

Abstract
The coronavirus disease 2019 pandemic has made deployment of an effective vaccine a global health priority. We evaluated the protective activity of a chimpanzee adenovirus-vectored vaccine encoding a prefusion stabilized spike protein (ChAd-SARS-CoV-2-S) in challenge studies with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and mice expressing the human angiotensin-converting enzyme 2 receptor. Intramuscular dosing of ChAd-SARS-CoV-2-S induces robust systemic humoral and cell-mediated immune responses and protects against lung infection, inflammation, and pathology but does not confer sterilizing immunity, as evidenced by detection of viral RNA and induction of anti-nucleoprotein antibodies after SARS-CoV-2 challenge. In contrast, a single intranasal dose of ChAd-SARS-CoV-2-S induces high levels of neutralizing antibodies, promotes systemic and mucosal immunoglobulin A (IgA) and T cell responses, and almost entirely prevents SARS-CoV-2 infection in both the upper and lower respiratory tracts. Intranasal administration of ChAd-SARS-CoV-2-S is a candidate for preventing SARS-CoV-2 infection and transmission and curtailing pandemic spread.
Keywords: vaccine, SARS-CoV-2, COVID-19, mucosal immunity, IgA, intranasal, protection, pathogenesis, antibody, T cells
Graphical Abstract
Highlights
-
•
Chimpanzee adenoviral vaccines encoding stabilized S induce neutralizing Abs
-
•
Chimpanzee adenoviral vaccines protect against SARS-CoV-2 infection and pneumonia
-
•
Intranasal vaccine delivery generates robust mucosal B and T cell responses
-
•
Intranasal ChAd-SARS-CoV-2 prevents upper and lower respiratory tract infection
Intranasal or intramuscular immunization of ChAd-SARS-CoV-2, a chimpanzee adenoviral vaccine encoding stabilized spike protein, prevents SARS-CoV-2 lung infection and pneumonia in mice. In particular, intranasally delivered ChAd-SARS-CoV-2 uniquely prevents both upper and lower respiratory tract infections, potentially protecting against SARS-CoV-2 infection and transmission.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a positive-sense, single-stranded RNA virus that was first isolated in late 2019 from patients with severe respiratory illness in Wuhan, China (Zhou et al., 2020b). SARS-CoV-2 is related to two other highly pathogenic respiratory viruses, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). SARS-CoV-2 infection results in a clinical syndrome, coronavirus disease 2019 (COVID-19), that can progress to respiratory failure (Guan et al., 2020) and also present with cardiac pathology, gastrointestinal disease, coagulopathy, and a hyperinflammatory syndrome (Cheung et al., 2020; Mao et al., 2020; Wichmann et al., 2020). The elderly, immunocompromised, and those with certain co-morbidities (e.g., obesity, diabetes, and hypertension) are at greatest risk of death from COVID-19 (Zhou et al., 2020a). Virtually all countries and territories have been affected with more than twenty-seven million infections to date, hundreds of thousands of deaths, and a case-fatality rate estimated at ∼4%. The extensive morbidity, mortality, and destabilizing socioeconomic consequences of COVID-19 highlight the urgent need for deployment of an effective SARS-CoV-2 vaccine to mitigate the severity of infection, curb transmission, end the pandemic, and prevent its return.
The SARS-CoV-2 RNA genome is approximately 30,000 nt in length. The 5′ two-thirds encode nonstructural proteins that enable genome replication and viral RNA synthesis. The remaining one-third encode structural proteins, such as spike (S), envelope, membrane, and nucleoprotein (NP), that form the spherical virion and accessory proteins that regulate cellular responses. The S protein forms homotrimeric spikes on the virion and engages the cell-surface receptor angiotensin-converting enzyme 2 (ACE2) to promote coronavirus entry into human cells (de Wit et al., 2016; Letko et al., 2020). The SARS-CoV and SARS-CoV-2 S proteins are cleaved sequentially during the entry process to yield S1 and S2 fragments, followed by further processing of S2 to yield a smaller S2′ protein (Hoffmann et al., 2020). The S1 protein includes the receptor binding domain (RBD), and the S2 protein promotes membrane fusion. The structure of a soluble, stabilized prefusion form of the SARS-CoV-2 S protein was solved by cryoelectron microscopy, revealing considerable similarity to the SARS-CoV S protein (Wrapp et al., 2020). This form of the S protein is recognized by potently neutralizing monoclonal antibodies (Cao et al., 2020; Pinto et al., 2020; Zost et al., 2020) and could serve as a promising vaccine target.
Release of the SARS-CoV-2 genome sequence spurred immediate development of vaccine candidates that principally targeted the viral S protein (Burton and Walker, 2020). Multiple platforms have been developed to deliver the SARS-CoV-2 S protein, including DNA plasmid, lipid nanoparticle encapsulated mRNA, inactivated virion, and viral-vectored vaccines (Graham, 2020). Several vaccines have entered clinical trials to evaluate safety, and some have advanced to trials assessing immunogenicity and efficacy. Most vaccines advanced to human testing without substantive efficacy data in animals (Diamond and Pierson, 2020). This circumstance occurred in part because vaccine design and development outpaced the generation of accessible pre-clinical disease models of SARS-CoV-2 infection and pathogenesis.
Adenovirus (Ad)-based vaccines against coronaviruses have been evaluated previously. A single dose of a chimpanzee Ad-vectored vaccine encoding the S protein of MERS-CoV protected mice from infection (Munster et al., 2017), reduced virus shedding, and enhanced survival in camels (Alharbi et al., 2019) and was safe and immunogenic in humans in a phase 1 clinical trial (Folegatti et al., 2020a). A human Ad-based vaccine expressing a MERS S1-CD40L fusion protein also was protective in mice (Hashem et al., 2019). An Ad-based SARS-CoV vaccine expressing the S protein prevented pneumonia in ferrets after challenge and was highly immunogenic in rhesus macaques (Kobinger et al., 2007). A chimpanzee Ad vector (Y25, a simian Ad-23; Dicks et al., 2012) encoding the SARS-CoV-2 S protein (ChAdOx1 nCoV-19) is currently under evaluation in humans as a single intramuscular injection (NCT04324606). In rhesus macaques, this vaccine protects against lung infection and pneumonia, but not against upper respiratory tract infection and virus shedding (van Doremalen et al., 2020).
Here, we develop a different chimpanzee Ad (simian Ad-36)-based SARS-CoV-2 vaccine (ChAd-SARS-CoV-2-S) encoding a prefusion stabilized spike (S) protein after introducing two proline substitutions in the S2 subunit (Pallesen et al., 2017). Intramuscular administration of ChAd-SARS-CoV-2-S induced robust systemic humoral and cell-mediated immune responses. One or two vaccine doses protected against lung infection, inflammation, and pathology after SARS-CoV-2 challenge of mice that transiently express the human ACE2 (hACE2) receptor. Despite the induction of high levels of neutralizing antibody, neither dosing regimen completely protected against SARS-CoV-2 infection, as substantial levels of viral RNA were still detected in the lung. In comparison, a single intranasal dose of ChAd-SARS-CoV-2-S induced high levels of neutralizing antibody and anti-SARS-CoV-2 immunoglobulin A (IgA) and conferred virtually complete protection against infection in both the upper and lower respiratory tracts in mice expressing hACE2 receptor after adenoviral vector delivery or as a transgene. Thus, intranasal immunization of ChAd-SARS-CoV-2-S has the potential to control infection at the site of inoculation, which should prevent both virus-induced disease and transmission.
Results
Chimpanzee Ad-Vectored Vaccine Induces Robust Antibody Responses against SARS-CoV-2
We constructed two replication-incompetent ChAd vectors based on a simian Ad-36 virus. The ChAd-SARS-CoV-2-S vector encodes the full-length, codon optimized sequence of SARS-CoV-2 S protein as a transgene, including the ectodomain, transmembrane domain, and cytoplasmic domain (GenBank: QJQ84760.1), and is stabilized in prefusion form by two proline substitutions at residues K986 and V987 (Pallesen et al., 2017; Wrapp et al., 2020). The ChAd-control has no transgene. The S protein transgene is controlled transcriptionally by a cytomegalovirus promoter. To make the vector replication incompetent and enhance packaging capacity, we replaced the E1A/B genes and introduced a deletion in the E3B gene, respectively (Figure 1 A). To confirm that the S protein was expressed and antigenically intact, we transduced 293T cells and confirmed binding of a panel of 22 neutralizing monoclonal antibodies against the S protein by flow cytometry (Figure 1B).
Figure 1.

Immunogenicity of ChAd-SARS-CoV-2-S
(A) Diagram of transgene cassettes: ChAd-control has no transgene insert; ChAd-SARS-CoV-2-S encodes for SARS-CoV-2 S protein with the two indicated proline mutations.
(B) Binding of ChAd-SARS-CoV-2-S transduced 293 cells with anti-S mAbs. (Left) Summary: +, ++, +++, and - indicate <25%, 25%–50%, >50%, and no binding, respectively. MFI, mean fluorescence intensity. (Right) Representative flow cytometry histograms of two experiments are shown.
(C–F) Four-week-old female BALB/c mice were immunized via intramuscular route with ChAd-control or ChAd-SARS-CoV-2-S and boosted 4 weeks later. Antibody responses in sera of immunized mice at day 21 after priming or boosting were evaluated. An ELISA measured anti-S and RBD IgG and IgA levels ([D] and [E]), and an FRNT determined neutralization activity (F). Data are pooled from two experiments (n = 15–30).
(G and H) Cell-mediated responses were analyzed at day 7 post-booster immunization after re-stimulation with an S protein peptide pool (Table S1). Splenocytes were assayed for IFNγ and granzyme B expression in CD8+ T cells and granzyme B only in CD4+ T cells by flow cytometry (G). A summary of frequencies and numbers of positive cell populations is shown in (H) (n = 5). Bars indicate median values, and dotted lines are the limit of detection (LOD) of the assays.
(I) Spleens were harvested at 7 days post-boost, and SARS-CoV-2 spike-specific IgG+ antibody-secreting cell (ASC) frequency was measured by ELISPOT (n = 13).
(J) CD8+ T cells in the lung were assayed for IFNγ and granzyme B expression by flow cytometry after re-stimulation with an S protein peptide pool (n = 4–5).
(K) Lung CD8+ T cells also were assayed for expression of CD69 and CD103.
(L) Spleens were harvested at 7 days post-boosting, and SARS-CoV-2 spike-specific IgA+ ASC frequency was measured by ELISPOT (n = 4–5).
(M and N) SARS-CoV-2 S- (M) and RBD-specific (N) IgG and IgA levels in BAL fluid were determined by ELISA (n = 4–5).
Bars and columns show median values, and dotted lines indicate the limit of detection (LOD) of the assays. For (D), (F), (H), (I), and (J): Mann-Whitney test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant. See Figure S1, Figure S2, and S3 and Table S1.
To assess the immunogenicity of ChAd-SARS-CoV-2-S, groups of 4-week-old BALB/c mice were immunized by intramuscular inoculation with 1010 virus particles of ChAd-SARS-CoV-2-S or ChAd-control. Some mice received a booster dose 4 weeks later. Serum samples were collected 21 days after primary or booster immunization (Figure 1C), and IgG responses against purified S and RBD proteins were evaluated by ELISA. Whereas ChAd-SARS-CoV-2-S induced high levels of S- and RBD-specific IgG, low if any levels were detected in the ChAd-control-immunized mice (Figure 1D). However, intramuscular injection of ChAd-SARS-CoV-2-S failed to induce S- or RBD-specific IgA in serum (Figure 1E). We next functionally characterized serum antibody responses by assaying neutralization of infectious SARS-CoV-2 using a focus-reduction neutralization test (FRNT) (Case et al., 2020). As expected, serum from ChAd-control-immunized mice did not inhibit SARS-CoV-2 infection after primary immunization or boosting. In contrast, serum from ChAd-SARS-CoV-2-S-vaccinated animals neutralized SARS-CoV-2 infection, and boosting enhanced this inhibitory activity (geometric mean titers [GMTs] of 1/240 and 1/719, respectively; Figures 1F, S1 A, and S1B).
