

Abstract
Diarylmethanes are cardinal scaffolds by virtue of their unique structural feature including the presence of a benzylic CH2 group that can be easily functionalized to generate a variety of fascinating molecules holding immense importance in pharmaceutical, agrochemical, and material sciences. While the originally developed protocols for benzylic C−H functionalization in diarylmethanes employing base‐mediated and metal‐catalyzed strategies are still actively used, they are joined by a new array of metal‐free conditions, offering milder and benign conditions. With the recent surge of interest towards the synthesis of functionalized diarylmethanes, numerous choices are now available for a synthetic organic chemist to transform the benzylic C−H bond to C−C or C−X bond offering the synthesis of any molecule of choice. This review highlights benzylic methylene (CH2) functionalizations of diaryl/heteroarylmethanes utilizing various base‐mediated, transition‐metal‐catalyzed, and transition‐metal free approaches for the synthesis of structurally diverse important organic molecules, often with a high chemo‐, regio‐ and enantio‐selectivity. This review also attempts to provide analysis of the scope and limitations, mechanistic understanding, and sustainability of the transformations.
Keywords: Diarylmethanes, Benzylic C−H functionalization
This review highlights benzylic methylene (CH2) functionalizations of diaryl/heteroarylmethanes utilizing various base‐mediated, transition‐metal‐catalyzed, and transition‐metal free approaches for the synthesis of structurally diverse important organic molecules, often with a high chemo‐, regio‐ and enantio‐selectivity.
1. Introduction
Diarylmethane scaffold has attracted much attention due to its distinctive structural, chemical and physical properties, and wide range of applications in pharmaceutical, agrochemical, and material sciences.
The scaffold has represented as nearly the ideal starting material for the synthesis of complex natural products, agrochemicals, and active pharmaceuticals. Diarylmethanes functionalized at the benzylic position display important pharmacophoric activities as demonstrated in drugs, such as fexofenadine, cetirizine, letrozole, ebastine, etc.1 Diarymethane moiety is also serving as an essential backbone for many active pharmaceutical molecules that are in pipeline aiming at developing anti‐malarial, anti‐proliferative, antiviral drugs. Importantly, the various marketed drugs containing functionalized diarylmethanes could also be repurposed for potential treatment of COVID‐19. Consequently, its functionalization at the benzylic position has caught attention to organic chemists for developing methodologies for the synthesis of variously functionalized diarylmethanes for over a century. It is likely that the field will grow exponentially due to upsurge in the importance of diarylmethane motifs in both biological and chemical fields1 (Figure 1). Diarylmethanes based molecular architectures have also played a pivotal role in the development of supramolecular chemistry through understanding of various molecular self‐assemblies and recognition processes.2
Figure 1.
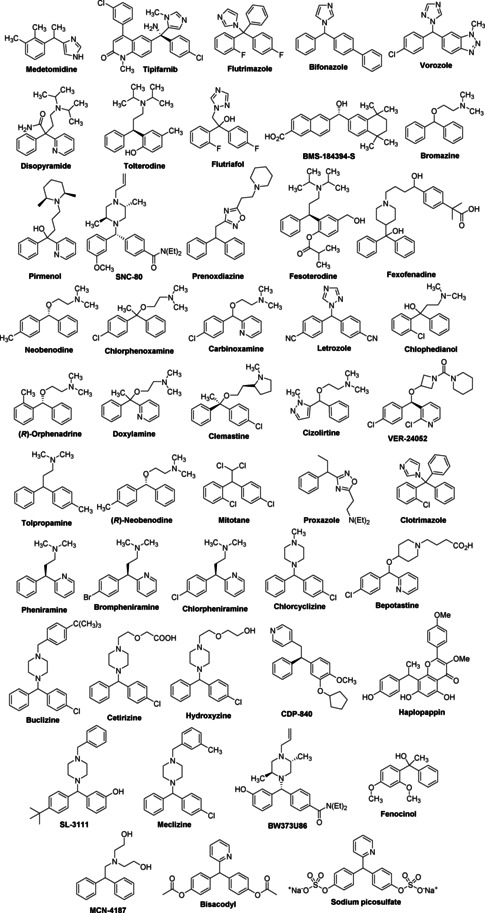
Representatives of biologically active diarylmethanes.
Conventionally, the benzylic C−H bond is much more reactive than the simple methyl C−H bonds, as it is one of the strongest aliphatic bonds. The enhanced reactivity of benzylic positions can be attributed to the low bond‐dissociation energy for benzylic C−H bonds (90 Kcal/mol) than that of methyl C−H bond (105 Kcal/mol). Moreover, the presence of neighboring aryl or heteroaryl rings stabilizes the benzyl radical. The benzylic C−H functionalization is reportedly achieved by cross coupling of aryl or alkenyl halides with a substrate under optimal catalytic conditions.3 However, over the years, direct functionalization of benzylic bonds has emerged as an area of significant interest to researchers.4 Direct functionalization of C−H bond offered high atom economy along with time and cost efficiency over conventional couplings. However, it has been compounded with issues of selectivity, functional group compatibility, and inclination toward over oxidation.
Direct functionalization of benzylic C−H bond of diaryl/heteroarylmethane could be broadly achieved via three approaches viz., 1) deprotonation of diaryl/heteroaylmethane utilizing strong bases to produce a carbanion followed by a subsequent nucleophilic substitution or addition reaction5 2) transition‐metal‐catalyzed deprotonative cross‐coupling processes (DCCP);6 and 3) direct oxidative coupling reactions or radical pathway resulting in the formation of benzylic radical7 (Scheme 1).
Scheme 1.
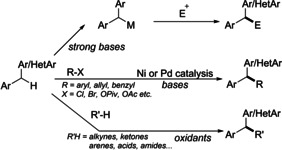
Approaches for functionalization of benzylic C−H bond.
The field of radical chemistry, an integral part of organic synthesis, has emerged long back in 1980s and till date radical reactions have continued to serve as keen research area on uncovering new ways to utilize radicals efficiently and selectively in synthetic planning. Radicals in general are highly reactive and short‐lived intermediates that react with utmost organic molecules including solvents. Primarily, the perception of radical reactions being non‐selective and uncontrollable had restrained scientists from using radicals in organic synthesis. However, the viewpoint has gradually changed with proliferating insights into the principle factors governing the radical reactions and hence this field of radical chemistry has conquered a major part of organic synthesis following which a couple of reports and reviews have become a paramount part of literature.8
Several years ago, an excellent review9 appeared in the literature, which covered the synthesis of diarylmethanes. The synthesis of diarylmethanes has largely been accessed through Friedel‐Crafts alkylation of benzyl alcohols with arenes, metal‐catalyzed cross coupling of aryl halides with benzyl nucleophiles, metal‐catalyzed cross coupling of benzyl halides with aryl nucleophiles and C−C bond formation between tosylhydrazones and aryl boronic acids. However, to the best of our knowledge, there has been no review covering the direct benzylic methylene (CH2) functionalization of diarylmethanes. The current review will attempt to cover the literature on direct functionalization of diarylmethanes that have appeared in the last 20 years. Our own contribution10 and continued interest in the benzylic C−H functionalization motivated us to learn the current development in benzylic functionalization on diarylmethanes.
With the perspective of much of the recent works on benzylic methylene functionalizations on diarylmethanes, this review highlights the recent advances on C−C and C−X bond (X=N, O) formation at the benzylic methylene position of diarylmethanes together with an emphasis on the scope and limitations, underlying different mechanisms. However, the discussion involving direct synthesis of diarylmethanes functionalized at the benzylic position and intramolecular benzylic functionalizations on diarylmethanes is beyond the scope of this review.
2. Base‐mediated C‐H functionalization of diarylmethanes
2.1. C−C bond formation
Functionalization of benzylic C−H in diarylmethane in the form of C−C bond covers the major portion of various diversified scaffolds that are important structural frameworks in pharmaceuticals. Triarylmethanes are the ones among those frameworks.9 Due to their special photochemical and photophysical properties and use in pharmaceuticals and polymers, their synthesis has attracted considerable attention.11 Provided the nucleophiles are generated in situ after deprotonation using base, they can be divided as “hard” nucleophiles or unstable nucleophiles (pKa >25) and “soft” nucleophiles or stable nucleophiles (pKa <25) depending on the pKa's of their conjugate acids. Nucleophiles, derived from diarylmethane derivatives with pKa's ranging from 25–33,12 make them significantly active towards variety of reactions especially the C−C bond formation reactions using sp3 hybridized carbon. In 2015, Cao, et al reported a practical and convenient approach for the C(sp3)−H arylation of diarylmethanes with various fluoroarenes in the presence of LDA at room temperature in the synthesis of triaryl/heteroarylmethanes13 (Scheme 2). The reaction proceeds via a base‐mediated aromatic nucleophilic substitution.
Scheme 2.
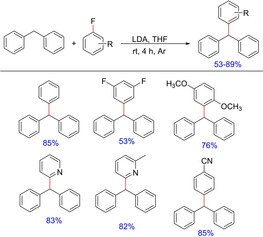
Synthesis of triarylmethanes using LDA‐mediated conditions.
In 2016, Kobayashi and group reported an interesting report stating regio‐ and stereoselective substitution of allylic and propargylic phosphates on to diarylmethane under metal‐free base‐catalyzed conditions yielding products in good to excellent yields (Scheme 3).5b It also highlights the presence of a Boc group for Pd‐catalyzed allylic substitution, whereas Pd‐free substitution can take place using BuLi in the case of an indispensable phosphate as the leaving group. The protocol demonstrates the formation of regioselective substitutions which is highly controlled by the substituents (Me vs. CH2OTBS) at the α‐ and γ‐positions of the allylic partner. As esters other than phosphate required prolonged reaction time, the substrate scope was largely exemplified using phosphates. The protocol proved to be successful with both diaryl‐ and aryl heteroarylmethane. However diversification utilizing various aromatic allylic partners remains unexplored.
Scheme 3.
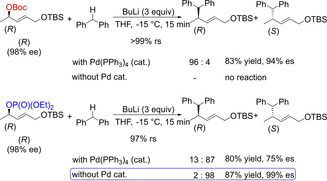
Base‐catalyzed regioselective and stereoselective substitution onto diarylmethane.
Another important transformation mediated by BuLi is reported by Reddy and group, wherein they have demonstrated the asymmetric synthesis of α‐(diarylmethyl)alkyl amines.14 Lithiation of diarylmethanes followed by diastereoselective addition to chiral N‐tert‐butanesulfinylimines generates enantioselective α‐(diphenylmethyl) phenylamine derivatives. The product can be easily converted to optically pure free amines in the presence of mild acidic conditions via the cleavage of the sulfinyl group (Scheme 4). The authors have demonstrated a good substrate scope with a wide variety of N‐tert‐butanesulfinylimines. However, the scope of different diarylmethanes is limited to unsubstituted diarylmethane and 4‐methyl substituted diarylmethane. Interestingly, in case of 4‐methoxy substituted diarylmethane, the addition of N‐tert‐butanesulfinylimines takes place onto the arene ring and not at the benzylic position. Although with a limited substrate scope, the report, however demonstrates an important asymmetric addition of diarylmethane anion to Ellman's imines for the direct synthesis of enantioselective products.
Scheme 4.
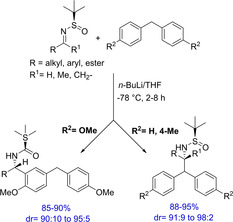
BuLi‐mediated asymmetric synthesis of α‐(Diarylmethyl) alkyl amines
The incorporation of difluoromethylene (−CF2−) and difluoromethyl (−CF2H) groups into benzylic C−H of diarylmethanes leads to substantial alteration into their physical, chemical and physiological properties, which could lead to the formation of some important pharmaceutical leads. Although a variety of fluorinating agents are available, Cao et alin 2017 carried out siladifluoromethylation of diarylmethanes using Ruppert‐Prakash reagent (TMSCF3). The method involved facile C(sp3)−H bond siladifluoromethylation of diarylmethanes with the reagent in the presence of LDA and HMPA at room temperature via cleavage of a C−F bond. The base facilitates the deprotonation of diarylmethane, while HMPA as an additive or co‐solvent is reported to have profound impact on yield, rate and selectivity of the reaction (Scheme 5).15 (Please delete the space in between in galley)
Scheme 5.
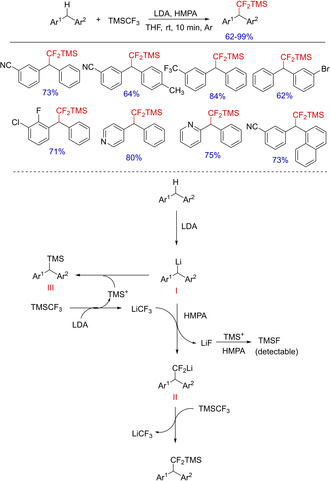
HMPA promoted siladifluoromethylation of diarylmethanes with Ruppert‐Prakash reagent.
3. Metal‐catalyzed benzylic C‐H functionalization of diarylmethanes
3.1. Iron‐catalyzed functionalization
Iron being the most abundant and non‐toxic transition metal plays an important role in chemical as well as biological sciences and shows its remarkable catalytic activity to facilitate many organic transformations. Catalytic direct C−H transformations have attracted a considerable interest since the early 1970s. Especially, iron‐catalyzed direct C−H transformations are among them, which have stimulated rapid development in the past several years.16 Due to its low cost and proved catalytic efficiency, iron has emerged as an important catalyst in a variety of benzylic C−H functionalizations. Only selected examples are presented in the following section.
