

Abstract
Catalytic C–H bond functionalization has become an important tool for organic synthesis. Metalloenzymes offer a solution to one of the foremost challenges in this field, site-selective C–H functionalization, but they are only capable of catalyzing a subset of the C–H functionalization reactions known to small molecule catalysts. To overcome this limitation, metalloenzymes have been repurposed by exploiting the reactivity of their native cofactors toward substrates not found in nature. Additionally, new reactivity has been accessed by incorporating synthetic metal cofactors into protein scaffolds to form artificial metalloenzymes. The selectivity and activity of these catalysts has been tuned using directed evolution. This review covers the recent progress in developing and optimizing both repurposed and artificial metalloenzymes as catalysts for selective C–H bond functionalization.
Introduction
Selective carbon-hydrogen (C–H) bond functionalization has the potential to greatly simplify the synthesis of organic molecules [1,2]. Ideally, in place of complex functional group manipulation sequences that often require the use of protecting groups and redox adjustments, C–H bonds could be directly used as handles to forge carbon-carbon and carbon-heteroatom bonds. This simplification could obviate the need for substrate prefunctionalization, improve atom economy, and enable late-stage diversification of complex molecules [3]. While the ubiquity of C–H bonds gives rise to these possibilities, it also necessitates the development of catalysts capable of functionalizing specific C–H bonds [4]. Most recent progress toward this end has focused on organometallic complexes that coordinate to a functional group on the substrate proximal to the desired site of functionalization [5,6]. Selective C–H functionalization in the absence of directing groups [7] is also possible using catalysts that can discriminate subtle steric [8], electronic [9], or stereoelectronic [10,11] properties of different C–H bonds.
Despite impressive advances in catalytic C–H bond functionalization, controlling catalyst selectivity, and perhaps more importantly tuning the selectivity of a given catalyst to functionalize different C–H bonds remains challenging [12]. The synthetic power of C–H functionalization thus remains best illustrated by natural product biosyntheses [13], which frequently involve enzyme-catalyzed functionalization of unactivated C–H bonds on both simple and complex molecular frameworks [14] (Figure 1). The biosynthesis of the diterpenoid taxol [15] (Figure 1a) alone has inspired multiple accounts of how methodology might ultimately achieve the levels of catalytic efficiency and selectivity that enzymes can impart to chemical synthesis [16,17]. At the same time, extensive effort has been devoted to engineering enzymes with altered selectivity or substrate scope [18]. The molecular recognition employed by different enzymes to enable selective catalysis has proven remarkably amenable to directed evolution approaches [19], and a number of enzymes have been evolved to functionalize specific C–H bonds on non-native substrates [14].
Figure 1.
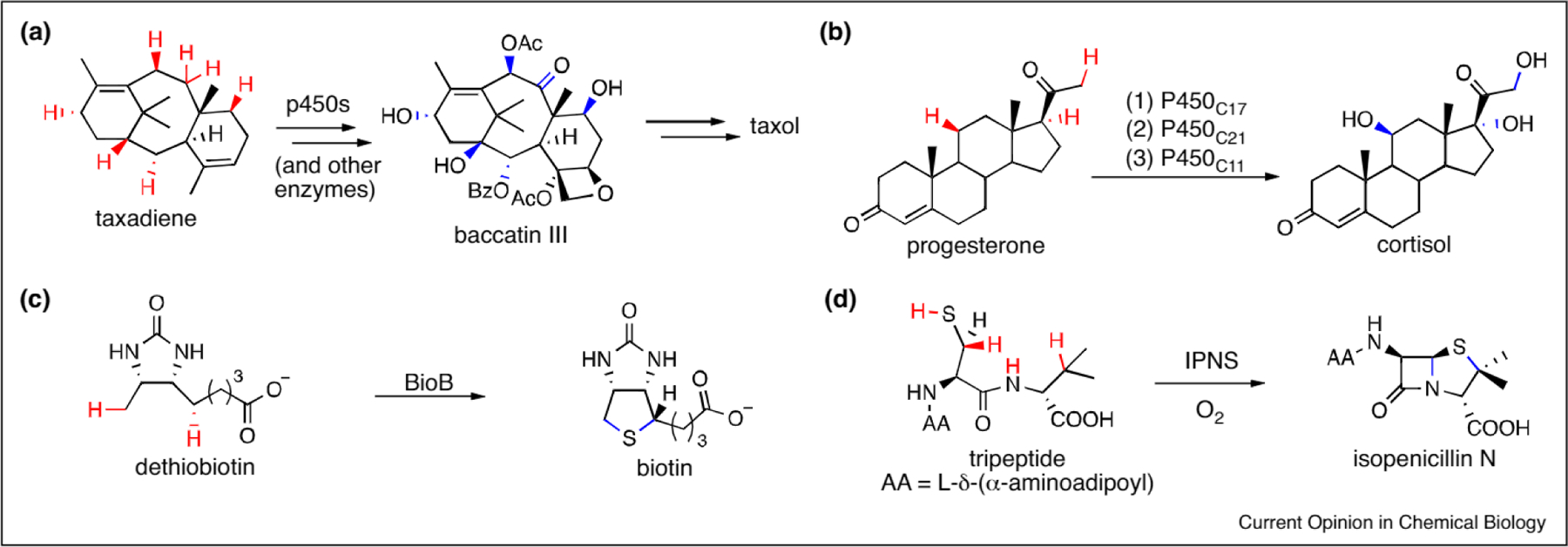
(a) Abridged biosynthetic pathway of the taxol precursor baccatin III from taxadiene [15]. (b) The biosynthesis of cortisol from progesterone through three P450-catalyzed hydroxylations [20]. (c) The radical-based synthesis of biotin by BioB, a [2Fe-2S]- and S-adenosylmethionine-dependent enzyme [14]. (d) The double cyclization to form isopenicillin N catalyzed by IPNS [14].
