

Abstract
Hsp90 is a highly conserved molecular chaperone important for the activity of many client proteins. Hsp90 has an N-terminal ATPase domain (N), a middle domain (M) that interacts with clients and a C-terminal dimerization domain (C). “Closing” of dimers around clients is regulated by ATP binding, co-chaperones, and post-translational modifications. ATP hydrolysis coincides with release of mature client and resetting the reaction cycle. Humans have two Hsp90s: hHsp90α and hHsp90β. Although 85% identical, hHsp90β supports Hsp90 function in yeast much better than hHsp90α. Determining the basis of this difference would provide important insight into functional specificity of seemingly redundant Hsp90s, and the evolution of eukaryotic Hsp90 systems and clientele. Here, we found host co-chaperones Sba1, Cpr6 and Cpr7 inhibited hHsp90α function in yeast, and we identified mutations clustering in the N domain that considerably improved hHsp90α function in yeast. The strongest of these rescuer mutations accelerated nucleotide-dependent lid closing, N–M domain docking, and ATPase. It also disrupted binding to Sba1, which prolongs the closed state, and promoted N–M undocking and lid opening. Our data suggest the rescuer mutations improve function of hHsp90α in yeast by accelerating return to the open state. Our findings imply hHsp90α occupies the closed state too long to function effectively in yeast, and define an evolutionarily conserved region of the N domain involved in resetting the Hsp90 reaction cycle.
Keywords: Hsp90, Hsp82, Sba1, Hsp90 alpha, Hsp90 beta
Introduction
Heat shock protein 90 (Hsp90) is a highly conserved molecular chaperone that is essential in eukaryotic cells [1]. Hsp90 is required for the function and stability of many client proteins, including kinases, transcription factors, metabolic enzymes, and transporters. It acts through a highly coordinated ATP-dependent cycle of client binding and release [2]. This reaction cycle is in turn regulated by a cohort of conserved co-chaperones that bind at specific sites and to specific nucleotide and conformational states of Hsp90 [3,4]. Hsp90 functions as a dimer that undergoes radical conformational rearrangements that are influenced by its interactions with nucleotides, co-chaperones, and clients. It has an amino-terminal (N) ATPase domain connected by a charged linker to a middle (M) domain where clients bind, and a carboxy-terminal (C) dimerization domain [1,5-8]. Upon ATP binding, Hsp90 dimers shift from an “open” state, where the N domains of the two protomers are separated, to the “closed” state, where the N domains are in direct contact [5,9,10]. In cells, there is an equilibrium of open and closed states. Three defined movements occur concurrently when ATP binds: a lid structure re-orients to cover the ATP binding pocket; the N and M domains physically dock, which positions the catalytic arginine in the M domain into the ATP binding pocket for hydrolysis; and the N domains come together and swap their N-terminal β-strands [5,11]. Only in this closed conformation does ATP hydrolysis occur [12,13]. The exact timing of ATP hydrolysis in each protomer and how it relates to client maturation and resetting of the cycle, i.e., re-opening of the Hsp90 dimer, is unclear and the subject of intense study.
Both Saccharomyces cerevisiae and humans have two isoforms of Hsp90, called Hsc82 and Hsp82 in yeast and hHsp90α and hHsp90β in humans [14,15]. Hsc82 and hHsp90β are constitutively expressed and abundant under optimal conditions. Levels of Hsp82 and hHsp90α are normally low but induced to high levels under stress. There are minor structural distinctions between Hsc82 and Hsp82, but their interactomes in vivo are essentially identical, with slight differences under stress conditions [14]. The sequence identity between hHsp90α and hHsp90β (85%) is lower than between yeast Hsp90s (97%). Perhaps accordingly, the functional differences of human Hsp90s are more widely documented. For example, hHsp90β is more important for certain immune responses than Hsp90α [16], and in colorectal cancer cells, hHsp90α seems to be more important for metastasis, while hHsp90β inhibits cancer cell differentiation [17]. Furthermore, hHsp90α and hHsp90β have distinct roles in wound healing [18,19]. The high level of evolutionary conservation of the eukaryotic Hsp90 system, coupled with the ease of genetic manipulations in yeast, makes S. cerevisiae an ideal model to study Hsp90 function.
Two laboratories independently showed that hHsp90β supports viability of yeast much better than hHsp90α [20,21], while a third group did not observe this difference [22,23]. Scroggins et al. showed that acetylation of hHsp90α lysine 294 is important for overall Hsp90 function [20]. Acetylation mimic substitutions at this site improve the ability of hHsp90α to support yeast growth, suggesting hHsp90α is not being adequately acetylated in yeast cells for proper function. It remains unclear whether the acetylation of lysine 294 or a conformational change resulting from the modification is crucial for its function. Synoradzki et al. used hHsp90α/β hybrid proteins to study functional differences of hHsp90β and hHsp90α and found a specific region of the M domain of hHsp90α (that does not include lysine 294) conferred the slow growth phenotype [21]. This region of hHsp90α also mediates increased interaction with the co-chaperone Aha1 compared to hHsp90β. However, deleting Aha1 exacerbates the slow growth of cells expressing hHsp90α showing the increased interaction between Aha1 and hHsp90α is not responsible for the slow growth phenotype [21]. It is possible that the region of the hHsp90α M domain they identified impacts function in yeast by altering the conformation equilibrium in a way that promotes Aha1 interaction yet is incompatible with yeast clients.
In order to more fully understand the mechanism underlying the different abilities of human Hsp90s to support growth of yeast, we searched for mutations that rescued hHsp90α's inability to support yeast viability. Because of the reported differences in hHsp90α function in yeast, we also monitored its function in different strains, and we identified host co-chaperones that modulate hHsp90α function in yeast. We identified novel mutations of conserved residues in a discreet region of the N domain that expanded the ability of hHsp90α to function in different yeast backgrounds. These mutations affected yeast and human Hsp90 isoforms similarly, pointing to a conserved functional element. Genetic and biochemical characterization of these mutants revealed they act by pushing Hsp90 dimers to the open state. Our findings highlight this region within the N domain of Hsp90 as a key regulatory locus that promotes the resetting of the Hsp90 reaction cycle.
Results
The ability of human Hsp90α to complement loss of yeast Hsp90 is influenced by strain background
We discovered that hHsp90α, unlike hHsp90β, was unable to complement essential yeast Hsp90 functions in our strains (Figure 1(a)) [24]. This inability is in contrast to previous studies from two groups that reported weak complementation by hHsp90α compared to hHsp90β [20,21]. These studies used the W303 or SEY6210 genetic backgrounds. Our strains, derived from 779-6A [25], are more closely related to S288C than to W303, which differ by one or more residues in 799 protein sequences [26]. Two reports from a third group did not observe differences between the ability of hHsp90α and hHsp90β to support growth in yeast [22,23]. They used a hybrid strain that also had deletions of nine ABC transporters. Differences in the strain background were clearly influencing the ability of hHsp90α to support growth in place of yeast Hsp90.
Figure 1.
