

Abstract
Objective
Giant cell arteritis (GCA) is a large‐vessel vasculitis that primarily affects the aorta and its branches. Extracranial branches of the carotid artery are frequently affected; however, intracranial involvement in GCA is rare.
Methods
A retrospective medical record review was performed to identify all patients with intracranial GCA (IC‐GCA) from January 1996 through May 2018.
Results
Nine patients with IC‐GCA were included (78% male; mean age, 72.1 years [SD: 7.9]). All patients met ACR criteria for GCA. The median time from onset of GCA to intracranial involvement was 0.6 months (interquartile range: 0.1‐5.1). All patients had neurologic symptoms, 89% had an ischemic cerebrovascular event. Transient or permanent vision loss was frequent (56% of patients). IC‐GCA was diagnosed by cranial imaging in all nine patients. Intracranial vasculitis most commonly affected the internal carotid artery (100%), followed by the vertebral artery (67%), posterior cerebral artery (67%), middle cerebral artery (44%), anterior cerebral artery (33%), and posterior inferior cerebral artery (11%). Intracranial vessel stenosis was present in 89%, occlusion in 33%, wall thickening in 33%, and dilation in 11%.
All patients received glucocorticoids. Additional therapeutic agents included cyclophosphamide (67%) and tocilizumab (22%). Despite treatment, five patients had rapid deterioration and mortality. Comparing IC‐GCA patient survival to the expected rates from the US population, the standardized mortality ratio (95% CI) for IC‐GCA was 58.1 (18.9‐135.6).
Conclusion
Although rare, IC‐GCA is associated with significant morbidity and mortality. It occurs predominantly in men and presents with ischemic cerebrovascular events. Current treatment strategies appear to be of limited efficacy for IC‐GCA.
INTRODUCTION
Giant cell arteritis (GCA) is a large‐vessel vasculitis that predominantly affects women of Northern European descent over the age of 50 years (1). Extracranial branches of the carotid artery are primarily affected; however, systemic large‐vessel involvement is also frequent (2).
GCA can lead to significant morbidity; in addition, aortic aneurysm, visual deficit, and cerebrovascular events are the most feared complications. Ischemic cerebrovascular accidents (CVAs) have been reported in 3.5% to 7.2% of patients with GCA (3, 4); potential risk factors for CVAs in this population include advanced age, accelerated atherosclerosis, hypercoagulability, as well as increased cardiovascular risk factors related to glucocorticoid use (5). In the setting of intracranial involvement, the risk of CVA and other neurologic deficits increases significantly (5). Intracranial involvement in GCA (IC‐GCA) is rare with only about 50 cases reported in the literature.
With the goal to expand the understanding of this subgroup of patients, we report a case series of nine patients with IC‐GCA treated at Mayo Clinic, Rochester, Minnesota, and describe their characteristics, treatment, and clinical outcomes.
METHODS
Study design
A retrospective medical record review was performed at the Mayo Clinic, Rochester, Minnesota, to identify all patients with IC‐GCA from January 1,1996, through May 31, 2018. The protocol was approved by the Mayo Clinic Institutional Review Board (IRB# 17‐010061). Study data were abstracted and collected using an electronic database (RedCap) hosted at Mayo Clinic.
Patients
A computer‐assisted medical record search was performed by cross‐referencing the respective International Classification of Disease (ICD)‐9 and ICD‐10 codes for GCA (codes 446.5 and M31.6, respectively) with cerebral arteritis (codes 437.4 and I67.7, respectively). A retrospective review of the medical records was performed to confirm the diagnosis and identify demographic, clinical, laboratory, radiographic, treatment, and outcomes of the patients with IC‐GCA, both at baseline and on subsequent follow‐up visits. The cohort of patients with confirmed IC‐GCA was compared to a previously described cohort of patients with biopsy‐proven GCA (6).