Figure S1.

ChAd-SARS-CoV-2-S Vaccine Induces Neutralizing Antibodies as Measured by FRNT, Related to Figure 1
Four-week old female BALB/c mice were primed or primed and boosted with ChAd-control or ChAd-SARS-CoV-2-S via intramuscular route. A-B. Serum samples from ChAd-control or ChAd-SARS-CoV-2 vaccinated mice were collected at day 21 after priming (A) or boosting (B) and assayed for neutralizing activity by FRNT. Serum neutralization curves corresponding to individual mice are shown for the indicated vaccines (n = 15-30 per group). Each point represents the mean of two technical replicates. (C). An ELISA measured anti-SARS-CoV-2 NP IgG responses in paired sera obtained 5 days before and 8 days after SARS-CoV-2 challenge of ChAd-control or ChAd-SARS-CoV-2-S mice vaccinated by an intramuscular route (n = 5: ∗∗p < 0.01; ∗∗∗p < 0.001; paired t test). Dotted lines represent the mean IgG titers from naive sera.
Vaccine-Induced Memory CD8+ T Cell and Antigen-Specific B Cell Responses
Because optimal vaccine immunity is often composed of both humoral and cellular responses (Slifka and Amanna, 2014), we measured the levels of SARS-CoV-2-specific CD4+ and CD8+ T cells after vaccination. 4-week-old BALB/c mice were immunized with ChAd-SARS-CoV-2-S or ChAd-control and boosted 3 weeks later. To assess the vaccine-induced SARS-CoV-2-specific CD4+ and CD8+ T cell responses, splenocytes were harvested 1 week after boosting and stimulated ex vivo with a pool of 253 overlapping 15-mer S peptides (Table S1). Subsequently, quantification of intracellular interferon γ (IFNγ) and granzyme B expression was determined by flow cytometry. After peptide re-stimulation ex vivo, splenic CD8+ T cells expressed IFNγ and both splenic CD4+ and CD8+ T cells expressed granzyme B in mice immunized with ChAd-SARS-CoV-2-S, but not the ChAd-control vector (Figures 1G, 1H, and S2 ). To assess the antigen-specific B cell responses, splenocytes were harvested and subjected to an ELISPOT analysis with S protein. The ChAd-SARS-CoV-2-S vaccine induced S-protein-specific IgG antibody-secreting cells in the spleen, whereas the control vaccine did not (Figure 1I).
Figure S2.

Gating Strategy for Analyzing T Cell Responses, Related to Figure 1
Four-week old female BALB/c mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S and boosted four weeks later. T cell responses were analyzed in splenocytes at day 7 post-boost. Cells were gated for lymphocytes (FSC-A/SSC-A), singlets (SSC-W/SSC-H), live cells (Aqua-), CD45+, CD19- followed by CD4+ or CD8+ cell populations expressing IFNγ or granzyme B.
To assess whether intramuscular vaccination could induce mucosal immune responses in the lungs, mice were immunized with either ChAd-SARS-CoV-2-S or ChAd-control and boosted similarly 4 weeks later. Lungs were harvested 1 week post-boosting, and T cells were analyzed by flow cytometry. Notably, we failed to detect an increase in IFNγ or granzyme B producing CD8+ T cells in the lungs of ChAd-SARS-CoV-2-S vaccinated mice after re-stimulation ex vivo with a pool of S peptides (Figure 1J) or CD103+CD69+CD8+ cells that could represent lung-resident memory T cells (Figure 1K). In the spleen, we also failed to detect antibody-secreting plasma cells producing IgA against the S protein after immunization with ChAd-SARS-CoV-2-S (Figure 1L). Moreover, we detected low S-specific IgG and no S-specific IgA or RBD-specific IgG or IgA antibodies in bronchoalveolar lavage (BAL) fluid of immunized mice (Figures 1M and 1N). Thus, although intramuscular vaccination with ChAd-SARS-CoV-2-S produced strong systemic adaptive immune responses against SARS-CoV-2, it induced minimal mucosal immune response.
Intramuscular Immunization with ChAd-SARS-CoV-2-S Vaccine Protects against SARS-CoV-2 Infection in the Lung
We tested the protective activity of the ChAd vaccine in a recently developed SARS-CoV-2 infection model, wherein BALB/c mice express hACE2 in the lung after intranasal delivery of a vectored human Ad (Hu-Ad5-hACE2; Hassan et al., 2020). Endogenous mouse ACE2 does not support viral entry (Letko et al., 2020), and this system enables productive SARS-CoV-2 infection in the mouse lung. 4-week-old BALB/c mice first were immunized via an intramuscular route with ChAd-control or ChAd-SARS-CoV-2-S vaccines. Approximately 30 days later, mice were administered 108 plaque-forming units (PFUs) of Hu-Ad5-hACE2 and anti-Ifnar1 monoclonal antibody (mAb) via intranasal and intraperitoneal routes, respectively. We administered a single dose of anti-Ifnar1 mAb to enhance lung pathogenesis in this model (Hassan et al., 2020). We confirmed the absence of cross-immunity between the ChAd and the Hu-Ad5 vector. Serum from ChAd-immunized mice did not neutralize Hu-Ad5 infection (Figures S3 A and S3B).
Figure S3.

Impact of Pre-existing ChAd Immunity on Transduction of Mice with Hu-AdV5-hACE2, Related to Figures 1 and 2
Four-week old female BALB/c mice were primed or primed and boosted. Serum samples were collected one day prior to Hu-AdV5-hACE2 transduction. Neutralizing activity of Hu-AdV5-hACE2 in the sera from the indicated vaccine groups was determined by FRNT after prime only (A) or prime and boost (B). Each symbol represents a single animal; each point represents two technical repeats and bars indicate the range. A positive control (anti-Hu-Adv5 serum) is included as a frame of reference.
5 days after Hu-Ad5-hACE2 transduction, mice were challenged via intranasal route with 4 × 105 focus-forming units (FFUs) of SARS-CoV-2 (Figure 2 A). At 4 days post-infection (dpi), the peak of viral burden in this model (Hassan et al., 2020), mice were euthanized and lungs, spleen, and heart were harvested for viral burden and cytokine analysis. Notably, there was no detectable infectious virus in the lungs of mice immunized with ChAd-SARS-CoV-2-S as determined by plaque assay, whereas high levels were present in mice vaccinated with ChAd-control (Figure 2B). Using primers that target a sequence within the N gene, we also detected no measurable genomic or subgenomic viral RNA in the heart and spleen and lower levels of viral RNA in the lungs of ChAd-SARS-CoV-2-S-vaccinated animals compared to mice receiving the ChAd-control vector (Figure 2C). In situ hybridization staining for viral RNA in lungs harvested at 4 dpi revealed a substantial decrease of SARS-CoV-2 RNA in pneumocytes of animals immunized with ChAd-SARS-CoV-2-S compared to the ChAd-control (Figure 2D). A subset of immunized animals was euthanized at 8 dpi, and tissues were harvested for evaluation. At this time point, viral RNA levels again were lower or absent in the lung and spleen of ChAd-SARS-CoV-2-S-immunized mice compared to the control ChAd vector (Figure 2C). Collectively, these data indicate that a single intramuscular immunization with ChAd-SARS-CoV-2-S results in markedly reduced, but not abrogated, SARS-CoV-2 infection in the lungs of challenged mice.
Figure 2.

Protective Efficacy of Intramuscularly Delivered ChAd-SARS-CoV-2-S against SARS-CoV-2 Infection
(A) Scheme of vaccination and challenge. Four-week-old BALB/c female mice were immunized ChAd-control or ChAd-SARS-CoV-2-S. Some mice received a booster dose of the homologous vaccine. On day 35 post-immunization, mice were challenged with SARS-CoV-2 as follows: animals were treated with anti-Ifnar1 mAb and transduced with Hu-AdV5-hACE2 via an intranasal route 1 day later. Five days later, mice were challenged with 4 × 105 focus-forming units (FFUs) of SARS-CoV-2 via the intranasal route.
(B and C) Tissues were harvested at 4 and 8 dpi for analysis. Infectious virus in the lung was measured by plaque assay (B), and viral RNA levels were measured in the lung, spleen, and heart at 4 and 8 dpi by qRT-PCR (C; n = 3–7).
(D) Viral RNA in situ hybridization using SARS-CoV-2 probe (brown color) in the lungs harvested at 4 dpi. Images show low- (top; scale bars, 100 μm) and medium- (middle; scale bars, 100 μm) power magnification with a high-power magnification inset (representative images from n = 3 per group).
(E) Fold change in gene expression of indicated cytokines and chemokines from lung homogenates at 4 dpi was determined by qRT-PCR after normalization to Gapdh levels and comparison with naive unvaccinated, unchallenged controls (n = 7).
(F and G) Mice that received a prime-boost immunization were challenged on day 35 post-booster immunization. Tissues were collected at 4 dpi for analysis. Infectious virus in the lung was determined by plaque assay (F), and viral RNA was measured in the lung, spleen, and heart using qRT-PCR (G; n = 6–7).
(B, C, and E–G) Columns show median values, and dotted lines indicate the LOD of the assays. For (B), (C), (E), (F), and (G): Mann-Whitney test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See Figure S3.
We next assessed the effect of the vaccine on lung inflammation and disease. Several proinflammatory cytokines and chemokine mRNA levels were lower in the lung tissues of animals immunized with ChAd-SARS-CoV-2-S compared to ChAd-control, including interleukin (IL)-1β, CXCL10, IFNβ, IFNλ, IL-6, and CXCL11 (Figure 2E). Moreover, mice immunized with ChAd-control vaccine and challenged with SARS-CoV-2 showed evidence of viral pneumonia characterized by immune cell accumulation in perivascular and alveolar locations, vascular congestion, and interstitial edema. In contrast, animals immunized with ChAd-SARS-CoV-2-S showed a marked attenuation of the inflammatory response in the lung that develops in the ChAd-control-immunized mice (Figure 3 ). Thus, immunization with Ch-Ad-SARS-CoV-2 decreases both viral infection and the consequent lung inflammation and injury associated with SARS-CoV-2 infection.
Figure 3.

Single-Dose Intramuscular Vaccination with ChAd-SARS-CoV-2-S Protects Mice against SARS-CoV-2-Induced Inflammation in the Lung
Four-week-old female BALB/c mice were immunized with ChAd-control and ChAd-SARS-CoV-2-S and challenged following the scheme described in Figure 2. Lungs were harvested at 8 dpi. Sections were stained with hematoxylin and eosin and imaged at 40× (left; scale bar, 250 μm), 200× (middle; scale bar, 50 μm), and 400× (right; scale bar, 25 μm) magnifications. Each image is representative of a group of 3 mice.
We then assessed for improved protection using a prime-boost vaccine regimen. BALB/c mice were immunized via an intramuscular route with ChAd-control or ChAd-SARS-CoV-2-S and received a homologous booster dose 4 weeks later. At day 29 post-boost, mice were treated with a single dose of anti-Ifnar1 antibody followed by Hu-Ad5-hACE2 and then challenged with SARS-CoV-2 5 days later. As expected, the prime-boost regimen protected against SARS-CoV-2 challenge with no infectious virus detected in the lungs (Figure 2F). Although marked reductions of viral RNA in the lung, spleen, and heart were detected at 4 dpi, residual levels of viral RNA still were present, suggesting protection was not complete, even after boosting (Figure 2G).