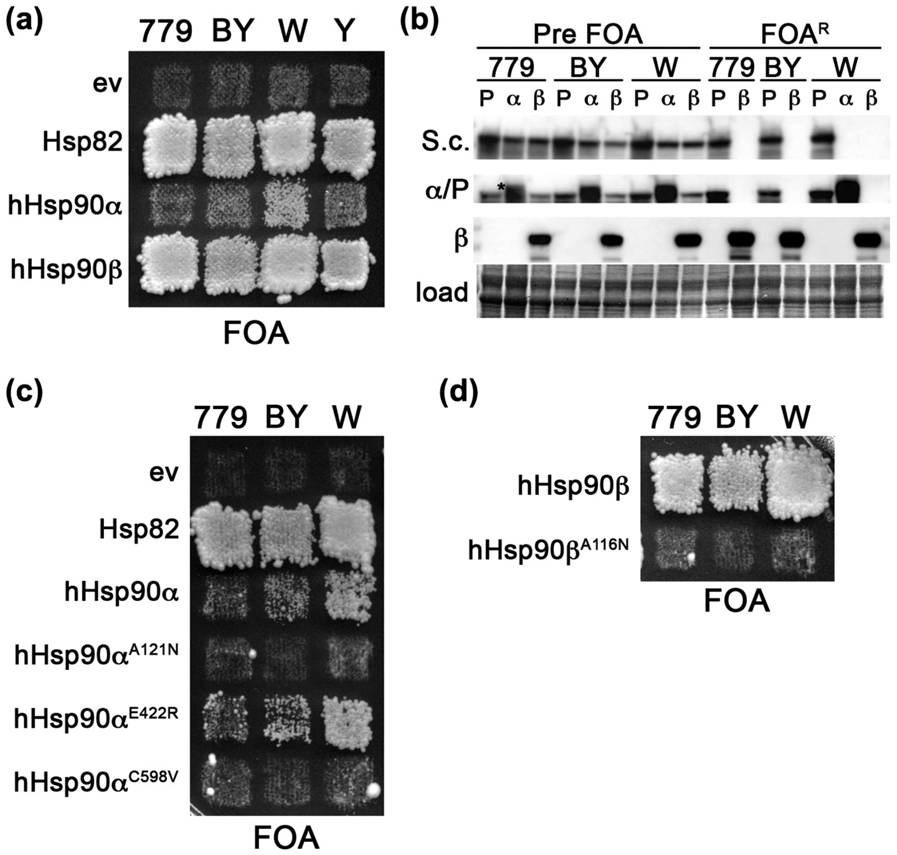
Complementation by hHsp90α is influenced by the yeast strain background. (a) FOA plate selecting for complementation by human Hsp90s in hsc82Δ/hsp82Δ strains 779-6A (MR318, 779), BY4741 (MR1075, BY), W303 (MR1088, W), and YPH499 (OR102, Y). Each expresses no Hsp90 (ev) or Hsp82, hHsp90α or hHsp90β as indicated. Extent of growth of cells on FOA reflects ability of the Hsp90 to complement yeast Hsp90 function. (b) Western blots showing relative expression levels of Hsp82 (P), hHsp90α (α) or hHsp90β (β) in primary transformants (Pre FOA) and in cells recovered from FOA (FOAR) in 779-6A (779), BY4741 (BY), and W303 (W). Hsp90-specific antibodies used are: S.c., recognizes yeast but not human Hsp90; α/P, recognizes hHsp90α (slightly higher MW band denoted with an asterisk) and Hsp82; and β, recognizes only hHsp90β. A portion of the membrane stained with amido black is shown as a loading control (load). (c) FOA plate as in (a) with strains expressing indicated mutants of hHsp90α with Hsp82 and empty vector (ev) controls. (d) FOA plate as in (a) with strains expressing wild-type hHsp90β or hHsp90βA116N. This image was cropped from the same plate in (c).
In an effort to reconcile these differences, we assessed the ability of hHsp90α to complement in different strain backgrounds. We deleted the HSC82 and HSP82 genes in strains W303 [26], BY4741 [27], and YPH499 [28], in a manner similar to our 779-6A-derived strain [29] (see Table 1 and Materials and Methods). BY4741 is a direct descendant of S288C and was used to construct the systematic gene deletion, GFP- and TAP-tagged strain collections [27]. YPH499 shares several marker alleles with 779-6A and is considered to be “congenic” to S288C [28]. All of the hsc82Δ/hsp82Δ strains harbored a parental URA3-marked centromeric plasmid (pMR62, see Table 2) that expresses the HSP82 open reading frame under the control of the HSC82 promoter, in order to support viability and serve as a platform to test for complementation of yeast Hsp90 function (see Materials and Methods).
Table 1.
Yeast strains used in this study
| Strain | Background; relevant genotypea | Reference |
|---|---|---|
| MR318 | 779-6Ab; hsc82::KanMX, hsp82::KanMX | [29] |
| 1670 | 779-6A; hsc82::KanMX, hsp82::KanMX, sti1::HIS3 | [24] |
| MR908 | 779-6A; hsc82::KanMX, hsp82::KanMX, sba1::ZEOR | [24] |
| MR1048 | 779-6A; hsc82::KanMX, hsp82::KanMX, sti1::HIS3, sba1::ZEOR | [24] |
| MR1075 | BY4741c; hsc82::HphMX, hsp82::NatMX | This study |
| MR1077 | BY4741; hsc82::HphMX, hsp82::NatMX, sti1::KanMX | This study |
| MR1079 | BY4741; hsc82::HphMX, hsp82::NatMX, aha1::KanMX | This study |
| MR1081 | BY4741; hsc82::HphMX, hsp82::NatMX, sba1::KanMX | This study |
| MR1083 | BY4741; hsc82::HphMX, hsp82::NatMX, ppt1::KanMX | This study |
| MR1097 | BY4741; hsc82::HphMX, hsp82::NatMX, cp6::KanMX | This study |
| MR1099 | BY4741; hsc82::HphMX, hsp82::NatMX, cpr7::KanMX | This study |
| MR1101 | BY4741; hsc82::HphMX, hsp82::NatMX, hch1::KanMX | This study |
| MR1088 | W303d; hsc82::HphMX, hsp82::NatMX | This study |
| OR102 | YPH499e; hsc82::HphMX, hsp82::NatMX | This study |
Table 2.
Plasmids used in this study
| Plasmid | Backbone | Description | Reference |
|---|---|---|---|
| pMR62 | pRS316 | CEN, URA3, PHSC82::HSP82::TCYC1 | [29] |
| p414-GPD | pRS314 | CEN, TRP1, PGPD::MCS::TCYC1; empty vector | [54] |
| pMR325 | p414-GPD | HSP82 | This study |
| pMR423 | p414-GPD | HSP90AA1 (hHsp90α) | This study |
| pMR365 | p414-GPD | HSP90AB1 (hHsp90β) | 24] |
| pMR349 | pRS315 | CEN, LEU2, PHSC82::HiS6-HSP82::TCYC1 | [24] |
| pMR62L | pRS315 | CEN, LEU2, PHSC82::HSP82::TCYC1 | [29] |
| pSK59 | pET28 | HSP82 | [24] |
| pSK92 | pET28 | AHA1 | [24] |
| pMR363 | pET28 | SBA1 | This study |
We performed the Hsp90 complementation assay in our four hsc82Δ/hsp82Δ strains and found hHsp90α was able to complement yeast Hsp90 function best in the W303 background (Figure 1(a)), as evident by growth on plates containing FOA. With regard to the ability and degree of human Hsp90s to complement essential Hsp90 functions, our W303 hsc82Δ/hsp82Δ strain behaved identically to one originally constructed in the Lindquist lab, called ΔPCLD82a [20,30] (see below and Figure 1(a)). Nonetheless, W303 cells expressing hHsp90α grew much slower than identical cells expressing Hsp82 or hHsp90β (Figure 1(a)), which is in line with previous observations [20,21]. Very slight growth was observed in patches of the BY4741 and YPH499 cells expressing hHsp90α, but these cells were not useful due to their extremely slow growth when recovered from FOA. 779-6A cells expressing hHsp90α failed to grow on FOA (Figure 1(a)). As expected, hHsp90β supported normal growth of all the strains (Figure 1 (a)). Since expression level exerts a strong influence on Hsp90 function [31,32], we analyzed the steady-state abundance of the various Hsp90 isoforms in the different strains. Western analysis showed that the differences in the ability of human Hsp90s to complement and the range of abilities of hHsp90α to complement in the different backgrounds were not related to differences in expression levels (Figure 1(b), Supplemental Figure 1A). Thus, the strain background exerted a strong influence on whether hHsp90α could complement essential yeast Hsp90 functions, and in all strains hHsp90β supported growth better than Hsp90α.
Since hHsp90α supported growth to varying degrees in the four strains, ranging from no growth in 779-6A, to extremely slow growth in BY4741, to slow growth in W303, we used these three strains in our genetics experiments, where practical, in order to assess impacts on general Hsp90 function and control for any effects due to the genetic background.
Recently, we showed that non-lethal mutations in Hsp82 that become lethal when the co-chaperone Sti1 (Hop in humans) is absent cluster into distinct regions that define two classes of Sti1-dependent mutations [24]. One class (SdN) is at the site of Hsp70 interaction in the M domain [33], and the other (SdC) is at the junction of the M and C domains. The SdN mutants require Sti1 for efficient interaction with Hsp70 and the SdC mutants require a separate function of Sti1 to facilitate Hsp90 dimer closing [24]. Unlike Hsp82, wild-type hHsp90β cannot support yeast growth when Sti1 is absent. This Sti1 dependency is rescued by the substitution E414R, which is analogous to E402R of Hsp82 that enhances Hsp70 binding. hHsp90β therefore behaves like an SdN mutant that needs Sti1 to interact effectively with Hsp70 in yeast [24].
Because wild-type hHsp90β depends on Sti1 in order to support yeast viability, we wondered if the inability of wild-type hHsp90α to function in our strain could be due to a dependency on Sti1 for either Hsp70 binding (SdN) or dimer closure (SdC). Such a dependency on Sti1 would have to be more severe than that of hHsp90β, since hHsp90α does not support yeast growth well even in STI1+ cells. We therefore introduced substitutions into hHsp90α that were analogous to the Hsp82 SdN suppressor (E422R in hHsp90α), or the SdC suppressors A107N or A577V (A121N or C598V in hHsp90α, respectively) (Table 3). None of these three substitutions allowed hHsp90α to support growth of 779-6A (Figure 1(c)). However, in BY4741 and W303, the E422R substitution marginally improved growth (Figure 1(c)). Abundance of the mutant hHsp90α proteins was similar to wild-type hHsp90α, indicating the failure to complement was not due to reduced expression (Supplemental Figure 1C). Thus, the inability of wild-type hHsp90α to function in our strain probably is not related to a dependency on Sti1.