Statistical methods
Data were summarized using descriptive statistics. Categorical variables were described using percentages and numbers; quantitative variables were described using means and standard deviations or medians and interquartile ranges. Comparisons between the IC‐GCA and biopsy‐proven GCA cohorts were performed using Chi‐square and rank sum tests. Survival rates after the diagnosis of IC‐GCA were estimated using the Kaplan‐Meier (KM) method and were subsequently compared with the expected survival rates in the white US population obtained from age‐, sex‐, and calendar year–specific life tables. Median length of follow‐up was computed using the reverse KM estimator (7). This is a more robust method that utilizes the KM method with the event indicator reversed so that censoring becomes the outcome. Analyses were performed using SAS version 9.4 (SAS Institute Inc) and R version 3.4.2 (R Foundation for Statistical Computing).
RESULTS
Demographics
One hundred and eighty‐five patients underwent direct medical chart review of which nine patients were identified as having confirmed IC‐GCA (Table 1). Seven of nine (78%) patients were male. The mean [±SD] age was 72.1 years [±7.9], and all were white. The median time from onset of GCA to intracranial diagnosis was 0.6 months (interquartile range: 0.1‐5.1). The median length of time to follow‐up after IC‐GCA diagnosis was 12.9 months.
Table 1.
Description of patient’s characteristics and presentation
|
Age IC‐GCA Dx/Sex |
Large‐Vessel Imaging | Timing of IC‐GCA Dx | Clinical Course | Outcome |
|---|---|---|---|---|
| 74 M | Not available | IC‐GCA diagnosed 5 days after GCA |
Presentation features: Three mo h/o HA, ST, JC, amaurosis fugax; exam with Horner syndrome; TAB positive Initial imaging: MRI/MRA with concentric thickening and VWE of the precavernous and supraclinoid right ICA, bilateral ICA, and ECA but no stroke Initial treatment: GC 1 mg/kg with taper and TCZ 162 mg SQ/wk IC‐GCA presentation: Two months later on 20 mg/d prednisone and TCZ, patient developed dysarthria and imbalance. Repeat MRI/MRA with progressive narrowing and VWE of right ICA, proximal and distal cavernous sinus segments, and a new subacute infarcts of the right frontal white matter and right basal ganglia Disease course: Pulse dose MP 1000 mg × 3 days followed by GC 1 mg/kg/d with taper, IV CTX × 5 months with transition to AZA. Pred tapered off after 1 y and patient remained on 2 mg/kg/d AZA. |
Alive, mild dysarthria persisted |
| 65 F | Not available | IC‐GCA diagnosed 5 months after GCA |
GCA presentation features: Two mo h/o PMR symptoms, HA; TAB positive Initial treatment: GC 1 mg/kg with taper IC‐GCA presentation: Five months later on 15 mg/d prednisone, patient developed tinnitus Initial imaging: MRI/MRA with high‐grade stenosis and enhancement involving the carotid siphons and proximal intracranial segments of the VAs. No stroke was identified. Disease course: Pulse dose MP 500 mg followed by GC 1 mg/kg/d, PO CTX uptitrated to 100 mg/d; prednisone tapered down to a stable 7.5 mg/d dose |
Alive, tinnitus persisted |
| 79 M | No large‐vessel involvement | IC‐GCA diagnosed 10 years after GCA |
GCA presentation features: HA, ST, PMR symptoms; TAB positive Initial treatment: GC 1 mg/kg tapered to a stable 5 mg/d prednisone IC‐GCA presentation: AMS 10 years after GCA dx Initial imaging: MRI: bilateral paramedian thalamic and rostral midbrain infarcts. Cerebral angiogram: narrowing of multiple cerebral arteries including the PCA, MCA bilaterally, and common carotid arteries Disease course: Pulse dose MP 1000 mg × 5 days. The patient experienced progressive neurologic decline and was transitioned to comfort care. |
Deceased 10 days after IC‐GCA Dx |
| 79 M | Not available | IC‐GCA diagnosed 5 weeks after GCA diagnosis |
