

Abstract
Recombinant vesicular stomatitis virus (VSV)-fusion and hemagglutinin (FH) was developed by substituting the promiscuous VSV-G glycoprotein (G) gene in the backbone of VSV with genes encoding for the measles virus envelope proteins F and H. Hybrid VSV-FH exhibited a multifaceted mechanism of cancer-cell killing and improved neurotolerability over parental VSV in preclinical studies. In this study, we evaluated VSV-FH in vitro and in vivo in models of hepatobiliary and pancreatic cancers. Our results indicate that high intrahepatic doses of VSV-FH did not result in any significant toxicity and were well tolerated by transgenic mice expressing the measles virus receptor CD46. Furthermore, a single intratumoral treatment with VSV-FH yielded improved survival and complete tumor regressions in a proportion of mice in the Hep3B hepatocellular carcinoma model but not in mice xenografted with BxPC-3 pancreatic cancer cells. Our preliminary findings indicate that VSV-FH can induce potent oncolysis in hepatocellular and pancreatic cancer cell lines with concordant results in vivo in hepatocellular cancer and discordant in pancreatic cancer without the VSV-mediated toxic effects previously observed in laboratory animals. Further study of VSV-FH as an oncolytic virotherapy is warranted in hepatocellular carcinoma and pancreatic cancer to understand broader applicability and mechanisms of sensitivity and resistance.
Keywords: hepatobiliary cancer, measles virus, oncolytic viral therapy, pancreatic cancer, vesicular stomatitis virus, hepatocellular carcinoma, cholangiocarcinoma
Graphical Abstract
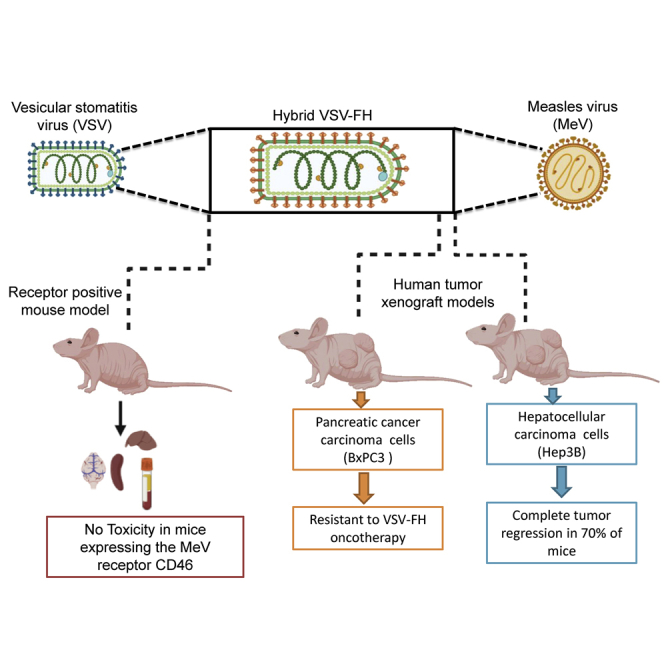
Oncolytic vesicular stomatitis virus (VSV)-FH is a hybrid virus with shared attributes of VSV and measles virus (MV). Herein, we present novel experiments, including safety pharmacology assessment of VSV-FH in receptor-positive animals and efficacy in hepatobiliary and pancreatic cancer (HBPC) models which, to our knowledge, have not been previously described.
Introduction
Oncolytic vesicular stomatitis virus (VSV) has engendered keen enthusiasm in the field of human cancer therapy.1, 2, 3 Several VSV-derived vectors have been extensively evaluated as virotherapies and vaccine vehicles.3, 4, 5, 6, 7, 8, 9, 10, 11, 12 However, successful clinical translation of VSV has been hindered by a number of factors, including neurovirulence, liver toxicity, and variable sensitivity of malignant tumors to VSV oncotherapy.3,4,13, 14, 15, 16, 17, 18
Several studies have attempted to remedy these limitations, with varying degrees of success.19, 20, 21 However, additional investigations and new approaches are needed before VSV-based virotherapy can be used clinically to treat human cancers.