Table 3.
Effects of Hsp90 mutations
| Substitution |
Effect |
Reference |
||
|---|---|---|---|---|
| Hsp82 | hHsp90α | hHsp90β | ||
| A107N | A121N | A116N | Stabilizes lid closure, promotes N domain dimerization | [11,13,22,24,34,35] |
| E402R | E422R | E414R | Increases Hsp70 binding | [24] |
| A577V | C598V | Promotes N domain dimerization | [24,36,37] | |
| A131T | A145T | A140T | Promotes dimer closing, and return to the open state | This study |
| A152V | A166V | A161V | Promotes dimer closing, and return to the open state | This study |
We also observed that either A121N or C598V destroyed the ability of hHsp90α to support viability of BY4741 and W303 (Figure 1(c)). These results are reminiscent of the finding that combining these two SdC suppressors in Hsp82 (A107N and A577V) is lethal [24]. They also are in line with the notion that hHsp90α is “too closed” for proper function in yeast, as SdC suppressor mutations, such A107N, increase the propensity to advance to the closed state [13] and would exacerbate this defect. Taken together, our observations can be inferred to suggest that imbalance in the cycle toward occupying the closed state can be detrimental to Hsp90 function in yeast.
In line with this idea, the negative effect of increasing dimer closure by A121N was not unique to hHsp90α, as the analogous substitution in hHsp90β, A116N, likewise destroyed its ability to complement in all strain backgrounds (Figure 1(d)). Taken together, these results suggest that hHsp90β supports yeast viability better than hHsp90α because it is just below a threshold of being “too closed.” They further reflect conservation of function among the yeast and human Hsp90s since the same substitution had similar effects on all three Hsp90s.
Deleting SBA1, CPR6, or CPR7 improves the ability of hHsp90α to complement yeast Hsp90 function in BY4741
Co-chaperones can regulate the Hsp90 reaction cycle positively or negatively. For example, Sti1 inhibits Hsp90 ATPase in vitro [38], while Aha1 greatly accelerates it [39,40]. To test if host co-chaperones were affecting the ability of hHsp90α to function in yeast, we assessed complementation in BY4741 strains with deletions of co-chaperone genes (Figure 2(a)). On FOA plates, we observed confluent growth in patches of cpr6Δ, cpr7Δ, and sba1Δ cells that was slightly, but reproducibly, greater than the growth of the control strain. Thus, hHsp90α was able to complement yeast Hsp90 function better in cells lacking either Cpr6, Cpr7, or Sba1, suggesting these co-chaperones inhibit hHsp90α function in yeast.
Figure 2.

Sba1, Cpr6, and Cpr7 inhibit hHsp90α function in yeast. (a) FOA plate as in Figure 1(a) using strain BY4741 with deletions of the Hsp90 co-chaperones indicated to the left and expressing Hsp82, hHsp90α or an empty vector control (ev). “BY” denotes the parental strain with no co-chaperone deletions. (b) Growth curves (see Materials and Methods) of wild-type (circles), sba1Δ (squares), cpR6Δ (triangles), and cpr7Δ (inverted triangles) BY4741 cells expressing Hsp82 (black) or hHsp90α (green). Data points are averages of three biological replicates and error bars are the standard deviation.
We measured growth of wild-type, cpr6Δ, cpr7Δ, and sba1Δ BY4741 cells recovered from the FOA plates in liquid cultures, which can reveal subtle variations that are not apparent when cells are grown on plates [32] (Figure 2(b)). Deleting CPR7 reduced growth of cells expressing Hsp82 (Figure 2(b)), in line with previous reports [41]. For cells expressing hHsp90α, deleting SBA1 or CPR7 improved growth noticeably, while deleting CPR6 did so but less effectively (Figure 2(b)). Deleting SBA1 improved function of hHsp90α enough to allow it to support growth of 779-6A (see below). Thus, deleting SBA1, CPR7, and to a lesser extent CPR6 improved function of hHsp90α in yeast.
In a reciprocal experiment, we found that increased expression of Sti1, Aha1, Hch1, or Sba1 failed to allow hHsp90α to complement yeast Hsp90 function in 779-6A cells (Supplemental Figure 2). These results suggest that abundance of these factors is not limiting, although they do not address how well they can cooperate with hHsp90α.
Novel mutations in a discreet region of the N domain improved the ability of hHsp90α to complement yeast Hsp90
In order to determine why hHsp90α functioned so poorly, we performed a forward genetic screen to identify alterations in hHsp90α that restored its function more completely. Using the most stringent background 779-6A (see Materials and Methods), we isolated the A166V substitution five times and A166T once. We also isolated A27T, A145T, V148A, and S165L. We refer to these mutations as α rescuers. All of the mutated residues cluster on the same edge of the central β-sheet that comprises the core of the N domain, opposite the nucleotide binding site (Figure 3(a)). This region of the N domain is highly conserved (Figure 3(b)). The mutated residues are at or near the loops between the strands of the β-sheet. Since cells expressing hHsp90αA145T and hHsp90αA166V grew fastest, they were selected for further study.
Figure 3.

Mutations in conserved residues of the N-terminal domain rescue hHsp90α function in yeast. (a) Cartoon of the hHsp90α N domain (green) in the apo, open lid state (pdb: 5j2v). Rescuer mutations identified as enabling hHsp90α to complement yeast Hsp90 function (A27, A145, V148, S165, and A166) are highlighted as maroon spheres. Lid residue alanine 121 is highlighted as orange spheres to show relative position of the α rescuer mutant cluster. (b) Alignment of Hsp82, hHsp90α, and hHsp90β regions of the N domain corresponding to hHsp90α amino acids 121 to 172. Residues A121 (orange circle), A145 (maroon square), and A166 (maroon triangle) are denoted. (c) FOA plate with strains 779-6A (779), BY4741 (By), and W303 (W) expressing the α rescuer mutants hHsp90αA145T and hHsp90αA166V. Papillae in patches of 779-6A cells with empty vector or hHsp90α are not evidence of complementation (see Materials and Methods). (d) Growth curves of cells recovered from FOA plate shown in (c): 779-6A (left), BY4741 (center), and W303 (right) expressing Hsp82 (black circles), hHsp90αA145T (maroon squares), hHsp90αA166V (maroon triangles), or hHsp90α (green circles). Data points are averages of three biological replicates and error bars are standard deviation. (e) W303 expressing the indicated Hsp90s were grown overnight in liquid YPAD, serially diluted, and spotted onto YPAD plates or YPAD supplemented with 1M sorbitol (right). The plates were incubated at the indicated temperatures for 4 (15 °C), 3 (20 °C), or 2 days (30 °C, 37 °C, and 37 °C + sorbitol).
In order to assess whether the effect of the α rescuers was general or specific to 779-6A in which they were selected, we tested for their ability to improve hHsp90α function in BY4741 and W303. For both BY4741 and W303, cells expressing hHsp90αA145T and hHsp90αA166V grew much faster than those expressing wild-type hHsp90α on FOA plates (Figure 3(c)). We next measured the growth rates of these cells in liquid culture; however, 779-6A cells expressing wild-type hHsp90α was not included as it was not viable. Cells of all backgrounds expressing the α rescuer mutants had lag times and logarithimic growth phases that resembled cells expressing Hsp82 more than hHsp90αWT (Figure 3(d)). Thus, while the α rescuer mutation function was not complete, it was strong and independent of strain background, reflecting functional conservation of these residues.
Since hHsp90α alanine 166 is conserved across Hsp90 isoforms, we determined its importance for general Hsp90 function by comparing hHsp90αA166V with analogous substitutions in hHsp90β (A161V) and Hsp82 (A152V) (Table 3). We used the W303 background in order to compare wild-type and mutant Hsp90s side by side. We observed no differences in growth of cells expressing Hsp82WT or Hsp82A152V at any temperature (Figure 3(e)). As observed above, cells expressing hHsp90α grew very slowly at optimal temperature (30 °C). Cells expressing hHsp90α or hHsp90β were cold sensitive (15 and 20 °C), and this phenotype was rescued by A166V or A161V, respectively. hHsp90β supported growth at 37 °C, but hHsp90α did not. hHsp90βA161V and hHsp90αA166V both failed to support viability at 37 °C. Thus, the α rescuer mutation in hHsp90α or hHsp90β improved Hsp90 function at low temperature but inhibited it at high temperature. In all instances, high temperature growth defects were suppressed by osmotic support (1M sorbitol) in the growth medium, indicating that the temperature sensitivity was not caused by a general loss of Hsp90 activity due to the mutations reducing protein stability at the high temperature [42].