In general, directed evolution requires some initial level of activity that can be optimized using simple in vitro assays [19]; however, a substantial range of reactions developed by chemists, particularly those catalyzed by transition metal complexes, find no analogue in nature. The absence of different reactions may have resulted for any number of reasons, but the lack of appropriate substrates, the low abundance of precious metals, and the air and water sensitivity of many ligands and metal complexes likely contributed. Expanding the scope of enzymatic catalysis to enable abiological reactions has therefore been achieved by providing natural enzymes with the necessary substrates (enzyme repurposing) [21•], or incorporating the synthetic metal catalysts into enzymes (artificial metalloenzymes) [22•]. Importantly, catalysts developed using both approaches can then be improved using the same directed evolution strategies previously discussed [23•]. Moreover, these catalysts provide novel platforms to investigate how precise control over the environment surrounding a metal ion or complex, its secondary coordination sphere, can impact its reactivity [24].
This review will cover recent advances in unactivated C–H bond functionalization catalyzed by repurposed and artificial metalloenzymes. It is intended to be an introduction to these catalysts and the unique opportunities they afford, including the use of directed evolution to overcome challenges associated with selective C–H functionalization. To ensure concise coverage of the topic, detailed analysis of chemical mechanisms and alternative reactions catalyzed by these metalloenzymes will not be discussed. Readers are directed to the many excellent reviews cited throughout this article for a complete discussion of these topics.
Repurposed metalloenzymes
The ability of enzymes to catalyze C–H bond functionalization and many other transformations often requires non-proteinogenic cofactors, including redox active organic compounds, transition metal complexes, and metal ions [25]. The reactivity of these species has inspired extensive efforts to create small molecules that mimic these cofactors, either structurally or functionally [26]. While efforts toward both of these ends have proven successful, the reactivity and selectivity of these species is typically far less than that of the corresponding enzymes. In short, cofactors are a necessary but not sufficient component of metalloenzyme reactivity. Researchers have therefore begun exploring the reactivity of native cofactors in their protein hosts [21•]. The surprisingly high activity of these repurposed enzymes toward non-native reactions illustrates the potential benefits of exploiting enzyme complexity rather than relying on reductionist cofactor-centric approaches.
Selective oxidation of aliphatic C–H bonds by cytochromes P450, several examples of which occur in the aforementioned taxol biosynthesis (Figure 1a) [15], have long inspired efforts to create metalloporphyrin analogues of the heme cofactors in these enzymes [27]. In its native context, the heme cofactor mediates oxygen activation to form a reactive FeIV-oxo species (1, Figure 2a) that can hydroxylate aliphatic C–H bonds via an ‘oxygen rebound’ mechanism. Studies on these enzymes and on porphyrin models have shown that Fe-oxo, -peroxo, and -superoxo intermediates can also be accessed via shunt pathways using a range of oxidants [26]. Similarly, suitable nitrene and carbene precursors led to the discovery that metalloporphyrins could catalyze nitrene and carbene insertion into C–H bonds via shunt-like mechanisms [28,29].
Figure 2.
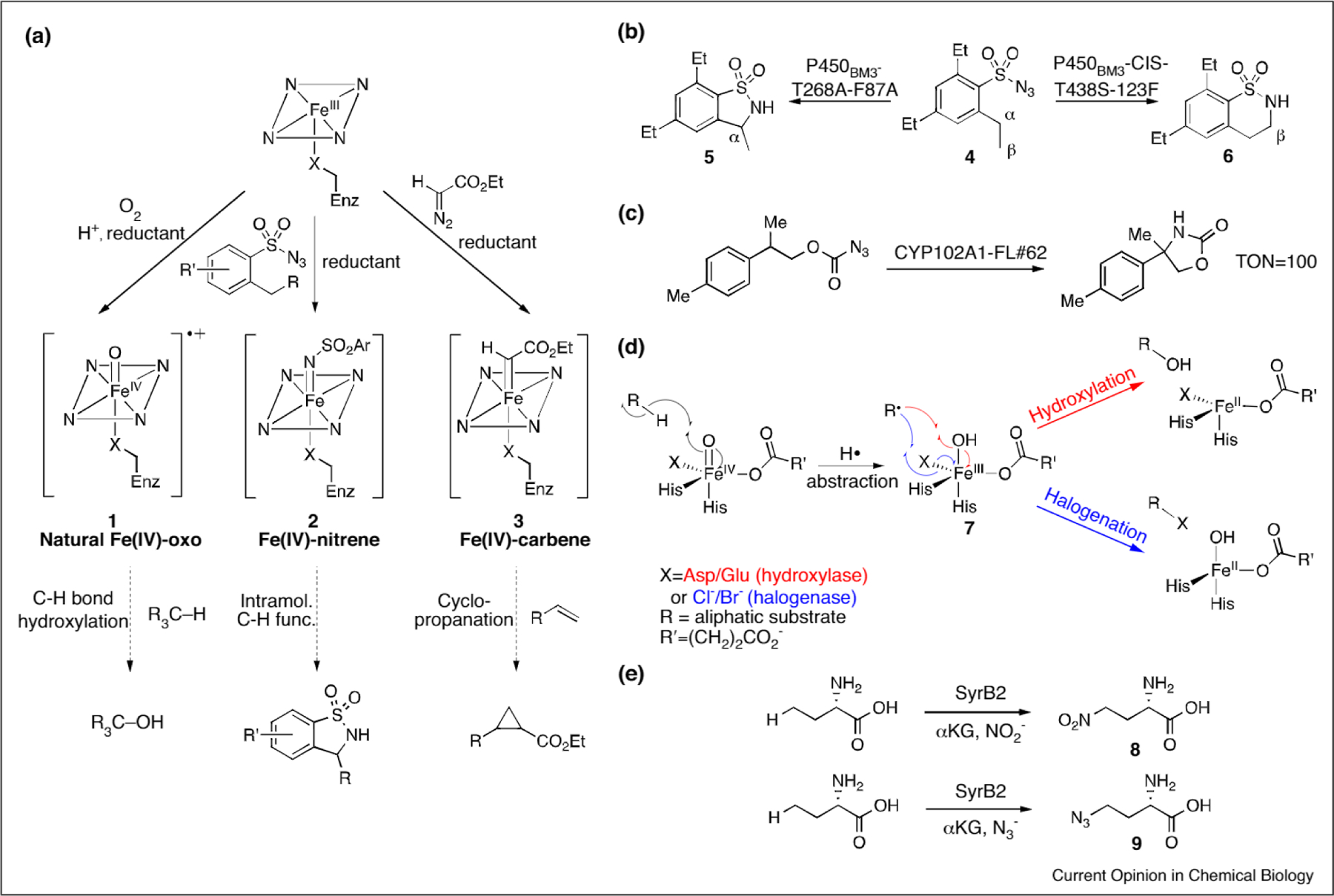