3.1.1. C−C bond formation
Radical cross‐coupling reactions have emerged as a powerful tool in the formation of new C−C bond. However, being non‐selective these pose potential challenges in various chemical reactions. Substantial product selective radical‐radical coupling could occur only if a persistent radical and a transient radical are generated at comparable rates according to persistent radical effect, overcoming the inevitable homocoupling of either of the two radicals. Li and co‐workers, in 2007, reported a ferrous chloride‐mediated selective oxidative cross‐coupling of benzylic C−H bond with 1,3‐dicarbonyl compounds to form the coupled product via radical cross‐coupling mechanism (Scheme 6).7a
Scheme 6.
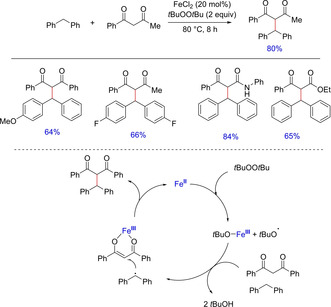
FeCl2‐mediated oxidative cross‐coupling reaction.
Under the optimized conditions, both more activated diarylmethanes and less activated cyclic substrates coupled effectively with 1,3‐dicarbonyl compounds to give the desired product in good yields. It is noteworthy that cross‐coupling with unsymmetrical dicarbonyl compounds gave the desired product in a 1 : 1 mixture of diastereomers. The authors proposed the reaction to proceed via a radical mechanism offering direct C−C bonds formation from C−H bonds under mild reaction conditions using inexpensive iron as a catalyst.
Transition metal‐catalyzed cross coupling of activated alkenes with aryl halide, i. e. Heck reaction has been used as one of the powerful tools for the formation of C−C bond.16b Among the other first row transition metals, iron and copper received much attention due to their high abundance, low price, catalytic efficiency and low toxicity.Zhang et al. in 2009 reported a Heck type iron‐catalyzed direct olefination of C(sp3)−H with 1‐aryl vinyl acetate (Scheme 7). [17] Although the authors have demonstrated sp3C−C bond formation via iron‐catalyzed benzylic C−H activation, however, acrylate derivatives that usually show high reactivity in traditional Heck reaction were found to be inefficient under these conditions. The reaction was also inhibited when substituted styrenes and electron rich olefins such as 1‐hexene, n‐butyl vinyl ether and 3,4‐dihydropyran were used. Although, this methodology of direct olefination finds its own identity, the low efficiency and limited substrate scope could make this transformation less attractive. Based on intermolecular isotopic competitive studies, the authors proposed both radical as well as ionic pathways for this transformation. The byproduct obtained during the reaction i. e. a dimerized product of diphenyl methane (I) supports the radical pathway. However, the possibility of an ionic pathway is also not ruled out as another byproduct (tert‐butoxymethylene)dibenzene (II) is also formed.
Scheme 7.
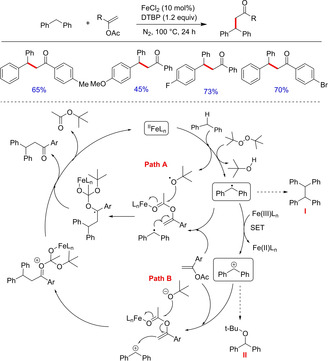
Iron‐catalyzed Heck‐type direct olefination.
In 2009, Li et al.reported an iron‐catalyzed cross‐coupling reaction, which involved simultaneous activation of two different C−H bonds: benzylic C(sp3)−H bond and C(sp2)−H bond in electron‐rich arene18 (Scheme 8). The methodology precisely demonstrated the role of electronic character of aromatic substrates in governing the high chemoselectivity. The homo‐coupled diarylmethane obtained during the preliminary mechanistic investigation unveiled the reaction to involve a single electron transfer oxidation of diarylmethanes.
Scheme 8.
Iron‐catalyzed cross‐coupling to form C(sp2)−C(sp3) bond.
3.1.2. C−X bond formation (X=Heteroatom)
Metal‐mediated C−N bond formation via C−H activation is an important methodology for the synthesis of various valuable nitrogen containing scaffolds16a including carboxamides and sulfonamides. Although a couple of reports are available on amidation of an unactivated sp3C−H bonds via a free‐radical mechanism utilizing metal catalysts such as rhodium, ruthenium, manganese, silver and copper; Wang and group explored an efficient intermolecular iron‐catalyzed amidation of benzylic sp3C−H bonds in the presence of NBS in 20087c (Scheme 9). The authors screened several iron salts wherein FeCl2 was found to be the most efficient one. The catalytic cycle involves an interesting intermediate (B) formed from the reaction of N‐bromocarboxamide (A) or N‐bromosulfonamide (A) and the iron salt. This intermediate resembles chloramines‐T, bromamines‐T and tosyloxy‐carbamates that are used as alternative nitrene sources. The intermediate (B) can be transferred into an iron‐nitrene complex (C), which further reacts with the benzylic C−H to give the functionalized product with concomittant removal of the iron.
Scheme 9.
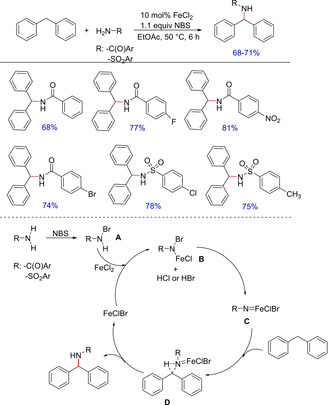
Iron‐catalyzed amidation of benzylic sp3C−H bond for the synthesis of carboxamides and sulfonamides.
Functionalized imidazole derivatives have also been of great interest to organic chemists due to their wide applications as precursors of N‐heterocyclic carbenes, ionic liquids and in drug synthesis.19 Traditionally, nucleophilic substitution with alkyl halides is the most common method for the preparation of N‐alkylated imidazole derivatives. Xia and group in 2011 developed an iron‐catalyzed direct C−N bond formation between benzylic substrates and imidazole.20 The methodology was applicable to a wide range of benzylic substrates and benzimidazole derivatives. However, the protocol was incompatible with imidazole itself. The author postulated the reaction to involve a single electron transfer from diarylmethane radical followed by nucleophilic reaction of diphenylmethane cation with benzimidazole (Scheme 10).
Scheme 10.
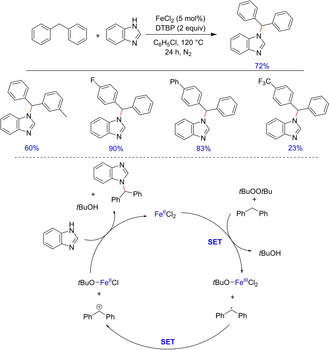
Iron‐catalyzed cross‐coupling of diarylmethanes and benzimidazoles.
Whitehead and Bentley introduced sulfoximine groups in organic chemistry in early 1950s.21 The numerous properties of its N‐alkylated derivatives had drawn the attention of organic chemists over the years.21 The preparation of N‐alkylated derivatives has been quite tedious and only a few efficient routes are known till date. In the year 2014, Cheng et al. reported an efficient iron‐catalyzed hetero‐cross‐dehydrogenative coupling for the formation of C−N bond between sulfoximines and diarylmethanes22 (Scheme 11). Although the reaction showed a good tolerance to various functional groups, the authors did not explain regioselectivity of the N‐alkylated sulfoximine product. The suppression of N‐alkylated product in the presence of a radical inhibitor, TEMPO demonstrated the reaction to follow a radical pathway.
Scheme 11.
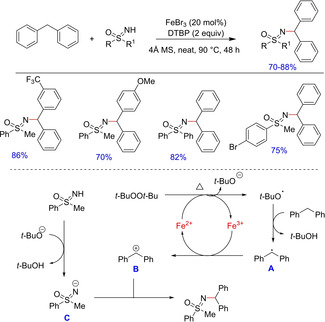
Iron‐catalyzed hetero‐cross dehydrogenative coupling.
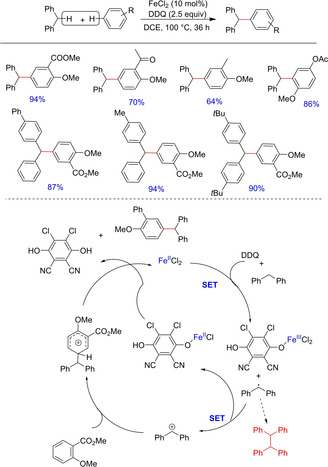
In 2016, Shi and his group described an iron‐catalyzed reaction between diarylmethanes and terminal alkynes in the presence of NCS and DDQ to the preparation of chloroalkenyl derivative of diarylmethanes (Scheme 12).23 The various polysubstituted alkenyl halides were prepared. The reaction was carried out in the presence of NCS, which served as the source of chlorine. The authors investigated a couple of substrates and reported that only E‐selective products were dominantly observed in most of the cases while in others a prominent E/Z ratio was observed. The transformation was established to proceed through a single‐electron transfer process with benzyl cations as key intermediates.
Scheme 12.
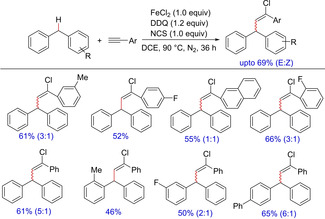
Iron promoted chlorobenzylation through benzylic C(sp3)−H functionalization.
3.2. Copper‐catalyzed functionalization
Copper being a versatile reagent has been extensively explored in a variety of coupling reactions, especially in oxidative couplings. Copper catalysts usually work via a single electron transfer mechanism and are proposed to serve as one‐electron oxidant. The C−H functionalization using copper salts has gained a considerable attention since long because of easy availability, stability and low cost of these salts. Copper‐mediated transformations via a single electron transfer process are utilized for the synthesis of various pharmaceutically active scaffolds containing C−C or C‐heteroatom bond formations,24 among which benzylic C−H functionalization reactions are discussed below.
3.2.1. C−C bond formation
In 2008, Borduas and Powell developed an inexpensive copper catalyst‐oxidant system for coupling of a wide range of benzylic C−H bonds with various 1,3‐dicarbonyl compounds in the absence of any added solvent.25 They have explored reactivity of various diketones of varying electronic properties. Although the method yields no significant quantities of over oxidized products, however, selectivity of C−H bond remained undefined. On the basis of kinetic isotope studies, the authors proposed the reaction mechanism to involve a benzylic hydrogen abstraction followed by a Lewis or Brønsted acid catalyzed nucleophilic substitution (Scheme 13).
Scheme 13.
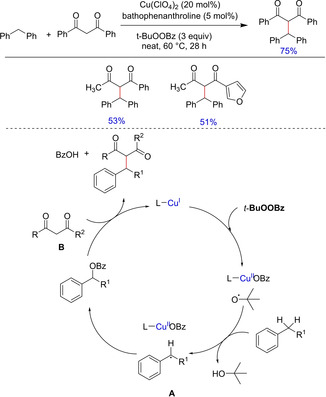
Copper‐catalyzed C−C bond formation via in‐situ substitution.
Traditionally, alkynylation has been known using elimination, substitution of benzylic alcohols/halides or through copper‐catalyzed Sonagashira reaction. In 2010, Correia and Li reported a novel methodology for alkynylation by copper‐catalyzed cross‐dehydrogenative coupling of alkynes C(sp)−H and benzylic C(sp3)−H bonds in the presence of DDQ26 (Scheme 14).The methodology was applicable to substituted diphenylmethane although limited to phenylacetylenes. The use of DDQ with a metal catalyst depicted a single electron transfer (SET) demonstrating the reaction to undergo a radical pathway.
Scheme 14.
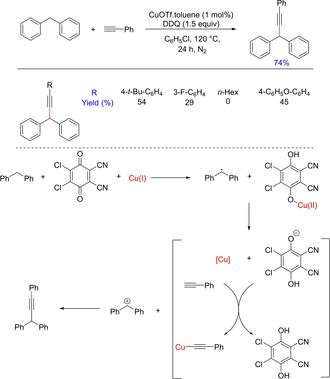
Functionalization of diphenylmethanes via a cross‐dehydrogenative Sonogashira coupling.
3.2.2. C−X bond formation (X=Heteroatom)
C‐Heteroatom bond formation using copper catalysis can be traced back to 1970s. In 1977, Sprecher and Zuberbühler reported a copper‐catalyzed autoxidation of substituted bisimidazole methane which mimicked the monooxygenase‐catalyzed reactions (Scheme 15).27 The report entirely focuses on the presence of copper catalyst and oxygen for benzylic C−H oxidation to C=O, which serves as a system fulfilling the basic requirements for internal monooxygenase.
Scheme 15.

Copper‐catalyzed benzylic C−H oxidation.
C−H amidation methodologies usually proceed through transition metal‐nitrene (imido) intermediates28 and has been worked out with variety of metals such as rhodium, ruthenium and manganese. With an alternative to metal‐nitene based amidation strategies, Katsuki and co‐workers first disclosed copper‐catalyzed benzylic and allylic amidation reactions29 in 1997. Because of several limitations, the synthetic utility of the reaction was limited. The limitations are overcome by Pelletier and Powell, who reported another copper‐catalyzed amidation strategy in 2006 (Scheme 16).30 The authors successfully demonstrated a copper‐catalyzed intermolecular sulfamidation strategy which was capable of coupling both primary and secondary sulfonamides with a range of hydrocarbon species.
Scheme 16.
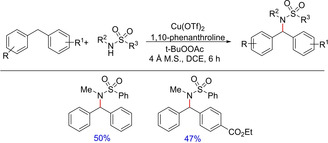
Copper‐catalyzed sulfamidation through benzylic C−H functionalization.