A152V decreased interaction of Hsp82 with Sba1
The ability of hHsp90α to support viability in sba1Δ cells, but not in cells overexpressing co-chaperones, suggested the α rescuer mutations impaired interaction with host co-chaperones. Unfortunately, unlike yeast Hsp90s, the addition of a hexa-histidine (His6) tag to the amino terminus of hHsp90α was deleterious to its function in yeast, precluding its use for testing in vivo binding directly. Therefore, we used the analogous A152V mutation in Hsp82 for in vivo protein interaction experiments. We treated portions of lysates with the non-hydrolyzable ATP analog AMP-PNP to trap Hsp90 in the closed conformation in order to detect interaction between Hsp90 and Aha1 or Sba1 [43]. Western analysis of proteins co-purifying with Hsp82 revealed that A152V dramatically reduced interaction with Sba1, while interactions with Sti1, Hsp70, and Aha1 were unaffected (Figure 4(a)). Thus, the α rescuer mutation disrupted an interaction with Sba1, which coincides with our finding that depleting Sba1 improved hHsp90α function in yeast cells (Figure 2(a)).
Figure 4.

α Rescuer mutation reduced interaction of Hsp82 with Sba1. (a) Pull-downs of His6-tagged Hsp82WT (WT) or Hsp82A152V (AV) from lysates with (+) or without (−) AMP-PNP were probed with antibodies specific to Hsp82, Sti1, Hsp70, Aha1, or Sba1. Untagged Hsp82 (nt) served as a control. Inputs on the left show equal loading. (b) Coomassie-stained SDS-PAGE gel of a representative chemical crosslinking experiment of purified wild-type or mutant Hsp82 as indicated, with and without Sba1 and ATP. The first four lanes after the MW marker are un-crosslinked proteins. Location of the high molecular weight Hsp82:Sba1 complex (SPPS), low molecular weight Hsp82:Sba1 complex (PPS), Hsp82 dimer (PP), and non-complexed Sba1 are indicated by arrows on the right. (c) Left, quantification of crosslinked products in the boxed area of the gel from (b). Right, quantification of un-complexed Sba1 in crosslinking reactions as determined by comparison of Sba1 bands in reactions lacking or containing ATP. Values are the average of three independent experiments and error bars are the standard deviation.
This disruption of Sba1 binding is consistent with alanine 152 residing near the Sba1 binding site (pdb: 2cg9, [5]). To gauge the extent that mutations impaired a direct interaction, we assessed binding of Sba1 to Hsp82WT, Hsp82A107N, and Hsp82A152V using chemical crosslinking of purified proteins (Figure 4(b) and (c)). In the absence of nucleotide, we observed crosslinked Hsp82 dimers but no complexes with Sba1, as expected. In the presence of ATP, Hsp82 formed two complexes with Sba1 that migrated slower than Hsp82 dimers. We presume these complexes contain an Hsp82 dimer with either one (PPS) or two (SPPS) Sba1 molecules (Figure 4(b)). The A107N substitution resulted in formation of 1.7-fold more of the PPS complex, but had little effect on formation of the SPPS complex (Figure 4(c), left panel). There was also approximately half the amount of un-complexed Hsp82 dimers (PP) in reactions containing Hsp82A107N (Figure 4(c), left panel). These observations are in line with previous studies showing Hsp82A107N binds more Sba1 than Hsp82WT [24,34]. Compared to Hsp82WT, Hsp82A152V formed similar amounts of PPS but approximately 2.5-fold less SPPS complex (Figure 4(b) and (c), left panel). Accordingly, there was more un-crosslinked Sba1 in reactions with Hsp82A152V than Hsp82WT (Figure 4(c), right panel). Essentially identical results were obtained using the non-hydrolyzable ATP analog AMP-PNP (data not shown). Thus, the α rescuer homolog A152V reduced binding to Sba1 overall and specifically inhibited formation of SPPS. The results of the crosslinking experiments are consistent with earlier findings that Hsp82 binds Sba1 asymmetrically [44], and suggest A152V inhibits binding of the second Sba1. Together with our finding that Hsp82A152V binds much less Sba1 in lysates, these results suggest the PPS complex is less stable and represents a transitional complex.
The α rescuer mutation increased intrinsic Hsp90 ATPase
Sba1 preferentially binds the closed, ATP-bound form of Hsp90 [5]. Indeed, interaction with Sba1 is used as an assay for the propensity of Hsp90 mutants to be in the closed state. For example, A107N and A577V increase binding to Sba1 because they promote N domain dimerization, or closing [24,34]. Increased N domain dimerization also explains the higher intrinsic rates of ATPase of Hsp82A107N and Hsp82A577V compared wild-type [13,24]. Because A152V inhibited formation of the high molecular weight complex with Sba1, we considered it might also reduce intrinsic ATPase. We measured kinetics of ATP hydrolysis for Hsp82A152V, using wild-type Hsp82 and Hsp82A107N as controls. Our values for the apparent KM and VMAX of ATP binding for Hsp82WT and Hsp82A107N (Figure 5(a) and Table 4) aligned closely with previous reports [24,38]. Hsp82A152V had a VMAX 2.4-fold higher than wild-type (Table 4) and a similar apparent KM for ATP binding as Hsp82A107N, which was 2.8-fold less than wild-type (Table 4). Thus, the A152V α rescuer mutation in Hsp82 enhanced ATP hydrolysis and apparent affinity for ATP compared to wild-type. These results were somewhat surprising since mutations that result in higher intrinsic ATPase, such as A107N and A577V, have been found to bind more Sba1 in vivo.
Figure 5.

α Rescuer mutation increased Hsp82 intrinsic ATPase. (a) Kinetics of ATP hydrolysis of purified Hsp82WT (black squares), Hsp82A152V (maroon triangles), and Hsp82A107N (orange circles) in increasing concentrations of ATP. Data points are averages of three independent experiments and error bars are standard deviation. Data were fitted to Michaelis–Menten kinetics (solid lines). (b) Kinetics of ATP hydrolysis of hHsp90αWT (green squares), hHsp90αA166V (maroon triangles), and hHsp90αA121N (orange circles) as in (a). Data points are averages of three independent experiments and error bars are standard deviation. Data were fitted to Michaelis–Menten kinetics (lines). (c) Aha1-stimulated ATP hydrolysis rates of Hsp82WT (black) or Hsp82A152V (maroon) with the indicated fold concentrations of Aha1 over Hsp82 (1 μM dimer). Values are the average of three experiments and error bars are standard deviation.
Table 4.
ATPase enzyme kinetic values
| Hsp82 | Apparent KM (μM) | VMAX (μmol/min) |
|---|---|---|
| Wild-type | 161 ± 41 | 0.36 ± 0.05 |
| A107N | 52 ± 3 | 1.75 ± 0.03 |
| A152V | 63 ± 3 | 0.87 ± 0.03 |
We then measured ATPase rates of purified hHsp90αWT, hHsp90αA121N1, and hHsp90αA166V (Figure 5(b)). A166V increased ATPase, but not as much as A121N. These results are very similar to those of the analogous substitutions A152V and A107N in Hsp82, which again demonstrates functional conservation of the residues.
In agreement with our finding that A152V had little effect on Aha1 binding (Figure 4(a)), A152V had no significant impact on the stimulation of Hsp82 ATPase by Aha1 (Figure 5(c)).
Sti1 is required for rescue of hHsp90α function by depleting Sba1, but not by α rescuer mutations
The α rescuer mutation blocked Sba1 binding, but hHsp90αA166V improved growth of yeast better than loss of Sba1. This difference suggested that the α rescuer mutations were doing something beyond inhibiting interaction with Sba1 to improve hHsp90α function in yeast. Because of the central roles of Sti1 in the Hsp90 client binding cycle, we considered the additional effects might involve Sti1. We tested this idea by assessing if Sti1 was required for rescue of hHsp90α function by the α rescuers or by depletion of Sba1. In cells lacking Sti1, the ability of the α rescuer mutants to improve hHsp90α function was unchanged (Figure 6(a)). However, hHsp90α was unable to support growth of cells lacking both Sba1 and Sti1. Thus, the α rescuers affect hHsp90α function in a manner beyond simply reducing interaction with Sba1.
Figure 6.