(a) Native and non-native reactions catalyze by heme-containing proteins, (b) P450-catalyzed nitrene insertion into the α (left) or β (right) C–H bond of a sulfonylazide substrate, (c) P450-catalyzed nitrene insertion into the benzylic position of a carbonazidate substrate, (d) Divergent radical rebound mechanism by αKG-dependent hydroxylases (red) and halogenases (blue), (e) Nitration (top) and azidation (bottom) catalyzed by the repurposed FeII- and αKG-dependent halogenase SyrB2.
Armed with this knowledge, Svastits et al. established more than thirty years ago that cytochromes P450 can catalyze both intramolecular and intermolecular nitrene insertion into C–H bonds by providing iminoiodinane substrates to purified microsomal enzymes [30]. Remarkably, no further investigations of similar reactions were reported until McIntosh et al. [31] and later Fasan and co-workers [32,33] showed that engineered P450s can catalyze analogous nitrene insertion chemistry using sulfonylazide substrates (Figure 2b). The FeIV-nitrene reactive intermediate 2 was found to insert into a proximal C–H bond of sulfonylazide 4 to form a cyclized benzosultam (5, Figure 2b). Axial heme ligation played an important role in the success of these nitrene insertion reactions; native cytochrome P450bm3, which contains an axial cysteine residue, achieved a turnover number (TON) of 2.1 while the corresponding serine mutant gave a TON of 32. Serine ligation both eliminated oxygenase activity and increased the FeIII-to-FeII reduction potential of heme, facilitating its reduction by NADPH to the ferrous state required for the desired nitrene insertion chemistry [34•]. A regiodivergent variant (Figure 2b) that functionalized an unactivated primary C–H bond to form 6 over 5 was identified via targeted mutagenesis of the BM3 active site [35••]. Substantially expanding the substrate scope of P450-catalyzed nitrene insertion, Singh et al. demonstrated that carbonazidates could also be used as nitrene precursors to generate oxazolidinone products following intramolecular C–H insertion (Figure 2c) [36]. Though enantioselectivity was not observed, the engineered cytochrome P450 CYP102A1-FL#62 provided TON up to 100.
The unique coordination environments of non-heme iron-dependent metalloenzymes have also been exploited to catalyze non-native reactions. FeII α-ketoglutarate (αKG)-dependent oxygenases, for example, contain an active site FeII center ligated by two histidine residues and either an aspartate or a glutamate residue. These enzymes catalyze C–H hydroxylation via a rebound pathway analogous to that described for heme enzymes, but involving intermediate 7 (Figure 2d). Interestingly, the active sites of structurally related αKG-dependent halogenases contain either an alanine or a glycine residue in place of the aforementioned aspartate or glutamate residues in hydroxylases, leaving an open coordination site that is occupied by a chloride or bromide anion. Rebound can then occur to give the corresponding halogenated products, as shown in Figure 2d. Initial attempts to repurpose hydroxylases as halogenases by opening up the coordination site by mutating aspartic/glutamic acid to alanine proved unsuccessful, perhaps caused by the substrate positioning and halide stabilizing capability of natural halogenase active sites [37,38]. While endowing halogenase activity to a hydroxylase has been challenging, repurposing the SyrB family of αKG-dependent halogenases to incorporate non-halogen functionality was reported by Matthews et al. [39]. In this work, nitrate or azide anions took the place of the natural ligands and successfully produced the ‘rebound’ products 8 and 9, respectively (Figure 2e). Though yields were initially less than 10% for both substrates, targeted mutagenesis of active site residues increased the yield of 8 to 52% and 9 to greater than 20%.