A number of successful viral engineering strategies have recently emerged to improve the safety of VSV-derived virotherapies, including substituting VSV-G glycoprotein (G) with envelope glycoproteins of heterologous viruses and displaying tumor-targeting ligands on VSV-G, which can confer tumor selectivity.4,20,22, 23, 24, 25 Among these is recombinant VSV-fusion and hemagglutinin (FH), a hybrid virus with shared attributes of VSV and measles virus (MV), which has potential for treating hepatobiliary and pancreatic cancers (HBPCs).25,26 VSV-FH was engineered by substituting the VSV-G protein gene, which causes neurovirulence, with the F and H genes of the Edmonston vaccine-strain MV (Ed-MV). In addition to having the potential to abrogate neurovirulence, VSV-FH utilizes Ed-MV receptor CD46 (membrane cofactor protein) for selective viral entry, as well as a syncytial/necrotic mechanism of cell killing from the expression of H (MV-H) and F (MV-F) proteins.25,26 Although no reports from ongoing clinical studies indicate that VSV-based vectors are able to cross the human blood-brain barrier and propagate in the brain, treatment with VSV-FH will allow for mitigation of neurotoxicity, because most normal neuronal cells lack surface expression of CD46,27, 28, 29, 30 which VSV-FH uses for cellular entry by virtue of its oncolytic MV attribute.22,25,26 VSV-FH has a robust and rapid lytic cycle attributable to the VSV platform and a preferential tropism for CD46-overexpressed cells from the MV platform. CD46 is ubiquitously expressed in all nucleated cells; however, its overexpression has been reported in human tumor cells and tissues, including HBPCs.31, 32, 33, 34, 35, 36, 37
We have previously shown that hybrid VSV-FH mediates a higher degree of human tumor selectivity, which is strongly associated with the number of CD46 molecules expressed on target cells.26 We also demonstrated that the multifaceted mechanisms of cancer-cell killing by VSV-FH are enhanced because of its inherent fusogenic activity.25,26 Furthermore, we showed that treatment with VSV-FH has the added benefit of being well tolerated and less neurotoxic in laboratory rodents than parental wild-type (WT)-VSV, which causes severe encephalitis in laboratory animals.25,26
HBPCs have extremely high rates of mortality and are increasing in incidence.38 Most patients are diagnosed at advanced stages, and the efficacy of systemic therapies remains modest.39, 40, 41 Therefore, there is an urgent need to develop more effective therapeutic approaches for these patients.
To determine whether VSV-FH could be an alternative therapeutic modality for HBPC patients who do not respond to treatment based on oncolytic VSV, we evaluated VSV-FH in a panel of CD46-overexpressing HBPC cell lines and in xenograft models. Furthermore, we established efficacy and safety profiles for this promising new vector, which will support its development as an oncotherapy to treat HBPCs. These preliminary results are anticipated to form the basis for first-in-human studies using this novel engineered oncolytic virus in patients with advanced HBPCs.
Results
VSV-FH-Enhanced Green Fluorescent Protein (EGFP) Infectivity in HBPC Cells
As previously described, the oncolytic virus VSV-FH-EGFP was generated by substitution of the VSV-G protein gene (necessary for the entry into target cells) with Ed-MV, MV-H, and MV-F genes.42 Figures 1A–1C show a schematic representation of the genome of VSV-FH-EGFP and the genomes of parental VSV and VSV-interferon (IFN)-β-sodium iodide symporter (NIS), which is a recombinant VSV currently in a clinical trial of primary and metastatic liver cancers (ClinicalTrials.gov: NCT01628640). Vectors that expressed either mouse or human IFN-β (mIFN-β or hIFN-β, respectively) and the human NIS gene were also used in this study.14 All HBPC cell lines had surface expression of CD46 molecules (Figure 1D) with variable expression levels.
Figure 1.

Schematic Representation of Viral Vector Genomes
(A) Parental VSV genome. (B) VSV-FH-EGFP. The VSV-G gene was replaced with genes encoding for the Edmonston strain MV-F and MV-H proteins and EGFP. (C) VSV-m/hIFN-β-NIS, containing the genes encoding for mouse or human IFN-β and NIS. (D) VSV-FH receptor expression levels (CD46) in a panel of gastrointestinal cancer cells. Expression of CD46 was measured by flow cytometry using a phycoerythrin-conjugated anti-human CD46 antibody (filled histogram) or an isotype control antibody (unfilled histogram). EGFP, enhanced green fluorescence protein; F, fusion; H, hemagglutinin; h, human; IFN, interferon; m, mouse; MV, measles virus; NIS, sodium iodide symporter; PE-A, phycoerythrin antibody; VSV, vesicular stomatitis virus.