In 2008, Chang et al. developed an efficient protocol for selective C−H functionalization of hydrocarbons.31 The group demonstrated the use of N‐Hydroxyphthalimide (NHPI) as a catalyst, which could be used for selective oxidation of various benzylic and allylic substrates in the presence of CuCl as co‐catalyst to give the corresponding alcohols, ketones or carboxylic acids (Scheme 17). Interestingly, it was observed that the yield of the reaction significantly declined in the absence of PhI(OAc)2 depicting its importance in the reaction along with CuCl. The mechanism postulated the generation of phthalimide N‐oxyl (PINO) radical from NHPI, which actually acted as an active catalytic species responsible for the reaction.
Scheme 17.
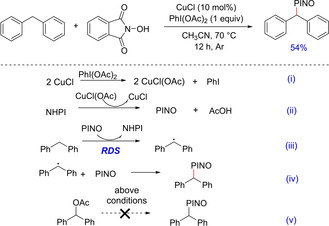
Use of N‐Hydroxyphthalimide for selective C−H functionalization.
Enantioselective synthesis has always been a demand in organic synthesis. Likewise, sp3C−H activation based asymmetric C−C bond formation is currently needed. In 2010, Gong and group carried out an enantioselective oxidative cross coupling reactions between indolylmethyl C−H bonds and 1,3‐dicarbonyl compounds using a chiral Lewis acid (Scheme 18).32 The group has mainly focused on DDQ oxidized coupling reactions. However, the lead role was played by the chiral ligands, such as chiral bis(oxazoline) and others, which were investigated for high yields as well as high enantioselectivities. The group proposed the reaction to proceed through a conjugate addition of nucleophile to the vinylogous iminium ion providing oxindole derivatives.
Scheme 18.
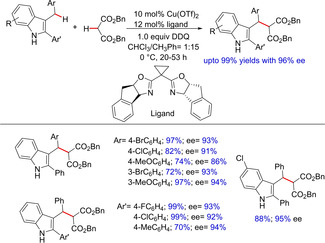
Copper‐catalyzed enantioselective C−H functionalization.
In 2011, Chiba and co‐workers demonstrated a copper‐catalyzed benzylic C−H oxygenation of carbonitriles and Grignard reagents via N−H imine intermediate under an oxygen atmosphere33 (Scheme 19). The reaction involved two steps performed in one‐pot: 1) addition of Grignard reagents to carbonitriles to form N−H imines, and 2) benzylic C−H oxygenation (C=O bond formation) triggered by 1,5‐hydrogen atom transfer with transient iminyl copper species.
Scheme 19.
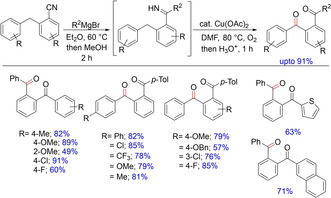
Copper‐catalyzed benzylic C−H oxygenation.
Another example of direct transformation of diarylmethane via transition‐metal‐catalyzed C−H activation was exemplified by Jiao and group in 2011. They established a protocol involving copper‐catalyzed direct nitrogen implantation into hydrocarbon molecules via C−H and C−C bond cleavages and subsequent C−N bond formation that aided the synthesis of 1,5‐disubstituted tetrazoles (Scheme 20).34 The report mainly included the synthesis of tetrazoles from 1,3‐diphenylprop‐1‐enes and azides; only a few examples involving diarylmethanes were investigated. The plausible pathway for product formation involves an initial transformation via single electron transfer to generate allyl radical (A), which is further oxidized to the corresponding allyl cation (B). Subsequent reactions generate allyl azide mixtures (C and C’) by a [3,3]‐sigmatropic rearrangement, which is further oxidized to allyl azide cation (D) and subsequently undergoes isomerization to generate intermediate E. Highly chemoselective aryl migration from carbon atom to nitrogen atom generates another intermediate F, which upon subsequent nucleophilic addition and cyclization reactions with another azide gives the desired product. Regioisomers are formed when unsymmetrical substrates were employed.
Scheme 20.
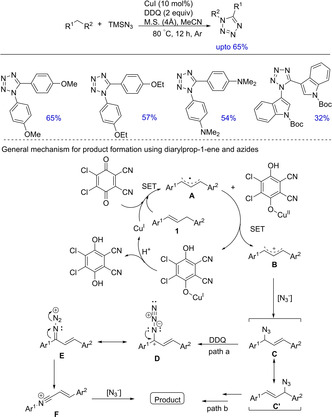
Copper‐catalyzed direct transformation of diarylmethanes into tetrazoles.
The combination of copper complex and visible light photocatalyst proved to be appealing. Greaney and group, in 2016, developed a protocol employing photoredox‐catalyzed azidation chemistry using Zhdankin azidoiodinane reagent and the Sauvage catalyst Cu(dap)2Cl (dap= 2,9‐bis(p‐anisyl)‐1,10‐phenanthroline), which reacted with the benzylic C(sp3)−H bond to form C−N bond (Scheme 21).35 Although the authors have demonstrated the use of this protocol mainly for alkyl arylmethanes, however, it also worked well with diarlymethanes. The propagation of the reaction is proposed to follow a radical mechanism.
Scheme 21.
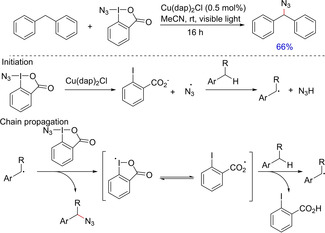
Copper‐assisted photoredox‐catalyzed azidation of benzylic C−H for C−N bond formation.
Synthesis of azides has always been in demand, since these are important, versatile organic intermediates36 and can be readily converted into different N‐heterocycles.37 For this reason, several efforts have been demonstrated for specific sp3C−H azidation (one such example is included above; Scheme 21). Stahl and group came up with another such important transformation involving site‐selective copper‐catalyzed azidation of benzylic C−H bonds.38 Although the authors have demonstrated a wide substrate scope for benzylic C−H azidation, most of the examples are demonstrated employing alkylarenes. Only a few substrates exemplify benzylic C−H azidation of diarylmethanes (Scheme 22). The mechanism of the reaction is believed to follow an ionic pathway wherein initial hydrogen atom transfer generates a benzylic radical, which reacts with a CuII‐azide intermediate to furnish the corresponding product.
Scheme 22.
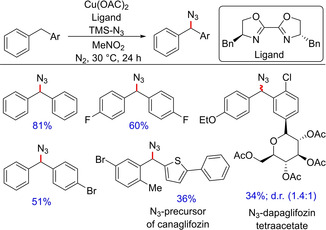
Copper‐catalyzed azidation of benzylic C−H bonds
In 2017, Wang and co‐workers established new amination reagents for copper‐catalyzed benzylic C(sp3)−H amination.39 Interestingly, these electrophilic hydroxylamine‐based amination reagents (RSO2NH−OAc) could be synthesized and stored on gram scale eliminating the need to form in‐situ reagents and avoiding potential explosion hazards during the reaction. Various arene substrates were well tolerated under the developed conditions. However, the benzylic substrate was required in excess during the reaction (Scheme 23). The detailed mechanistic insight of the reaction suggested to proceed through two subsequent radical catalytic cycles with Ph−CH2(NTsOAc) as a major intermediate to produce the product. It was then demonstrated that the excess amount of benzylic substrate was required to counter the formation of a radical addition product in the second catalytic cycle. The authors have also reported other mechanistic features of the reaction, such as the role of a bidentate ligand to improve the reactivity of the catalytic system. It was observed that the presence of a perfluorinated, highly electron‐withdrawing aryl groups onto a bidentate ligand (substituted diimine ligand shown in scheme 23) is the most activating substitution on the ligand because of its stability under the reaction conditions. Moreover, the counter anions in the catalyst also influenced the catalytic activity with Cu(BF4)2 ⋅ 6H2O being the most efficient one.
Scheme 23.
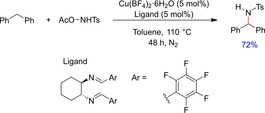
Copper‐catalyzed Benzylic C(sp3)−H amination.
In 2018, Kermani and group developed an inexpensive and readily available catalyst‐ligand‐oxidant system for the synthesis of N‐Alkyl hydrazines via C−H functionalization of benzylic substrates with dialkyl azodicarboxylates (Scheme 24).40 The selective mono‐amination of the substrates was achieved due to the phenanthroline ligand. Although the exact role of copper remained unveiled, the authors have precisely demonstrated the variation of substrate in terms of electronic and steric effects on reaction outcome. The kinetic isotopic study showed that the abstraction of sp3C−H hydrogen is involved in the reaction, which was supported by the product suppression in the presence of radical scavenger TEMPO.
Scheme 24.
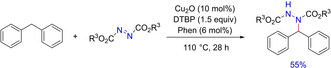
Benzylic C(sp3)−H amination via stable catalyst‐ligand‐oxidant system.
Copper‐catalyzed enantioselective arylation of benzylic C−H bonds via radical relay pathway was described by Liu and group in 2019.41 Although the method provides an excellent enantioselectivity, still the substrate scope for benzylic C−H arylation revolves much around arylalkanes. Only one example demonstrating enantioselective benzylic C−H arylation of diarylmethanes is included giving the product in 37% yield with moderate enantioselectivity (80 : 20 er) (Scheme 25). Chiral bisoxazoline ligand that bears an acetate group was used in the reaction, which was likely to play a key role in both reactivity and enantioselectivity of the reaction.
Scheme 25.
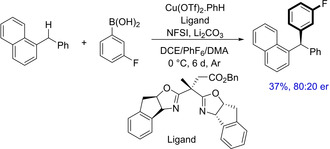
Copper‐catalyzed enantioselective arylation of benzylic C−H bonds.
A very recent paper by Stahl and group describes interesting modifications of copper catalysis.42 The group has demonstrated selective benzylic C−H cross‐coupling reactions with diverse partners for the synthesis of different C(sp3)−O, −N, <C‐<yC coupled products. The reaction occurs by a site‐selective transformation via Cu‐catalyzed C−H fluorination followed by nucleophilic substitution (Scheme 26). Hydrogen‐bond donors or Lewis acids are employed to tune the reactivity of the fluorinated compounds, which could be then used without isolation. The authors have mostly demonstrated the utility of this reaction by C(sp3)−H functionalization of aryl alkyl systems and only two examples of diarylmethane as the substrate. No examples employing aryl/heteroaryl systems as substrates are included. Moreover, the yields of the finally C−H cross‐coupled products are low to moderate.
Scheme 26.
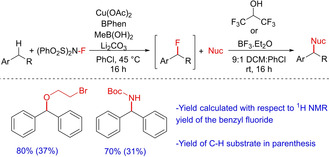
Copper‐catalyzed C−H functionalization viaC−H fluorination.
3.3. Cobalt‐catalyzed functionalization
In recent years, direct and selective synthesis of amine derivatives proved to be an intriguing task. However, the metal‐catalyzed nitrene insertion into C−H bond with suitable nitrene sources outstood the other known intermolecular C−H amination and proved to be a promising approach. In 2010, Lu et al.reported the first cobalt‐catalyzed nitrene insertion of sp3 C−H bond utilizing a carbonyl azide (Scheme 27).43 Upon screening of various metalloporphyrins, Co(TPP) proved to be a competent catalyst with 2,2,2‐trichloroethoxycarbonyl azide (TrocN3). Under the optimized condition, cyclic benzylic C−H substrates, ethyl benzene and its para‐brominated derivatives, naphthalene and its derivatives and challenging substrates like ethyl phenylacetate gave the corresponding amine products in good to moderate yields. The use of water in the reaction increased TrocNH2 of TrocN3, the common side product and reduced the yield of desired aminated product. The authors proposed the reaction to undergo metalloradical pathway including metallo‐nitrene intermediate. However, no experimental evidence was provided to support the formation of these intermediates.
Scheme 27.
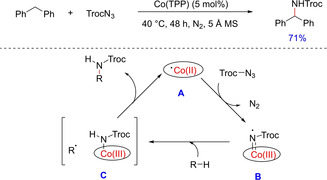
Cobalt‐ catalyzed nitrene insertion to benzylic C−H bond.
In 2011, Ye along with his group came up with another cobalt‐catalyzed benzylic C(sp3)−H functionalization and developed an inexpensive catalyst/oxidant system for the direct amination of benzylic C−H via dehydrogenative coupling with unmodified primary and secondary amides including sulfonamides, carboxamides and carbamatesas the amine source (Scheme 28).44
Scheme 28.
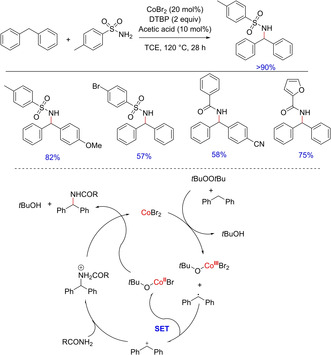
Dehydrogenative amination of benzylic C−H bond.
Notably, the electronic effect played a crucial role in the reaction. The presence of an electron‐withdrawing group either associated with sulfonamide or on the phenyl ring of diphenylmethane declined the yield due to reduced electron density on nitrogen and destabilization of the corresponding benzyl cation. The authors proposed the reaction to involve benzylic radical intermediate, thereby rationalizing the reaction to proceed via radical pathway.
3.4. Palladium‐catalyzed functionalization
Palladium‐catalyzed cross‐coupling reaction is an interesting area in organic synthesis. Not only direct conversion of sp2C−H bonds into sp2 C(sp2)−C bonds are widely investigated, direct functionalization of sp3C−H bonds to C−C or C−X bond formation is also pronounced using palladium catalysis. Oshima et al.in 2007 reported a palladium‐catalyzed direct arylation of aryl(azaaryl)methanes with aryl halides in the presence of a base involving benzylic C−H functionalization yielding triarylmethanes (Scheme 29).45 The authors have reported a wide substrate scope with products forming in moderate to good yields. However, electron‐deficient substrates remained unreacted under the optimized condition.