A Single Intranasal Immunization with ChAd-SARS-CoV-2-S Induces Protective Immunity against SARS-CoV-2 in Mice Expressing hACE2 Delivered by an Adenoviral Vector
Mucosal immunization through the nasopharyngeal route can elicit local immune responses, including secretory IgA antibodies that confer protection at or near the site of inoculation of respiratory pathogens (Neutra and Kozlowski, 2006). To assess the immunogenicity and protective efficacy of mucosal vaccination, 5-week-old BALB/c mice were inoculated via intranasal route with 1010 viral particles of ChAd-control or ChAd-SARS-CoV-2-S (Figure 4 A). Serum samples were collected at 4 weeks post-immunization to evaluate humoral immune responses. Intranasal immunization of ChAd-SARS-CoV-2-S, but not ChAd-control, induced high levels of S- and RBD-specific IgG and IgA (Figures 4B and 4C) and SARS-CoV-2-neutralizing antibodies (GMT of 1/1,574) in serum (Figures 4D and S4 A). Antibodies from mice immunized with ChAd-SARS-CoV-2-S equivalently neutralized a recombinant, luciferase-expressing variant of SARS-CoV-2 encoding a D614G mutation in the S protein (Figure S4B); this finding is important, because many circulating viruses contain this substitution, which is associated with greater infectivity in cell culture (Korber et al., 2020). We also assessed the SARS-CoV-2-specific antibody responses in BAL fluid of immunized mice. BAL fluid from ChAd-SARS-CoV-2-S-, but not ChAd-control-, vaccinated mice showed high levels of S- and RBD-specific IgG and IgA antibodies (Figures 4E and 4F), including those with neutralizing activity (Figures 4G and S4C).
Figure 4.

Immune Responses after Intranasal Immunization of ChAd-SARS-CoV-2-S
(A) Scheme of experiments. 5-week-old BALB/c female mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal route.
(B–D) Antibody responses in sera of immunized mice at 1 month after priming were evaluated. An ELISA measured SARS-CoV-2 S- and RBD-specific IgG (B) and IgA levels (C), and a FRNT determined neutralization activity (D). Data are pooled from two experiments with n = 10–25 mice per group.
(E–J) Mice that received a booster dose were sacrificed 1 week later to evaluate mucosal and cell-mediated immune responses. SARS-CoV-2 S- and RBD-specific IgG (E) and IgA levels (F) in BAL fluid were determined by ELISA. Neutralizing activity of BAL fluid against SARS-CoV-2 was measured by FRNT (G). CD8+ T cells in the lung were assayed for IFNγ and granzyme B expression by flow cytometry after re-stimulation with an S protein peptide pool (H). CD8+ T cells in the lung also were phenotyped for expression of CD103 and CD69 (I). SARS-CoV-2 spike-specific IgG+ and IgA+ ASC frequency in the spleen harvested 1 week post-boost was measured by ELISPOT (J). Data for mucosal and cell-mediated responses are pooled from two experiments (E–I: n = 7–9 per group; J: n = 5 per group).
(B–J) Bars and columns show median values, and dotted lines indicate the LOD of the assays. Mann-Whitney test: ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See Figure S4 and Table S1.
Figure S4.

Intranasal Inoculation of ChAd-SARS-CoV-2-S Induces Neutralizing Antibodies as Measured by FRNT and Protects against SARS-CoV-2 Replication, Related to Figures 4 and 5
Five-week old female BALB/c mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal inoculation route. Serum samples collected one month after immunization were assayed for neutralizing activity by FRNT. Mice were boosted at day 30 after priming and were sacrificed one week later to evaluate immune responses. (A) Serum samples from ChAd-control or ChAd-SARS-CoV-2-S vaccinated mice were tested for neutralizing activity with SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 (n = 8-10 per group). (B) Serum samples from ChAd-SARS-CoV-2-S vaccinated mice were tested for neutralization of recombinant luciferase-expressing SARS-CoV-2 viruses (wild-type (left) and D614G variant (middle)). (Right) Paired EC50 values are indicated (n = 5; n.s. not significant, paired t test). (C) BAL fluid was collected from ChAd-control or ChAd-SARS-CoV-2-S vaccinated mice, and neutralization of SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 was measured using a FRNT assay (n = 8-10 per group). Each point represents the mean of two technical replicates. (D) On day 35 post-immunization, mice were challenged via intranasal route with 4 × 105 FFU of SARS-CoV-2 five days after Hu-AdV5-hACE2 transduction and anti-Ifnar1 mAb treatment as described in Figure 2. Tissues were collected at 4 dpi for viral burden measurements. Genomic RNA (ORF1a) levels were determined in lungs and nasal turbinates (two experiments, n = 6-9; ∗∗∗∗p < 0.0001; Mann-Whitney test). Columns show median values, and dotted lines indicate the LOD of the assays.
To assess T cell responses activated via mucosal immunization, mice were vaccinated via intranasal route with either ChAd-SARS-CoV-2-S or ChAd-control and boosted similarly 4 weeks later. Lungs were harvested 1 week post-boosting, and T cells were analyzed by flow cytometry. Re-stimulation ex vivo with a pool of S peptides resulted in a marked increase in IFNγ and granzyme B producing CD8+ T cells in the lungs of mice that received the ChAd-SARS-CoV-2-S vaccine (Figure 4H). Specifically, a population of IFNγ-secreting, antigen-specific CD103+CD69+CD8+ T cells in the lung was identified (Figure 4I), which is phenotypically consistent with vaccine-induced resident memory T cells (Takamura, 2017). In the spleen, we detected antibody-secreting plasma cells producing IgA or IgG against the S protein after intranasal immunization with ChAd-SARS-CoV-2-S (Figure 4J). Of note, we observed an ∼5-fold higher frequency of B cells secreting anti-S IgA than IgG.
We evaluated the protective efficacy of the ChAd vaccine after single-dose intranasal immunization. At day 30 post-vaccination, mice were administered 108 PFUs of Hu-Ad5-hACE2 and anti-Ifnar1 mAb as described above. 5 days later, mice were challenged with 4 × 105 FFUs of SARS-CoV-2. At 4 and 8 dpi, lungs, spleen, heart, nasal turbinates, and nasal washes were harvested and assessed for viral burden. Intranasal delivery of the ChAd-SARS-CoV-2-S vaccine demonstrated remarkable protective efficacy as judged by an absence of infectious virus in the lungs (Figure 5 A) and almost no measurable viral RNA in the lung, spleen, heart, nasal turbinates, or nasal washes, as determined with subgenomic/genomic (N gene) or genomic only (ORF1a gene) TaqMan probes (Figures 5B and S4D). The very low viral RNA levels in the lung and nasal turbinates at 4 dpi may reflect the input, non-replicating virus, as similar amounts were measured at this time point in C57BL/6 mice lacking hACE2 receptor expression (Hassan et al., 2020). Cytokine and chemokine mRNA levels in lung homogenates also were substantially lower in mice immunized with the ChAd-SARS-CoV-2-S than the ChAd-control vaccine (Figure 5C), with residual expression likely due to the human Ad vector hACE2 delivery system. Finally, histopathological analysis of lung tissues from animals vaccinated with ChAd-SARS-CoV-2-S by intranasal route and challenged with SARS-CoV-2 showed minimal, if any, perivascular and alveolar infiltrates at 8 dpi compared to the extensive inflammation observed in ChAd-control-vaccinated animals (Figure 5D).
Figure 5.

Single-Dose Intranasal Immunization with ChAd-SARS-CoV-2-S Protects against SARS-CoV-2 Infection
Five-week-old BALB/c female mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal route. On day 35 post-immunization, mice were challenged as follows: animals were treated with anti-Ifnar1 mAb and transduced with Hu-AdV5-hACE2 via the intranasal route 1 day later. Five days later, mice were challenged intranasally with 4 × 105 FFUs of SARS-CoV-2.
(A–C) Tissues and nasal washes were collected at 4 and 8 dpi for analysis. Infectious virus in the lung was measured by plaque assay (A). Viral RNA levels in the lung, spleen, heart, nasal turbinates, and nasal washes were measured at 4 and 8 dpi by qRT-PCR (B). Fold change in gene expression of indicated cytokines and chemokines was determined by qRT-PCR, normalized to Gapdh, and compared to naive controls in lung homogenates at 4 dpi (C; 2 experiments, n = 6–9; median values are shown). Columns show median values, and dotted lines indicate the LOD of the assays.
(D) Lungs were harvested at 8 dpi. Sections were stained with hematoxylin and eosin and imaged at 40× (left; scale bar, 250 μm), 200× (middle; scale bar, 50 μm), and 400× (right; scale bar, 25 μm) magnifications. Each image is representative of a group of 3 mice.
(E) An ELISA measured anti-SARS-CoV-2 NP IgM (left) and IgG (right) antibody responses in paired sera obtained 5 days before and 8 days after SARS-CoV-2 challenge of ChAd-control or ChAd-SARS-CoV-2-S mice vaccinated by an intranasal route (n = 6). Dotted lines represent the LOD of the assay. Dashed lines indicate the mark for a 4-fold increase of pre-boost IgM and IgG levels.
For (A)–(C): Mann-Whitney test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; for (E): ∗∗p < 0.01, ∗∗∗∗p < 0.0001; paired t test. See Figures S4 and S5.
To begin to determine whether sterilizing immunity might be achieved with intranasal delivery of ChAd-SARS-CoV-2-S, we measured anti-NP antibodies at 8 dpi and compared them to responses from 5 days before SARS-CoV-2 infection. Because the NP gene is absent from the vaccine vector, induction of humoral immune responses against NP after SARS-CoV-2 exposure (Ni et al., 2020) suggests viral protein translation and active infection. After SARS-CoV-2 challenge, anti-NP antibody responses were detected in ChAd-control mice vaccinated by an intranasal route or ChAd-control and ChAd-SARS-CoV-2-S mice vaccinated by an intramuscular route (Figures 5E and S1C). Remarkably, none of the mice immunized with ChAd-SARS-CoV-2-S via an intranasal route showed significant increases in anti-NP antibody responses after SARS-CoV-2 infection (Figures 5E and S5 ). Combined with our virological analyses, these data suggest that a single intranasal immunization of ChAd-SARS-CoV-2-S induces robust mucosal immunity, which prevents SARS-CoV-2 infection in the upper and lower respiratory tracts of mice expressing the hACE2 receptor.
Figure S5.

SARS-CoV-2 NP-Specific IgM and IgG Antibody Responses following SARS-CoV-2 Challenge, Related to Figure 5
Five-week-old BALB/c female mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal route. One month later, mice were transduced with Hu-AdV5-ACE2 and challenged with SARS-CoV-2 as described in Figure 5. An ELISA measured anti-SARS-CoV-2 NP IgM and IgG responses in paired sera obtained 5 days before and 8 days after SARS-CoV-2 challenge. Serum ELISA curves corresponding to individual mice are shown for the indicated vaccines or control naive BALB/c mice (n = 6 per group).
A Single Intranasal Immunization with ChAd-SARS-CoV-2-S Prevents SARS-CoV-2 Infection in the Upper and Lower Respiratory Tracts of hACE2 Transgenic Mice
Recently, we and others established a more stringent, lethal SARS-CoV-2 challenge model (Rathnasinghe et al., 2020; Winkler et al., 2020) in transgenic C57BL/6 mice that have eight inserted copies of the hACE2 gene driven by the K18 cytokeratin epithelial cell promoter (McCray et al., 2007). Within 1 week of SARS-CoV-2 inoculation by an intranasal route, K18-hACE2 mice develop severe lung infection and inflammation, immune cell infiltration, and compromised respiratory function that results in death. As an independent test of the efficacy of intranasal administration of ChAd-SARS-CoV-2-S, we assessed its immunogenicity and protective efficacy in K18-hACE2 mice.