To evaluate the susceptibility of CD46high HBPC cells to VSV-FH infection, confluent cell monolayers were infected with VSV-FH-EGFP at a multiplicity of infection (MOI) of 0.1 and incubated for 24 or 48 h. As shown by observable GFP-positive cells, all HBPC cell lines were susceptible to infection with VSV-FH, and crystal violet staining performed 48 h after infection revealed differences in the strength of the virus-induced cytopathic effect (CPE) depending on the cell line infected (Figure 2). Hepatocellular carcinoma (HCC) cell lines (Huh7 and Hep3B) exhibited extensive CPE compared with mock-infected monolayers, whereas the cell lines HepG2 (HCC) and HuCCT1 (biliary tract cancer) were less sensitive to infection with VSV-FH (Figure 2).
Figure 2.

VSV-FH-EGFP Infectivity in a Panel of Hepatobiliary and Pancreatic Cancer Cell Lines
The columns show 5 × 105 cells infected with VSV-FH-EGFP at a multiplicity of infection (MOI) of 0.1 or mock infected. Left 2 columns, representative images of VSV-FH-EGFP-infected cells taken with epifluorescence microscopy, 24 and 48 h after infection with an MOI of 0.1. Right 2 columns, representative images of infected or mock-infected cells fixed at 48 h, stained with crystal violet (scale bar, 200 μm). EGFP, enhanced green fluorescent protein; F, fusion; H, hemagglutinin; VSV, vesicular stomatitis virus.
Cell-Killing Activity of VSV-FH-EGFP
To further investigate the difference in the infectivity and CPE of VSV-FH, monolayers of HBPC were infected at different MOIs (1, 0.1, and 0.01), and cell viability was measured 72 h after infection. The results showed that bile duct cancer cells were more resistant to the VSV-FH-induced cytotoxicity, as manifested by 60% to 80% of cell viability at an MOI of 1 (Figure 3A). In contrast, high cytotoxicity was observed in HCC cell lines and the pancreatic ductal adenocarcinoma (PDAC) cell line BxPC-3 (Figure 3A). A similar phenotype was observed when cells were infected with VSV-mIFN-β-NIS or VSV-hIFN-β-NIS, although liver and pancreatic cancer cells were more sensitive to VSV-mIFN-β-NIS. Indeed, VSV-hIFN-β-NIS was less efficient for killing these cancer cells, reducing cell viability in the range of 10% to 40%, 72 h after exposure. To determine the kinetics of infectious viral particle (VSV-FH) production, viral titers were calculated from the collected supernatant of infected cells with an MOI of 0.1 at 24, 48, and 72 h after infection (Figure 3B). Most infected cell lines reached their maximum yield 24 h after infection, and the titer remained at similar levels during the 3 days of analysis. The 50% tissue-culture infective dose (TCID50)/mL range was between 1 × 104 and 5 × 106 in the cancer cells tested, confirming that live VSV-FH was produced but exhibited different cell-killing activity in HBPC models (Figure 3A).
Figure 3.

Cytotoxicity of VSV-FH in Hepatobiliary and Pancreatic Cancer Cell Lines
(A) 1.2 × 104 cells were mock infected or infected with the indicated recombinant virus at an MOI of 0.1. The percentage of cells alive was determined 72 h after infection by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assays. The average (and error bars) of 3 independent experiments is shown. (B) Indicated cell lines were infected with VSV-FH-EGFP. Viral particles present in the supernatant at 24, 48, or 72 h after infection were determined by titration in Vero cells. The average log (and error bars) of the 3 independent experiments is plotted. EGFP, enhanced green fluorescence protein; F, fusion; H, hemagglutinin; h, human; IFN, interferon; M, mouse; MOI, multiplicity of infection; NIS, sodium iodide symporter; TCID50, 50% tissue culture infective dose; VSV, vesicular stomatitis virus.