Scheme 29.
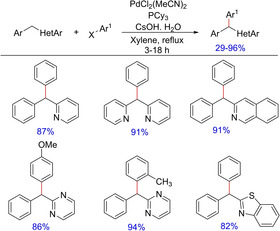
Palladium‐catalyzed synthesis of triarylmethanes.
Trost and co‐workers reported a palladium‐catalyzed regio‐, diastereo‐, and enantioselective benzylic allylations employing 2‐substituted pyridines. Initially the authors used 2‐methylpyridines,46 and later in 2009, hypothesized the analogous reaction using higher order 2‐substituted pyridines.47 The reaction was carried out using palladium catalysis with the aid of a base and a Lewis acid. The presence of the Lewis acid provides both diastero‐ and enantiocontrol after coordinating with the pyridyl nitrogen. The benzylic deprotonation provides a nucleophile that exists as a single geometric isomer because of the steric demands imposed by the Lewis acid (Scheme 30). Although the report exemplifies a wide substrate scope, however the reaction mechanism and the role of lithium aggregates remain unexplored. Also, the entire substrate scope relies on the pivalate ester and the effect of other groups remains unexplored.
Scheme 30.
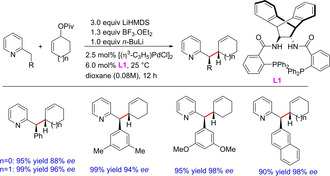
Palladium‐catalyzed enantioselective benzylic allylation.
Palladium‐catalyzed direct benzylation of azoles with benzyl carbonates is another interesting transformation reported by Miuara and group.48 The authors have successfully demonstrated direct aromatic sp2C−H benzylation of different substituted azoles using benzyl carbonates yielding corresponding diarylmethane derivatives. The product further undergoes sp3C−H benzylation with same or different benzyl carbonates to furnish the corresponding benzylated diarylmethanes without the addition of any external base (Scheme 31).
Scheme 31.
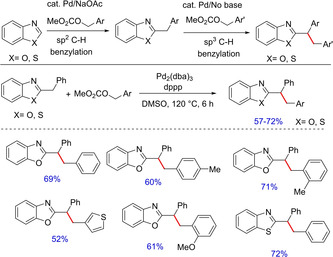
Palladium‐catalyzed direct benzylation of azoles using benyl carbonates.
In 2010, Patrick J. Walsh and co‐workers reported a palladium‐catalyzed cross‐coupling of tricarbonylchromium‐activated benzyllithiums for the synthesis of polyarylated methanes (Scheme 32).49 The reported method complements the Friedel‐Craft approach and involves the activation of the benzylic protons of [(η6‐arene)Cr(CO)3] and in situ generation of benzyllithium with the aid of LiN(SiMe3). Subsequently, it directly participates in the palladium‐catalyzed cross‐coupling to generate polyarylated products including unsymmetrically substituted triarylmethanes. Later, the group came up with another palladium‐catalyzed allylation reaction of toluene‐derived pro‐nucleophiles activated by tricarbonyl‐chromium to facilitate the access of α‐2‐propenyl benzyl motifs (Scheme 33).6a The group successfully demonstrated the reaction of a variety of cyclic and acyclic allylic electrophiles with in situ generated (η6‐C6H5CH2Ph)Cr(CO)3 nucleophiles. The reaction mixture was then exposed to light and air to afford metal‐free diphenylmethane derivatives in one‐pot tandem fashion. The Xantphos/Palladium catalyst system proved to be a hit and was used as the general catalyst system for this class of reactions.
Scheme 32.
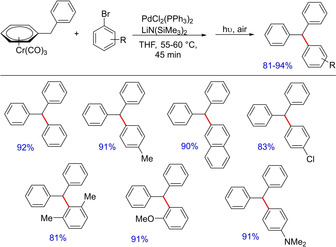
Palladium‐catalyzed synthesis of polyarylated methanes.
Scheme 33.
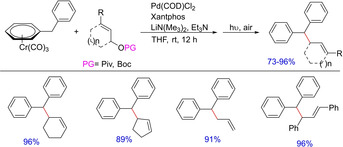
Palladium‐catalyzed allylation reactions of toluene derived pronucleophiles.
Again in 2012, the group came up with another palladium‐catalyzed deprotonative cross‐coupling process (DCCP) for intermolecular arylation of unactivated C(sp3)−H bonds in the absence of a directing group.50 The authors successfully demonstrated palladium‐catalyzed C(sp3)−H arylation of diarylmethanes at room temperature and synthesized a variety of sterically and electronically diverse aryl and heteroaryl containing triarylmethanes. In the subsequent years, they demonstrated the effect of additives on DCCP6b and soon introduced NiXantphos as the deprotonatable chelating aryldiphosphine ligand for room temperature palladium‐catalyzed coupling of aryl halides, especially aryl chlorides.51 The Pd‐NiXantphos catalyst system (Scheme 34) proved to be momentous and had more positive impact on DCCP over other mono‐ and bidentate ligands. Also, the catalyst system along with aryl chloride exhibited remarkable chemoselectivity with various heteroaryl groups possessing sensitive functional groups.
Scheme 34.
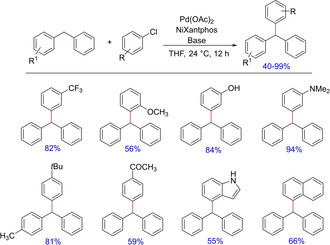
Palladium‐catalyzed deprotonative cross‐coupling reactions for intermolecular benzylic C−H arylation.
Continuing with the development on allylic substitution, the group further reported a palladium‐catalyzed synthesis of diallylated derivatives of dirarylmethanes with a quaternary centers, wherein the scope of “soft” nucleophiles derived from diarylmethanes and heterocyclic derivatives have been manifested (Scheme 35).6c Mechanistic studies showed that the nucleophile derived from diphenylmethane undergoes an external attack on π‐allyl palladium species under the reported optimized conditions. This unexpected observation indicated that diarylmethane derivatives behave as “soft” or stabilized nucleophiles.
Scheme 35.
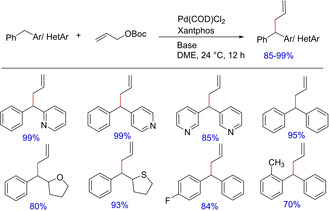
Palladium‐catalyzed benzylic allylation.
Further extension to their previous work on the synthesis of triarylmethanes using Pd‐NiXantphos catalyst system, the group developed a protocol for arylation of diarylmethanes followed by subsequent air oxidation to yield triarylmethanols (Scheme 36).6d The report is well furnished with a one‐pot tandem arylation/oxidation of diarylmethane derivatives for the convenient synthesis of triarylmethanols bearing different aryl and heteroaryl groups with both electron donatingas well as sterically hindered groups. However, the study with electron releasing effects was not investigated. The method extends the reactivity of the compounds with less acidic sp3C−H and can be further utilized for diversification.
Scheme 36.
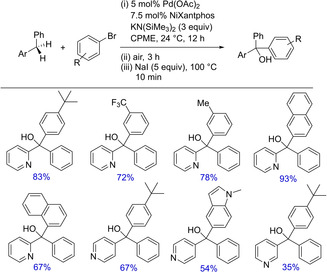
Pd/NiXantphos‐catalyzed synthesis of triaylmethanes and subsequent oxidation.
In another report by Walsh and group in 2017, palladium‐catalyzed functionalization of benzylic C−H of diaryl(heteroaryl)methanes was discussed.52 Using the protocol, the authors demonstrated the introduction of various aryl groups onto the benzylic C−H to get tri‐ and tetraaryl(heteroaryl)methanes in good to excellent yields (Scheme 37).
Scheme 37.
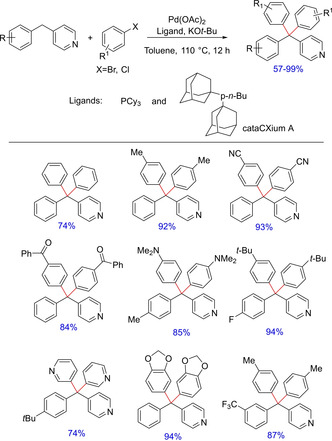
Palladium‐catalyzed synthesis of tri and tetraaryl/heteroarylmethanes.
3.5. Other metal‐catalyzed functionalizations including dual catalysts
Oxidation of alkyl and cycloalkylarenes with TBHP under bismuth catalysis was demonstrated by Barrett and group in 2005 (Scheme 38).53 The authorslargely demonstrated the use benzylic compounds that are oxidized to the corresponding benzylic ketones or carboxylic acids in the presence of bismuth and picolinic acid in pyridine and acetic acid.Only one example of a diarylmethane compound containing methylene functional group is appended in the report. Because of the poor solubility of bismuth salts, the study with bismuth catalysis is limited.
Scheme 38.
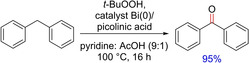
Bismuth‐catalyzed benzylic oxidation.
In year 2010, Li and Correia, reported an oxidative alkylation of benzylic C−H bonds with 1,3‐dicarbonyl compounds employing a catalytic reagent system comprising of NHPI, FeCl2, and CuCl and oxygen as the terminal oxidant (Scheme 39).54 Although the authors have demonstrated the success of this methodology with a few diphenylmethane derivatives, compatibility with other diaryl/heteroarylmethane scaffolds remains unexplored. Also, the exact role of CuCl in the reaction remains undefined. The authors postulate the possibility of multiple reaction pathways, one stating benzyl radical as the reactive intermediate and the other in which benzhydrol coordinates with iron enolate intermediate giving the desired product; but no evidence in favor of any conclusion is provided.
Scheme 39.
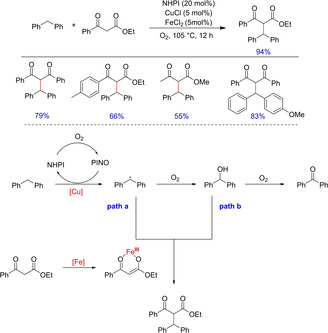
Functionalization of diphenylmethanes with 1,3‐dicarbonyl compounds.
An efficient and facile synthesis of unsymmetrical triaryl(hetero‐aryl)methanes by RhIII‐catalyzed C(sp3)−H arylation of diaryl(hetero‐aryl)methanes was reported by Glorius and group in 2015.55 The authors have demonstrated the reaction of triarylboroxines with Cp*RhIII‐activated C(sp3)−H bonds for the synthesis of C(sp3)‐aryl bonds. Originally, the protocol focuses on the selective β‐arylation of 2‐alkylpyridines through direct functionalization of an unactivated C(sp3)−H bond, which was then extended to the synthesis of unsymmetrical triaryl(hetero‐aryl)methane derivatives using substituted 8‐benzylquinolines asone of the substrates (Scheme 40). The mechanism of reaction was presumed to follow the general sequence, i. e. coordination followed by transmetalation/reductive elimination and subsequent functionalization. Interestingly, the products were obtained from substrates that could potentially undergo β‐hydride elimination.
Scheme 40.
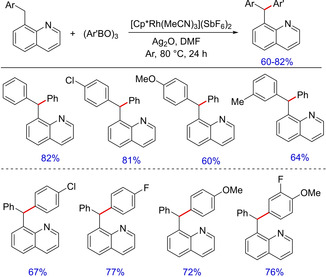
Rhodium‐catalyzed arylation of aryl/heteroaryl methane.
Subsequently, in 2016, the group reported another important transformation witnessing a Rh(I)/NHC*‐catalyzed site‐ and enantioselective functionalization of C(sp3)−H bonds towards the synthesis of chiral triarylmethanes.56 With the change in the catalytic system (i. e. from Cp*RhIII‐catalyzed reaction to a Rh(I)/NHC*‐catalyzed reaction), enantioselective arylation of benzylic C−H bonds could be achieved (Scheme 41). Interestingly, no enantioselectivity was observed in case of 2‐benzylpyridine, whereas a modest enantioselectivity (82 : 18 er) was observed with 8‐benylquinolines. This could be due to the formation of a more rigid metal complex with 8‐benzylquinolines than 2‐benzylpyridine, which is likely to induce asymmetry. Mechanistic studies reveal an intramolecular C(sp3)−H activation by the newly designed chiral NHCs leading to a defined chiral environment. These newly designed chiral NHCs along with Rh(PPh3)3Cl have been utilized to demonstrate the synthesis of diverse triarylmethanes with good enantioselectivities.
Scheme 41.
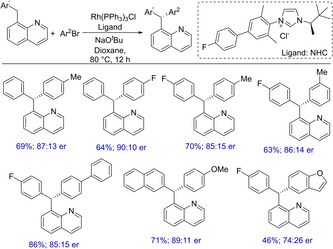
Rhodium‐catalyzed enantioselective functionalization of diarylmethane for the synthesis of chiral triarylmethane.
In 2015, Patrick J. Walsh and group explored the use of nickel as an efficient catalyst system for benzylic C−H functionalization. The authors earlier demonstrated the use of palladium catalyst in benzylic C−H functionalization with NiXantphos as a ligand51 and subsequently with nickel as the catalyst for such functionalization with varying diarylmethanes and aryl halides (Scheme 42).57 The report witnesses a wide substrate scope with good conversions and excellent yields. On comparison, it was found that a Ni/NiXantphos catalyst system was far better than Ni/Xantphos and Pd/Xantphos in terms of conversion due to poor solubility of Pd/Xantphos catalyst system in common organic solvents.
