

Cardio-oncology has blossomed as a new clinical and research subspecialty in cardiovascular medicine due to the explosion of novel cancer therapies, which have dramatically changed the natural course of cancer but are also associated with cardiovascular and metabolic sequelae.1 Nevertheless, the intersection of cardiology and oncology is increasingly recognized to extend beyond toxic effects of cancer or cancer therapies on the cardiovascular system. Common risk factors, including genetic ones, can predispose to both cancer and heart disease.2, 3 Furthermore, recent data also suggest that heart failure and cardiac ischemic insults may potentiate cancer progression (FIGURE).4 In this issue of Circulation, Avraham and colleagues demonstrate that early hypertrophic cardiac remodeling, in the absence of heart failure, enhances tumor growth and metastasis, further highlighting the cross-talk between cardiovascular disease and cancer.5
The world of cardio-oncology.
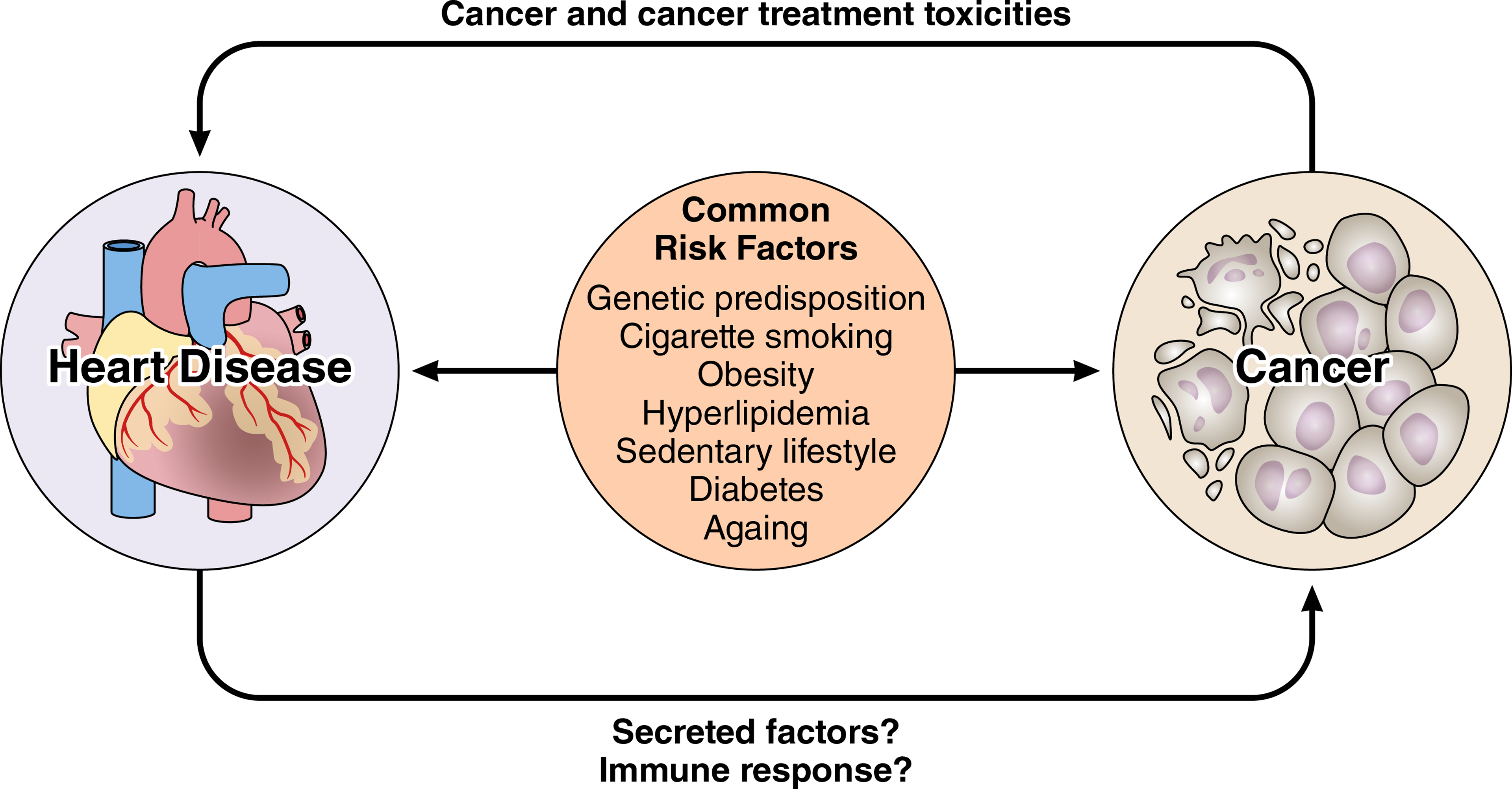
The intersection between cancer and cardiovascular disease extends beyond cancer and cancer treatment effects on the cardiovascular and cardiometabolic systems. There is a growing appreciation of common risk factors that predispose patients to both cancer and cardiovascular disease. Emerging data also suggest the concept of “reverse cardio-oncology” whereby heart disease potentiates cancer.