α Rescuer mutation bypassed need for Sti1. (a) FOA plate as in Figure 1(a) with 779-6A (779) and isogenic sti1Δ, sba1Δ, and sti1Δ sba1Δ (ΔΔ) cells expressing the indicated hHsp90α (90α) mutants with Hsp82, hHsp90α, and empty vector (ev) controls. (b) As in (a) with sti1Δ cells expressing the indicated hHsp90β (90β) mutants. (c) 779-6A (STI1) or isogenic sti1Δ cells expressing the indicated Hsp82 alleles recovered from FOA were grown overnight, serially diluted, and spotted onto YPAD plates. The plates were incubated at 30 °C (left) or 37 °C (right) and imaged after 3 days.
It was possible that elevated ATPase was connected to this other function affected by the α rescuer. If the mechanism of α rescuer function was a combination of increased ATPase and inhibition of Sba1 binding, then hHsp90αA121N might support viability of sba1Δ cells. We found that hHsp90αA121N failed to complement in sba1Δ cells (Figure 6(a)). These results suggest that the way α rescuers affect Hsp90 function is more complex than combining increased ATP hydrolysis with a block in Sba1 binding. They also suggest that the degree of shift toward the closed state caused by A121N is not overcome by depletion of Sba1.
Since the α rescuers did not require Sti1 to improve hHsp90α function, we predicted that the analogous mutations in hHsp90β (see Table 3) would rescue its Sti1 dependence. The α rescuer mutations in hHsp90β rescued Sti1 dependence to a degree equal to or better than E414R (Figure 6(b)), which we showed earlier rescued hHsp90β Sti1 dependence by increasing binding to Hsp70 [24]. We then tested whether the α rescuer mutations in Hsp82 suppressed the temperature sensitivity of sti1Δ cells and found Hsp82A152V supported growth of sti1Δ cells at 37 °C (Figure 6(c)). Taken together, these results suggest that the α rescuer mutations generally bypass the need for Sti1 in the Hsp90 reaction cycle.
A152V alters dynamics of Hsp90 nucleotide-dependent conformational rearrangements
Hsp90 dimers undergo radical, nucleotide-dependent conformational changes upon ATP binding: 1) a lid-like structure closes over the ATP-binding pocket, 2) the N domain docks onto the M domain (which positions the catalytic arginine in the M domain into contact with the nucleotide), and 3) the N domains of the two protomers make physical contact and swap their amino-terminal β-strands (Figure 7(a)). These movements seem to happen concurrently resulting in the “closing” of Hsp90 dimers. We investigated the effect of the α rescuer mutation on these movements using fluorescence quenching through photoinduced electron transfer (PET; Figure 7(a)) [11,45].
Figure 7.

N–M domains of Hsp82A152V dock and undock faster than wild-type. (a) Cartoon representing the N–M domain docking PET fluorescence experiment, with location of the N, M and C domains denoted on left. Addition of AMP-PNP to fluorescently labeled proteins causes N–M domain docking resulting in quenching of fluorescence (red flash) due to proximity of the label to an introduced tryptophan residue (W). Numbered arrows denote (1) AMP-PNP-induced lid closing over the ATP binding pocket, (2) N–M domain docking, and (3) β-strand swapping as described in the text. (b) N–M docking experiment comparing Hsp82WT (black circles), Hsp82A152V (maroon triangles), and Hsp82A107N (orange squares). AMP-PNP was added at T = 10 min. (c) Cartoon representing the N–M domain undocking experiment. Addition of ADP to fluorescently labeled proteins preincubated with AMP-PNP results in increased fluorescence due to undocking, which increases the distance between the label and an introduced tryptophan residue in the open state. (d) N–M undocking experiment where ADP was added at a 10-fold increase over AMP-PNP comparing Hsp82WT (black circles), Hsp82A152V (maroon triangles), and Hsp82A107N (orange squares). ADP was added at T = 10 min. (e) N–M undocking experiment as in (d) except ADP as was added at concentrations equal to (circles), 2.5-fold (squares), or 5-fold (triangles) of the AMP-PNP concentration. In all experiments, data points are averages of three independent experiments, error bars are the standard deviation, and data were fitted to a single exponential decay function (solid lines). Insets show decay constants (min−1 ± SD)
We first measured docking of the N and M domains of Hsp82WT, Hsp82A107N, and Hsp82A152V (Figure 7(b)). As shown earlier, A107N increased the rate of N–M docking 7-fold compared to wild-type (Figure 7(b)) [11]. Also in line with that study, the fluorescence signal plateaued at a lower intensity in reactions with Hsp82A107N compared to Hsp82WT. These data indicate that at equilibrium a higher proportion of Hsp82A107N dimers are in the docked state. The A152V mutation also enhanced N–M docking, with a rate 4-fold higher than wild-type. However, Hsp82A152V plateaued at an intensity similar to wild-type (Figure 7(b)). Thus, Hsp82A152V reached a wild-type equilibrium, but got there faster.
In similar experiments that measured rates of lid closing (Supplemental Figure 3A) [11], we found Hsp82A107N and Hsp82A152V were 6- and 5.5-fold higher than Hsp82WT, respectively, and all three fluorescence signals plateaued at comparable intensities (Supplemental Figure 3B). Thus, A152V enhanced Hsp82 lid closing dynamics similarly to A107N.
The similar behavior of Hsp82A107N and Hsp82-A152V in N–M docking and lid closing does not explain the different physiological effects that A107N and A152V have in both yeast and human Hsp90s. It is possible that A152V affects rearrangements that increase the rate of dimer opening. To test this idea, we modified the PET assay to measure rates of N–M undocking by titrating ADP into Hsp82 proteins that were first docked by pre-incubating with AMP-PNP. Fluorescence should unquench as the molecules undock (Figure 7(c)). Upon addition of 10-fold excess of ADP, all of the Hsp82 proteins returned to an undocked state, with fluorescence intensities similar to untreated control proteins (Figure 7(d)). Hsp82-A152V undocked three times faster than Hsp82WT, while Hsp82A107N dimers undocked approximately half as fast as Hsp82WT (Figure 7(d)). Thus, A152V and A107N have opposing effects on N–M undocking, which probably underlie their functional differences in vivo.
We used lower but increasing concentrations of ADP (1, 2.5, and 5-fold molar excess) to more accurately measure the kinetics of N–M undocking. We found Hsp82A152V N- and M-domains undocked at an average rate that was 2-fold faster than wild-type, yet both proteins reached a similar plateau at a given ratio of nucleotides (Figure 7(e)). In contrast, the proportion of closed Hsp82A107N dimers that undocked at the lower ADP concentrations was much lower than Hsp82WT and Hsp82A152V under the same conditions (Figure 7(e)). However, at the lower concentrations of ADP, Hsp82A107N reached equilibrium faster than Hsp82WT or Hsp82A152V (Figure 7(e)). Similar results were observed in analogous experiments that measured the lid opening in response to ADP titration (Supplemental Figure 3C, D and E).
Therefore, under these conditions, A107N, which stabilizes nucleotide binding, promoted and stabilized the closed state. The α rescuer homolog A152V similarly promoted closing, but it also enhanced the return to the open state. Thus, the α rescuer shifts the open/closed equilibrium of Hsp82 dimers toward the open state relative to wild-type, while A107N pushed the equilibrium toward the closed state (Figure 8). Taken together, our data suggest the α rescuers allow hHsp90α to function in yeast cells as the only source of Hsp90 by increasing the propensity of dimers to open.
Figure 8.

Equilibrium model for Hsp90 function in yeast. Note sizes of the arrows. The open/closed equilibrium of yeast Hsp90 (balanced arrows, upper left panel) is optimal for proper chaperoning of yeast Hsp90 clients. This equilibrium of human Hsp90α is shifted toward closed (bigger top arrow, upper right panel) compared to yeast Hsp90 and is thus poorly suited for function in yeast. It supports growth weakly and only in certain strain backgrounds. Mutations that further drive the equilibrium toward the closed state (even bigger top arrow, lower left panel), such as A121N, exacerbate this dynamic and are lethal for Hsp90α in all yeast strains. The α rescuer mutation A166V pushes the equilibrium of Hsp90α toward the open conformation (heavy, but balanced arrows, lower right panel), restoring an open/closed equilibrium that supports yeast viability.
Discussion
Using a forward genetics approach, we isolated novel mutations in conserved residues clustered in the hHsp90α N domain that considerably improved hHsp90α function in yeast. We found that the strongest α rescuer mutation A152V promoted lid closing over the ATP binding pocket, N–M docking and intrinsic ATPase, which are indicative of Hsp90 dimer closing. These effects are similar to those caused by A107N. However, A107N also stabilizes the closed lid [13], which we show slows lid opening and N–M undocking. Unlike A107N, A152V accelerated N–M undocking, which is indicative of dimer opening, and reduced Sba1 binding. Not surprisingly, A121N in hHsp90α (homologous to A107N) had the opposite physiological effect as the α rescuers: it destroyed the ability of hHsp90α to function in yeast. These differences from A107N suggest that the mechanism for the rescue involves facilitating return of Hsp90 to the open state and concomitant release of clients. Our findings support a model in which the function of hHsp90α in yeast is very weak because it spends too much time in the closed state (Figure 8).