4-week-old K18-hACE2 mice were inoculated via an intranasal route with 1010 viral particles of ChAd-control or ChAd-SARS-CoV-2-S (Figure 6 A). Serum samples were collected at 4 weeks post-immunization, and humoral immune responses were evaluated. Intranasal immunization of ChAd-SARS-CoV-2-S, but not ChAd-control, induced high levels of S- and RBD-specific IgG and IgA (Figures 6B and 6C) and neutralizing antibodies (GMT of 1/1,424) in serum (Figures 6D and S6 ). At day 30 post-vaccination, K18-hACE2 mice were challenged via an intranasal route with 103 FFUs of SARS-CoV-2. At 4 dpi, lungs, spleen, heart, nasal turbinates, and nasal washes were harvested and assessed for viral burden. Similar to that seen in BALB/c mice transiently expressing hACE2, intranasal immunization of ChAd-SARS-CoV-2-S conferred remarkable protection as judged by an absence of infectious virus in the lungs (Figure 6E), no measurable viral subgenomic/genomic RNA in the lungs or hearts, and very low levels of viral RNA in the nasal turbinates or washes (Figure 6F). Consistent with these results, after SARS-CoV-2 challenge, we observed no induction of cytokine and chemokine mRNA (e.g., CCL2, CXCL1, CXCL10, CXCL11, IL-6, IFNβ, and IFNλ) in the lung homogenates of ChAd-SARS-CoV-2-S-immunized compared to naive mice, whereas ChAd-control-vaccinated mice sustained high levels (Figure 6G). Collectively, these data suggest that a single intranasal immunization of ChAd-SARS-CoV-2-S induces robust systemic and mucosal immunity that blocks SARS-CoV-2 infection in the upper and lower respiratory tract of highly susceptible K18-hACE2 transgenic mice.
Figure 6.

Immunogenicity and Protective Efficacy after Intranasal Immunization of ChAd-SARS-CoV-2-S in K18-hACE2 Mice
(A) Scheme of experiments. Four-week-old K18-hACE2 female mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal route.
(B–D) Antibody responses in sera of immunized mice at 4 weeks after priming were evaluated. An ELISA measured SARS-CoV-2 S-and RBD-specific IgG (B) and IgA levels (C), and a FRNT determined neutralization activity (D; n = 7).
(E–G) At 1 month post-immunization, mice were challenged with 103 FFUs of SARS-CoV-2. Tissues and nasal washes were harvested at 4 dpi. Infectious virus in lungs was measured using plaque assay (E). Viral RNA levels in the lung, spleen, heart, nasal turbinates, and nasal washes were measured at 4 dpi by qRT-PCR (F). Fold change in gene expression of indicated cytokines and chemokines was determined by qRT-PCR, normalized to Gapdh, and compared to naive controls in lung homogenates at 4 dpi (G; n = 7). Boxes indicate median values, and dotted lines indicate the LOD of the assays.
For (B)–(G): Mann-Whitney test: ∗∗p < 0.01; ∗∗∗p < 0.001. See Figure S6.
Figure S6.

Intranasal Inoculation of ChAd-SARS-CoV-2-S Induces Neutralizing Antibodies in K18-hACE2 Mice, Related to Figure 6
Four-week old female K18-hACE2 mice were immunized with ChAd-control or ChAd-SARS-CoV-2-S via an intranasal inoculation route. Serum samples collected four weeks after immunization were assayed for neutralizing activity by FRNT (n = 7 per group). Each point represents the mean of two technical replicates.
Discussion
In this study, we evaluated intramuscular and intranasal administration of a replication-incompetent ChAd vector as a vaccine platform for SARS-CoV-2. Single-dose immunization of a stabilized S-protein-based vaccine via an intramuscular route induced S- and RBD-specific binding as well as neutralizing antibodies. Vaccination with one or two doses protected mice expressing human ACE2 against SARS-CoV-2 challenge, as evidenced by an absence of infectious virus in the lungs and substantially reduced viral RNA levels in lungs and other organs. Mice immunized with ChAd-SARS-CoV-2-S also showed markedly reduced if not absent lung pathology, lung inflammation, and evidence of pneumonia compared to the control ChAd vaccine. However, intramuscular vaccination of ChAd-SARS-CoV-2-S did not induce IgA responses or confer sterilizing immunity, as evidenced by detectable viral RNA levels in the lung and induction of anti-NP antibody responses. Mice immunized with a single dose of the ChAd-SARS-CoV-2-S via an intranasal route also were protected against SARS-CoV-2 challenge. Intranasal vaccination, however, generated robust IgA and neutralizing antibody responses that likely protected against both upper and lower respiratory tract SARS-CoV-2 infection and inhibited infection of both wild-type and D614G variant viruses. The very low levels of genomic viral RNA in upper airway tissues and absence of serological response to NP in the context of challenge strongly suggests that at least some animals receiving a single intranasal dose of ChAd-SARS-CoV-2-S likely achieved sterilizing immunity.
Although several vaccine candidates (e.g., lipid-encapsulated mRNA, DNA, inactivated, and viral vectored) rapidly advanced to human clinical trials in an expedited effort to control the pandemic (Amanat and Krammer, 2020), few studies have demonstrated efficacy in pre-clinical models. Rhesus macaques immunized with two or three doses of a DNA plasmid vaccine encoding full-length SARS-CoV-2 S protein induced neutralizing antibody in sera and reduced viral burden in BAL and nasal mucosa fluids (Yu et al., 2020). Three immunizations over 2 weeks with purified, inactivated SARS-CoV-2 induced neutralizing antibodies and, depending on the dose administered, provided partial or complete protection against infection and viral pneumonia in rhesus macaques (Gao et al., 2020). Finally, two intramuscular doses of a lipid-encapsulated, mRNA-based vaccine against S protected against SARS-CoV-2 infection in nasal swabs and BAL fluid of rhesus macaques (Corbett et al., 2020). One limitation of these challenge models is that rhesus macaques develop lower viral burden and mild interstitial pneumonia after SARS-CoV-2 infection compared to some humans and other non-human primate species (Chandrashekar et al., 2020; Munster et al., 2020). Our study in hACE2-expressing mice shows that a single intramuscular or intranasal dose of ChAd-SARS-CoV-2-S vaccine confers substantial and possibly complete protection against viral replication, inflammation, and lung disease.
Our approach supports the use of non-human Ad-vectored vaccines against emerging RNA viruses, including SARS-CoV-2. Previously, we showed the efficacy of single-dose or two-dose regimens of a gorilla Ad encoding the prM-E genes of Zika virus (ZIKV) in several mouse challenge models, including in the context of pregnancy (Hassan et al., 2019). Others have evaluated ChAd or rhesus macaque Ad vaccine candidates against ZIKV and shown efficacy in mice and non-human primates (Abbink et al., 2016; López-Camacho et al., 2018). A different ChAd encoding the wild-type SARS-CoV-2 S protein (ChAdOx1) is currently in clinical trials in humans (NCT04324606) and a phase 1/2 trial showed neutralizing antibody responses against SARS-CoV-2 in 91% and 100% participants after one- or two-dose immunization regimens (Folegatti et al., 2020b). Studies in rhesus macaques with ChAdOx1 suggest that a single intramuscular dose protects against infection in the lung, but not in the upper respiratory tract (van Doremalen et al., 2020). None of the vaccines evaluated against SARS-CoV-2 has shown evidence of immune enhancement in any pre-clinical or clinical study, which has been a theoretical risk based on studies with other human and animal coronaviruses (de Alwis et al., 2020; Diamond and Pierson, 2020). Indeed, and in contrast to data with SARS-CoV vaccines or antibodies (Bolles et al., 2011; Liu et al., 2019; Weingartl et al., 2004), we did not observe enhanced infection, immunopathology, or disease in animals immunized with ChAd encoding SARS-CoV-2 S proteins or administered passively transferred monoclonal antibodies (Alsoussi et al., 2020; Hassan et al., 2020).
ChAd-SARS-CoV-2-S induced SARS-CoV-2-specific CD8+ T cell responses, including a high percentage and number of IFNγ- and granzyme-expressing cells after ex vivo S protein peptide re-stimulation. The induction of CD8+ T cell responses by the ChAd-SARS-CoV-2-S vaccine is consistent with reports with other simian Ad vectors (Douglas et al., 2010; Hodgson et al., 2015; Reyes-Sandoval et al., 2010). ChAd vaccine vectors not only overcome issues of pre-existing immunity against human adenoviruses but also have immunological advantages because they do not induce exhausted T cell responses (Penaloza-MacMaster et al., 2013).
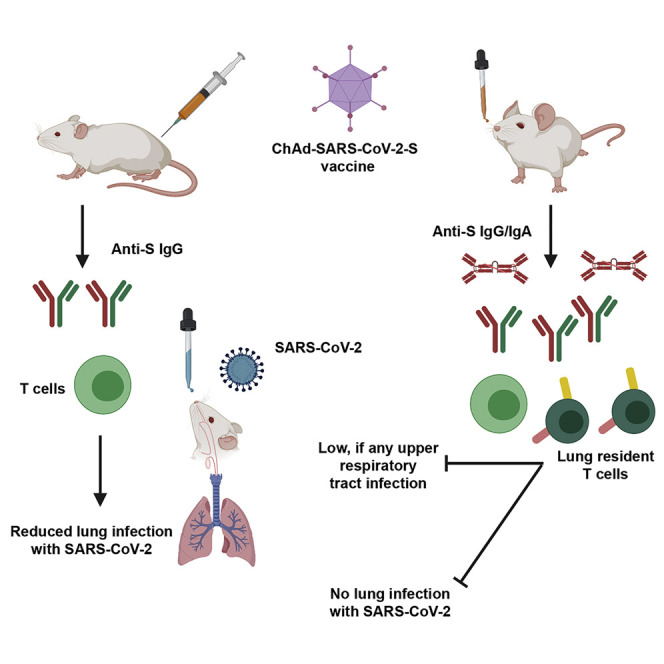
A single intranasal dose of ChAd-SARS-CoV-2-S conferred superior immunity against SARS-CoV-2 challenge, more so than one or two intramuscular immunizations of the same vaccine and dose (Figure 7 ). Additionally, one intranasal dose of ChAd-SARS-CoV-2-S prevented upper and lower respiratory tract infection and inflammation by SARS-CoV-2 in highly susceptible K18-hACE2 transgenic mice (Rathnasinghe et al., 2020; Winkler et al., 2020). Given that the serum-neutralizing antibody responses were relatively comparable, we hypothesize the greater protection observed after intranasal delivery was because of the mucosal immune responses generated. Indeed, high levels of anti-SARS-CoV-2 IgA were detected in serum and lung, and B cells secreting IgA were detected in the spleen only in mice vaccinated via an intranasal route. Moreover, intranasal, but not intramuscular, vaccination induced SARS-CoV-2-specific CD8+ T cells in the lung, including CD103+CD69+ cells, which are likely of a resident memory phenotype. Future antibody passive transfer and T cell depletion studies can assess the relative contribution of each immune arm to protection and establish more precisely the mechanistic basis for the enhanced protection conferred by intranasal delivery of ChAd-SARS-CoV-2-S.
Figure 7.