Cancer and heart disease represent major public health burdens and the two most common causes of death in the world. While previously viewed as distinct disease entities, cardiovascular disease and cancer often occur in the same individual. Indeed, cardiovascular risk factors such as tobacco, diabetes and hyperlipidemia have been associated with an increase in cancer risk.1, 3 Genetic entities such as clonal hematopoiesis of indeterminate potential (CHIP) - somatic mutations in hematopoietic stem cells previously recognized as a major risk factor for hematological malignancies - have now emerged as potent risk factors for cardiovascular events.2 Fundamental biological mechanisms such as chronic inflammation that contribute to both conditions have been proposed to drive these associations. However, demonstrating potential links between existing cardiovascular disease and subsequent malignancy can be challenging clinically. For example, lead-time bias, whereby diagnosis time of cancer is advanced by screening may occur among heart failure patients as a result of more frequent visits to a health care provider.6
Laboratory platforms provide a more objective and biologically dissectible means of addressing the hypothesis that cardiac injury potentiates tumor. In 2018, in groundbreaking experiments, De Boer and colleagues utilized an elegant genetic mouse model prone to precancerous intestinal tumors.4 Subjecting these mice to myocardial infarction resulted in an increased number and size of intestinal polyps. Heterotopic heart transplantation, whereby an infarcted or sham-operated heart was transplanted into a recipient mouse also led to increased tumor number, volume, and proliferation ruling out hemodynamic impairment as a simple explanation. A number of candidate secreted proteins were identified that were elevated in mice subjected to myocardial infarction, as well as patients with heart failure, which promoted tumorigenesis in vitro. The new study by Avraham and colleagues further supports cardiac remodeling as an acute pathological stressor that accelerates cancer via the secretion of pro-tumorigenic factors.5
Avraham and colleagues used transverse aortic constriction (TAC) in mice, which simulates pressure overload-induced cardiac hypertrophy, to investigate whether pathological cardiac remodeling can alter cancer growth and progression. TAC was found to enhance tumor growth in two mouse syngeneic tumor models, a breast orthotopic cancer model (Mouse mammary tumor virus-polyomavirus middle T-antigen; MMTV-PyMT) and a lung cancer model (Lewis Lung Carcinoma; LLC). TAC resulted in increased volume of tumor implants, with higher numbers of proliferating tumor cells, as well as enhanced metastatic colonization compared to tumors grown in mice following either a sham surgery or no operation. Temporal delay in tumor implantation following TAC, corresponding to greater cardiac hypertrophy, resulted in larger growth of tumors suggesting a correlation between hypertrophied remodeling and tumor growth. To determine if the host immune system played a role in TAC-associated increased tumor growth, experiments were repeated in immunodeficient NOD/SCID mice, which lack T and B lymphocytes, and have reduced natural killer cell function; however, the differential effects of tumor growth following TAC persisted excluding a significant role for the adaptive immune system. Notably, sera derived from TAC-operated mice enhanced cancer cell proliferation in vitro, suggesting the role of a secreted molecule in mediating tumor growth.
Transcriptomic profiling of the hearts from TAC-operated mice identified a number of candidate factors upregulated in cardiac tissue that were previously recognized to be pro-tumorigenic, including connective tissue growth factor (CTgF) and Periostin.7 The authors showed that CTgF and Periostin were also increased in the sera of TAC-operated mice, compared to mice that underwent sham surgery, independent of cancer. Periostin is a non-structural matricellular protein that regulates key aspects of tumor biology, including proliferation, invasion, and dissemination to pre-metastatic niches in distant organs.8 Exogenous addition of Periostin was sufficient to increase PyMT and LLC cell proliferation in vitro, and depletion of Periostin from the sera of TAC-mice abrogated its proliferative effects on PyMT cells. While these in vitro studies showed that Periostin is a necessary component in sera from TAC-operated mice to drive cancer cell replication, it is unclear whether the concentration used represents the physiological condition in vivo following TAC procedure. Furthermore, in vitro tumor proliferation assays cannot recapitulate the complex tumor microenvironment, which includes both immune and stromal-vascular cells, and no studies of Periostin inhibition in vivo were performed to validate its causal role. Future studies will be needed to investigated whether Periostin, either by itself or by collaborating with other secreted factors, promotes cardiac remodeling-associated tumorigenesis in vivo.