Artificial metalloenzymes
The examples noted above show the great potential of repurposing natural enzymes to catalyze non-native reactions. Efforts to evolve enzymes for these non-native reactions are in their infancy, leaving open the question of exactly how far their reactivity can be pushed. For example, while heme proteins can catalyze oxygenation via 2, amination via 3, and cyclopropanation via 4, C–H insertion of carbenes has not been reported. The expansion of reactivity may ultimately be limited not by mutable aspects of the host protein, but by the intrinsic chemical reactivity of cofactors. For example, C–H insertion of carbenes catalyzed by FeIII-porphyrins requires high reaction temperatures (>80°C) in neat substrate [28,29], and even under these forcing conditions, complex product mixtures typically result. Perhaps directed evolution can rectify these reactivity problems; perhaps not. Even if reactivity is not inherently limiting, however, a given active site may not accommodate particular substrates caused by their size or other physical properties.
If evolution or genome mining efforts cannot overcome the limitations of specific metal centers or active sites, however, appropriate metal cofactors can instead be incorporated into suitable protein scaffolds. The resulting artificial metalloenzymes could then serve as starting points for subsequent evolution efforts to improve activity and selectivity of catalysts for C–H bond functionalization. While enzyme repurposing has focused on catalyzing synthetic reactions using enzymes, efforts to engineer artificial metalloenzymes are motivated by possibility of improving control over the reactivity and selectivity of synthetic catalysts [22•,40]. The profound influence that protein scaffolds have over the reactivity of cofactors in native metalloenzymes {vide supra) raises the question of just how much better small molecule catalysts could become within an artificial metalloenzyme. Though some of the earliest investigations into ArMs occurred some 40 years ago [41,42], interest in the area has accelerated in recent years, bringing with it new and unique catalysts for C–H bond functionalization.
Metalloprotein scaffolds
The active sites of metalloenzymes, which evolved to bind metal cofactors and small molecule substrates, make natural candidates for ArM formation using synthetic cofactors structurally analogous to the native cofactor. This approach was taken by Oohora etal. [43••] to construct the first ArM that catalyzed C(sp3)–H bond functionalization. Horse heart myoglobin (Mb) was reconstituted with a Mn-porphycene cofactor (Figure 3a) to form the ArM Mb⊂Mn-porphycene. While Mb containing the native Fe-protoporphyrin IX cofactor (Fe-PPIX, Figure 3a) does not have any hydroxylase activity, reconstituting Mb⊂Mn-porphycene led to an ArM that hydroxylated C(sp3)-H bonds in the presence of H2O2. The catalyst, which the authors suggest proceeds through a rebound mechanism involving a MnV-oxo intermediate, achieved a TON of 13. Importantly, free Mn-porphycene and Mb reconstituted with Mn-PPIX or Fe-porphycene gave no desired product. The authors suggest the axial ligation by a histidine residue is required for reactivity of Mb⊂Mn-porphycene. The reactivity conferred to the unreactive metalloenzyme demonstrates the impact that the identity of the metal and cofactor has on an enzyme, properties that can be finely tuned in ArMs.
Figure 3.
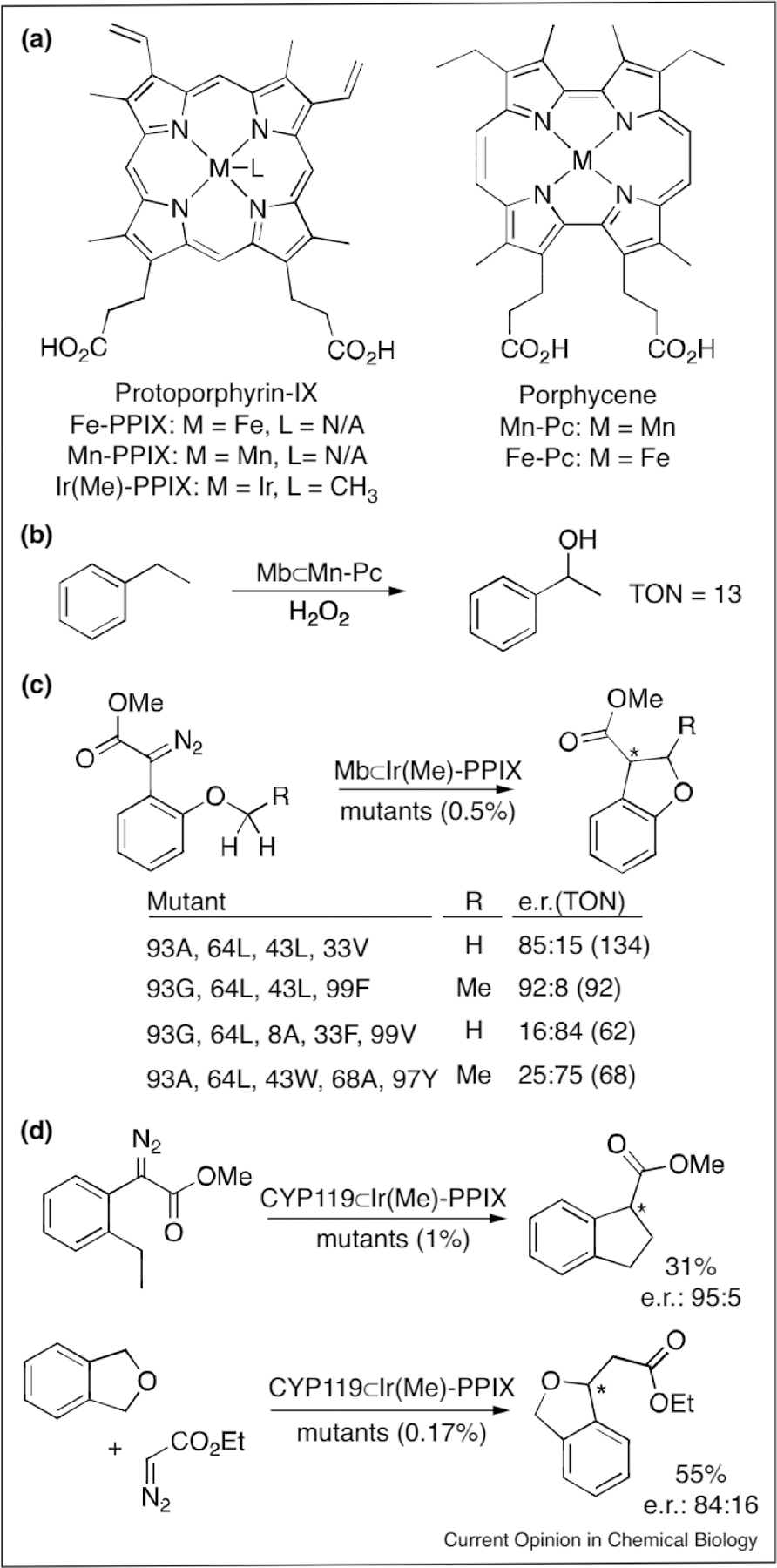
(a) The structure of protoporphyrin-IX (PPIX), porphycene and associated complexes, (b) Benzylic oxygenation catalyzed by Mb⊂Mn-porphycene. (c) The mutations and resulting catalytic profile of selected variants of Mb⊂lr(Me)-PPIX for the reaction shown. (d) Intramolecular and intermolecular C–H insertion catalyzed by CYP119⊂lr(Me)-PPIX.