Comparison of Immunogenicity of Single-Dose Administration of ChAd-SARS-CoV-2-S by Intramuscular and Intranasal Delivery Routes
(Upper panel) After intramuscular inoculation, ChAd-SARS-CoV-2 vaccine transduces antigen-presenting cells (APCs) at the site of injection in muscle tissues. APCs migrate to lymphoid tissues where antigen-specific CD8+T cells become activated, proliferate, and produce IFNγ and granzyme B. Antigen-specific B cells proliferate, some of which become plasmablasts and plasma cells that secrete anti-S IgG. After SARS-CoV-2 challenge, activated CD8+ T cells migrate to the lungs to control infection and anti-S IgG neutralizes virus particles. Intramuscular vaccination protects against lower, but not upper, airway infection and does not efficiently induce mucosal immunity. (Lower panel) After intranasal inoculation, ChAd-SARS-CoV-2 transduces APCs in the upper respiratory tract. APCs then migrate to bronchial- or mucosal-associated lymphoid tissues to present antigens to lymphocytes, including B and T cells. After SARS-CoV-2 challenge, activated CD8+ T cells migrate to the lungs, secrete cytokines, and attack virus-infected cells. Some CD8+ T cells likely adopt a tissue-resident memory phenotype (CD103+ CD69+), enabling them to reside in the lung (or upper airway) and respond more rapidly after re-encountering cognate antigen (SARS-CoV-2 S peptides). The activated B cell becomes responsive after intranasal vaccination produces cells that secrete anti-SARS-CoV-2-S IgG or IgA, the latter of which neutralizes virus within the upper and lower respiratory tracts. The mucosal immunity generated by intranasal inoculation of ChAd-SARS-CoV-2 likely controls infection at the point of initiation in the upper respiratory tract.
To our knowledge, none of the SARS-CoV-2 vaccine platforms currently in clinical trials use an intranasal delivery approach. There has been great interest in using intranasal delivery for influenza A virus vaccines because of their ability to elicit local humoral and cellular immune responses (Calzas and Chevalier, 2019). Indeed, sterilizing immunity to influenza A virus re-infection requires local adaptive immune responses in the lung, which is optimally induced by intranasal and not intramuscular inoculation (Dutta et al., 2016; Laurie et al., 2010). Although there are concerns with administering live-attenuated viral vaccines via an intranasal route, subunit-based or replication-incompetent vectored vaccines are promising for generating mucosal immunity in a safer manner, especially given advances in formulation (Yusuf and Kett, 2017).
Limitations of Study
Although a single intranasal administration of ChAd-SARS-CoV-2-S protected against SARS-CoV-2 replication in lungs, we note some limitations in our study. We performed most challenge studies in mice transduced with hACE2 using a human Ad5 vector, which could be complicated by vector immunity. Notwithstanding this possibility, we confirmed an absence of heterologous neutralizing serum antibody induced by one or two doses of ChAd vector against the Hu-Ad5 and, more importantly, detected high levels of SARS-CoV-2 infection in the ChAd-control-vaccinated mice after challenge. Moreover, we reproduced the level of protection by ChAd-SARS-CoV-2-S in K18-hACE2 transgenic mice. Immunization and challenge studies with hamsters and non-human primates are planned to confirm the extent of protective mucosal immunity conferred by ChAd-SARS-CoV-2-S in mice and expand upon our understanding of how the route of inoculation impacts vaccine-mediated protection. Moreover, studies in humans are needed to assess for cross-immunity between the ChAd vector (simian Ad-36) and adenoviruses circulating in humans. Although we expect this will be much less than that observed with Hu-Ad5 vaccine vectors (Zhu et al., 2020), low levels of pre-existing immunity against simian adenoviruses could affect vaccine efficacy in selected populations with non-human primate exposures (e.g., zoo workers, veterinarians, and laboratory workers at primate centers). Finally, studies must be conducted to monitor immune responses over time after intranasal vaccination with ChAd-SARS-CoV-2-S to establish the durability of the protective response.
In summary, our studies establish that immunization with ChAd-SARS-CoV-2-S induces both neutralizing antibody and antigen-specific CD8+ T cell responses. Although a single intramuscular immunization of ChAd-SARS-CoV-2-S confers protection against SARS-CoV-2 infection and inflammation in the lungs, intranasal delivery of ChAd-SARS-CoV-2-S induces mucosal immunity, provides superior protection, and possibly promotes sterilizing immunity, at least in mice that transiently or stably express the hACE2 receptor. We suggest that intranasal delivery of ChAd-SARS-CoV-2-S is a promising platform for preventing SARS-CoV-2 infection, disease, and upper airway transmission and thus warrants clinical evaluation in humans.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SARS2-01, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-02, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-07, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-11, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-12, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-16, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-18, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-20, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-21, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-22, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-23, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-29, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-31, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-32, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-34, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-38, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-39, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-50, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-55, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-58, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-66, anti-SARS-CoV-2 mAb | This paper | N/A |
| SARS2-71, anti-SARS-CoV-2 mAb | This paper | N/A |
| Goat anti-mouse IgG, human ads-HRP | Southern Biotech | Cat # 1030-05; RRID: AB_2619742 |
| CR3022, anti-SARS-CoV-2 mAb | Yuan et al., 2020 | N/A |
| Goat anti-mouse IgG, human ads-BIOT | Southern Biotech | Cat # 1030-08; RRID: AB_2794296 |
| Goat anti-mouse IgA-BIOT | Southern Biotech | Cat # 1040-08; RRID: AB_2794374 |
| Goat anti-mouse IgM-BIOT | Southern Biotech | Cat # 1021-08; RRID: AB_2794242 |
| HRP-conjugated goat anti-human IgG | Thermo Fisher | 62-8420; RRID: AB_2533962 |
| MAR1-5A3, anti-Ifnar1 mAb | Leinco | I-401; RRID: AB_2491621 |
| Anti-HuAdV5-hexon antibody | Novus Biologicals | Cat # NB600413; RRID: AB_10002549 |
| Streptavidin-HRP | Vector Laboratories | Cat # SA-5004; RRID: AB_2336509 |
| Peroxidase Streptavidin | Jackson ImmunoResearch | Cat # 016-030-084; RRID: AB_2337238 |
| Alexa Fluor 647 anti-IFNγ | BD BioScience | Cat # 557735; RRID: AB_396843 |
| Anti-mouse CD16/32 | eBioSceince | Cat # 14-0161-85; RRID: AB_467134 |
| BUV395 anti-CD45 | BD BioSciences | Cat # 564279; RRID: AB_2651134 |
| PE anti-CD44 | BioLegend | Cat # 103024; RRID: AB_493687 |
| PerCP/Cy5.5 anti-CD8b | BioLegend | Cat # 126610; RRID: AB_2260149 |
| APC/Cy7 anti-CD19 | BioLegend | Cat # 115530; RRID: AB_830707 |
| Fixable Aqua dead cell stain | Invitrogen | Cat # L34965; |
| BV 605 anti-TNFα | BioLegend | Cat # 506329; RRID:AB_11123912 |
| BV421 anti-CD4 | BioLegend | Cat # 100437; RRID: AB_10900241 |
| FITC anti-CD103 | BioLegend | Cat # 121419; RRID: AB_10709438 |
| BV711 anti-CD69 | BioLegend | Cat # 104537; RRID: AB_2566120 |
| PE anti-granzyme B | Invitrogen | Cat # GRB04; RRID: AB_2536538 |
| PE/Cy5 anti-CD4 | BioLegend | Cat # 100409; RRID: AB_312694 |
| Virus and bacterial Strains | ||
| SARS-CoV-2 (strain 2019 n-CoV/USA_WA1/2020) | CDC/BEI Resources | NR52281 |
| ChAd-SARS-CoV-2-S | This paper | N/A |
| ChAd-Control | This paper | N/A |
| AdV-hACE2 | This paper | N/A |
| E. coli strain BJ5183 | Agilent | Cat # 200154 |
| BL21(DE3)-RIL E. coli | Invitrogen | Cat # C601003 |
| Experimental Models: Cell Lines | ||
| Vero CCL-81 | ATCC | CCL-81; RRID: CVCL_0059 |
| Vero E6 | ATCC | CRL-1586; RRID: CVCL_0574 |
| HEK293 | ATCC | CRL-1573; RRID: CVCL_0045 |
| HEK293T | ATCC | CRL-3216; RRID: CVCL_0063 |
| Expi293F cells | Invitrogen | Cat # A14527 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat#000664; RRID: IMSR_JAX:000664 |
| Mouse: BALB/c | Jackson Laboratory | Cat#000651; RRID: IMSR_JAX:000651 |
| Mouse: B6.Cg-Tg(K18-ACE2)2Prlmn/J | Jackson Laboratory | Cat# 034860; RRID: IMSR_JAX:03486 |
| Oligonucleotides | ||
| SARS-CoV-2 N F: 5′-ATGCTGCAATCGTGCTACAA-3′ | This paper | N/A |
| SARS-CoV-2 N R: 5′-GACTGCCGCCTCTGCTC-3′ | This paper | N/A |
| SARS-CoV-2 N Probe: 5′-/56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/-3′ | This paper | N/A |
| SARS-CoV-2 ORF1a F: 5′- TTCAGTTGACTTCGCAGTGG −3′ | This paper | N/A |
| SARS-CoV-2 ORF1a R: 5′- GGACGGGTTTGAGTTTTTCA −3′ | This paper | N/A |
| SARS-CoV-2 ORF1a Probe: 5′-/56-FAM/AACTAACAT/ZEN/CTTTGGCACTGTTT/3IABkFQ | This paper | N/A |
| SARS-CoV-2 RNA ISH probe (S gene) | Advanced Cell Diagnostics | Cat# 4848561 |
| Gapdh TaqMan Primer/Probe set | IDT | Mm.PT.39a.1 |
| Ifng TaqMan Primer/Probe set | IDT | Mm.PT.58.41769240 |
| Il6 TaqMan Primer/Probe set | IDT | Mm.PT.58.10005566 |
| Il1b TaqMan Primer/Probe set | IDT | Mm.PT.58.41616450 |
| Tnfa TaqMan Primer/Probe set | IDT | Mm.PT.58.12575861 |
| Cxcl10 TaqMan Primer/Probe set | IDT | Mm.PT.58.43575827 |
| Ccl2 TaqMan Primer/Probe set | IDT | Mm.PT.58.42151692 |
| Ccl5 TaqMan Primer/Probe set | IDT | Mm.PT.58.43548565 |
| Cxcl11 TaqMan Primer/Probe set | IDT | Mm.PT.58.10773148.g |
| Ifnb TaqMan Primer/Probe set | IDT | Mm.PT.58.30132453.g |
| Ifnl(2/3) TaqMan Primer/Probe set | Thermo Fisher | Mm04204156_gH |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | v10 |
| GraphPad Prism | GraphPad | v 8.2.1 |
| Biorender | biorender.com | N/A |
| Recombinant DNA | ||
| pABVec6W | Davis et al., 2019 | N/A |
| Simian Ad36 vector | Penn Vector Core | Roy et al., 2011 |
| pSAd36-S | This paper | N/A |
| pAd36-Control | This paper | N/A |
| pAd-hACE2 | This paper | N/A |
| AdEasy AdV vector system | Agilent technologies | Cat# 240009 |
| PCR-II topo | Invitrogen | Cat# 451245 |
Resource Availablity
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael S. Diamond (diamond@wusm.wustl.edu).
Materials Availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact author. This includes mice, antibodies, viruses, vaccines, proteins, and peptides. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and Code Availability
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.
Experimental Model and Subject Details
Viruses and cells
Vero E6 (CRL-1586, American Type Culture Collection (ATCC), Vero CCL81 (ATCC), and HEK293 cells were cultured at 37°C in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES pH 7.3, 1 mM sodium pyruvate, 1X non-essential amino acids, and 100 U/ml of penicillin–streptomycin
SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 was obtained from the Centers for Disease Control and Prevention (a gift from Natalie Thornburg). The virus was passaged once in Vero CCL81 cells and titrated by focus-forming assay (FFA) on Vero E6 cells. The recombinant luciferase-expressing full-length SARS-CoV-2 reporter virus (2019 n-CoV/USA_WA1/2020 strain) has been reported previously (Zost et al., 2020), and the D614G variant will be described elsewhere (R. Baric, manuscript in preparation). All work with infectious SARS-CoV-2 was performed in Institutional Biosafety Committee approved BSL3 and A-BSL3 facilities using appropriate positive pressure air respirators and protective equipment.
