar]
- 15. Higuchi M, Suzuki H, Shio Y, et al. Successfully resected intrathoracic low-grade fibromyxoid sarcoma. Gen Thorac Cardiovasc Surg 2010; 58: 348–351. [DOI] [PubMed] [Google Scholar]
- 16. Tominaga Y, Eguchi T, Shiina T, et al. An intrathoracic low-grade fibromyxoid sarcoma arising from the chest wall with massive pleural effusion. Ann Thorac Cardiovasc Surg 2014; 20: 509–512. [DOI] [PubMed] [Google Scholar]
- 17. Folpe AL, Lane KL, Paull G, et al. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 2000; 24(10): 1353–1360. [DOI] [PubMed] [Google Scholar]
- 18. Bejarano PA, Padhya TA, Smith R, et al. Hyalinizing spindle cell tumor with giant rosettes—a soft tissue tumor with mesenchymal and neuroendocrine features. An immunohistochemical, ultrastructural, and cytogenetic analysis. Arch Pathol Lab Med 2000; 124(8): 1179–1184. [DOI] [PubMed] [Google Scholar]
- 19. Arbajian E, Puls F, Magnusson L, et al. Recurrent EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol 2014; 38: 801–808. [DOI] [PubMed] [Google Scholar]
- 20. Doyle L, Moller E, Dal Cin P, et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol 2011; 35: 733–741. [DOI] [PubMed] [Google Scholar]
- 21. Thway K, Ng W, Benson C, et al. DOG1 expression in low-grade fibromyxoid sarcoma: a study of 11 cases, with molecular characterization. Int J Surg Pathol 2015; 23(6): 454–460. [DOI] [PubMed] [Google Scholar]