Key et al. later developed an ArM capable of C(sp3)–H bond functionalization using mutants of Physeter macrocephalus myoglobin reconstituted with an IrIII-protoporphyrin IX (Ir(Me)-PPIX, Figure 3a) [44]. This cofactor performed the best in a screen of nine metal(ligand)-PPIX and eight different axially-coordinating residues, with WT-Mb⊂Ir(Me)-PPIX catalyzing the reaction shown in Figure 3c (R = H) with yields up to 50% and an e.r. of 1:1. Targeted mutagenesis was then used to engineer a catalyst with improved activity and selectivity for both product enantiomers (Figure 3c). In a follow-up study [45••], the same group used a thermophilic P450 (CYP119) from Sulfolobus solfataricus as the scaffold supporting the same cofactor, Ir(Me)-PPIX. A similar mutagenesis approach was used to improve the catalytic activity of this ArM, engineering catalysts capable of an e.r. of 97:3 (582 TON) and of 96.5:3.5 (120 TON). Kinetic studies on this library of ArMs indicated that the most efficient variant achieved kinetics on par with a similar class of natural metalloenzymes (kcat = 45.8 min−1, KM = 0.17 mM, and kcat/KM = 269 min−1 mM−1). The capacity for rate acceleration caused by substrate binding by an enzyme scaffold illustrates the unique potential of these hybrid catalysts compared to their small molecule analogues. These ArMs also catalyzed a greater scope of C–H functionalization reactions, including less activated C(sp3)-H bonds and intermolecular functionalization (Figure 3d).
These examples show that metalloproteins reconstituted with unnatural metal-porphyrins provide an excellent platform for ArM engineering. Interestingly, both reactions are hypothesized to proceed through intermediates analogous to those in the repurposed heme metalloenzymes previously discussed. The proposed MnV-oxo species is comparable to the FeIV-oxo intermediate 1, and the IrIII-carbene is similar in structure to the FeIV-carbene 3, though the electronic characteristics of these intermediates are likely distinct. Regardless of the identity of the metal, each of these reactions proceeds through a metal-atom double bond with electrophilic character [46,47].
Non-metalloprotein scaffolds
Developing ArMs from non-metalloenzyme scaffolds presents an opportunity to sample a broader scope of active sites and metal cofactors beyond those that already exist in native metalloproteins. This could provide more general starting points for evolving selective catalysts, but it also necessitates selection or development of protein scaffolds that can accommodate the highly reactive metal fragments used in C–H functionalization. The very first example of ArM-catalyzed C–H bond functionalization was reported by Hyster et al. [48••] using the streptavidin (Sav) scaffold. Biotinylated metal cofactors, such as 10 (Figure 4a), are tightly bound by Sav, providing a straight-forward process for ArM formation. RhIII-catalyzed aromatic C–H functionalization reactions had been previously explored, though efforts to achieve enantioselectivity were hindered by difficulties introducing asymmetric ligands caused by a lack of coordination sites available on Rh during catalysis [48••]. The secondary coordination sphere provided by the Sav ArM proved powerful however, as WT-Sav⊂10 catalyzed the coupling of an arene C–H bond to an olefin forming a dihydroisoquinoline (Figure 4b) with a 46% yield, 9:1 regioisomeric ratio, and a 75:25 enantiomeric ratio (major regioisomer) of products in acetate buffer. To avoid the need for added acetate and to potentially accelerate the reaction, which proceeds via concerted metalation/deprotonation (Figure 4c), aspartate and glutamate residues were introduced at position 112 in the active site. The resulting ArM provided improved conversion (95%) and selectivity (r.r. of 19:1 and e.r. of 91:9). Increased rates compared to WT were also observed for the rationally designed mutants, which also had up to 92-fold higher activity than free 10. While regioselectivity was achieved due to coordination of the substrate to the metal before C–H bond cleavage, asymmetry was enabled by the secondary coordination sphere of the enzymatic scaffold.