Mouse experiments
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381-01). Virus inoculations were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering.
Female BALB/c (catalog 000651) and K18-hACE2 C57BL/6 (catalog 034860) mice were purchased from The Jackson Laboratory. Four to five-week-old animals were immunized with 1010 viral particles (vp) of ChAdV-empty or ChAd-SARS-CoV-2-S in 50 μl PBS via intramuscular injection in the hind leg or via intranasal inoculation. Subsets of immunized animals were boosted four weeks after primary immunization using the same route used for primary immunization. Vaccinated BALB/c mice (10 to 11-week-old) were given a single intraperitoneal injection of 2 mg of anti-Ifnar1 mAb (MAR1-5A3 (Sheehan et al., 2006), Leinco) one day before intranasal administration of 108 PFU of Hu-AdV5-hACE2. Five days after Hu-AdV5 transduction, mice were inoculated with 4 × 105 FFU of SARS-CoV-2 by the intranasal route. K18-hACE2 mice were challenged on day 30 after immunization with 103 PFU of SARS-CoV-2 via an intranasal route. Animals were euthanized at 4 or 8 dpi, and tissues were harvested for virological, immunological, and pathological analyses.
Method Details
Construction of chimpanzee adenovirus vectors
Simian Ad36 vector (ChAd) (Roy et al., 2011) was obtained from the Penn Vector Core of the University of Pennsylvania. The ChAd genome was engineered with deletions in the E1 and E3B region (GenBank: FJ025917.1; nucleotides 455-3026 and 30072-31869, respectively). A modified human cytomegalovirus major immediate early promoter sequence was incorporated in place of the E1 gene in reverse orientation on the complementary DNA strand. CMV modification included an addition of two copies of the tet operator 2 (TetO2) sequence (Hillen and Berens, 1994) inserted in tandem (5′-TCT CTA TCA CTG ATA GGG AGA TCT CTA TCA CTG ATA GG GA-3′) between the TATA box and the mRNA start (GenBank: MN920393, nucleotides 174211-174212). SARS-CoV-2 S (GenBank MN908947.3; and two proline substitutions at residues K986 and V987 that stabilize the prefusion form of S (Pallesen et al., 2017; Wrapp et al., 2020)) were cloned into a unique PmeI site under the CMV-tetO2 promoter control in pSAd36 genomic plasmid to generate pSAd36-S. In parallel, a pSAd36-control carrying an empty CMV-tetO2 cassette with no transgene also was generated. The pSAd36-S and pSAd-control plasmids were linearized with PacI restriction enzyme to liberate viral genomes for transfection into T-Rex 293-HEK cells (Invitrogen). The rescued replication-incompetent ChAd-SARS-CoV-2-S and ChAd-Control vectors were scaled up in 293 cells and purified by CsCl density-gradient ultracentrifugation. Viral particle concentration in each vector preparation was determined by spectrophotometry at 260 nm as described (Maizel et al., 1968).
Construction of a human adenovirus vector expressing human ACE2
Codon-optimized hACE2 sequences were cloned into the shuttle vector (pShuttle-CMV, Addgene 240007) to generate pShuttle-hACE2. pShuttle-hACE2 was linearized with PmeI and subsequently cotransformed with the HuAdv5 backbone plasmid (pAdEasy-1 vector; Addgene 240005) into E. coli strain BJ5183 to generate pAdV5-ACE2 by homologous recombination as described (He et al., 1998). The pAdEasy-1 plasmid containing the HuAdV5 genome has deletions in E1 and E3 genes. hACE2 is under transcriptional control of a cytomegalovirus promotor and is flanked at its 3′ end by a SV40 polyadenylation signal. The pAd-hACE2 was linearized with PacI restriction enzyme before transfection into T-Rex 293 HEK cells (Invitrogen) to generate HuAdv5-hACE2. Recombinant HuAdv5-hACE2 was produced in 293-HEK cells and purified by CsCl density-gradient ultracentrifugation. The viral titer was determined by plaque assay in 293-HEK cells.
In situ RNA hybridization and histology
RNA in situ hybridization was performed using RNAscope 2.5 HD (Brown) (Advanced Cell Diagnostics) according to the manufacturer’s instructions. Left lung tissues were collected at necropsy, inflated with 10% neutral buffered formalin (NBF), and thereafter immersion fixed in 10% NBF for seven days before processing. Paraffin-embedded lung sections were deparaffinized by incubating at 60°C for 1 h, and endogenous peroxidases were quenched with H2O2 for 10 min at room temperature. Slides were boiled for 15 min in RNAscope Target Retrieval Reagents and incubated for 30 min in RNAscope Protease Plus reagent prior to SARS-CoV2 RNA probe (Advanced Cell Diagnostics 848561) hybridization and signal amplification. Sections were counterstained with Gill’s hematoxylin and visualized by brightfield microscopy. Some lung sections were processed for histology after hematoxylin and eosin staining.
SARS-CoV-2 neutralization assays
Heat-inactivated serum samples were diluted serially and incubated with 102 FFU of SARS-CoV-2 for 1 h at 37°C. The virus-serum mixtures were added to Vero cell monolayers in 96-well plates and incubated for 1 h at 37°C. Subsequently, cells were overlaid with 1% (w/v) methylcellulose in MEM supplemented with 2% FBS. Plates were incubated for 30 h before fixation using 4% PFA in PBS for 1 h at room temperature. Cells were washed and then sequentially incubated with anti-SARS-CoV-2 CR3022 antibody (Yuan et al., 2020) (1 μg/mL) and a HRP-conjugated goat anti-human IgG (Sigma) in PBS supplemented with 0.1% (w/v) saponin (Sigma) and 0.1% BSA. TrueBlue peroxidase substrate (KPL) was used to develop the plates before counting the foci on a BioSpot analyzer (Cellular Technology Limited). For neutralization experiments with luciferase expressing SARS-CoV-2, serum samples were diluted 3-fold starting at 1:50 and mixed with 85 PFU of each recombinant virus (wild-type and D614G). Vero E6 cells plated in clear-bottom black-walled 96-well plates (Corning) were inoculated with serum-virus mixtures, and cells were cultured at 37°C for 48 h. Subsequently, cells were lysed and luciferase activity was measured using the Nano-Glo Luciferase Assay System (Promega) according to the manufacturer’s specifications.
Hu-AdV5 neutralization assays
One day prior to Hu-AdV5-hACE2 transduction, serum samples were collected from mice immunized intramuscularly with ChAd-Control or ChAd-SARS-CoV-2-S. Sera were heat-inactivated and serially diluted prior to incubation with 102 FFU of Hu-AdV5 for 1 h at 37°C. The virus-serum mixtures were added to HEK293 cell monolayers in 96-well plates and incubated for 1 h at 37°C. Cells were then overlaid with 1% (w/v) methylcellulose in MEM supplemented with 5% FBS. Plates were incubated at 37°C for 48 h before fixation with 2% PFA in PBS for 1 h at room temperature. Subsequently, plates were washed with PBS and incubated overnight at 4°C with biotinylated anti-HuAdV5-hexon antibody (2 μg/mL; Novus Biologicals NB600413) diluted in permeabilization buffer (PBS supplemented with 0.1% (w/v) saponin and 0.1% BSA). Plates were washed again and incubated with streptavidin-HRP (1:3000; Vector Laboratories SA-5004) in permeabilization buffer for 30 min at room temperature. After a final wash series, plates were developed using TrueBlue peroxidase substrate (KPL) and foci were counted on a BioSpot analyzer (Cellular Technology Limited).
Protein expression and purification
Purified RNA from the 2019-nCoV/USA-WA1/2020 SARS-CoV-2 strain was reverse transcribed into cDNA and used as the template for recombinant gene cloning. A full-length SARS-CoV-2 NP was cloned into pET21a with a hexahistidine tag and recombinantly expressed using BL21(DE3)-RIL E. coli in Terrific Broth (bioWORLD). Following overnight induction with isopropyl β-d-1-thiogalactopyranoside (Goldbio) at 25°C, cells were lysed in 20 mM Tris-HCl pH 8.5, 1 M NaCl, 5 mM β-mercaptopethanol, and 5 mM imidazole for nickel-affinity purification. Following elution in the prior buffer supplemented with 500 mM imidazole, the protein was purified to homogeneity using size exclusion and, in some cases, cation exchange chromatography. SARS-CoV-2 RBD and S ectodomain (S1/S2 furin cleavage site was disrupted, double proline mutations were introduced into the S2 subunit, and foldon trimerization motif was incorporated) were cloned into pFM1.2 with a C-terminal hexahistidine or octahistidine tag, transiently transfected into Expi293F cells, and purified by cobalt-charged resin chromatography (G-Biosciences) as previously described (Alsoussi et al., 2020).
ELISA
Purified antigens (S, RBD, or NP) were coated onto 96-well Maxisorp clear plates at 2 μg/mL in 50 mM Na2CO3pH 9.6 (70 μL) overnight at 4°C. Coating buffers were aspirated, and wells were blocked with 200 μL of 1X PBS + 0.05% Tween-20 + 1% BSA + 0.02% NaN3 (Blocking buffer, PBSTBA) either for 1 h at 37°C or overnight at 4°C. Heat-inactivated serum samples were diluted in PBSTBA in a separate 96-well polypropylene plate. The plates then were washed thrice with 1X PBS + 0.05% Tween-20 (PBST), followed by addition of 50 μL of respective serum dilutions. Sera were incubated in the blocked ELISA plates for at least 1 h at room temperature. The ELISA plates were again washed thrice in PBST, followed by addition of 50 μL of 1:2000 anti-mouse IgG-HRP (Southern Biotech Cat. #1030-05) in PBST or 1:10000 of biotinylated anti-mouse IgG, anti-mouse IgM, or anti-mouse IgA in PBSTBA (SouthernBiotech). Plates were incubated at room temperature for 1 h, washed thrice in PBST, and then 1:5000 dilution of streptavidin-HRP (ThermoFisher) was added to wells. Following a 1 h incubation at room temperature, plates were washed thrice with PBST and 50 μL of 1-Step Ultra TMB-ELISA was added (ThermoFisher Cat. #34028). Following a 12 to 15-min incubation, reactions were stopped with 50 μL of 2 M sulfuric acid. The absorbance of each well at 450 nm was read (Synergy H1) within 2 min of addition of sulfuric acid. Optical density (450 nm) measurements were determined using a microplate reader (Bio-Rad). The endpoint serum dilution was calculated with curve fit analysis of optical density (OD) values for serially diluted sera with a cut-off value set to three times the background signal.
ELISpot assay
MultiScreen-HA filter 96-well plates (Millipore) plates were pre-coated with 3 μg/ml of SARS-CoV-2 S protein overnight at 4°C. After rinsing with PBST, plates were blocked for 4 h at 37°C with culture medium (RPMI, 10% FBS, penicillin-streptomycin, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 10 mM HEPES, and 50 mM β-mercaptoethanol). Single cell suspensions of splenocytes in culture medium were added to the S protein coated plates and incubated at 37°C and 5% humidified CO2 for 4 h. After washing with PBS and PBST, plates were incubated with biotinylated anti-IgG or anti-IgA (Southern Biotech) followed by incubation with streptavidin conjugated horseradish peroxidase (Jackson ImmunoResearch), each for 1 h at room temperature. After additional washes with PBS, 3-amino-9-ethylcarbazole (Sigma) substrate solution was added for spot development. The reaction was stopped by rinsing with water. Spots were counted using a Biospot plate reader (Cellular Technology).