Scheme 42.
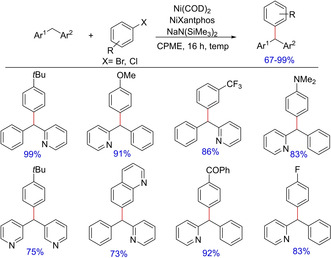
Nickel‐catalyzed benzylic C−H functionalization.
In the meanwhile, the group has also demonstrated nickel‐catalyzed asymmetric allylic alkylation with soft diarylmethane nucleophiles with high ee values.58 While the earlier reports with nickel catalysis were limited to hard nucleophiles, this report demonstrates the reliable solution to the limitation using Ni(COD)2 in the presence of DPPF as the ligand employing soft nucleophiles (Scheme 43). The report includes use of various Boc protected allylic alchols and several diaryl‐ or diheteroarylmethanes. The protection of OH group other than Boc has not been demonstrated.
Scheme 43.
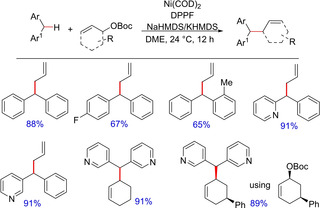
Nickel‐catalyzed benzylic allylation
In 2017, Dong et al.developed a Mn‐catalyzed methodology for the synthesis of wide range of secondary amides by amination of benzylic C(sp3)−H bonds with nitriles (Scheme 44).59 In this method, manganese (III) acetate acted as Lewis acid catalyst and its interaction with DDQ significantly increased efficiency and selectivity, whereas TFA accelerated hydrolysis of nitrilium cation to amide. The methodology was compatible with various arylmethane derivatives such as ethylbenzene, tetrahydronaphthalene, and indane, also applicable to primary, secondary, tertiary nitriles. Interestingly, the strained cyclopropane carbonitrile was also well tolerated. Kinetic isotopic studies suggested that a C−H bond cleavage is involved to form a benzyl radical, which was supported by suppression of product in the presence of radical scavengers like BHT and TEMPO.
Scheme 44.
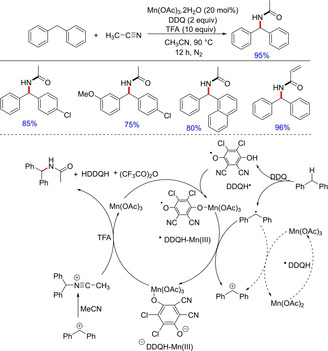
C−N bond formation using manganese catalysis.
Recently, in 2018, Walsh and group demonstrated another protocol which utilizes non‐activated aryl fluorides for C(sp3)−H arylation.60 Their initial attempts utilizing the (NHC)Pd‐ and (NHC)Ni‐ based catalyst systems were limited to aryl bromides and chlorides, however, proved ineffective for transformations involving aryl fluorides. Therefore, the authors invented a new catalyst system involving Ni(COD)2 and IMes to mediate C−F bond activation, which promoted the synthesis of triarylmethanes (Scheme 45). The catalyst system is well tolerated by multiple substrates except for a few heterocycles, which ended up with multiple products. The use of IMes as the ligand was crucial to the success of transformation, as it facilitates the oxidative addition step of the catalytic cycle owing to its strong σ‐donor ability. Also, this method could find distinctive applications in the presence of multiple aryl halides for selective transformations.
Scheme 45.
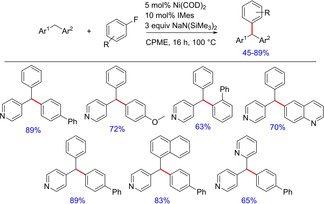
Synthesis of triarylmethanes using nickel and NHC catalyst system.
4. Metal‐free oxidative benzylic CyC‐>H functionalization of diarylmethanes
Transition‐metal‐catalyzed coupling reactions have made a significant progress since the emergence of organometallic chemistry. The extensive variations and modifications have enabled these coupling reactions to find wide applications in organic chemistry as well as in pharmaceutical chemistry. Although transition‐metal catalysis has gained a lot of importance, often it suffers from the inherent limitations of the catalytic systems. These limitation may include (i) many transition metal catalysts are very expensive and the ligands used to support the catalytic system are even more expensive, (ii) transition metals exert variable toxicity causing removal of even a trace amount costly and challenging, (iii) many transition metal catalysts are sensitive to moisture and oxygen requiring additional precaution, (iv) special additives and co‐catalysts are required in many cases to promote the efficiency and selectivity of transformations, (v) transition‐metal catalysis also rarely meets the demands of sustainable chemistry.61 Thus, studies on metal‐free benzylic (sp3)C−H functionalizations of diarylmethanes are of great significance. The following discussion will cover selected examples of these reactions.
4.1. Visible light and photoredox‐catalyzed functionalization
Photocatalysis, which involves environmentally harmonious, ecologically clean and safe, sustainable energy, is an emerging area towards sustainable chemistry. It also furnishes the advantage of metal‐free coupling which is the demand of the time.62 Pyridines are widely used as building blocks in preparation of various chiral ligands and functional materials with photo‐ or electrochemical properties. In 2013, Hoshikawa and Inoue reported a metal‐free photochemical methodology for direct 4‐pyridination of C(sp3)−H bond employing benzophenone and 4‐cyanopyridine in aqueous acetonitrile at ambient temperature (Scheme 46).63 This methodology postulated high chemoselectivity especially at benzylic C(sp3)−H bond, proficient compatibility with various polar and halogen functionalities, and high efficiency in single step formation of hindered linkages between carbo‐skeletons and pyridine. The authors proposed the reaction to proceed via a radical based ipso‐substitution followed by radical‐radical coupling.
Scheme 46.
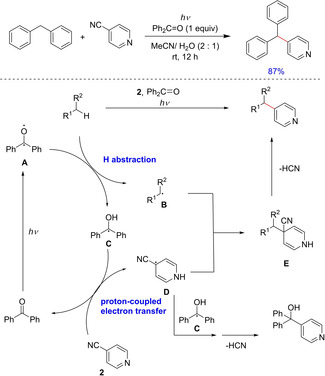
Photochemical metal‐free direct 4‐pyridination.
Another report involving visible‐light‐mediated mono‐ and difluorination of benzylic CH2 utilizing ketone based catalysts (e. g.; 9‐fluorenones and xanthones) surfaced in 2013 by Chen et al.64 featuring an operationally simple procedure with wide functional group tolerance (Scheme 47). The mechanistic studies revealed that both visible light and catalyst were required for the C−H fluorination, while a non‐metal radical C−H abstraction is involved in the rate‐limiting step.
Scheme 47.
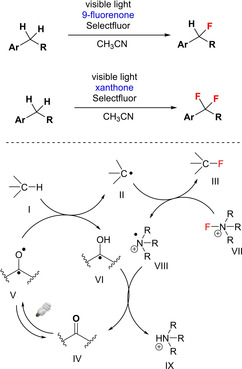
Photocatalytic benzylic C−H fluorination.
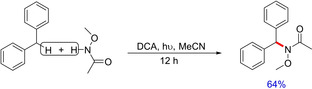
In another report, Pandey and Laha demonstrated a direct amination of benzylic CH2 bond via visible‐light‐mediated one‐electron photoredox oxidation affording the corresponding mono‐aminated products in good to excellent yields (Scheme 48)65 The cross‐dehydrogenative coupling proposedly involved the benzylic C−H radical formation via hydrogen atom transfer to an aminyl radical followed by one electron oxidation of benzylic radical to form benzylcarbocation. The aminyl radical was generated from amine by one‐electron oxidation in the presence of 9,10‐dicyanoanthracene (DCA) at the singlet excited state. The nucleophilic interception of benzylcarbocation by amine furnished the benzylaminated product.
Scheme 48.
Visible‐light‐catalyzed direct benzylic C(sp3)−H amination.
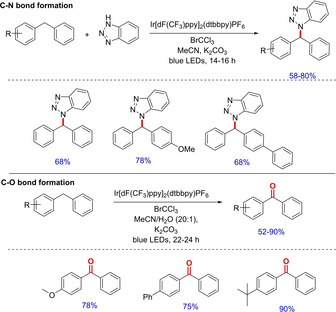
The group further developed a visible‐light‐mediated highly regioselective benzylic C–H bond functionalization leading to C–N and C–O bond formation (Scheme 49).66 The method is quite feasible for incorporating different azoles at the benzylic position for the generation of different heteroaromatics. Also, the same protocol is extended to afford direct benzylic oxidation of various alkyl aryls to corresponding carbonyl compounds viavisible‐light‐photoredox catalysis using Ir[dF(CF3)ppy]2(dtbbpy)PF6. Single electron transfer (SET) from electron rich aromatics to highly electron deficient Ir(IV), in‐situ generated, to form the arene radical cation followed by interception with the nucleophile gave the desired product.
Scheme 49.
Photocatalytic benzylic C(sp3)−H bond functionalization.
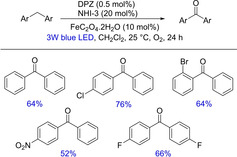
In 2017, Jiang and co‐workers disclosed a photoredox, visible‐light‐driven cooperative catalysis between dicyanopyrazine‐derived chromophore (DPZ) and N‐hydroxyimide facilitating the aerobic oxygenation of a series of benzylic sp3C−H bonds (Scheme 50).67 The method provided a novel approach to access valuable diarylketones from diarylmethanes and diarylmethanols in moderate to excellent yields. The mechanistic insights determined an underlying plausible SET pathway.
Scheme 50.
Aerobic oxygenation of benzylic C(sp3)−H bond.
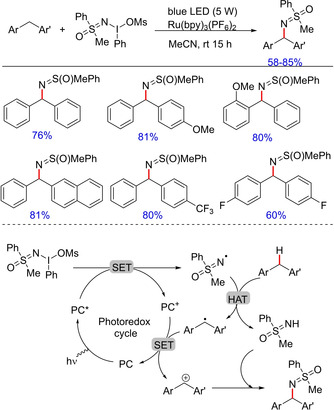
Sulfoximines are important class of compounds with broad applications in medicinal and agrochemistry68 and N‐functionalized sulfoximines have always been of great interest to organic chemists.21 Iron‐catalyzed sulfoximidation of benzylic C−H bond of diarylmethanes was reported by Bolm et al in 2014.22 Subsequently, in 2018, the group developed an efficient photocatalytic sulfoximidation of benzylic C−H bonds.69 The protocol offers a wide substrate scope and mild reaction conditions employing a sulfoximidoyl‐containing hypervalentiodine(III) reagent for the generation of a variety of functionalized diarylmethanes (Scheme 51). The generation of a nitrogen‐centered sulfoximidoyl radical via an electron‐transfer process is proposed in the mechanism, which upon subsequent hydrogen atom abstraction from diraylmethanes form the corresponding diarylmethane radical. Further electron transfer from the carbon‐centered radical forms a benzylic carbocation, which is trapped by the sulfoximine nucleophile.
Scheme 51.
Photocatalyzed synthesis of functionalized sulfoximines.
Another important photocatalyzed transformation is reported by Scheidt and group in 2019, wherein an enantioselective hydroxylation of benzylic C−H has been demonstrated using a one‐flask method via photoredox/enzymatic catalysis.70 It is interesting to note that the photocatalyzed pathway is responsible for the generation of the ketone product, which is then reduced enzymatically by a KRED using NAD(P)H as the hydride source providing the corresponding enantioselective alcohol (Scheme 52). The authors have presented an integrated substrate‐guided oxidation process with enzymatically‐enforced enantioselectivity to generate wide substrate scope ranging from aryl‐alkyl alcohols to diarylmethanols, γ,δ‐lactones, α‐hydroxy esters and 1,2‐amino alcohols.
Scheme 52.
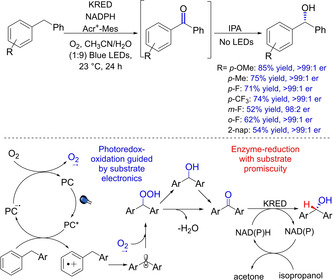
Photoredox/enzymatic catalysis for the enantioselective hydroxylation of benzylic C−H.
4.2. Oxidant‐mediated functionalization
In 2009, Fan and group developed a transition metal free direct amination of C(sp3)−H bond in diraylmethanes with amines employing iodobenzene diacetate and iodine as the active oxidant system (Scheme 53).71 The authors demonstrated the use of various amines including chloro amines, amino alcohol, amino ester, sulfonamides and N‐sulfonylimine. However, the reaction is highly regioselective with only aromatic sulfonamides. The reaction is proposed to undergo in‐situ halide substitution on diarylmethanes followed by nucleophilic displacement by amine to form the product.
Scheme 53.
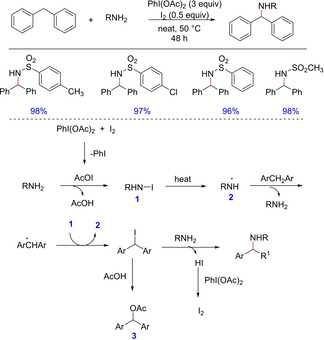
Metal‐free C−N bond formation via in‐situ halide substitution.
In 2012, Minakata et al.reported a transition metal free intermolecular benzylic C−H bond amination utilizing an inexpensive oxidant system of chloramine‐T and I2 (Scheme 54).72 Apart from acyclic n‐alkyl substituted benzenes and ethylbenzene derivatives, the protocol was applicable for chemoselective amination of adamantane to N‐protected form of memantine, which is an active therapeutic agent for moderate to severe Alzheimer disease.According to significant retardation of the reaction in presence of TEMPO, the authors proposed a radical pathway for this transformation.