Figure 4.
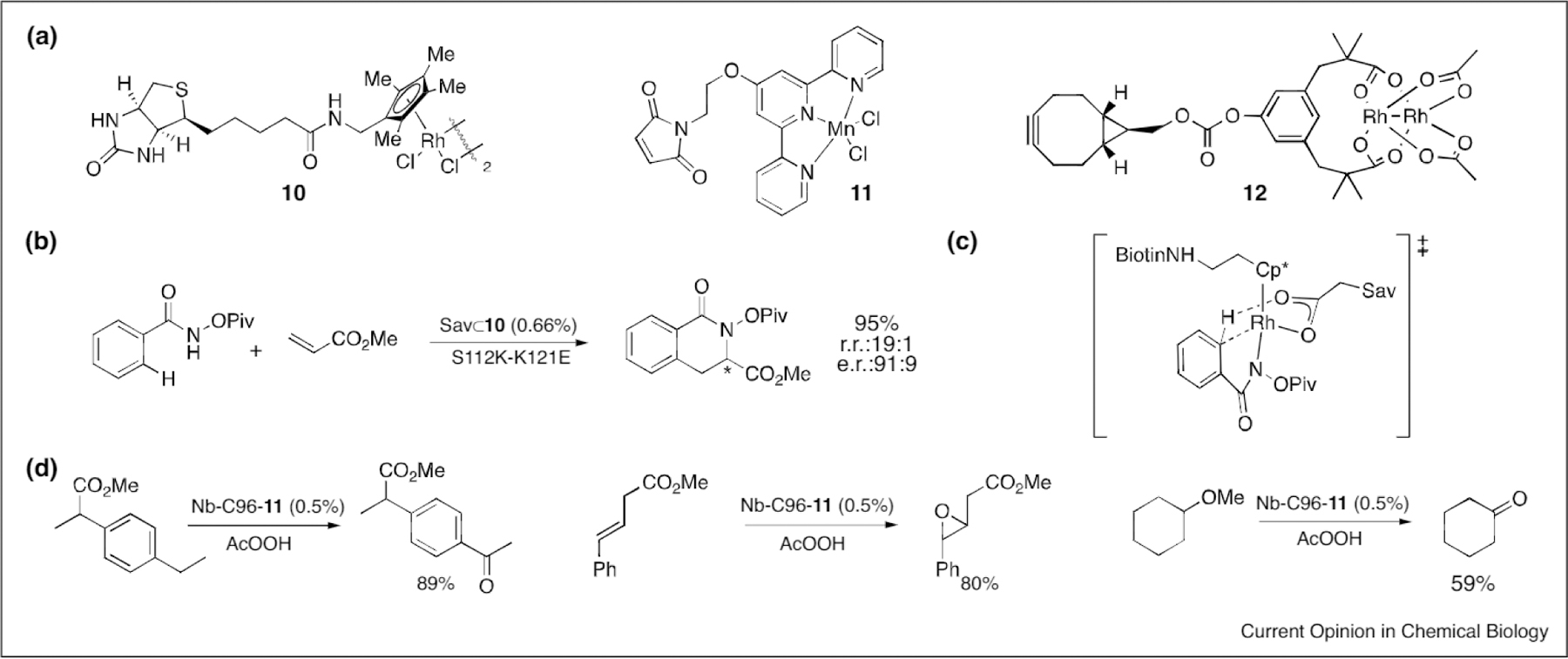
(a) Cofactors used to form ArMs for C–H functionalization, (b) Example C–H functionalization catalyzed by Sav⊂10. (c) Proposed transition state in the C–H activation/deprotonation step, (d) Selected oxygenation reactions catalyzed by Nb-C96-11.
Oxygenation of C–H bonds using a non-metalloenzyme scaffold was reported by Zhang et al. using an Mn-terpyridine ArM [49]. A cysteine mutant (C96) of apo-nitrobindin (Nb) was used a scaffold to which maleimide-substituted cofactor 11 (Figure 4a) was covalently linked. The ArM was then used to catalyze a variety of reactions, such as benzylic oxygenation, epoxidation of olefins, and oxidative cleavage of cyclohexyl methyl ether (Figure 4d). Unfortunately, the selectivity in each of these reactions was identical to that observed by the cofactor alone. This finding highlights a key challenge facing efforts to use non-metalloproteins for ArM formation: the scaffold must either serendipitously orient cofactor and substrate in a manner suitable for selective catalysis, or it must be engineered to do so. Toward this end, Yang et al. explored the incorporation of 11 and various other cofactors, including dirhodium cofactor 12 (Figure 4a) into a range of different protein scaffolds [50]. This led to the finding that a prolyl oligopeptidase based ArMs (POP-12) can catalyze highly enantioselective carbene insertion reactions using donor-acceptor carbenes [51••]. To date, carbene insertion reactions using such carbene precursors have not been reported in either repurposed heme enzymes or artificial metalloenzymes, showing how non-native cofactors can enable novel reactions even in cases where similar reactivity has been established. Efforts to optimize the C–H insertion activity of POP-12 is underway.