Measurement of viral burden
SARS-CoV-2 infected mice were euthanized using a ketamine and xylazine cocktail, and organs were collected. Tissues were weighed and homogenized with beads using a MagNA Lyser (Roche) in 1 mL of Dulbecco’s Modified Eagle’s Medium (DMEM) containing 2% fetal bovine serum (FBS). RNA was extracted from clarified tissue homogenates using MagMax mirVana Total RNA isolation kit (Thermo Scientific) and the KingFisher Flex extraction system (Thermo Scientific). SARS-CoV-2 RNA levels were measured by one-step quantitative reverse transcriptase PCR (qRT-PCR) TaqMan assay as described previously (Hassan et al., 2020). SARS-CoV-2 nucleocapsid (N) and Open Reading Frame 1a (ORF1a) specific primers and probe sets were used: (N: F Primer: ATGCTGCAATCGTGCTACAA; R primer: GACTGCCGCCTCTGCTC; probe: /56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/; ORF1a: F Primer: TTCAGTTGACTTCGCAGTGG; R primer: GGACGGGTTTGAGTTTTTCA; probe: /56-FAM/AACTAACAT/ZEN/CTTTGGCACTGTTT/3IABkFQ) (Integrated DNA Technologies). Viral RNA was expressed as (N) or (ORF1a) gene copy numbers per milligram on a log10 scale. For some samples, viral titer was determined by plaque assay on Vero E6 cells.
Cytokine and chemokine mRNA measurements
RNA extracted from lung homogenates was DNase-treated and used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Scientific) with the addition of RNase inhibitor according to the manufacturer’s protocol. Cytokine and chemokine expression was determined using TaqMan Fast Universal PCR master mix (Thermo Scientific) with commercial primers/probe sets specific for IFN-γ (IDT: Mm.PT.58.41769240), IL-6 (Mm.PT.58.10005566), IL-1β (Mm.PT.58.41616450), CXCL10 (Mm.PT.58.43575827), CCL2 (Mm.PT.58.42151692), CCL5 (Mm.PT.58.43548565), CXCL11 (Mm.PT.58.10773148.g), IFN-β (Mm.PT.58.30132453.g), and IFNλ−2/3 (Thermo Scientific Mm04204156_gH) and results were normalized to GAPDH (Mm.PT.39a.1) levels. Fold change was determined using the 2-ΔΔCt method comparing treated mice to naive controls.
Plaque assay
Vero-Furin cells were seeded in 12-well plates one day prior to inoculation. Lung homogenates were serially diluted in DMEM supplemented with 2% FBS, HEPES, and penicillin-streptomycin and incubated on cells for 1 hr at 37°C. After 1 h incubation, cells were overlaid with 1% (w/v) methylcellulose in MEM with 2% FBS and incubated again at 37°C. At 3 dpi, overlays were removed, and plates were fixed with 4% PFA for 20 min at room temperature. After removal of PFA, plates were stained with 0.05% (w/v) crystal violet in 20% methanol for 10-20 min. Crystal violet was removed, and plates were rinsed with water or PBS. Plaques were counted manually to determine infectious virus titer.
Peptide restimulation and intracellular cytokine staining
Splenocytes from intramuscularly vaccinated mice were incubated in culture with a pool of 253 overlapping 15-mer SARS-CoV-2 S peptides (Grifoni et al., 2020) for 12 h at 37°C before a 4 h treatment with brefeldin A (BioLegend, 420601). Following blocking with FcγR antibody (BioLegend, clone 93), cells were stained on ice with CD45 BUV395 (BD BioSciences clone 30-F11); CD44 PE-Cy7, CD4 PE-Cy5, CD8b PreCP-Cy5.5, and CD19 APC-Cy7 (BioLegend clones, IM7, GK1.5, YTS156.7.7, and 6D5, respectively), and Fixable Aqua Dead Cell Stain (Invitrogen, L34966). Stained cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBiosciences, 00-5523). Subsequently, intracellular staining was performed with anti-IFN-γ Alexa 647 (BD Biosciences, clone XMG1.2), anti-TNFα BV605 (BioLegend, clone MP6-XT22), and anti-GrB PE (Invitrogen, GRB04). Lungs from immunized mice were harvested and digested for 1 h at 37°C in digestion buffer consisting of RPMI media supplemented with (167 μg/ml) of Liberase DH (Sigma) and (100 μg/ml) of DNase I (Sigma). Lung cells were incubated at 37°C with the pool of 253 overlapping 15-mer SARS-CoV-2 S peptides described above in the presence of brefeldin A for 5 h at 37°C. Lung cells then were stained as described above except that CD4-BV421 (BioLegend clone GK1.5) replaced the CD4-PE-Cy5, no CD19 staining was included, and CD103-FITC and CD69-BV711 (BioLegend clones, 2E7, and, H1.2F3, respectively) were added. Analysis was performed on a BD LSRFortessa X-20 cytometer, using FlowJo X 10.0 software.
Flow cytometry-based antigen characterization
HEK293T cells were seeded at 106 cells/well in 6-well plates 24 h prior to transduction with ChAd-SARS-CoV-2-S (MOI of 5). After 20 h, cells were harvested, fixed and permeabilized using Foxp3 Transcription Factor Staining Buffer Set (Thermo Fisher), and stained for viral antigen after incubation with the following anti-SARS-CoV-2 neutralizing murine mAbs: SARS2-01, SARS2-02, SARS2-07, SARS2-11, SARS2-12, SARS2-16, SARS2-18, SARS2-20, SARS2-21, SARS2-22, SARS2-23, SARS2-29, SARS2-31, SARS2-32, SARS2-34, SARS2-38, SARS2-39, SARS2-50, SARS2-55, SARS2-58, SARS2-66, and SARS2-71 (L.A.V. and M.S.D., unpublished data). H77.39 (Sabo et al., 2011), an isotype-matched anti-HCV E2 mAb was used as a negative control. Cells were washed, incubated with Alexa Fluor 647 conjugated goat anti-mouse IgG (Thermo Fisher), and analyzed by flow cytometry using a MACSQuant Analyzer 10 (Miltenyi Biotec). The percentage of cells positive for a given mAb was compared with cells stained with a saturating amount of an oligoclonal mixture of anti-SARS-CoV-2 mAbs.
Quantification and Statistical Analysis
Statistical significance was assigned when P values were < 0.05 using Prism Version 8 (GraphPad). Tests, number of animals (n), median values, and statistical comparison groups are indicated in the Figure legends. Analysis of anti-S, anti-RBD, neutralization titers, and ELISPOT values in mice after vaccination was performed using a Mann-Whitney test. Differences in viral titers or chemokine and cytokine levels after SARS-CoV-2 infection of immunized mice were determined using a Mann-Whitney test. Anti-nucleoprotein antibody levels were compared using a paired t test.
Acknowledgments
This study was supported by NIH contracts and grants (75N93019C00062, 75N9301900065, R01 AI127828, R01 AI130591, R01 AI149644, and R35 HL145242), Center for Structural Genomics of Infectious Diseases (HHSN272201400018C, and HHSN272201200026C), and the Defense Advanced Research Projects Agency (HR001117S0019). B.T.M. is supported by F32 AI138392, E.S.W. is supported by T32 AI007163, and J.B.C. is supported by a Helen Hay Whitney Foundation postdoctoral fellowship. We thank Sean Whelan, Susan Cook, and Jennifer Philips for facilitating studies with SARS-CoV-2; James Earnest for performing cell culture; and the Pulmonary Morphology Core at Washington University School of Medicine for tissue sectioning. Some figures were created with BioRender software.
Author Contributions
A.O.H. designed experiments and performed intranasal inoculations of adenovirus. I.P.D., E.K., A.O.H., and D.T.C. designed and generated the ChAd constructs. J.B.C. propagated the SARS-CoV-2 stocks and performed plaque assays. A.L.B. designed the qRT-PCR assays. A.O.H. evaluated cell-mediated immune responses. A.O.H., N.M.K., E.S.W., S.S., B.T.M., R.E.C., L.E.G., and J.M.F. performed clinical analysis, histopathological studies, and viral burden analyses. B.T.M. performed in situ hybridization. A.S. and Y.J.H. designed and performed experiments with recombinant strains of SARS-CoV-2. A.S.K. performed flow cytometry analysis experiments. S.P.K. and M.J.H. analyzed the tissue sections for histopathology. A.W.W. performed the ChAd-immune serum neutralization assays. D.W. generated the T cell peptide pools. L.A.V.B. provided SARS-CoV-2 monoclonal antibodies. H.Z., Y.-N.D., M.M., L.J.A., and D.H.F. designed and produced the recombinant S, RBD, and N proteins. I.B.H., J.E.D., and B.K.S. performed ELISAs, and J.B.C. and R.E.C. performed neutralization assays. R.S.B. designed experiments and secured funding. A.O.H. and M.S.D. wrote the initial draft, with the other authors providing editorial comments.
Declaration of Interests
M.S.D. is a consultant for Inbios, Vir Biotechnology, and NGM Biopharmaceuticals and on the Scientific Advisory Board of Moderna. The Diamond laboratory has received unrelated funding support from Moderna, Vir Biotechnology, and Emergent BioSolutions. M.S.D., D.T.C., A.O.H., and I.P.D. have filed a disclosure with Washington University for possible development of ChAd-SARS-CoV-2. M.J.H. is a member of the DSMB for AstraZeneca and founder of NuPeak Therapeutics. The Baric laboratory has received unrelated funding support from Takeda, Pfizer, and Eli Lilly.
Published: August 19, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.026.