Scheme 54.
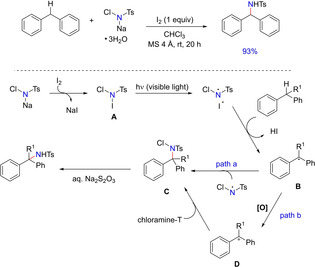
Oxidative C−N bond formation via iodine.
In the same year, Moriyama and group described a simple yet important transformation involving direct benzylic oxidation of diraylmethanes via C−H bond abstraction using alkali metal bromides and oxidants under mild conditions (Scheme 55).73 The authors have very nicely demonstrated the effect of solvent and other reaction conditions (thermal or photochemical) on the reaction pathways. However, the yields are comparable in both thermal and photochemical reactions. The methodology can be utilized for not only diarylmethanes, but also can be further extended to alkylarenes.
Scheme 55.
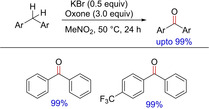
Benzylic oxidation of diarylmethanes using alkali metal bromide and oxidants.
2,3‐Dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ) is one of the most commonly used oxidants in organic chemistry. In 2012, Lei et al. reported an efficient DDQ‐catalyzed methodology for oxidative C−O coupling of benzylic C(sp3)−H with various carboxylic acids in the presence of MnO2 as terminal oxidant (Scheme 56).74 The C−O oxidative coupling was compatible with variety of carboxylic acids such as long‐chain aliphatic, aromatic and vinyl carboxylic acids. The methodology does not work with 1‐benzyl‐4‐nitrobenzene indicating the involvement of a radical that could be possibly inhibited by the nitro group. Interestingly, when a catalytic amount of DDQ was used for‐weakly acidic carboxylic acids, only a limited conversion was observed. To enhance the conversion, trifluoroacetic acid was used, which probably caused the increased oxidation capacity of MnO2, facilitating the regeneration of DDQ. The use of radical scavenger TEMPO and kinetic isotopic studies suggested the C−H bond dissociation to be the rate limiting step in the transformation.
Scheme 56.
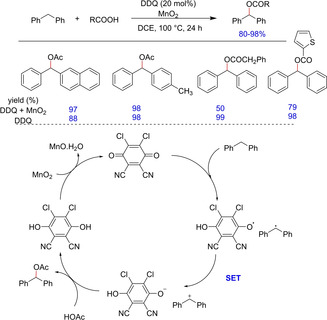
Oxidative C−O coupling via DDQ.
In 2012, another methodology employing metal free conditions for C−O bond formation utilizing benzyl substrates and various substituted carboxylic acids was developed by Yu and group.75 The reaction involves the use of tetrabutylammonium iodide as a catalyst and tert‐butyl hydroperoxide as the co‐oxidant, demonstrating a radical pathway for benzylic C−H esterification (Scheme 57). Although, the report unveils only limited examples of diarylmethanes, a variety of alkyl arylmethanes have been successfully functionalized with different carboxylic acids. The method is also suitable for the carboxyl protected N‐Boc amino acids, which could be an efficient metal free procedure for amino acid protection.
Scheme 57.
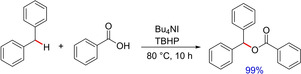
TBHP/TBAI‐mediated C−O bond formation.
The use of DDQ for benzylic functionalization was also explored by Liu and group in 2014 for environment friendly and efficient synthesis of functionalized diarylmethanes.76 The products were further transformed into polysubstituted 1H‐indenes via a radical‐initiated two C(sp2)−C(sp2) and C(sp2)−C(sp3) bond formation (Scheme 58).
Scheme 58.
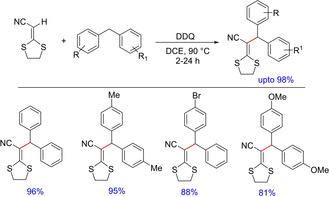
DDQ‐mediated functionalization of benzylic C−H bond.
In 2015, Liu and group developed a transition metal free oxidative C−N coupling of benzylic C−H bonds using DDQ as the only oxidant (Scheme 59).77 A series of sulfonamide and carboxamide substrates smoothly underwent this transformation. Interestingly, when electron‐deficient anilines were employed, diphenylmethyl imine was obtained instead of the desired C−N coupling product implying over‐oxidation of the active aminated product. Kinetic isotopic studies and TEMPO experiment suggested the transformation to involve a radical intermediate.
Scheme 59.
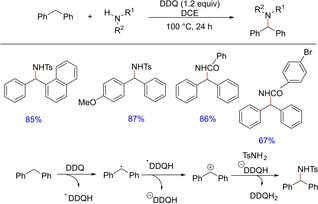
Transition metal free oxidative C−N coupling.
Following the use of DDQ alone as well as in combination with other oxidants74, 76, 77 for benzylic C−H functionalizations, Shen and group in 2015 came up with another strategy involving catalytic amounts of DDQ/tert‐Butyl nitrite in the presence of acetic acid under aerobic conditions for the synthesis of various diarylketones from corresponding diarylmethanes (Scheme 60).78 The fate of the reaction is a radical pathway. In the presence of DDQ, diarylmethane radical is generated, which is transformed into the corresponding acetate derivative, which is further converted to the desired diarylketone in the presence of DDQ and water. Beside diarylmethanes, the authors also have successfully demonstrated the use of aryl/heteroarylmethanes.
Scheme 60.
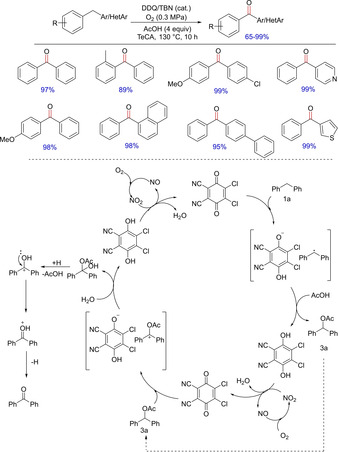
Synthesis of diarylketones using DDQ.
The combination of TBHP/TBAI catalyst system has been widely utilized for a variety of benzylic C−H functionalizations in diarylmethanes.79 Utilizing the same catalytic system, Li and group had only a limited success for the oxidative amination of benzylic methylene group in diarylmethanes.79b After optimization of the reaction conditions, the group obtained another set of reagents including potassium iodide as the pre‐catalyst and TBHP as the terminal oxidant for effective transformation (Scheme 61). The optimized condition was well tolerated by diarylmethanes containing both electron‐donating and electron‐withdrawing groups to give the corresponding benzylated amines in moderate to good yields. It is observed that the yields of the aminated products are affected by the substitution on the phenyl ring. Diarylmethane with substitution close to the reaction site gives the corresponding coupled products in relatively lower yields.
Scheme 61.
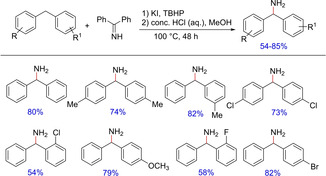
Oxidant‐mediated C−N bond formation.
Over the years, 1,2,4‐triazole derivatives have drawn considerable attention from medicinal chemists due to their increasing biological activities such as antimicrobial, anti‐inflammatory, anticancer, antiviral etc. Despite their potential activities, only limited economical/ cost‐effective methods are known for their synthesis. In 2016, Abebe and group reported an efficient metal free and cost‐effective catalyst‐oxidant system comprising of TBAI/TBHP for the construction of C−N bond via cross dehydrogenative coupling of 1H‐1,2,4‐triazoles and diarylmethanes.80 The significant suppression of the desired product in presence of TEMPO and BHT illustrates the reaction to proceed via a radical pathway (Scheme 62).
Scheme 62.
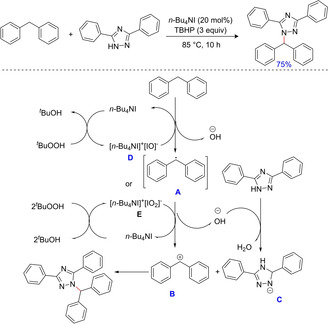
Cross‐dehydrogenative coupling of 1H‐1,2,4‐triazoles with methyl arenes.
Following a variety of photoredox catalysis and other metal‐free approaches for specific sp 3 benzylic oxidation, Xu and group in 2017 supplemented another metal‐free approach for the synthesis of diaryl/heteroarylketones from diaryl(heteroaryl)methanes (Scheme 63) utilizing TBHP.81 The reaction proceeds at high temperatures via a radical mechanism. Decreased reaction temperature causes a detrimental effect on the rate of oxidation causing low yield probably due to the reduced rate of radical generation through thermal decomposition.
Scheme 63.
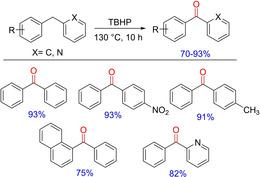
TBHP‐mediated synthesis of diaryl/heteroarylmethanes
4.3. Ionic liquid‐mediated functionalization
In 2015, Liu and co‐workers successfully demonstrated the use of heterocyclic ionic liquid in a non‐classical catalysis for direct oxidative amination via activation of benzylic C−H bonds for the synthesis of substituted and functionalized N‐alkylated azoles under metal‐free, mild, and green conditions. The catalyst system includes recycled and reused ionic liquid 1‐butylpyridinium iodide ([Bpy]I) as a catalyst and TBHP as an oxidant (Scheme 64).82 This metal‐free catalytic system is suitable for the oxidative coupling reactions between a wide range of azoles and benzyl substrates. The mechanism revealed that the benzyl radical was liable to be oxidized by active iodine species, which thereby underwent nucleophilic reaction with the anionic species formed in‐situ to afford the desired product.
Scheme 64.
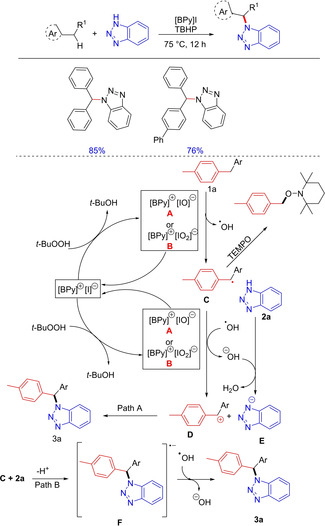
Ionic liquid‐catalyzed oxidative C−N bond formation.
4.4. Others
In 2012, Inoue and co‐workers developed a chemoselective method for direct intermolecular functionalization of C(sp3)−H to C(sp3)‐N bond by employing a reagent system using N‐hydroxyphthalimide (NHPI) as an oxyl radical precursor and azodicarboxylate both as an oxidant and as a radical acceptor.83 The protocol proved to be powerful and efficient for chemoselective C−H functionalization of benzylic, propargylic and aliphatic substrates. Their methodology displayed a wide range of substrate scope and good functional group compatibility incorporating protected alcohols, amines, and carboxylic acids, cyanides and bromides. Further conversion of the product hydrazines to the corresponding carbamates and amines served as a unique tool for the economical synthesis of complex amine substituted natural products and pharmaceuticals (Scheme 65).
Scheme 65.
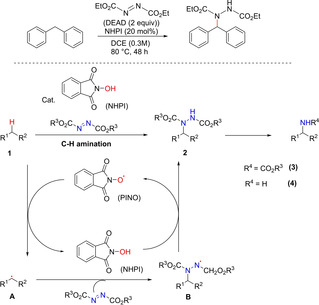
N‐ hydroxyphthalimide‐mediated C−N bond formation.
In 2014, Zhu and group came up with another simple yet important methodology for the efficient synthesis of diarylketones from diarylmethanes in the presence of NBS (Scheme 66).84 The protocol utilizes both sunlight irradiation and heating for radical initiation and water serves as the source of oxygen. The optimized condition shows a good substrate scope, compatible with both electron‐donating and electron‐withdrawing groups on the aryl ring. Interestingly, diarylmethanes bearing methoxyl or ethoxyl group at the para‐position of the benzene ring afforded mono‐bromo substituted ketones in moderate yields indicating dual role of NBS i. e. a radical initiator as well as a brominating agent.
Scheme 66.
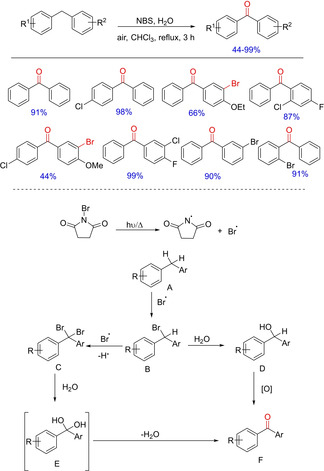
NBS‐catalyzed synthesis of diarylketones.
An efficient (hetero)benzylic sp3C−H oxidation method for the synthesis of diaryl/heteroarylketones was developed by Wang and group in 2016.85 The protocol employs inexpensive potassium tert‐butoxide as the promoter and proceeds under mild conditions using oxygen as the oxidant (Scheme 67). The optimized condition works well with electron‐donating groups, whereas a slight decrease in the chemical yields of the oxidation products was observed with electron‐deficient groups present in the substrate. In case of ortho‐substituted substrates, reduced reactivity and longer reaction time was required for the formation of the oxidized product. The reaction follows a radical pathway involving interactions between the complex of KOtBu with 18‐crown‐6 and DMF followed by electron transfer and radical generation. The generated radical then reacts with O2 to form hydroperoxide, which loses water molecule to generate the desired ketone. The authors have also demonstrated the utility of this strategy by gram‐scale synthesis of biologically important heteroaryl ketones.
Scheme 67.