GCA presentation features: HA, amaurosis fugax, and dizziness; TAB positive Initial treatment: Pulse dose MP 1000 mg × 3 days followed by GC 1 mg/kg/d with a taper and MTX 12.5 mg PO/wk. IC‐GCA presentation: Five weeks later while on 40 mg/d prednisone and MTX 12.5 mg/wk, patient developed blurred vision, vertigo, and right hemiparesis. Initial imaging: MRI with subacute infarcts in the right cerebellar peduncle and left paramedian medulla. MRA: occluded right VA with high‐grade stenosis of left VA and basilar artery Disease course: Rapid neurologic decline, was made comfort care and passed 3 days after diagnosis |
Deceased 3 days after IC‐GCA Dx |
| 76 F | Bilateral subclavian stenosis; other large vessels were normal | IC‐GCA diagnosed 3 weeks after GCA diagnosis |
GCA presentation features: Right vision loss, upper extremity claudication; TAB nondiagnostic (inadequate sample) Chest CTA with bilateral subclavian stenosis Initial treatment: Pulse dose MP 1000 mg followed by GC 1 mg/kg/d with taper IC‐GCA presentation: Three weeks later while on 60 mg/d prednisone, patient developed right hemiparesis. Initial imaging: Angiogram with multiple irregularities in the anterior, posterior circulation, and branches of ECA and ICA bilaterally. Severe stenosis in the right carotid siphon at area of takeoff of the right ophthalmic artery. Severe left vertebrobasilar stenosis, ischemic infarct Disease course: Patient underwent stenting of left vertebrobasilar artery. Pulse dose MP 1000 mg IV × 3 days followed by GC 1 mg/kg/d and IV CTX × 5 months; was later transitioned to MTX 15 mg/wk. |
Alive at 9 months of follow‐up. Had full neurologic recovery |
| 66 M | No large‐vessel involvement | IC‐GCA diagnosed 6 months after GCA diagnosis |
GCA presentation features: HA, ST, JC, weight loss, right vision loss (central retina artery occlusion); TAB positive Initial treatment: Pulse dose MP 1000 mg followed by GC 1 mg/kg/d IC‐GCA presentation: Six months later while on 20 mg/d prednisone, patient experienced AMS, aphasia, and right upper extremity weakness. Initial imaging: MRI/MRA with multiple acute infarcts throughout the left cerebellar hemisphere, both posterior occipital lobes, both posterior parietal lobes, and a few small foci of infarct in both frontal lobes. Angiogram with high‐grade stenosis involving both cavernous ICAs Disease course: Pulse dose MP 1000 mg × 3 days followed by GC 1 mg/kg and IV CTX. Patient had progressive neurologic decline. |
Deceased 10 weeks following IC‐GCA diagnosis |
| 78 M | Thickening of descending thoracic, abdominal aorta, SMA, left renal artery, and bilateral common iliac arteries | IC‐GCA diagnosed 1 month after GCA diagnosis |
GCA presentation features: Lower extremity claudication; TAB positive Initial treatment: GC 1 mg/kg/d IC‐GCA presentation: One month later while on 40 mg/d prednisone, patient developed AMS. Initial imaging: MRI/MRA infarct in the corpus callosum and in the left corona radiate; occlusion of the pericallosal arteries, distal left ICA, and right A1 segment; irregularity of the major cerebral vessels and in most of the right MCA branches Disease course: Pulse dose MP 1000 mg × 3 days with plans to initiate IV CTX, but patient had progressive neurologic decline and was placed on comfort care. |
Deceased 2 weeks following IC‐GCA diagnosis |
| 79 M | No large‐vessel involvement | GCA and IC‐GCA diagnosed concomitantly |
GCA presentation features: Weight loss and PMR symptoms, amaurosis fugax with subsequent bilateral vision loss, cognitive dysfunction, hallucinations, vision deficit, and gait instability; TAB positive Initial imaging: MRI with infarctions in bilateral occipital and parietal lobes, corona radiata (right > left), and both cerebral hemispheres; MRA with high‐grade stenosis and VWE of the cavernous/paraclinoid ICA and reduced flow in distal MCA vessels bilaterally and bilateral PCA vessels Disease course: Pulse dose MP 1000 mg × 3 days followed by GC 1 mg/kg/d and IV CTX. Died from neurologic complications 3 months after |
Deceased 3 months following IC‐GCA diagnosis |
| 59 M | Bilateral femoropopliteal and tibial vessel thickening; other large vessels were normal | IC‐GCA diagnosed 1 month after GCA diagnosis |