Conclusion and outlook
Repurposed and artificial metalloenzymes have shown their potential as tunable catalysts for selective C–H bond functionalization. Given the range of C–H functionalization reactions catalyzed by small molecule analogues of metalloenzyme cofactors [27–29], repurposing native metalloenzymes [21•] to catalyze these same reactions has the potential to significantly expand the scope of biocatalytic C–H functionalization. An even broader range of C–H functionalization reactions can perhaps be expected to succumb to ArM catalysis given the diversity of synthetic C–H functionalization catalysts that could be incorporated into protein scaffolds [22•]. While relatively well-known enzymes and proteins have been repurposed and used for ArM formation to date, genome mining [52] and computational design [53] could be used to significantly expand the range of sequence space that has been interrogated for both approaches, leading to an even broader range of new C–H functionalization catalysts. Directed evolution of both repurposed and artificial metalloenzymes will then permit rapid optimization of these catalysts [19], ultimately enabling the remarkable levels of selectivity and activity exhibited by natural metalloenzymes [15] (Figure 1) for synthetic reactions.
Acknowledgements
This material is based upon work supported by, or in part by, the U. S. Army Research Laboratory and the U. S. Army Research Office under grant number W911NF-14-1-0334, The David and Lucile Packard Foundation, and the NSF under the CCI Center for Selective C–H Functionalization (CHE-1205646). D. M. Upp was funded by an NIH Chemistry and Biology Interface Training Grant (T32 GM008720).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Chen DYK, Youn SW: C–H activation: a complementary tool in the total synthesis of complex natural products. Chem Eur J 2012, 18:9452–9474. [DOI] [PubMed] [Google Scholar]
- 2.White MC: Adding aliphatic C–H bond oxidations to synthesis. Science 2012, 335:807–809. [DOI] [PubMed] [Google Scholar]
- 3.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW: The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem Soc Rev 2016, 45:546–576. [DOI] [PubMed] [Google Scholar]
- 4.Bergman RG: Organometallic chemistry: C–H activation. Nature 2007, 446:391–393. [DOI] [PubMed] [Google Scholar]
- 5.Engle KM, Mei TS, Wasa M, Yu JQ: Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc Chem Res 2012, 45:788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neufeldt SR, Sanford MS: Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc Chem Res 2012, 45:936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartwig JF, Larsen MA: Undirected, homogeneous C–H bond functionalization: challenges and opportunities. ACS Cent Sci 2016, 2:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartwig JF: Borylation and silylation of C–H bonds: a platform for diverse C–H bond functionalizations. Acc Chem Res 2012, 45:864–873. [DOI] [PubMed] [Google Scholar]
- 9.O’Hara F, Blackmond DG, Baran PS: Radical-based regioselective C–H functionalization of electron-deficient heteroarenes: scope, tunability, and predictability. J Am Chem Soc 2013, 135:12122–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gormisky PE, White MC: Catalyst-controlled aliphatic C–H oxidations with a predictive model for site-selectivity. J Am Chem Soc 2013, 135:14052–14055. [DOI] [PubMed] [Google Scholar]
- 11.Davies HML, Morton D: Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes. Chem Soc Rev 2011, 40:1857. [DOI] [PubMed] [Google Scholar]
- 12.Andorfer MC, Park HJ, Vergara-Coll J, Lewis JC: Directed evolution of RebH for catalyst-controlled halogenation of indole C–H bonds. Chem Sci 2016:1–10. [DOI] [PMC free article] [PubMed]
- 13.Coelho PS, Lewis JC, Arnold FH: Synthetic biology approaches for organic synthesis In Comprehensive Organic Synthesis. Edited by Knochel P, Molander GA. Elsevier Ltd.; 2014:390–420. [Google Scholar]
- 14.Lewis JC, Coelho PS, Arnold FH: Enzymatic functionalization of carbon-hydrogen bonds. Chem Soc Rev 2011, 40:2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaspera R, Croteau R: Cytochrome P450 oxygenases of taxol biosynthesis. Phytochem Rev 2006, 5:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walji AM, MacMillan DWC: Strategies to bypass the taxol problem. Enantioselective cascade catalysis, a new approach for the efficient construction of molecular complexity. Synlett 2007, 12:1477–1489. [Google Scholar]
- 17.Ishihara Y, Baran P: Two-phase terpene total synthesis: historical perspective and application to the Taxol® problem. Synlett 2010:1733–1745.
- 18.Lewis JC, Arnold FH: Catalysts on demand: selective oxidations by laboratory-evolved cytochrome P450 BM3. Chim Int J Chem 2009, 63:309–312. [Google Scholar]
- 19.Romero PA, Arnold FH: Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 2009, 10:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanukoglu I: Steroidogenic enzymes: structure function, and role in regulation of steroid hormone biosynthesis. J Steroid Biochem Mol Biol 1992, 43:779–804. [DOI] [PubMed] [Google Scholar]
- 21.•.Prier CK, Arnold FH: Chemomimetic biocatalysis: exploiting the synthetic potential of cofactor-dependent enzymes to create new catalysts. J Am Chem Soc 2015, 137:13992–14006.Provides a nice overview of catalysis using repurposed enzymes.
- 22.•.Lewis JC: Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal 2013, 3:2954–2975.Provides a nice overview of catalysis using artificial metalloenzymes.
- 23.•.Hyster TK, Ward TR: Genetic optimization of metalloenzymes: enhancing enzymes for non-natural reactions. Angew Chem Int Ed 2016, 55:7344–7357.Outlines progress made toward evolving artificial metalloenzymes.