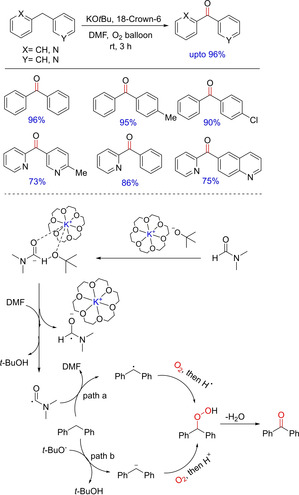
Oxidative benzylic C−H functionalization.
Another method for the synthesis of diarylketones was reported by Li and group in 2017 involving metal‐free oxygenation of benzylic sp3C−H bond by base using an O2‐ promoted process (Scheme 68).86 Although other strong bases also gave the desired results, the efficiency was comparatively low. The substrate scope of the reaction is optimum. While the presence of electron‐withdrawing groups increased the reactivity towards oxidation and selectivity, the introduction of the electron‐donating substituents decreased the reaction efficiencies. The increased acidity of the benzylic C−H bond by electron‐withdrawing group could be the reason for such reactivity pattern. Unlike the electronic effect, the steric effect does not have much influence on the reaction. The reaction is proposed to undergo an anion‐radical oxidation process.
Scheme 68.
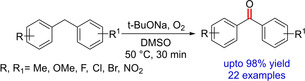
Base‐mediated oxidative benzylic C−H functionalization.
Although many reports, as mentioned above, are available involving base‐catalyzed direct functionalization of the benzylic C−H bond of diarylmethanes, all these methods involve stoichiometric amount of a strong base or an oxidant. Recently in 2018, Guan and group described a method involving a catalytic amount of potassium zincate complex for benzylic C−H bond addition of diarylmethanes to styrenes and conjugated dienes (Scheme 69).87 The bridging structure of the potassium zincate complex generated from potassium benzyl and zinc amide plays a critical role in the catalytic alkylation reaction thereby showing good activity as well as chemoselectivity. The optimized condition shows a wide substrate scope employing various diarylmethanes as well as differently substituted styrene and dienes. Interestingly, para‐halogen substituted diarylmethane greatly inhibited the alkylation reaction. Control experiments conducted to understand the inhibitory effect of a para halogen suggested the possibility of coordination of chlorobenzene to the potassium‐zincate complex, thereby inhibiting its activity. Although the catalytic application of the complex is insightful, the synergic interaction between zinc and potassium leading to the catalytic activity needs to be elucidated.
Scheme 69.
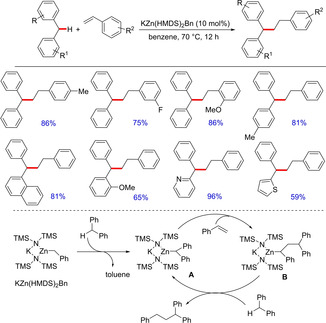
Synthesis of functionalized diarylmethanes through potassium‐zincate complex catalysis.
5. Conclusion and Outlook
A steady increase in the number of important pharmaceutical molecules containing a diarylmethane motif and molecules useful in material sciences functionalized at the benzylic position has generated a strong impetus towards the development of their chemistry. For example, several best selling drugs containing a diarylmethane motif include cetrizine, letrozole, bifonazole, peperomin B, tolterodine and lasofoxifene found useful for the treatment of different types of diseases. The diversity and utility of functionalized diarylmethanes has been very influential in spurring new developments towards the functionalization of benzylic CH2‐position.The present review discusses the recent developments in the chemistry of functionalization of diarylmethanes at the benzylic CH2‐position.The versatility in the methods of functionalization ranging from base‐mediated to metal‐catalyzed and further to metal free conditions is a result of innovation and improvement in the reaction conditions, which deserves much appreciation. However, their translational potential is rarely demonstrated. Nucleophilic substitution or addition reactions form the foundation for base‐mediated reactions. However, the chemo‐ and regio‐selectivity issues still persist probably due to intrinsic mechanistic limitations. These limitations have been conquered to a certain extent by metal‐catalyzed functionalizations, which offers flexible modifications due to parameters such as coordinating ligands, counter anions, oxidative states of metals etc. that can be tuned according to the requirements. Although transition‐metal‐catalyzed methodologies have dominated the organic synthesis for the last decade, a walk‐over by transition‐metal‐free approaches is the upcoming reality which has almost took over a majority of benzylic CH2 functionalizations. The endless pursuit of employing green and sustainable chemistry is the driving force for the development of new, highly efficient, metal‐free catalytic reaction systems for the functionalization of benzylic CH2 in diarylmethanes and other such systems. However, still there lies a scope for further development using the recent methods of chemical transformations such as photoredox catalysis and electrochemistry. Perhaps, some of these advances in the functionalization strategies would stand the test of time in terms of sustainability (use of metal‐free reaction conditions), compatibility (late‐stage functionalizations) and selectivity (enantioselectivity) etc. and may replace the original protocols. Incorporation of green chemistry features in the reported methods could also attract the pharmaceutical companies for in‐house practice and implementation. The demonstration of these strategies in gram scale is required to be able to translate them in API synthesis. Also, asymmetric benzylic C−H functionalization of diarylmethanes has been the subject of least investigation. Such important development is in high demand. For example, the best‐selling drug letrozole possesses a stereocenter at the benzylic position. Although an elegant synthesis of letrozole has been reported via benzylic C−H functionalization of the corresponding diarylmethane, whether the same protocol could be implemented in an asymmetric fashion remains a question; and challenges like these become the scope for new methodologies and strategies which could not only enhance the developments in the synthetic organic chemistry but also could notably benefit the pharmaceutical chemistry research. We hope that this review would serve the readers as an important guide towards various strategies for benzylic CH2 functionalizations and at the same time would be a lending hand towards development of further exciting expansions in this area, keeping in mind, the current challenges and the upcoming opportunities.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Upma Gulati completed M. S. (Pharm.) from the Department of Natural Products, NIPER S.A.S. Nagar in 2017. She is working as a Ph.D. scholar in the Department of Pharmaceutical Technology (Process Chemistry), NIPER S.A.S. Nagar since 2017. Her current research activities are focused on decarboxylation chemistry and approaches for benzylic functionalization.

Biographical Information
Radhika Gandhi completed M. Tech (Pharm.) from the Department of Pharmaceutical Technology (Process Chemistry), NIPER S.A.S. Nagar in 2018 under the supervision of Dr. Joydev K. Laha. Her research project involved benzylic C‐H amination at the late‐stage functionalization of drug intermediates

Biographical Information
Dr. Joydev K. Laha started his independent career at NIPER S.A.S. Nagar in July 2011. Prior to joining NIPER, Dr. Laha was employed in the Laboratory for Drug Discovery in Neurodegeneration (LDDN) at Harvard Medical School. Dr. Laha obtained a Ph.D. degree in organic chemistry from the National Chemical laboratory at Pune. He acquired several years of postdoctoral research experiences in synthetic organic chemistry and medicinal chemistry at the North Carolina State University and Mayo Clinic in the United States. Dr. Laha's current research interests include oxidative radical reactions largely using persulfates, understanding their mechanisms, and their applications to the synthesis of heterocycles and API (active pharmaceutical ingredient) synthesis.

Acknowledgements
Financial support from NIPER, S.A.S. Nagar, and CSIR, New Delhi is greatly acknowledged.
U. Gulati, R. Gandhi, J. K. Laha, Chem. Asian J. 2020, 15, 3135.
References
- 1.
- 1a. Nair V., Thomas S., Mathew S. C., Abhilash K. G., Tetrahedron 2006, 62, 6731–6747; [Google Scholar]
- 1b. Bolm C., Schmidt F., Stemmler R. T., Rudolph J., Chem. Soc. Rev. 2006, 35, 454–470; [DOI] [PubMed] [Google Scholar]
- 1c. Snape T. J., Ameen D., MedChemComm 2013, 4, 893–907; [Google Scholar]
- 1d. Rachwalski M., Wujkowska Z., Jarzyński S., Pieczonka A. M., Leśniak S., Tetrahedron: Asymmetry 2016, 27, 1238–1244; [Google Scholar]
- 1e. Anand R. V., Jadhav A. S., Org. Biomol. Chem. 2017, 15, 56–60. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ma J. C., Dougherty D. A., Chem. Rev. 1997, 97, 1303–1324; [DOI] [PubMed] [Google Scholar]
- 2b. Jasant A., Sherman J. C., Chem. Rev. 1999, 99, 931–968. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Kofron W. G., Mathew J., J. Org. Chem. 1976, 41, 114–116; [Google Scholar]
- 3b. Elz S., Kramer K., Pertz H. H., Detert H., terLaak A. M., Kühne R., Schunack W., J. Med. Chem. 2000, 43, 1071–1084; [DOI] [PubMed] [Google Scholar]
- 3c. Clausen D. J., Wan S., Floreancig P. E., Angew. Chem. Int. Ed. 2011, 50, 5178–5181; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5284–5287. [Google Scholar]
- 4. Bosque I., Chinchilla R., G-Gomez J. C., Guijarro D., Alonso F., Org. Chem. Front. 2020, 7, 1717–1742. [Google Scholar]
- 5.
- 5a. Fischer E., Larsen J., Christensen J. B., Fourmigué M., Madsen H. G., Harrit N., J. Org. Chem. 1996, 61, 6997–7005; [DOI] [PubMed] [Google Scholar]
- 5b. Kawashima H., Ogawa N., Saeki R., Kobayashi Y., Chem. Commun. 2016, 52, 4918–4921. [DOI] [PubMed] [Google Scholar]
- 6.For selected examples, see:
- 6a. Zhang J., Stanciu C., Wang B., Hussain M. M., Da C-S., Carroll P. J., Dreher S. D., Walsh P. J., J. Am. Chem. Soc. 2011, 133, 20552–20560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Bellomo A., Zhang J., Trongsiriwat N., Walsh P. J., Chem. Sci. 2013, 4, 849–857; [Google Scholar]
- 6c. Sha S., Zhang J., Carroll P. J., Walsh P. J., J. Am. Chem. Soc. 2013, 135, 17602–17609; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Mao J., Eberle K., Zhang J., Rodríguez-Escrich C., Xi Z., Pericàs M. A., Walsh P. J., Tetrahedron Lett. 2015, 56, 3604–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selected examples, see
- 7a. Li Z., Cao L., Li C., Angew. Chem. Int. Ed. 2007, 46, 6505–6507; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6625–6627; [Google Scholar]
- 7b. Liu X., Zhang Y., Wang L., Fu H., Jiang Y., Zhao Y., J. Org. Chem. 2008, 73, 6207–6212; [DOI] [PubMed] [Google Scholar]
- 7c. Wang Z., Zhang Y., Fu H., Jiang Y., Zhao Y., Org. Lett. 2008, 10, 1863–1866; [DOI] [PubMed] [Google Scholar]
- 7d. Pinter A., Sud A., Sureshkumar D., Klussmann M., Angew. Chem. Int. Ed. 2010,49, 5004–5007; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5124–5128; [Google Scholar]
- 7e. Baba H., Moriyama K., Togo H., Tetrahedron Lett. 2011, 52, 4303–4307. [Google Scholar]
- 8.For selected examples, see
- 8a. Jasperse C. P., Curran D. P., Fevig T. L., Chem. Rev. 1991, 91, 1237–1286; [Google Scholar]
- 8b. Chatgilialoglu C., Crich D., Komatsu M., Ryu I., Chem. Rev. 1999, 99, 1991–2070; [DOI] [PubMed] [Google Scholar]
- 8c. Bar G., Parsons A. F., Chem. Soc. Rev. 2003, 32, 251–263; [DOI] [PubMed] [Google Scholar]
- 8d. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2015,55, 58–102; [DOI] [PubMed] [Google Scholar]
- 8e. Yan M., Lo J. C., Edwards J. T., Baran P. S., J. Am. Chem. Soc. 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mondal S., Panda G., RSC Adv. 2014, 4, 28317–28358. [Google Scholar]
- 10.
- 10a. Laha J. K., Tummalapalli K. S. S., Nair A., Patel N., J. Org. Chem. 2015, 80, 11351–11359; [DOI] [PubMed] [Google Scholar]
- 10b. Laha J. K., Jethava K. P., Patel S., Org. Lett. 2015, 17, 5890–5893; [DOI] [PubMed] [Google Scholar]
- 10c. Laha J. K., Tummalapalli K. S. S., Jethava K. P., Org. Biomol. Chem. 2016, 14, 2473–2479. [DOI] [PubMed] [Google Scholar]
- 11. Nambo M., Crudden C. M., ACS Catal. 2015, 5, 4734–4742. [Google Scholar]
- 12. Bordwell F. G., Bares J. E., Bartmess J. E., Drucker G. E., Gerhold J., McCollum G. J., Puy M. V. D., Vanier N. R., Matthews W. S., J. Org. Chem. 1977, 42, 326–332. [Google Scholar]
- 13. Ji X., Huang T., Wu W., Liang F., Cao S., Org. Lett. 2015, 17, 5096–5099. [DOI] [PubMed] [Google Scholar]
- 14. Reddy L. R., Kotturi S., Waman Y., Patel C., Danidharia M., Shenoy R., J. Org. Chem. 2018, 83, 6573–6579. [DOI] [PubMed] [Google Scholar]
- 15. Ji X., Zhao X., Shi H., Cao S., Chem. Asian J. 2017, 12, 2794–2798. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Sun C-L., Li B-J., Shi Z-J., Chem. Rev. 2011, 111, 1293–1314; [DOI] [PubMed] [Google Scholar]
- 16b. Wang S-S., Yang G-Y., Catal. Sci. Technol. 2016, 6, 2862–2876. [Google Scholar]
- 17. Song C-X., Cai G-X., Farrell T. R., Jiang Z-P., Li H., Gan L-B., Shi Z-J., Chem. Commun. 2009, 0, 6002–6004. [DOI] [PubMed] [Google Scholar]
- 18. Li Y-Z., Li B-J., Lu X-Y., Lin S., Shi Z-J., Angew. Chem. Int. Ed. 2009, 48, 3817–3820; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3875–3878. [Google Scholar]
- 19.