VSV-FH Safety Studies in CD46 Transgenic Mice
To determine whether VSV-FH is causally associated with liver damage and liver toxicity in animal models, transgenic mice expressing murine IFN-α receptor and human CD46 (CD46 IFN-αR WT/WT), obtained from the Mayo Clinic Viral Toxicology Pharmacology Core (Mayo Clinic, Rochester, MN), were subjected to 2 intrahepatic injections of VSV-FH (5 × 107 and 5 × 108 TCID50/kg) or saline. Similar genetically modified mice lacking a functional murine type I IFN receptor have been previously used for toxicology studies of oncolytic MV30,43,44 and VSV-FH.30
Analyses of multiple toxicology parameters, including complete blood counts (CBCs), serum chemistry, and liver function tests, were performed at 2, 21, and 45 days after treatment. As shown in Figure 4A, there was a transitory decrease in the number of white blood cells, lymphocytes, and platelets in mice treated with the highest dose (5 × 108) at days 2 and 21 after initiation of the treatment. However, by day 45, the mice recovered, and the levels of these 2 parameters were similar to those of control mice. Additionally, mild increases that were not statistically significant compared with saline in alanine aminotransferase (ALT), aspartate aminotransferase (AST), granulocytes, and serum total bilirubin were also observed at necropsy (day 45) in some mice. Serum urea nitrogen levels were no different in VSV-FH-treated than control mice.
Figure 4.

Toxicity of VSV-FH Injected into the Liver of CD46 IFN-αR WT/WT Transgenic Mice
(A) No dramatic change occurred in the average values for white blood cells (WBCs), lymphocytes (LYMs), platelets (PLTs), or granulocytes (GRAs) 45 days after treatment. Although a slight, transient decrease in the number of WBCs and LYMs occurred in animals treated with the highest dose of VSV-FH (5 × 108 TCID50/kg) at post-treatment days 2 and 21, at day 45, the levels of these 2 parameters were similar to those of the control-treated mice. (B) Weight loss in the saline group was comparable to that of the virus-treated mice. (C) Significantly higher absorbance values were obtained at 450 nm in the ELISA test specific for VSV antibodies in samples obtained from VSV-FH-treated animals compared with saline controls. In contrast, greater absorbance was observed in tests performed to determine the presence of MV antibodies in samples obtained from mice treated with VSV-FH at 5 × 108 TCID50/kg compared with 5 × 107 TCID50/kg and with saline-treated animals. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; ELISA, enzyme-linked immunosorbent assay; F, fusion; H, hemagglutinin; MV, measles virus; SUN, serum urea nitrogen; TBIL, total bilirubin; TCID50, 50% tissue culture infective dose; VSV, vesicular stomatitis virus.
At necropsy (day 45), liver samples (left lateral lobes) stained with hematoxylin and eosin were reviewed by the Histology Core Facility at Mayo Clinic (Scottsdale, AZ, USA) and showed no evidence of virus-induced hepatic damage (data not shown). Body weight decreased slightly by day 2 post-treatment for all groups (including the saline control group), although body weight consistently increased from 4 days after injection until the end of the study (Figure 4B).
As shown in Figure 4C at day 45, the levels of VSV antibodies and MV antibodies at day 45 were similar at 5 × 107 TCID50/kg of VSV-FH but not in the cohort treated with the highest dose (5 × 108 TCID50/kg), where elevated MV antibodies were detected (p < 0.001).
Collectively, these data indicate that intrahepatic injection of VSV-FH did not elicit significant liver toxicity in the treated animals (100% survival) or result in impairment (Kaplan-Meier method, p < 0.05).