GCA presentation features: Two mo h/o lower extremity claudication, HA, ST; TAB positive Initial treatment: GC 1 mg/kg/d IC‐GCA presentation features: One month later while on 40 mg/d pred, patient developed word‐finding difficulties, unsteadiness, AMS, and seizures. Initial imaging: MRI with infarcts in right gyrus rectus, nucleus accumbens, precentral gyrus, right precuneus, and periatrial white matter. Angiogram with symmetric narrowing of both internal supraclinoid carotid arteries, the left VA distal to its dura entrance, and complete occlusion of the V4 proximal portion of the right VA distal to its dura entrance Disease course: Pulse dose MP 1000 mg × 5 days followed by GC 1 mg/kg and IV CTX. Patient had neurologic recovery and was discharged with plans to continue monthly CTX. Upon local follow‐up, he was transitioned to TCZ 162 mg SQ/wk. Two months later, he was readmitted with new infarcts in the right anterior‐inferior frontal lobe/operculum/insula, left parieto‐occipital region, and left corona radiata. Pulse dose MP 1000 mg × 2 and IV CTX were given; however, he had progressive neurologic decline and was placed on comfort care. |
Deceased 4 months after IC‐GCA diagnosis |
Abbreviation: AMS, altered mental status; AZA, azathioprine; CTX, cyclophosphamide; Dx, diagnosis; ECA, external carotid artery; F, female; GC, glucocorticoid; GCA, giant cell arteritis; HA, headache; h/o, history of; IC, intracranial; ICA, internal carotid artery; IV, intravenous; JC, jaw claudication; M, male; MCA, middle cerebral artery; MP, methylprednisolone; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; MTX, methotrexate; PCA, posterior cerebral artery; PMR, polymyalgia rheumatic; PO, by mouth; SQ, subcutaneous; ST, scalp tenderness; TAB, temporal artery biopsy; TCZ, tocilizumab; VA, vertebral artery; VWE, vessel wall enhancement.
Eight (89%) patients had treated hypertension, seven (78%) were current or former smokers, and six (67%) had dyslipidemia. None of the patients had a personal history of cardiovascular or peripheral vascular disease.
Clinical features
The most frequent symptoms at the time of GCA presentation were new‐onset headache (67%), temporal artery tenderness (44%), fatigue (44%), and scalp tenderness (44%). Polymyalgia rheumatica symptoms, anorexia, and unexplained weight loss were present in 33% of patients. Two patients (22%) also experienced jaw claudication.
All patients had neurologic symptoms at the time of IC‐GCA presentation, including altered mental status, word‐finding difficulties, and gait instability. Eight patients (89%) had ischemic cerebrovascular events. Transient or permanent vision loss was frequent, occurring in five (56%) patients.
Unilateral permanent vision loss occurred at the time of GCA diagnosis in two patients and bilateral permanent vision loss occurred at the time of IC‐GCA diagnosis in one patient who had previously experienced amaurosis fugax at GCA diagnosis.
Diagnostic studies
C‐reactive protein was elevated in seven (100%) of seven tested patients, and six (75%) of eight patients tested had elevated erythrocyte sedimentation rate (ESR). In five (56%) patients, the ESR was greater than 50 mm/h. Anemia was also frequent, identified in four (57%) of seven tested patients. All patients underwent temporal artery biopsy (TAB); TAB was positive for GCA in eight (89%) patients. One patient with negative TAB had clear evidence of large‐vessel GCA on thoracic computed tomography angiography (CTA).
IC‐GCA was diagnosed by central nervous system (CNS) imaging in all patients. Intracranial angiographic imaging modalities used included magnetic resonance angiography (89%), CTA (22%), and conventional cerebral angiography (22%). The most frequently affected arteries were the internal carotid artery (ICA; 100%), followed by the vertebral artery (67%), posterior cerebral artery (67%), middle cerebral artery (44%), anterior cerebral artery (33%), and posterior inferior cerebral artery (11%) (Figure 1). All the patients had bilateral involvement. Intracranial vessel stenosis was present in 89%, occlusion in 33%, dilatation in 11%, and wall thickening in 11%.
Figure 1.