Due to technical limitations, most biochemical characterization of the α rescuer mutation was done in the context of A152V in Hsp82. We recognize the caveat that the homologous A166V might not affect hHsp90α the same way. However, the A121N and A166V substitutions had very similar effects on hHsp90α ATPase as A107N and A152V in Hsp82, and effects of A166V on hHsp90α function in vivo were consistent across all three Hsp90s and the different strain backgrounds.
These observations demonstrate functional conservation of the roles of the residues in Hsp90 activity and support our experimental approach, which is not uncommon. They point to the region of the N domain where α rescuers cluster as representing a conserved structural element that seems to be involved in effecting structural changes that regulate dissociation of the N domains.
We showed earlier that combining two mutations in Hsp82 (A107N and A577V) that separately promote the closed state is lethal [24]. These data are in line with the idea that being too closed disrupts the ability of Hsp90 to support yeast growth, possibly due to inefficient client capture or release. Either of these mutations alone in hHsp90α (A121N or C598V) is lethal in yeast, which is consistent with their enhancing an already high propensity of hHsp90α to be closed. This propensity to be closed distinguishes hHsp90α and hHsp90β, but it seems to be a matter of degree. It is possible that this property of hHsp90α underlies its specialized roles in responding to stress in human cells.
Sba1 stabilizes the closed conformation of Hsp90 [5,34,46], and here we find depleting it partially rescued the ability of hHsp90α to support growth of yeast. Indeed, in the context of Hsp82, the α rescuer mutation inhibited Sba1 interaction, specifically formation of the high molecular weight complex containing two Sba1 molecules (SPPS). However, the mechanism of how α rescuers restore function is not merely by reducing Sba1 binding, because complementation by hHsp90α in sba1Δ cells was only partial and it required Sti1. Our data suggest that complete rescue by the α rescuers combines facilitating conformational changes that promote return to the open state with reduced Sba1 interaction, which also should contribute to that effect. However, our data do not exclude the possibility that the reduced formation of the higher order complex with Sba1 (SPPS) is a consequence of conformational or structural changes caused by A152V.
Other explanations for poor function of human Hsp90s in yeast could be related to differences in post-translational modifications, cooperation with co-chaperones or client interactions. The importance of post-translation modifications on Hsp90 regulation is well documented [47-51]. With particular relevance, optimal activity of hHsp90α in human cells was shown to depend on acetylation of lysine 294 [20,50]. Acetylation-mimic variants hHsp90αK294Q and hHsp90αK294A supported growth of W303 yeast cells better than wild-type hHsp90α [20]. In mammalian cells, these variants bound less clients and p23 (the mammalian homolog of Sba1). In light of our data, acetylation of lysine 294 might be improving function of hHsp90α in vivo by promoting the open state.
In contrast, K294R, a substitution that preserves positive charge but cannot be acetylated, impaired hHsp90α function in yeast. In mammalian cells hHsp90αK294R bound more FKBP52, a cyclophilin peptidyl proline isomerase related to Cpr6 and Cpr7, than wild-type or the acetylation mimic mutants. In line with this latter finding, we found depleting Cpr6 or Cpr7 in our strains allowed partial rescue of hHsp90α. Little is known of the structure of Cpr6-Hsp90 interaction, but Graf et al. showed Cpr6 binds to a conformation of Hsp82 that is different from that when Hsp82 is bound to Sti1 (open) or Sba1 (closed) [52]. They also concluded that Cpr6 did not stabilize the closed conformation of Hsp82. Taken together, the effects of lysine 294 modification are in general agreement with our model and point to a link in the ways α rescuer mutations and acetylation alter Hsp90 function. Beyond changes at lysine 294, other differences in post-translational modification landscapes may account, at least in part, for the influence that yeast strain background has on the ability of hHsp90α to function in yeast.
Others showed that poor function of hHsp90α in yeast cells maps to a region of the M domain [21]. Our findings also raise the interesting possibility that this region of hHsp90α stabilizes dimer closing. If true, then that would mean the α rescuer mutations bypass or otherwise overcome stabilization mediated by this region of the M domain.
While our data show that the α rescuer mutations can facilitate opening of Hsp90 dimers, the mechanistic underpinnings of this effect remain to be determined. Molecular dynamics or NMR studies may provide crucial insight into the mechanism of α rescuer function. Additionally, substitutions that cause defective dimer closing may also overcome hHsp90α's propensity to be in the closed state and partially rescue its function in yeast. Owing to the high level of evolutionary conservation, we predict that in human cells hHsp90αA166V would bind less p23 than wild-type hHsp90α. These speculations are testable subjects for future work.
Since the forward mutations identified in our screen are widely conserved, tightly clustered and have a specific effect on the reaction cycle, we infer that this region of the N domain serves as a type of switch. This switch could normally act to limit non-productive dimer opening. In the crystal structure of the Hsp82:Sba1 complex (pdb: 2cg9), the side-chain of alanine 152 is sandwiched between the side-chains methionine 16 of Hsp82 and aspartate 86 of Sba1. Methionine 16 is in the middle of the α-helix just downstream of the N-terminal β-strand that is swapped between protomers of closed Hsp82 [5]. This central location between Sba1 and a highly movable motif of Hsp82 might indicate the defect in the ability of Hsp82A152V to form the high-molecular-weight complex with Sba1 (SPPS) is a disruption of the Sba1 binding site. Another explanation for this defect may be that A152V disrupts conformational changes after binding of the first Sba1 molecule that are needed for efficient binding of the second Sba1. Regardless of the explanation, our data suggest the site where the α rescuer mutants are located is important for coordinating or stabilizing conformational rearrangements and Sba1 binding in response to nucleotides. Our findings implicate this region of the Hsp90 N domain as important for dimer opening and resetting of the reaction cycle.
Materials and Methods
Yeast strains, growth conditions, and plasmids
Yeast strains are listed in Table 1. 779-6A is described [25,33]. Gene knockouts were made according to established protocols [53] or were obtained from the yeast knockout collection (Thermo Fisher Open Biosystems). All knockouts were verified by PCR and western blotting, if antibodies were available.
To make the hsc82Δ and hsp82Δ mutations in the BY4741, W303, and YPH499 backgrounds, HSP82 was replaced with NatMX6 and HSC82 was replaced with HphMX6 in MATa and MATα strains, respectively. MATa hsp82Δ Nat strain was transformed with plasmid pMR62 (see below) then crossed to the MATα hsc82ΔHyg strain and diploids were selected, sporulated and dissected. The BY4741 hsc82Δ/hsp82Δ strain was backcrossed to BY4735 to combine the hsc82Δ/hsp82Δ mutations and plasmid pMR62 with the lys2Δ0 and trp1Δ63 markers to give strain MR1060. Strains that had hsc82Δ/hsp82Δ combined with co-chaperone deletions were made by genetic crosses of strain MR1060 with strains from the yeast knockout library (OpenBiosystems). Strain MR1075, which was used as the wild-type complementation strain in the BY4741 background, was a STI1+ meiotic spore clone derived from the diploid formed from MR1060 and BY4741 sti1ΔKan.
Yeast were grown on YPAD (1% yeast extract, 2% peptone, 100 mg/L adenine, 2% dextrose). Solid media contained 2% agar. Selective media were synthetic complete (SC) (Sunrise Scientific) lacking appropriate nutrients with 2% dextrose, 0.67% yeast nitrogen base, and 100 mg/L adenine. Counterselection media were identical except but contained 1 g/L 5'-fluoro-orotic acid (FOA). All cultures were grown at 30 °C for 3 days unless noted otherwise.
Plasmids are listed in Table 2. Plasmids pMR62, pMR62L, pMR349, pMR365, pSK59, and pSK92 are described [24,29]. Plasmid pMR325 is the HSP82 open reading frame cloned into p414-GPD [54] as a SpeI/XhoI fragment. Plasmid pMR423 is the human HSP90AA1 open reading frame (encoding hHsp90α) cloned into p414-GPD as a SpeI/XhoI fragment. Plasmid pMR363 is pET28 containing the SBA1 open reading frame cloned as a NheI/XhoI fragment. Restriction enzyme sites were added via PCR. Point mutations were introduced using site-directed mutagenesis and verified via sequencing.
Complementation assay
FOA-based counterselection assays to assess complementation of yeast Hsp90 function have been described [24]. Briefly, hsc82Δhsp82Δ strains that harbored HSP82 on a URA3 plasmid were transformed with TRP1 plasmids encoding various Hsp90s. If the Hsp90 protein expressed from the TRP1 plasmid complements loss of yeast Hsp90 function, then a population of cells within a colony or patch can lose the parental URA3-marked plasmid when grown on medium containing uracil. These cells are auxotrophic for uracil and resistant to 5-fluoro-orotic acid (FOA), a chemical that kills cells expressing Ura3. Thus, growth on FOA reports on complementation of yeast Hsp90 function.