- 19a. Hauel N. H., Nar H., Priepke H., Ries U., Stassen J. M., Wienen W., J. Med. Chem. 2002, 45, 1757–1766; [DOI] [PubMed] [Google Scholar]
- 19b. Velik J., Baliharova V., Fink-Gremmels J., Bull S., Lamka J., Skalova L., Res. Vet. Sci. 2004, 76, 95–108 ; [DOI] [PubMed] [Google Scholar]
- 19c. Benhamou L., Chardon E., Lavigne G., Bellemin-Laponnaz S., Cesar V., Chem. Rev. 2011, 111, 2705–2733 ; [DOI] [PubMed] [Google Scholar]
- 19d. Martins M. A. P., Frizzo C. P., Moreira D. N., Zanatta N., Bonacorso H. G., Chem. Rev. 2008, 108, 2015–2050. [DOI] [PubMed] [Google Scholar]
- 20. Xia Q., Chen W., Qiu H., J. Org. Chem. 2011, 76,7577–7582. [DOI] [PubMed] [Google Scholar]
- 21. Bizet V., Kowalczyk R., Bolm C., Chem. Soc. Rev. 2014, 43, 2426–2438. [DOI] [PubMed] [Google Scholar]
- 22. Cheng Y., Dong W., Wang L., Parthasarathy K., Bolm C., Org. Lett. 2014,16, 2000–2002. [DOI] [PubMed] [Google Scholar]
- 23. Shi J-L., Zhang J-C., Wang B-Q., Hu P., Zhao K-Q., Shi Z-J., Org. Lett. 2016, 18, 1238–1241. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Zhang C., Tang C., Jiao N., Chem. Soc. Rev. 2012, 41, 3464–3484; [DOI] [PubMed] [Google Scholar]
- 24b. Allen S. E., Walvoord R. R., Padilla-Salinas R., Kozlowski M. C., Chem. Rev. 2013, 113, 6234–6458; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Guo X-X., Gu D-W., Wu Z., Zhang W., Chem. Rev. 2015, 115, 1622–1651; [DOI] [PubMed] [Google Scholar]
- 24d. Wan J-P., Jing Y., Beilstein J. Org. Chem. 2015, 11, 2209–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borduas N., Powell D. A., J. Org. Chem. 2008,73,7822–7825. [DOI] [PubMed] [Google Scholar]
- 26. Correia C. A., Li C-J., Adv. Synth. Catal. 2010, 352, 1446–1450. [Google Scholar]
- 27. Sprecher C. A., Zuberbühler A. D., Angew. Chem. Int. Ed. 1977, 16, 189. [DOI] [PubMed] [Google Scholar]
- 28. Müller P., Fruit C., Chem. Rev. 2003, 103, 2905–2919. [DOI] [PubMed] [Google Scholar]
- 29. Kohmura Y., Kawasaki K.-i., Katsuki T., Synlett 1997, 12, 1456–1458. [Google Scholar]
- 30. Pelletier G., Powell D. A., Org. Lett. 2006, 8, 6031–6034. [DOI] [PubMed] [Google Scholar]
- 31. Lee J. M., Park E. J., Cho S. H., Chang S., J. Am. Chem. Soc. 2008, 130, 7824–7825. [DOI] [PubMed] [Google Scholar]
- 32. Guo C., Song J., Luo S-W., Gong L-Z., Angew. Chem. Int. Ed. 2010, 49, 5558–5562; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5690–5694. [Google Scholar]
- 33. Zhang L., Ang G. Y., Chiba S., Org. Lett. 2011, 13, 1622–1625. [DOI] [PubMed] [Google Scholar]
- 34. Chen F., Qin C., Cui Y., Jiao N., Angew. Chem. Int. Ed. 2011, 50, 11487–11491; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11689–11693. [Google Scholar]
- 35. Rabet P. T. G., Fumagalli G., Boyd S., Greaney M. F., Org. Lett. 2016, 18, 1646–1649. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Wang X., Huang B., Liu X., Zhan P., Drug Discovery Today 2016, 21, 118–132; [DOI] [PubMed] [Google Scholar]
- 36b. El-Sagheer A. H., Brown T., Acc. Chem. Res. 2012, 45, 1258–1267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36c. Gololobov Y. G., Kasukhin L. F., Tetrahedron 1992, 48, 1353–1406. [Google Scholar]
- 37. Bräse S., Gil C., Knepper K., Zimmermann V., Angew. Chem. Int. Ed. 2005, 44, 5188–5240; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5320–5374. [Google Scholar]
- 38. Suh S-E., Chen S-J., Mandal M., Guzei I. A., Cramer C. J., Stahl S. S., J. Am. Chem. Soc. 2020, 142, 11388–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang A., Venditto N. J., Darcy J. W., Emmert M. H., Organometallics 2017, 36, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Samzadeh-Kermani A., New J. Chem. 2018, 42, 4766–4772. [Google Scholar]
- 41. Zhang W., Wu L., Chen P., Liu G., Angew. Chem. Int. Ed. 2019, 58, 6425–6429. [DOI] [PubMed] [Google Scholar]
- 42. Vasilopoulos A., Golden D. L., Buss J. A., Stahl S. S., Org. Lett. 2020, doi: 10.1021/acs.orglett.0c02238. [Google Scholar]
- 43. Lu H., Subbarayan V., Tao J., Zhang X. P., Organometallics 2010, 29, 389–393. [Google Scholar]
- 44. Ye Y-H., Zhang J., Wang G., Chen S-Y., Yu X-Q., Tetrahedron 2011, 67, 4649–4654. [Google Scholar]
- 45. Niwa T., Yorimitsu H., Oshima K., Org. Lett. 2007, 9, 2373–2375. [DOI] [PubMed] [Google Scholar]
- 46. Trost B. M., Thaisrivongs D. A., J. Am. Chem. Soc. 2008, 130, 14092–14093. [DOI] [PubMed] [Google Scholar]
- 47. Trost B. M., Thaisrivongs D. A., J. Am. Chem. Soc. 2009, 131, 12056–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukai T., Hirano K., Satoh T., Miura M., Org. Lett. 2010, 12, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 49. McGrew G. I., Temaismithi J., Carroll P. C., Walsh P. J., Angew. Chem. Int. Ed. 2010, 122, 5673–5676. [Google Scholar]
- 50. Zhang J., Bellomo A., Creamer A. D., Dreher S. D., Walsh P. J., J. Am. Chem. Soc. 2012, 134, 13765–13772. [DOI] [PubMed] [Google Scholar]
- 51. Zhang J., Bellomo A., Trongsiriwat N., Jia T., Carroll P. J., Dreher S. D., Tudge M. T., Yin H., Robinson J. R., Schelter E. J., Walsh P. J., J. Am. Chem. Soc. 2014, 136, 6276–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang S., Kim B-S., Wu C., Mao J., Walsh P. J., Nat. Commun. 2017, 8, 14641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonvin Y., Callens E., Larrosa I., Henderson D. A., Oldham J., Burton A. J., Barrett A. G. M., Org. Lett. 2005, 7, 4549–4552. [DOI] [PubMed] [Google Scholar]
- 54. Correia C. A., Li C-J., Tetrahedron Lett. 2010, 51, 1172–1175. [Google Scholar]
- 55. Wang X., Yu D-G., Glorius F., Angew. Chem. Int. Ed. 2015, 54, 10280–10283; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10419–10422. [Google Scholar]
- 56. Kim J. H., Greßies S., Boultadakis-Arapinis M., Daniliuc C. G., Glorius F., ACS Catal. 2016, 6, 7652–7656. [Google Scholar]
- 57. Cao X., Sha S-C., Li M., Kim B-S., Morgan C., Huang R., Yang X., Walsh P. J., Chem. Sci. 2016, 7, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sha S-C., Jiang H., Mao J., Bellomo A., Jeong S. A., Walsh P. J., Angew. Chem. Int. Ed. 2016, 55, 1070–1074; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1082–1086. [Google Scholar]
- 59. Zhang Y., Dong J., Liu L., Liu L., Zhou Y., Yin S-F., Org. Biomol. Chem. 2017, 15, 2897–2901. [DOI] [PubMed] [Google Scholar]
- 60. Li J., Wu C., Zhou B., Walsh P. J., J. Org. Chem. 2018, 83, 2993–2999. [DOI] [PubMed] [Google Scholar]
- 61. Sun C-L., Shi Z-J., Chem. Rev. 2014, 114, 9219–9280. [DOI] [PubMed] [Google Scholar]
- 62. Schneider J., Matsuoka M., Takeuchi M., Zhang J., Horiuchi Y., Anpo M., Bahnemann D. W., Chem. Rev. 2014, 114, 9919–9986. [DOI] [PubMed] [Google Scholar]
- 63. Hoshikawa T., Inoue M., Chem. Sci. 2013, 4, 3118–3123. [Google Scholar]
- 64. Xia J-B., Zhu C., Chen C., J. Am. Chem. Soc. 2013, 135, 17494–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pandey G., Laha R., Angew. Chem. Int. Ed. 2015, 54, 14875–14879; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15088–15092. [Google Scholar]
- 66. Pandey G., Laha R., Singh D., J. Org. Chem. 2016, 81, 7161–7171. [DOI] [PubMed] [Google Scholar]
- 67. Liu X., Lin L., Ye X., Tan C-H., Jiang Z., Asian J. Org. Chem. 2017, 6, 422–425. [Google Scholar]
- 68.
- 68a. Lücking U., Angew. Chem. Int. Ed. 2013, 52, 9399–9408; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9570–9580; [Google Scholar]
- 68b. Frings M., Bolm C., Blum A., Gnamm C., Eur. J. Med. Chem. 2017, 126, 225–245. [DOI] [PubMed] [Google Scholar]
- 69. Wang H., Zhang D., Bolm C., Angew. Chem. Int. Ed. 2018, 57, 5863–5866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5965–5968. [Google Scholar]
- 70. Betori R. C., May C. M., Scheidt K. A., Angew. Chem. Int. Ed. 2019, 58, 16490–16494. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71. Fan R., Li W., Pu D., Zhang L., Org. Lett. 2009, 11, 1425–1428. [DOI] [PubMed] [Google Scholar]
- 72. Takeda Y., Hayakawa J., Yano K., Minakata S., Chem. Lett. 2012, 41, 1672–1674. [Google Scholar]
- 73. Moriyama K., Takemura M., Togo H., Org. Lett. 2012, 14, 2414–2417. [DOI] [PubMed] [Google Scholar]
- 74. Yi H., Liu Q., Liu J., Zeng Z., Yang Y., Lei A., ChemSusChem 2012, 5, 2143–2146. [DOI] [PubMed] [Google Scholar]
- 75. Feng J., Liang S., Chen S-Y., Zhang J., Fu S-S., Yu X-Q., Adv. Synth. Catal. 2012, 354, 1287–1292. [Google Scholar]
- 76. Wang H., Zhao Y-L., Li L., Li S-S., Liu Q., Adv. Synth. Catal. 2014, 356, 3157–3163. [Google Scholar]
- 77. Liu J., Zhang H., Yi H., Liu C., Lei A., Sci. China Chem. 2015,58,1323–1328. [Google Scholar]
- 78. Ma J., Hu Z., Li M., Zhao W., Hu X., Mo W., Hu B., Sun N., Shen Z., Tetrahedron 2015, 71, 6733–6739. [Google Scholar]
- 79.
- 79a. Wu X-F., Gong J-L., Qi X., Org. Biomol. Chem. 2014, 12, 5807–5817; [DOI] [PubMed] [Google Scholar]
- 79b. Huang H., Chen W., Xu Y., Li J., Green Chem. 2015, 17, 4715–4719. [Google Scholar]
- 80. Abebe H., Vidavalur S., Battula V. R., RSC Adv. 2016, 6, 82289–82293. [Google Scholar]
- 81. Tan J., Zheng T., Yu Y., Xu K., RSC Adv. 2017, 7, 15176–15180. [Google Scholar]
- 82. Liu W., Liu C., Zhang Y., Sun Y., Abdukadera A., Wang B., Li H., Ma X., Zhang Z., Org. Biomol. Chem. 2015, 13, 7154–7158. [DOI] [PubMed] [Google Scholar]
- 83. Amaoka Y., Kamijo S., Hoshikawa T., Inoue M., J. Org. Chem. 2012, 77, 9959–9969. [DOI] [PubMed] [Google Scholar]
- 84. He C., Zhang X., Huang R., Pan J., Li J., Ling X., Xiong Y., Zhu X., Tetrahedron Lett. 2014, 55, 4458–4462. [Google Scholar]
- 85. Wang H., Wang Z., Huang H., Tan J., Xu K., Org. Lett. 2016, 18, 5680–5683. [DOI] [PubMed] [Google Scholar]
- 86. Li J-S., Yang F., Yang Q., Li Z-W., Chen G-Q., Da Y-D., Huang P-M., Chao C., Zhang Y., Huang L-Z., Synlett 2017, 28, 994–998. [Google Scholar]
- 87. Liu Y-F., Zhai D-D., Zhang X-Y., Guan B-T., Angew. Chem. Int. Ed. 2018, 57, 8245–8249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8377–8381. [Google Scholar]