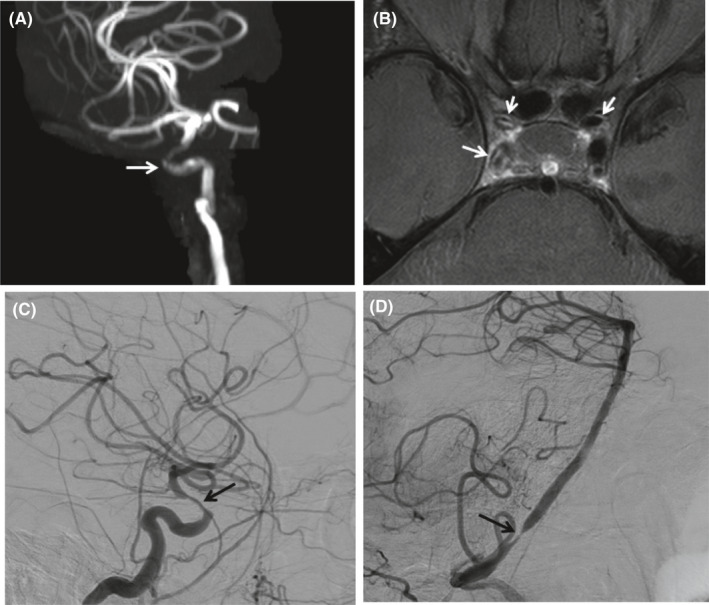
A, Three‐dimensional volume–rendered image of a three‐dimensional time‐of‐flight magnetic resonance angiogram demonstrates moderate stenosis of the right cavernous and supraclinoid internal carotid artery (ICA; white arrow) and, to a lesser extent, the left ICA (not shown). B, A proton density‐weighted axial high‐resolution vessel wall imaging examination with gadolinium demonstrates vessel wall thickening and high‐grade circumferential vessel wall enhancement consistent with inflammation of the distal horizontal petrous, cavernous, and supraclinoid right ICA as well as involved areas on the left (white arrows). C, Lateral conventional digital subtraction angiogram demonstrates moderate to severe stenosis of the right supraclinoid ICA (black arrow). D, Lateral conventional digital subtraction angiogram demonstrates focal severe stenosis of the right vertebral artery immediately above the entrance to the skull base (black arrow).
Treatment and outcomes
All patients received glucocorticoids. Three patients were treated with intravenous pulses of methylprednisolone preceding oral glucocorticoid therapy after GCA diagnosis. The mean starting dose of oral prednisone was 52.2 mg daily (range: 40‐60). Eight patients received pulse dose methylprednisolone following IC‐GCA diagnosis.
Following the diagnosis of IC‐GCA, additional therapeutic agents were administered as follows: cyclophosphamide (67% of patients) and tocilizumab (22% of patients). The two patients treated with tocilizumab also received cyclophosphamide. One patient was initiated on cyclophosphamide and was then transitioned to tocilizumab with a subsequent rapid decline and death. A second patient had disease progression while receiving tocilizumab and was then treated successfully with cyclophosphamide.
Despite treatment, outcomes for patients with IC‐GCA were poor. Only one patient had full neurologic recovery. The remaining eight patients had persistent neurologic symptoms. Five experienced a rapidly progressive course with subsequent demise. The one‐month survival rate was 44% (95% confidence interval [CI]: 21%‐92%). The primary cause of death was stroke.
Comparing IC‐GCA patient survival to the expected rates from the US population (Figure 2), the standardized mortality rate for IC‐GCA was 58.1 (95% CI: 18.9‐135.6).
Figure 2.