Oncolytic Activity of VSV-FH against HBPC Xenografts
Hep3B (HCC)- and BxPC-3 (PDAC)-derived xenografts were established in female athymic nude mice and treated with a single intratumoral administration of 1 × 107 TCID50 units of VSV-FH. Changes in tumor-bearing mice, such as tumor volume, weight loss, clinical signs, and survival, were monitored for 8 weeks (56 days). Surprisingly, in the BxPC-3 model, VSV-FH did not significantly reduce tumor growth or increase overall survival (p = 0.36; Figure 5B), even though oncolytic activity was observed in vitro (Figures 2 and 3A), and immunochemistry on tumor samples in the VSV-FH-treated group showed the presence of the virus in 70% of tumors as early as day 3 in BxPC-3 and Hep3B models (Figure 5A). In contrast in the Hep3B HCC model, a significant reduction in the average tumor volume was observed as soon as 7 days after treatment (p = 0.009; Figure 5C) in the VSV-FH-treated group (n = 10), followed by complete tumor regression in 7 mice (70%). The reduced tumor volume after treatment with VSV-FH in the Hep3B cohort resulted in a significant difference in overall survival (p = 0.01; Figure 5C), indicating effectiveness of treatment with VSV-FH.
Figure 5.

Efficacy of VSV-FH Intratumoral Treatment of Subcutaneous Tumors (Hep3B and BxPC-3) in Female Athymic Nude Mice
To investigate the oncolytic activity of VSV-FH, 1 × 107 infectious particles were injected intratumorally in mice with BxPC-3 or Hep3B tumors. (A) On day 3, post-treatment, tumor tissues were collected in the virus-treated groups for immunohistochemistry analysis by fluorescent anti-VSV staining to detect the presence of VSV-FH. (B and C) Tumor volumes and percent survival of female mice were monitored at different times after treatment. Tumor size at different days after injection (left) and survival curves (right) were plotted and analyzed for (B) BxPC-3, pancreatic adenocarcinoma cell line, and (C) Hep3B, hepatocellular carcinoma cell line. VSV, vesicular stomatitis virus; F, fusion; H, hemagglutinin.
Discussion
In this study, we evaluated whether treatment with oncolytic VSV-FH could trigger a potent cytotoxicity effect in HBPC cell lines in vitro and in vivo using animal models. We conducted a causality assessment in the context of potential VSV-FH-induced liver damage and toxicity in a CD46 IFN-αR WT/WT. This particular mouse strain has been shown to be susceptible to infections by viruses that use human CD46 as the entry receptor, including MV, VSV-FH, and some adenoviruses.30,45,46 Additionally, we compared cytotoxic effects of VSV-FH with that of another recombinant, neuro-attenuated VSV that is currently being evaluated in a clinical trial (VSV-hIFN-β-NIS).14 In this study, we showed that VSV-FH potently induced HBPC cell killing in vitro and did not cause any significant toxicity in a humanized mouse model. Strong in vivo antitumor efficacy of VSV-FH was observed in a mouse xenograft of human HCC but not in a mouse xenograft of human PDAC.
Several oncolytic VSVs have been evaluated preclinically in both hematologic and solid tumors and are now in early phases of clinical development, including studies for refractory liver cancer and other advanced solid tumors (ClinicalTrials.gov: NCT01628640, NCT02923466, NCT03120624, NCT03017820, NCT03865212, and NCT03647163). There remains a need for evaluation of novel vesiculovirus oncolytic vectors, given the attenuated nature of these vectors to avert the potential for neurotoxicity with VSV.3,13,17,18,47
Among the vectors recently developed to remedy these safety concerns, hybrid VSV-FH is particularly appealing because it is exclusively dependent on CD46 for viral entry and exhibits syncytial/necrotic destructive capabilities, which are attributes of oncolytic MV.25,26 More importantly, VSV-FH allows for mitigation of VSV-induced neurotoxicity because most normal neuronal cells lack expression of CD46 on their surface,27,28 which VSV-FH uses for cellular entry by virtue of its oncolytic MV attribute.22,25 The anticancer activity of VSV-FH has been established for other types of cancer, including multiple myeloma and ovarian cancer by using human epidermal growth factor receptor-2 (HER-2)-specific VSV-FH.25 In these models, VSV-FH showed promising oncolytic activity and improved survival. Nonetheless, these studies did not focus on the safety aspect of treatment with VSV-FH in a relevant animal model in a fashion that recapitulated locoregional oncolytic viral administration approaches currently used in clinical trial (ClinicalTrials.gov: NCT01628640).