While TAC is frequently used in the laboratory to study the adverse cardiac remodeling upon pressure overload, there is considerable variability in severity and time course to heart failure, depending on mouse strain, sex, follow-up time and severity of the surgical constriction. Initially developed by Rockman and colleagues as an in vivo murine model of myocardial cell hypertrophy9, the model progresses to ventricular dilatation and heart failure. Detailed characterization of the myocardium following TAC has revealed induction of chemokines and cytokines and limited inflammation, with subsequent fibrosis.10 In the current study, the cardiac hypertrophy clearly developed but remodeling did not include subsequent ventricular dilatation. In addition, both the innate and adaptive immune system contribute to pathology in heart failure models, which may promote cross-disease communication.11, 12 Although NOD/SCID mice were used in the current study to exclude the potential role of the immune system in cardiac remodeling-mediated tumor growth, these mice still maintain low NK cell activity and myeloid cell function, which may have contributed to the observed cancer phenotype.13 As myeloid cells have important roles in tumorigenesis in both the MMTV-PyMT and LLC orthotopic models used, further studies will be needed to investigate such a possibility. Indeed, work by one of our groups (KJM) shows that an incident myocardial infarction accelerates breast cancer outgrowth in mice, by epigenetic modification of monocytes in the bone marrow.14 Furthermore, additional testing of the effects of TAC in genetically engineered mouse models of cancer are needed to assess whether early cardiac remodeling affects transformation (de novo cancer formation) or merely the growth and metastasis of existing tumors.
To address the human disease relevance of the meticulous mouse experiments, the authors retrospectively assessed echocardiographic data of more than 80,000 patients, nearly 5000 of whom had a clinical diagnosis of aortic stenosis, and tried to find correlations with subsequent diagnosis of non-hematological malignancies between those with or without aortic stenosis. While the crude hazard ratio suggested an increased incidence rate of malignancy in patients with moderate or severe aortic stenosis compared to those with no aortic stenosis, once corrected for other confounders, such as smoking or diabetes, this correlation only held true in the younger population (40–60 years old). These data are suggestive, but they are preliminary, with the added caveat that TAC is at best a very exaggerated form of aortic stenosis. Prospective clinical studies using cardiac imaging will be needed to systematically address whether cardiac remodeling potentiates the risk of cancer in humans. Still, the authors’ results add to the growing dimension of “reverse cardio-oncology”.15 In 2020, the field of cardio-oncology continues to evolve with growing appreciation of interaction between cardiovascular disease and cancer.
Footnotes
Conflict of Interest/Disclosures
JM has served on advisory boards for Pfizer, Novartis, Bristol-Myers Squibb, Deciphera, Audentes Pharmaceuticals, Nektar, Ipsen, Takeda, Myokardia, AstraZeneca, TripleGene/Precisgen, Regeneron GlaxoSmithKline, Boston Biomedical, ImmunoCore, Janssen, and Myovant and is supported by R01 HL141466. QZ is a Cancer Prevention and Research Institute of Texas (CPRIT) Scholar and is supported by CPRIT (RR190058) and NCI R01CA211732. KJM is supported by the National Institutes of Health (R35HL135799 and P01HL131478).
References
- 1.Moslehi JJ. Cardiovascular Toxic Effects of Targeted Cancer Therapies. N Engl J Med. 2016;375:1457–1467. [DOI] [PubMed] [Google Scholar]
- 2.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meijers WC and de Boer RA. Common risk factors for heart failure and cancer. Cardiovasc Res. 2019;115:844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, Haubner BJ, Nagengast WB, Lyon AR, van der Vegt B, et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation. 2018;138:678–691. [DOI] [PubMed] [Google Scholar]
- 5.Avraham S, Abu-Sharki S, Shofti R, Haas T, Korin B, Kalfon R, Friedman T, Shiran A, Saliba W, Shaked Y and Aronheim A. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation. 2020[In Press]. [DOI] [PubMed] [Google Scholar]
- 6.Duffy SW, Nagtegaal ID, Wallis M, Cafferty FH, Houssami N, Warwick J, Allgood PC, Kearins O, Tappenden N, O’Sullivan E, et al. Correcting for lead time and length bias in estimating the effect of screen detection on cancer survival. Am J Epidemiol. 2008;168:98–104. [DOI] [PubMed] [Google Scholar]
- 7.Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D, Liu Y and Ouyang G. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep. 2020;30:793–806 e6. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Gonzalez L and Alonso J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front Oncol. 2018;8:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr. and Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia Y, Lee K, Li N, Corbett D, Mendoza L and Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumgarten G, Knuefermann P, Kalra D, Gao F, Taffet GE, Michael L, Blackshear PJ, Carballo E, Sivasubramanian N and Mann DL. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation. 2002;105:2192–2197. [DOI] [PubMed] [Google Scholar]
- 12.Suetomi T, Miyamoto S and Brown JH. Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am J Physiol Heart Circ Physiol. 2019;317:H877–H890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, Shultz LD, Kiyono H and Ishikawa F. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. J Immunol. 2012;188:6145–6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koelwyn G, Newman A, Afonso M, C S, Corr E, Brown E, Albers K, Yamaguchi N, Narke D, Schlegel P, Sharma M, et al. Myocardial Infarction Accelerates Breast Cancer via Innate Immune Reprogramming. Nat Med. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aboumsallem JP, Moslehi J and de Boer RA. Reverse Cardio-Oncology: Cancer Development in Patients With Cardiovascular Disease. J Am Heart Assoc. 2020;9:e013754. [DOI] [PMC free article] [PubMed] [Google Scholar]