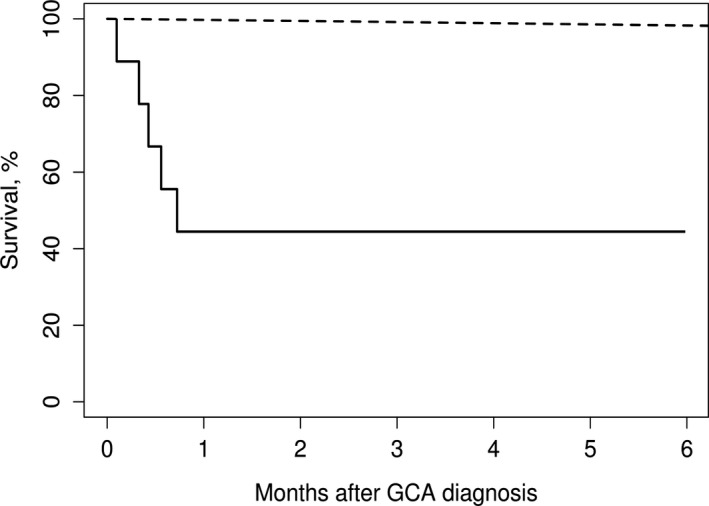
Survival of nine patients with intracranial giant cell arteritis (GCA) compared with expected rates from US population lifetables (observed: solid line; expected: dashed line).
Comparison with biopsy‐positive GCA cohort
When comparing patients with IC‐GCA with a cohort of patients with biopsy‐proven GCA (and no known intracranial vasculitis) it was noted that patients with IC‐GCA were more frequently male (78% versus 26%; P = 0.001), had more frequent cranial features, such as temporal artery tenderness (56% versus 16%; P = 0.002), permanent vision loss (33% versus 6%; P = 0.001), and neurologic symptoms (67% versus 7%; P < 0.001). Other presenting features and baseline inflammatory markers were similar in the two groups.
With regard to comorbidities, patients with IC‐GCA were more frequently current/former smokers (78% versus 41%; P = 0.027), diabetic (33% versus 9%; P = 0.014), and hypertensive (89% versus 47%; P = 0.014).
DISCUSSION
The major findings of the current case series study were a high incidence of neurologic deficits and poor prognosis despite treatment. On imaging, all patients had involvement of the intracranial ICA, whereas other intracranial arteries were variably affected and there was evidence of vessel wall inflammation when vessel wall imaging was performed. These findings are important because further characterization of this uncommon condition is useful to improve understanding of the clinical and imaging features as well as the prognosis.
The current study demonstrated neurologic symptoms in all patients and ischemic stroke in most. This is consistent with the prior case series and literature review by Alsolaimani et al. This group reported five patients with IC‐GCA and reviewed 42 additional cases in the literature, finding CVA reported in 38 (81%) of the 47 patients (5). In contrast, the reported incidence of CVA in GCA in general is only 3.5% to 7.2% (8, 9). This difference could be partially accounted for by selection bias as patients in our study and the Canadian study had concerning neurologic symptoms and suspected intracranial involvement. When cardiovascular comorbidities were compared between groups, patients with IC‐GCA more frequently had a history of smoking, diabetes, and hypertension; however, imaging findings were consistent with vasculitis rather than atherosclerotic disease. Furthermore, the current study indicates that vision loss and cranial symptoms occur more frequently in patients with IC‐GCA compared with patients with GCA and no intracranial involvement.
It is possible that the frequency of IC‐CGA is underestimated because of selection bias of patients with GCA and neurologic symptoms who undergo CNS imaging. Although the exact prevalence of IC‐GCA is unknown, a study by Siemonsen et al prospectively evaluated patients with suspected GCA, but no neurologic symptoms, with head MRI examinations (10). Fifty percent (10 of 20 patients) of those with confirmed GCA were found to have intradural vascular involvement compared with all patients in a cohort with neurologic deficits in the current study. The internal carotid and vertebral arteries were the most frequently affected in both studies. Twenty percent (2 of 10) of patients with IC‐GCA had parenchymal changes consistent with ischemic events, which is higher than the reported stroke rate in patients with GCA (generally 3%‐4%) (3, 4). The findings of Siemonsen et al suggest that CNS involvement in patients with GCA may be more prevalent than is currently recognized and is frequently asymptomatic. A larger prospective intracranial imaging study of patients with GCA could help clarify the rate of symptomatic and asymptomatic intracranial involvement.