Therefore, we conducted toxicopharmacology and efficacy studies with VSV-FH in pancreatic and hepatocellular cancer models to delineate a clear path for clinical translation as virotherapy for patients with advanced forms of these cancers. We found that all HBPC cell lines in this study were permissive to VSV-FH infection. However, infectious virus progeny recovered from infected HBPC cells showed a differential productive infection. Overall, infection with VSV-FH triggered significantly higher cytotoxicity (60%–90%) in HBPC in vitro compared with VSV-hIFN-β-NIS. Additionally, VSV-hIFN-β-NIS showed reduced cytotoxicity compared with VSV-mIFN-β-NIS. Because of the species-specific nature of mammalian antiviral mechanisms mediated by IFN-β, in some of the cell lines tested, hIFN-β shielded HBPC cells from lysis by VSV-hIFN-β-NIS, whereas mIFN-β had no protective effect, resulting in higher oncolysis associated with infection with VSV-mIFN-β-NIS. It should also be noted that whereas VSV-hIFN-β-NIS was more attenuated in vitro than VSV-FH, there may be advantages in vivo due to immunomodulatory effects of hIFN-β. In order to discern the impact of this duality, clinical evaluation of VSV-FH would need to be undertaken and compared to VSV-hIFN-β vectors, which are already in the clinic. Another facet that would need to be factored into the assessment would be the potential for VSV-FH to be neutralized by pre-existing immunity to measles in most patients.
High intrahepatic doses of VSV-FH in healthy transgenic mice expressing human CD46 were found to be manageable, safe, and tolerable. There was no statistically significant increase in liver enzyme levels (ALT, AST), granulocytes, and total bilirubin at the end of study. However there was a slight increase in alkaline phosphatase. In the absence of severe toxic effects, we deduced that these increases may not result in clinically significant sequelae and could have been confounded by blood hemolysis from the terminal cardiac puncture, which is known to interfere with the measurement of liver enzymes. No mice were euthanized because of adverse clinical signs, including hepatotoxicity, neurotoxicity, or loss of body weight.
Finally, we demonstrated that a single intratumoral dose of VSV-FH controlled the size and induced complete regression in 70% of mice in the HCC group (Hep3B), whereas xenografts in the PDAC model (BxPC-3) were resistant to treatment with VSV-FH. Because VSV-FH displayed strong cell-killing activity against BxPC-3 cells in vitro, we hypothesize that different factors, such as the presence of dense desmoplastic stroma, reduced vascularization, and ultimately diminished drug-delivery in PDAC,48,49 could have affected the in vivo oncolytic activity of VSV-FH in the BxPC-3 in vivo model, in contrast to the robust in vitro oncolytic activity. As such, in addition to evaluating VSV-FH in additional in vivo models for pancreatic cancer, future research to improve VSV-FH efficacy in pancreatic cancer models could focus on combining VSV-FH with agents that can reduce intratumoral perfusion pressures or encode stromal-depleting agents into the viral genome.50,51
In summary, we have shown that VSV-FH has preliminary evidence of oncolytic activity in HBPC models in cell culture and in a Hep3B cell line-derived xenograft HCC mouse model. In addition, VSV-FH did not stimulate any hepatotoxicity or neurotoxicity in CD46 receptor-positive mice, which warrants continued development in HCC. In PDAC, VSV-FH should be evaluated with combination approaches that can overcome vector delivery-limiting factors mediated by tumor-stroma interactions and effects of pre-existing immunity against MV in vaccinated patients.
Materials and Methods
Cell Lines
BHK-21, Vero, Huh7 (JCRB0403; JCRB Cell Bank, Tokyo, Japan), Hep3B (ATCC HB-8064; American Type Culture Collection [ATCC], Rockville, MD, USA), and HepG2 (ATCC HB-8065; ATCC) were grown in DMEM (Dulbecco’s modified Eagle’s medium; Gibco Life Technologies, Grand Island, NY, USA). BxPC-3 (ATCC CRL-1687; ATCC) and HuCCT1 (JCRB0425; JCRB Cell Bank) cells were cultured in RPMI-1640 medium (Corning Cellgro, Mediatech, Manassas, VA, USA). OZ cells (JCRB1032; JCRB Cell Bank) were cultured in William’s medium E (Gibco Life Technologies), and bile duct carcinoma EGI-1 cells (ACC-385; DSMZ, Braunschweig, Germany) were grown in advanced DMEM (Gibco Life Technologies). Media were supplemented with 10% heat-inactivated fetal bovine serum (Gibco Life Technologies) and 5 mL of 100 U/mL of penicillin-streptomycin (Corning Cellgro). All cell lines were maintained in a humidified atmosphere at 37°C and 10% CO2. All cell lines and reagents are available for purchase through the JCRB Cell Bank, ATCC, Corning Cellgro, and Gibco Life Technologies.