Occasionally we observed papillae in patches of 779-6A cells, but these colonies arose from single cells in a patch of millions that may have acquired a suppressor mutation or underwent a plasmid-to-plasmid recombination event. Likewise, papillae were also rarely observed in patches of cells that harbored empty TRP1 plasmids. Therefore, papillae were not considered to be evidence of complementation.
Western analysis
Cells were grown overnight in liquid YPAD and 20 OD600 units were harvested by centrifugation, washed with water and stored at −80 °C. Lysates were prepared by suspending cell pellets in 250 μl of lysis buffer (25 mM Hepes (pH 7.3), 100 mM NaCl, 2 mM MgCl2, 0.1% Tween-20, 2 mM PMSF, Roche Complete EDTA-free protease inhibitors). An equal volume of glass beads was added, and cells were mechanically lysed via two 40-s pulses in a bead beater. Lysates were cleared via centrifugation at 5000g for 3 min. Equal volumes of cleared lysate and 2× SDS-PAGE loading buffer (100 mM Tris–HCl (pH 7.5), 4% SDS, 100 mM DTT, 20% glycerol, bromphenol blue) were mixed, boiled, and run on 4%–12% Criterion TGX SDS-PAGE gels (Bio-Rad). Proteins were transferred to PVDF membranes and probed with appropriate antibodies. Hsp90-specific antibodies used were: ADI-SPA-840 (Enzo Life Sciences), recognizes hHsp90α and Hsp82; Anti-Hsc82 (Abcam ab30920), recognizes both yeast Hsp90 isoforms but neither human Hsp90 isoform; and H9010 (Enzo Life Sciences), recognizes only hHsp90β. The Hsp70 antibody used was ADI-SPA-kinetic values 757 (Enzo Life Sciences). Antibodies specific to Sti1 have been described [55]. Antibodies specific to Aha1 or Sba1 were raised in rabbits using whole purified proteins as antigen (Genscript).
Growth curves
Overnight cultures were diluted to OD600 = 0.1 to 0.2 in 1 ml of liquid YPAD in 24-well plates and incubated with shaking (300 rpm) at 30 °C for 24 h in a BMG Labtech Omega plate reader. The OD600 was measured every 20 min. All experiments were performed three times, each replicate from an individual primary transformant that was isolated after counterselection on FOA.
Genetic screen
To find novel mutations that rescued hHsp90α's ability to complement yeast Hsp90 function, a plasmid encoding hHsp90α under the control of the strong GPD promoter was first mutagenized via passage in E. coli strain XL-1 Red (Agilent Technologies) according to the manufacturer's instructions. The mutagenized pool was then used to transform strain MR318, and ~10,000 colonies were selected on 20 SC-Trp plates (containing uracil). After incubation for 3 days, the plates were replica-plated to identical medium containing FOA. FOA-resistant colonies were isolated after 1 and 4 days of incubation, and the plasmids were recovered and sequenced using standard techniques.
Pull-downs
Isolation of His6-Hsp82 complexes from yeast lysates and Western analysis has been described [24].
Protein purification
Expression and purification of His6-tagged wild-type and mutant Hsp82 and Aha1 proteins from Escherichia coli has been described [24]. Human Hsp90 proteins were purified identically to Hsp82, and Sba1 protein was purified identically to Aha1.
Chemical crosslinking
Purified wild-type or mutant Hsp82 (2 μM monomer) was incubated with and without Sba1 (1 μM) and/or ATP (2 mM) in 100 μl of 25 mM Hepes (pH 7.3), 100 mM NaCl, 5 mM MgCl2, 1 M trimethylamine N-oxide for 30 min at room temperature. Disuccinimidyl sulfoxide (DSSO, Thermo) was added to a final concentration of 2 mM, mixed, and incubated at room temperature for 30 min. Crosslinking was quenched by the addition of an equal volume of 2× SDS-PAGE loading buffer and boiled for 10 min prior to separation on 4%–20% Criterion TGX SDS-PAGE gels. Gels were stained with InstantBlue (Expedeon) according to the manufacturer's instructions. Band intensity was determined using Fiji [56].
ATPase measurements
ATPase measurements were performed essentially as described [45] except using a BMG Labtech Omega plate reader. All ATPase reactions were carried out at 30 °C and performed at least three times. Data were analyzed and fitted to Michaelis Menten kinetics using Prism 8 (GraphPad Software, San Diego, CA, USA).
PET measurements
Photon-induced electron transfer (PET) quenching of fluorescence experiments were performed as described [11,45] with some modifications. Hsp82 proteins (10 μM in 25 mM Tris–HCl (pH 7.5), 100 mM NaCl, 2 mM MgCl2, 10% glycerol) containing appropriate cysteine/tryptophan substitutions (S51C/A110W for lid closing, E192C/N298W for N–M docking) in wild-type, A107N or A152V backgrounds were labeled with 13 μM Atto-Oxa11 maleimide (Atto-Tec GmbH) supplemented with 100 μM TCEP at room temperature for 2 h. Unreacted dye was removed by two serial passages through Zeba desalting spin columns (Pierce) equilibrated with reaction buffer (25 mM Hepes (pH 7.3), 200 mM NaCl, 10 mM MgCl2).
The concentration of labeled protein was measured using a standard curve of free dye diluted in reaction buffer. Reactions (100 μl final volume) containing 4.5 μM unlabeled proteins (that did not have cysteine/tryptophan substitutions) and 0.15 μM labeled proteins in reaction buffer were incubated in black 96-well plates at 30 °C for at least 30 min prior to the initiation of the experiment. Fluorescence (620-nm excitation and 680-nm emission) intensities were measured in a BMG Labtech Omega plate reader every 20 s at 30 °C. The reaction was initiated at T = 10 min by the injection of adenylyl-imidodiphosphate (AMP-PNP, Roche) to a final concentration of 2 mM. In other reactions, the proteins as above were incubated with 2 mM AMP-PNP prior to the initiation of the reaction and adenosine diphosphate (ADP, Sigma) was injected at T = 10 min to a final concentration of 2, 5, 10, or 20 mM. All experiments were performed two to three times and the normalized, averaged data were fitted to an exponential decay function as described [11,45] using Prism 8 (GraphPad Software).
Supplementary Material
Acknowledgments
We are grateful to Dr. Len Neckers and Dr. Orna Cohen-Fix for insightful discussions and reagents, Dr. Coney Lin and Oscar Ramos for strain construction, and NIH colleagues for critical review of the manuscript. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney diseases.
Abbreviations used:
- Hsp90
heat shock protein 90 kD
- N
amino-terminal domain
- M
middle domain
- C
carboxy-terminal domain
- AMP-PNP
adenylyl-imidodiphosphate
- ABC
ATP-binding cassette
- SdN
Sti1-dependent mutation N-terminal proximal
- SdC
Sti1-dependent mutation C-terminal proximal
- TCEP
tris-(2-carboxyethyl)-phosphine
- FOA
5-fluoro-orotic acid
- OD600
optical density at 600 nm
- PMSF
phenylmethylsulfonyl fluoride
- PVDF
poly-vinylidene fluoride
- GPD
glycerol-phosphate dehydrogenase
- DSSO
disuccinimidyl sulfoxide
- PET
photon-induced electron transfer
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.06.015.