- 24.Lu Y, Berry SM, PfisterTD: Engineering novel metalloproteins: design of metal-binding sites into native protein scaffolds. Chem Rev 2001, 101:3047–3080. [DOI] [PubMed] [Google Scholar]
- 25.Ragsdale SW: Metals and their scaffolds to promote difficult enzymatic reactions. Chem Rev 2006, 106:3317–3337. [DOI] [PubMed] [Google Scholar]
- 26.Groves JT: High-valent iron in chemical and biological oxidations. J Inorg Biochem 2006, 100:434–447. [DOI] [PubMed] [Google Scholar]
- 27.Que L Jr, Tolman WB: Biologically inspired oxidation catalysis. Nature 2008, 455:333–340. [DOI] [PubMed] [Google Scholar]
- 28.Che C-M, Lo VK-Y, Zhau C-Y, Huang J-S: Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem Soc Rev 2011,40:1950–1975. [DOI] [PubMed] [Google Scholar]
- 29.Lu H, Zhang XP: Catalytic C–H functionalization by metalloporphyrins: recent developments and future directions. Chem Soc Rev 2011, 40:1899–1909. [DOI] [PubMed] [Google Scholar]
- 30.Svastits EW, Dawson JH, Breslow R, Gellman SH: Functionalized nitrogen atom transfer catalyzed by cytochrome P-450. J Am Chem Soc 1985, 107:6427–6428. [Google Scholar]
- 31.McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH: Enantioselective intramolecular C–H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew Chem Int Ed 2013, 52:9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh R, Bordeaux M, Fasan R: P450-catalyzed intramolecular sp 3C–H amination with arylsulfonyl azide substrates. ACS Catal 2014, 4:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bordeaux M, Singh R, Fasan R: Intramolecular C(sp3)H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg Med Chem 2014, 22:5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.•.Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM: A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat Chem Biol 2013, 9:485–487.A repurposed P450 with high activity toward cyclopropanation via a putative heme-carbene inermediate.
- 35.••.Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH: Enzyme-controlled nitrogen-atom transfer enables regiodivergent C–H amination. J Am Chem Soc 2014, 136:15505–15508.Site-selective C–H amination catalyzed by a repurposed P450.
- 36.Singh R, Kolev JN, Sutera PA, Fasan R: Enzymatic C(sp3)–H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal 2015, 5:1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM: Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci USA 2009, 106:17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulik HJ, Blasiak LC, Marzari N, Drennan CL: First-principles study of non-heme Fe(II) halogenase SyrB2 reactivity. J Am Chem Soc 2009, 131:14426–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matthews ML, Chang W, Layne AP, Miles LA, Krebs C, Bollinger JM: Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat Chem Biol 2014, 10:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinisch T, Ward TR: Latest developments in metalloenzyme design and repurposing. Eur J Inorg Chem 2015:3406–3418. [Google Scholar]
- 41.Yamamurua K, Kaiser ET: Studies on the oxidase activity of copper(II) carboxypeptidase A. J Chem Soc Chem Commun 1976:830–831. [Google Scholar]
- 42.Wilson ME, Whitesides GM: Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J Am Chem Soc 1978, 100:306–307. [Google Scholar]
- 43.••.Oohora K, Kihira Y, Mizohata E, Inoue T, Hayashi T: C(sp3)–H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J Am Chem Soc 2013, 135:17282–17285.First example of sp3 C–H functionalization catalyzed by an artificial metalloenzyme.
- 44.Key HM, Dydio P, Clark DS, Hartwig JF: Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534:534–537. [DOI] [PubMed] [Google Scholar]
- 45.••.Dydio P, Key HM, Nazarenko A, Rha JY-E, Seyedkazemi V, Clark DS, Hartwig JF: An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354:102–106.Carbene insertion into unactivated C–H bonds catalyzed by an artificial metalloenzyme.
- 46.Moreau Y, Chen H, Derat E, Hirao H, Bolm C, Shaik S: NR transfer reactivity of azo-compound I of P450 How does the nitrogen substituent tune the reactivity of the species toward C–H and C=C activation? J Phys Chem B 2007, 111:10288–10299. [DOI] [PubMed] [Google Scholar]
- 47.Khade RL, Fan W, Ling Y, Yang L, Oldfield E, Zhang Y: Iron porphyrin carbenes as catalytic intermediates: structures, mossbauer and NMR spectroscopic properties, and bonding. Angew Chem Int Ed 2014, 53:7574–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.••.Hyster TK, Knorr L, Ward TR, Rovis T: Biotinylated Rh(III) complexes in engineered streptavadin for accelerated asymmetric C–H activation. Science 2012, 338:500–503.Early example of C–H functionalization catalyzed by an artificial metalloenzyme.
- 49.Zhang C, Srivastava P, Ellis-Guardiola K, Lewis JC: Manganese terpyridine artificial metalloenzymes for benzylic oxygenation and olefin epoxidation. Tetrahedron 2014, 70:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H, Srivastava P, Zhang C, Lewis JC: A general method for artificial metalloenzyme formation through strain-promoted azide-alkyne cycloaddition. ChemBioChem 2014, 15:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.••.Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC: Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat Commun 2015, 6:7789.Details the formation of an artificial metalloenzyme for selective cyclopropanation via carbene transfer using a non-metalloenzyme scaffold.
- 52.Furuya T, Kino K: Genome mining approach for the discovery of novel cytochrome P450 biocatalysts. Appl Microbiol Biotechnol 2010, 86:991–1002. [DOI] [PubMed] [Google Scholar]
- 53.Munoz Robles V, Ortega-Carrasco E, Alonso-Cotchico L, Rodriguez-Guerra J, Lledos A, Marechal JD: Toward the computational design of artificial metalloenzymes: from protein-ligand docking to multiscale approaches. ACS Catal 2015, 5:2469–2480. [Google Scholar]