Supplemental Information
References
- Abbink P., Larocca R.A., De La Barrera R.A., Bricault C.A., Moseley E.T., Boyd M., Kirilova M., Li Z., Ng’ang’a D., Nanayakkara O. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science. 2016;353:1129–1132. doi: 10.1126/science.aah6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alharbi N.K., Qasim I., Almasoud A., Aljami H.A., Alenazi M.W., Alhafufi A., Aldibasi O.S., Hashem A.M., Kasem S., Albrahim R. Humoral immunogenicity and efficacy of a single dose of ChAdOx1 MERS vaccine candidate in dromedary camels. Sci. Rep. 2019;9:16292. doi: 10.1038/s41598-019-52730-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsoussi W.B., Turner J.S., Case J.B., Zhao H., Schmitz A.J., Zhou J.Q., Chen R.E., Lei T., Rizk A.A., McIntire K.M. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. J. Immunol. 2020;205:915–922. doi: 10.4049/jimmunol.2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolles M., Deming D., Long K., Agnihothram S., Whitmore A., Ferris M., Funkhouser W., Gralinski L., Totura A., Heise M., Baric R.S. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011;85:12201–12215. doi: 10.1128/JVI.06048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton D.R., Walker L.M. Rational vaccine design in the time of COVID-19. Cell Host Microbe. 2020;27:695–698. doi: 10.1016/j.chom.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzas C., Chevalier C. Innovative mucosal vaccine formulations against influenza A virus infections. Front. Immunol. 2019;10:1605. doi: 10.3389/fimmu.2019.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J.B., Rothlauf P.W., Chen R.E., Liu Z., Zhao H., Kim A.S., Bloyet L.M., Zeng Q., Tahan S., Droit L. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host Microbe. 2020 doi: 10.1016/j.chom.2020.06.021. Published July 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekar A., Liu J., Martinot A.J., McMahan K., Mercado N.B., Peter L., Tostanoski L.H., Yu J., Maliga Z., Nekorchuk M. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. 2020;369:812–817. doi: 10.1126/science.abc4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung E.W., Zachariah P., Gorelik M., Boneparth A., Kernie S.G., Orange J.S., Milner J.D. Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. JAMA. 2020 doi: 10.1001/jama.2020.10374. Published online June 8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett K.S., Flynn B., Foulds K.E., Francica J.R., Boyoglu-Barnum S., Werner A.P., Flach B., O’Connell S., Bock K.W., Minai M. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2024671. Published online July 28, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C.W., Jackson K.J.L., McElroy A.K., Halfmann P., Huang J., Chennareddy C., Piper A.E., Leung Y., Albarino C.G., Crozier I. Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell. 2019;177:1566–1582. doi: 10.1016/j.cell.2019.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alwis R., Chen S., Gan E.S., Ooi E.E. Impact of immune enhancement on Covid-19 polyclonal hyperimmune globulin therapy and vaccine development. EBioMedicine. 2020;55:102768. doi: 10.1016/j.ebiom.2020.102768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Pierson T.C. The challenges of vaccine development against a new virus during a pandemic. Cell Host Microbe. 2020;27:699–703. doi: 10.1016/j.chom.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicks M.D., Spencer A.J., Edwards N.J., Wadell G., Bojang K., Gilbert S.C., Hill A.V., Cottingham M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS ONE. 2012;7:e40385. doi: 10.1371/journal.pone.0040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas A.D., de Cassan S.C., Dicks M.D., Gilbert S.C., Hill A.V., Draper S.J. Tailoring subunit vaccine immunogenicity: maximizing antibody and T cell responses by using combinations of adenovirus, poxvirus and protein-adjuvant vaccines against Plasmodium falciparum MSP1. Vaccine. 2010;28:7167–7178. doi: 10.1016/j.vaccine.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A., Huang C.T., Lin C.Y., Chen T.C., Lin Y.C., Chang C.S., He Y.C. Sterilizing immunity to influenza virus infection requires local antigen-specific T cell response in the lungs. Sci. Rep. 2016;6:32973. doi: 10.1038/srep32973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folegatti P.M., Bittaye M., Flaxman A., Lopez F.R., Bellamy D., Kupke A., Mair C., Makinson R., Sheridan J., Rohde C. Safety and immunogenicity of a candidate Middle East respiratory syndrome coronavirus viral-vectored vaccine: a dose-escalation, open-label, non-randomised, uncontrolled, phase 1 trial. Lancet Infect. Dis. 2020;20:816–826. doi: 10.1016/S1473-3099(20)30160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folegatti P.M., Ewer K.J., Aley P.K., Angus B., Becker S., Belij-Rammerstorfer S., Bellamy D., Bibi S., Bittaye M., Clutterbuck E.A. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396:467–478. doi: 10.1016/S0140-6736(20)31604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Bao L., Mao H., Wang L., Xu K., Yang M., Li Y., Zhu L., Wang N., Lv Z. Development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369:77–81. doi: 10.1126/science.abc1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B.S. Rapid COVID-19 vaccine development. Science. 2020;368:945–946. doi: 10.1126/science.abb8923. [DOI] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem A.M., Algaissi A., Agrawal A.S., Al-Amri S.S., Alhabbab R.Y., Sohrab S.S., Almasoud A.S., Alharbi N.K., Peng B.-H., Russell M. A highly immunogenic, protective, and safe adenovirus-based vaccine expressing Middle East respiratory syndrome coronavirus S1-CD40L fusion protein in a transgenic human dipeptidyl peptidase 4 mouse model. J. Infect. Dis. 2019;220:1558–1567. doi: 10.1093/infdis/jiz137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Dmitriev I.P., Kashentseva E.A., Zhao H., Brough D.E., Fremont D.H., Curiel D.T., Diamond M.S. A gorilla adenovirus-based vaccine against Zika virus induces durable immunity and confers protection in pregnancy. Cell Rep. 2019;28:2634–2646.e4. doi: 10.1016/j.celrep.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Case J.B., Winkler E.S., Thackray L.B., Kafai N.M., Bailey A.L., McCune B.T., Fox J.M., Chen R.E., Alsoussi W.B. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell. 2020;182:744–753.e4. doi: 10.1016/j.cell.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T.C., Zhou S., da Costa L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen W., Berens C. Mechanisms underlying expression of Tn10 encoded tetracycline resistance. Annu. Rev. Microbiol. 1994;48:345–369. doi: 10.1146/annurev.mi.48.100194.002021. [DOI] [PubMed] [Google Scholar]
- Hodgson S.H., Ewer K.J., Bliss C.M., Edwards N.J., Rampling T., Anagnostou N.A., de Barra E., Havelock T., Bowyer G., Poulton I.D. Evaluation of the efficacy of ChAd63-MVA vectored vaccines expressing circumsporozoite protein and ME-TRAP against controlled human malaria infection in malaria-naive individuals. J. Infect. Dis. 2015;211:1076–1086. doi: 10.1093/infdis/jiu579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobinger G.P., Figueredo J.M., Rowe T., Zhi Y., Gao G., Sanmiguel J.C., Bell P., Wivel N.A., Zitzow L.A., Flieder D.B. Adenovirus-based vaccine prevents pneumonia in ferrets challenged with the SARS coronavirus and stimulates robust immune responses in macaques. Vaccine. 2007;25:5220–5231. doi: 10.1016/j.vaccine.2007.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie K.L., Carolan L.A., Middleton D., Lowther S., Kelso A., Barr I.G. Multiple infections with seasonal influenza A virus induce cross-protective immunity against A(H1N1) pandemic influenza virus in a ferret model. J. Infect. Dis. 2010;202:1011–1020. doi: 10.1086/656188. [DOI] [PubMed] [Google Scholar]
- Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Wei Q., Lin Q., Fang J., Wang H., Kwok H., Tang H., Nishiura K., Peng J., Tan Z. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4:e123158. doi: 10.1172/jci.insight.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Camacho C., Abbink P., Larocca R.A., Dejnirattisai W., Boyd M., Badamchi-Zadeh A., Wallace Z.R., Doig J., Velazquez R.S., Neto R.D.L. Rational Zika vaccine design via the modulation of antigen membrane anchors in chimpanzee adenoviral vectors. Nat. Commun. 2018;9:2441. doi: 10.1038/s41467-018-04859-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizel J.V., Jr., White D.O., Scharff M.D. The polypeptides of adenovirus. I. Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology. 1968;36:115–125. doi: 10.1016/0042-6822(68)90121-9. [DOI] [PubMed] [Google Scholar]
- Mao R., Qiu Y., He J.-S., Tan J.-Y., Li X.-H., Liang J., Shen J., Zhu L.-R., Chen Y., Iacucci M. Manifestations and prognosis of gastrointestinal and liver involvement in patients with COVID-19: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020;5:667–678. doi: 10.1016/S2468-1253(20)30126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCray P.B., Jr., Pewe L., Wohlford-Lenane C., Hickey M., Manzel L., Shi L., Netland J., Jia H.P., Halabi C., Sigmund C.D. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster V.J., Wells D., Lambe T., Wright D., Fischer R.J., Bushmaker T., Saturday G., van Doremalen N., Gilbert S.C., de Wit E., Warimwe G.M. Protective efficacy of a novel simian adenovirus vaccine against lethal MERS-CoV challenge in a transgenic human DPP4 mouse model. NPJ Vaccines. 2017;2:28. doi: 10.1038/s41541-017-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster V.J., Feldmann F., Williamson B.N., van Doremalen N., Pérez-Pérez L., Schulz J., Meade-White K., Okumura A., Callison J., Brumbaugh B. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature. 2020 doi: 10.1038/s41586-020-2324-7. Published online May 12, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutra M.R., Kozlowski P.A. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J., Wang N., Corbett K.S., Wrapp D., Kirchdoerfer R.N., Turner H.L., Cottrell C.A., Becker M.M., Wang L., Shi W. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA. 2017;114:E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaloza-MacMaster P., Provine N.M., Ra J., Borducchi E.N., McNally A., Simmons N.L., Iampietro M.J., Barouch D.H. Alternative serotype adenovirus vaccine vectors elicit memory T cells with enhanced anamnestic capacity compared to Ad5 vectors. J. Virol. 2013;87:1373–1384. doi: 10.1128/JVI.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Rathnasinghe R., Strohmeier S., Amanat F., Gillespie V.L., Krammer F., García-Sastre A., Coughlan L., Schotsaert M., Uccellini M. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. bioRxiv. 2020 doi: 10.1101/2020.07.06.190066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Sandoval A., Berthoud T., Alder N., Siani L., Gilbert S.C., Nicosia A., Colloca S., Cortese R., Hill A.V. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect. Immun. 2010;78:145–153. doi: 10.1128/IAI.00740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Medina-Jaszek A., Wilson M.J., Sandhu A., Calcedo R., Lin J., Wilson J.M. Creation of a panel of vectors based on ape adenovirus isolates. J. Gene Med. 2011;13:17–25. doi: 10.1002/jgm.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo M.C., Luca V.C., Prentoe J., Hopcraft S.E., Blight K.J., Yi M., Lemon S.M., Ball J.K., Bukh J., Evans M.J. Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J. Virol. 2011;85:7005–7019. doi: 10.1128/JVI.00586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan K.C., Lai K.S., Dunn G.P., Bruce A.T., Diamond M.S., Heutel J.D., Dungo-Arthur C., Carrero J.A., White J.M., Hertzog P.J., Schreiber R.D. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 2006;26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Amanna I. How advances in immunology provide insight into improving vaccine efficacy. Vaccine. 2014;32:2948–2957. doi: 10.1016/j.vaccine.2014.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura S. Persistence in temporary lung niches: a survival strategy of lung-resident memory CD8+ T cells. Viral Immunol. 2017;30:438–450. doi: 10.1089/vim.2017.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doremalen N., Lambe T., Spencer A., Belij-Rammerstorfer S., Purushotham J.N., Port J.R., Avanzato V.A., Bushmaker T., Flaxman A., Ulaszewska M. ChAdOx1nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature. 2020;30 doi: 10.1038/s41586-020-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingartl H., Czub M., Czub S., Neufeld J., Marszal P., Gren J., Smith G., Jones S., Proulx R., Deschambault Y. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J. Virol. 2004;78:12672–12676. doi: 10.1128/JVI.78.22.12672-12676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann D., Sperhake J.-P., Lütgehetmann M., Steurer S., Edler C., Heinemann A., Heinrich F., Mushumba H., Kniep I., Schröder A.S. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann. Intern. Med. 2020 doi: 10.7326/M20-2003. Published online May 6, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler E.S., Bailey A.L., Kafai N.M., Nair S., McCune B.T., Yu J., Fox J.M., Chen R.E., Earnest J.T., Keeler S.P. SARS-CoV-2 infection in the lungs of human ACE2 transgenic mice causes severe inflammation, immune cell infiltration, and compromised respiratory function. bioRxiv. 2020 doi: 10.1101/2020.07.09.196188. [DOI] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Tostanoski L.H., Peter L., Mercado N.B., McMahan K., Mahrokhian S.H., Nkolola J.P., Liu J., Li Z., Chandrashekar A. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science. 2020;369:806–811. doi: 10.1126/science.abc6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Wu N.C., Zhu X., Lee C.D., So R.T.Y., Lv H., Mok C.K.P., Wilson I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368:630–633. doi: 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf H., Kett V. Current prospects and future challenges for nasal vaccine delivery. Hum. Vaccin. Immunother. 2017;13:34–45. doi: 10.1080/21645515.2016.1239668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F.C., Li Y.H., Guan X.H., Hou L.H., Wang W.J., Li J.X., Wu S.P., Wang B.S., Wang Z., Wang L. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet. 2020;395:1845–1854. doi: 10.1016/S0140-6736(20)31208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zost S.J., Gilchuk P., Chen R.E., Case J.B., Reidy J.X., Trivette A., Nargi R.S., Sutton R.E., Suryadevara N., Chen E.C. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. 2020 doi: 10.1038/s41591-020-0998-x. Published online July 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.