References
- [1].Schopf FH, Biebl MM, Buchner J, (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol, 18, 345–360. [DOI] [PubMed] [Google Scholar]
- [2].Hoter A, El-Sabban ME, Naim HY, (2018). The HSP90 family: structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci, 19,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Biebl MM, Buchner J, (2019). Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb. Perspect. Biol, 11,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rohl A, Rohrberg J, Buchner J, (2013). The chaperone Hsp90: changing partners for demanding clients. Trends Biochem. Sci, 38, 253–262. [DOI] [PubMed] [Google Scholar]
- [5].Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, et al. , (2006). Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature, 440, 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Prodromou C, Roe SM, Piper PW, Pearl LH, (1997). A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat. Struct. Biol, 4, 477–482. [DOI] [PubMed] [Google Scholar]
- [7].Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, et al. , (2003). Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and co-chaperone interactions. Mol. Cell, 11, 647–658. [DOI] [PubMed] [Google Scholar]
- [8].Richter K, Muschler P, Hainzl O, Buchner J, (2001). Coordinated ATP hydrolysis by the Hsp90 dimer. J. Biol. Chem, 276, 33689–33696. [DOI] [PubMed] [Google Scholar]
- [9].Krukenberg KA, Bottcher UM, Southworth DR, Agard DA, (2009). Grp94, the endoplasmic reticulum Hsp90, has a similar solution conformation to cytosolic Hsp90 in the absence of nucleotide. Protein Sci, 18, 1815–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krukenberg KA, Street TO, Lavery LA, Agard DA, (2011). Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys, 44, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schulze A, Beliu G, Helmerich DA, Schubert J, Pearl LH, Prodromou C, et al. , (2016). Cooperation of local motions in the Hsp90 molecular chaperone ATPase mechanism. Nat. Chem. Biol, 12, 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Prodromou C, (2012). The “active life” of Hsp90 complexes. Biochim. Biophys. Acta, 1823, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Prodromou C, Panaretou B, Chohan S, Siligardi G, O’Brien R, Ladbury JE, et al. , (2000). The ATPase cycle of Hsp90 drives a molecular “clamp” via transient dimerization of the N-terminal domains. EMBO J, 19, 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Girstmair H, Tippel F, Lopez A, Tych K, Stein F, Haberkant P, et al. , (2019). The Hsp90 isoforms from S. cerevisiae differ in structure, function and client range. Nat. Commun, 10, 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sreedhar AS, Kalmar E, Csermely P, Shen YF, (2004). Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett, 562, 11–15. [DOI] [PubMed] [Google Scholar]
- [16].Kuo CC, Liang CM, Lai CY, Liang SM, (2007). Involvement of heat shock protein (Hsp)90 beta but not Hsp90 alpha in antiapoptotic effect of CpG-B oligodeoxynu-cleotide. J. Immunol, 178, 6100–6108. [DOI] [PubMed] [Google Scholar]
- [17].Milicevic Z, Bogojevic D, Mihailovic M, Petrovic M, Krivokapic Z, (2008). Molecular characterization of hsp90 isoforms in colorectal cancer cells and its association with tumour progression. Int. J. Oncol, 32, 1169–1178. [PubMed] [Google Scholar]
- [18].Guo J, Chang C, Li W, (2017). The role of secreted heat shock protein-90 (Hsp90) in wound healing—how could it shape future therapeutics? Expert Rev Proteomics, 14, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jayaprakash P, Dong H, Zou M, Bhatia A, O’Brien K, Chen M, et al. , (2015). Hsp90alpha and Hsp90beta together operate a hypoxia and nutrient paucity stress-response mechanism during wound healing. J. Cell Sci, 128, 1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, et al. , (2007). An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell, 25, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Synoradzki K, Bieganowski P, (2015). Middle domain of human Hsp90 isoforms differentially binds Aha1 in human cells and alters Hsp90 activity in yeast. Biochim. Biophys. Acta, 1853, 445–452. [DOI] [PubMed] [Google Scholar]
- [22].Millson SH, Prodromou C, Piper PW, (2010). A simple yeast-based system for analyzing inhibitor resistance in the human cancer drug targets Hsp90alpha/beta. Biochem. Pharmacol, 79, 1581–1588. [DOI] [PubMed] [Google Scholar]
- [23].Millson SH, Truman AW, Racz A, Hu B, Panaretou B, Nuttall J, et al. , (2007). Expressed as the sole Hsp90 of yeast, the alpha and beta isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90beta generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J, 274, 4453–4463. [DOI] [PubMed] [Google Scholar]
- [24].Reidy M, Kumar S, Anderson DE, Masison DC, (2018). Dual roles for yeast Sti1/Hop in regulating the Hsp90 chaperone cycle. Genetics, 209, 1139–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jung G, Masison DC, (2001). Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr. Microbiol, 43, 7–10. [DOI] [PubMed] [Google Scholar]
- [26].Ralser M, Kuhl H, Ralser M, Werber M, Lehrach H, Breitenbach M, et al. , (2012). The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: insights into ancestry and physiology of a laboratory mutt. Open Biol, 2, 120093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, et al. , (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast, 14, 115–132. [DOI] [PubMed] [Google Scholar]
- [28].Sikorski RS, Hieter P, (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Genest O, Reidy M, Street TO, Hoskins JR, Camberg JL, Agard DA, et al. , (2013). Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol. Cell, 49, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nathan DF, Lindquist S, (1995). Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol, 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jiang L, Mishra P, Hietpas RT, Zeldovich KB, Bolon DN, (2013). Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genet, 9, e1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wider D, Peli-Gulli MP, Briand PA, Tatu U, Picard D, (2009). The complementation of yeast with human or Plasmodium falciparum Hsp90 confers differential inhibitor sensitivities. Mol. Biochem. Parasitol, 164, 147–152. [DOI] [PubMed] [Google Scholar]
- [33].Kravats AN, Hoskins JR, Reidy M, Johnson JL, Doyle SM, Genest O, et al. , (2018). Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl. Acad. Sci. U. S. A, 115, E2210 E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zuehlke AD, Reidy M, Lin C, LaPointe P, Alsomairy S, Lee DJ, et al. , (2017). An Hsp90 co-chaperone protein in yeast is functionally replaced by site-specific posttranslational modification in humans. Nat. Commun, 8, 15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vaughan CK, Piper PW, Pearl LH, Prodromou C, (2009). A common conformationally coupled ATPase mechanism for yeast and human cytoplasmic HSP90s. FEBS J, 276, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, et al. , (2005). S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. U. S. A, 102, 8525–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, et al. , (2009). Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep, 10,1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee CT, Graf C, Mayer FJ, Richter SM, Mayer MP, (2012). Dynamics of the regulation of Hsp90 by the cochaperone Sti1. EMBO J, 31, 1518–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lotz GP, Lin H, Harst A, Obermann WM, (2003). Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem, 278, 17228–17235. [DOI] [PubMed] [Google Scholar]
- [40].Wolmarans A, Lee B, Spyracopoulos L, LaPointe P, (2016). The mechanism of Hsp90 ATPase stimulation by Aha1. Sci. Rep, 6, 33179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Duina AA, Marsh JA, Gaber RF, (1996). Identification of two CyP-40-like cyclophilins in Saccharomyces cerevisiae, one of which is required for normal growth. Yeast, 12, 943–952. [DOI] [PubMed] [Google Scholar]
- [42].Truman AW, Millson SH, Nuttall JM, Mollapour M, Prodromou C, Piper PW, (2007). In the yeast heat shock response, Hsf1-directed induction of Hsp90 facilitates the activation of the Slt2 (Mpk1) mitogen-activated protein kinase required for cell integrity. Eukaryot. Cell, 6, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Johnson JL, Halas A, Flom G, (2007). Nucleotide-dependent interaction of Saccharomyces cerevisiae Hsp90 with the cochaperone proteins Sti1, Cpr6, and Sba1. Mol. Cell. Biol, 27, 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ratzke C, Hellenkamp B, Hugel T, (2014). Four-colour FRET reveals directionality in the Hsp90 multicomponent machinery. Nat. Commun, 5, 4192. [DOI] [PubMed] [Google Scholar]
- [45].Mercier R, Wolmarans A, Schubert J, Neuweiler H, Johnson JL, LaPointe P, (2019). The conserved NxNNWHW motif in Aha-type co-chaperones modulates the kinetics of Hsp90 ATPase stimulation. Nat. Commun, 10, 1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li J, Soroka J, Buchner J, (2012). The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta, 1823, 624–635. [DOI] [PubMed] [Google Scholar]
- [47].Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, et al. , (2013). Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget, 4, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mollapour M, Neckers L, (2012). Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta, 1823, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Prodromou C, (2016). Mechanisms of Hsp90 regulation. Biochem. J, 473, 2439–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Scroggins BT, Neckers L, (2007). Post-translational modification of heat-shock protein 90: impact on chaperone function. Expert Opin. Drug Discovery, 2, 1403–1414. [DOI] [PubMed] [Google Scholar]
- [51].Woodford MR, Dunn D, Miller JB, Jamal S, Neckers L, Mollapour M, (2016). Impact of posttranslational modifications on the anticancer activity of Hsp90 inhibitors. Adv. Cancer Res, 129, 31–50. [DOI] [PubMed] [Google Scholar]
- [52].Graf C, Lee CT, Eva Meier-Andrejszki L, Nguyen MT, Mayer MP, (2014). Differences in conformational dynamics within the Hsp90 chaperone family reveal mechanistic insights. Front. Mol. Biosci, 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goldstein AL, McCusker JH, (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast, 15,1541–1553. [DOI] [PubMed] [Google Scholar]
- [54].Mumberg D, Muller R, Funk M, (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene, 156, 119–122. [DOI] [PubMed] [Google Scholar]
- [55].Song Y, Masison DC, (2005). Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein Sti1 (Hop1). J. Biol. Chem, 280, 34178–34185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. , (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.