Lastly, the findings of the current study indicate that patients with neurologic symptoms and intracranial involvement have a poor prognosis, even when treated with glucocorticoids and other immunosuppressive agents. Notably, two patients in our cohort had intracranial disease progression while receiving tocilizumab. To our knowledge, use of tocilizumab in patients with IC‐GCA has not been previously reported (11). A specific clinical or imaging factor that predicted prognosis was not identified in our patients, although the study cohort was small. Specifically, mortality was seen in over half the study subjects with a rapidly deteriorating course and much higher than expected mortality rate compared with the US population.
The limitations of our study include the retrospective design that prevents standardized collection of data and protocolized imaging. Further studies that prospectively evaluate the prevalence of intracranial involvement in patients with GCA and more effective therapeutic options are necessary.
Although rare, IC‐GCA is associated with significant morbidity and mortality. It occurs predominantly in men, presenting with ischemic cerebrovascular events. Current treatment strategies appear to be of limited efficacy for IC‐GCA.
AUTHOR CONTRIBUTIONS
All authors were involved in the preparation of the manuscript and have approved the manuscript and this submission. Dr. Sanchez‐Alvarez drafted the article, Drs. Koster, Lehman, and Warrington revised the article critically for important intellectual content, and all authors gave final approval of the version of the article to be published.
Study conception and design
Crowson, Warrington.
Acquisition of data
Sanchez‐Alvarez, Hawkins.
Analysis and interpretation of data
Sanchez‐Alvarez, Koster, Crowson, Warrington.
Drs. Sanchez‐Alvarez and Hawkins contributed equally to this work.
No potential conflicts of interest relevant to this article were reported.
REFERENCES
- 1. Crowson CS, Matteson EL, Myasoedova E, Michet CJ, Ernste FC, Warrington KJ, et al. The lifetime risk of adult‐onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum 2011;63:633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koster MJ, Matteson EL, Warrington KJ. Large‐vessel giant cell arteritis: diagnosis, monitoring and management. Rheumatology (Oxford) 2018;57 Suppl 2:ii32‐42. [DOI] [PubMed] [Google Scholar]
- 3. González‐Gay MA, Blanco R, Rodríguez‐Valverde V, Martínez‐Taboada VM, Delgado‐Rodriguez M, Figueroa M, et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum 1998;41:1497–504. [DOI] [PubMed] [Google Scholar]
- 4. Larivière D, Sacre K, Klein I, Hyafil F, Choudat L, Chauveheid M‐P, et al. Extra‐ and intracranial cerebral vasculitis in giant cell arteritis: an observational study. Medicine (Baltimore) 2014;93:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alsolaimani RS, Bhavsar SV, Khalidi NA, Pagnoux C, Mandzia JL, Tay KY, et al. Severe intracranial involvement in giant cell arteritis: 5 cases and literature review. J Rheumatol 2016;43:648–56. [DOI] [PubMed] [Google Scholar]
- 6. Labarca C, Koster MJ, Crowson CS, Makol A, Ytterberg SR, Matteson EL, et al. Predictors of relapse and treatment outcomes in biopsy‐proven giant cell arteritis: a retrospective cohort study. Rheumatol (Oxford) 2016;55:347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schemper M, Smith TL. A note on quantifying follow‐up in studies of failure time. Control Clin Trials 1996;17:343–6. [DOI] [PubMed] [Google Scholar]
- 8. Ungprasert P, Wijarnpreecha K, Koster MJ, Thongprayoon C, Warrington KJ. Cerebrovascular accident in patients with giant cell arteritis: a systematic review and meta‐analysis of cohort studies. Semin Arthritis Rheum 2016;46:361–6. [DOI] [PubMed] [Google Scholar]
- 9. Lo Gullo A, Koster MJ, Crowson CS, Makol A, Ytterberg SR, Saitta A, et al. Venous thromboembolism and cerebrovascular events in patients with giant cell arteritis: a population‐based retrospective cohort study. PloS One 2016;11:e0149579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siemonsen S, Brekenfeld C, Holst B, Kaufmann‐Buehler AK, Fiehler J, Bley TA. 3T MRI reveals extra‐ and intracranial involvement in giant cell arteritis. AJNR Am J Neuroradiol 2015;36:91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant‐cell arteritis. N Engl J Med 2017;377:317–28. [DOI] [PubMed] [Google Scholar]