Recombinant Viruses
VSV-FH and VSV-FH-EGFP were produced by removing the VSV-G gene from a plasmid containing the full-length genome of VSV Indiana strain (GenBank: J02428.1) and replacing it with MV-F and MV-H genes from the MV Ed-MV strain. Cloning, rescue, and production have been previously described for this virus.42 VSV-mIFN-β-NIS and VSV-hIFN-β-NIS are based on the VSV Indiana strain that expresses hIFN-β or mIFN-β proteins and the recombinant human NIS protein, as has been previously reported.14
Flow Cytometry
For flow cytometry, 4 × 105 cells were trypsinized, fixed with 4% paraformaldehyde (Hyclone Laboratories, Logan, UT, USA), and washed thrice using Dulbecco’s phosphate-buffered saline (DPBS; modified; GE Healthcare Life Sciences, Pittsburgh, PA, USA), 0.2% fetal bovine serum. Flow cytometry analyses were performed with phycoerythrin (PE)-labeled anti-human CD46 or PE-labeled control isotype (BD Biosciences Pharmingen, San Diego, CA, USA). The expression results are the sum of 3 independent experiments.
Susceptibility and Effect of VSV-FH in HBPC Cells
VSV-FH-EGFP was used to infect 5 × 105 cells per well of 6-well plates at an MOI of 0.1. Cells were incubated at 37°C until analysis. At 24 or 48 h after infection, pictures of GFP-positive cells were taken using an epifluorescence microscope. Cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet, 48 h after infection, to allow establishment of strong cell-fusion formation (syncytia), and pictures of representative areas were taken. These results are the sum of 3 independent experiments.
Viral Particle Production in HBPC Cells
To determine the production of VSV-FH infectious particles, 5 × 105 cells per well of 6-well plates were infected with VSV-FH-EGFP at an MOI of 0.1. At 24, 48, or 72 h after infection, an aliquot of the supernatant (∼100 μL) was collected and subjected to three freeze-thaw cycles before determination of viral titers using serial dilutions of the virus samples (i.e., TCID50) and titration in 96-well plates on Vero cells, as previously described.26 These assays were repeated 3 times with similar results.
Viability Assay for HBPC Cells after Infection with VSV-FH
For cell viability assays, 1.2 × 104 cells were treated in triplicate with the indicated virus at an MOI of 1 to 0.01 in wells of 96-well plates. At 72 h, supernatant was removed and replaced with fresh media. Cell viability was determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). These experiments were repeated 3 times with similar results.
Animal Studies
Under a Mayo Clinic Institutional Animal Care and Use Committee-approved protocol, we conducted the following in vivo evaluations.
Studies in CD46 Transgenic Mice
To assess potential toxicity, especially hepatotoxicity, 30 transgenic mice (15 female and 15 male) expressing CD46 IFN-αR WT/WT) in a pattern similar to that observed in humans in all organs, including the brain and the liver30 (Toxicology Laboratory, Mayo Clinic, Rochester, MN), received a single intrahepatic dose of VSV-FH (without GFP) or saline solution. Mice were tested for human CD46 expression and the IFN-WT genotype by polymerase chain reaction (PCR; IFN-αR WT/WT) before initiation of the study. Thirty anesthetized CD46 IFN-αR WT/WT mice (n = 10) were injected in the left lateral lobe of the liver with 30 μL of saline solution or 30 μL of VSV-FH at a dose of 5 × 107 or a higher dose of 5.0 × 108 infectious particles/kg. Body weight was constantly monitored and recorded during the course of the experiment. Mice were bled for downstream analysis at days 2, 21, or 45 days after treatment.
Efficacy of VSV-FH in HBPC Xenograft Models
To evaluate the antitumor efficacy of VSV-FH, 5 × 106 Hep3B or BxPC-3 cells were implanted subcutaneously in the right flank of approximately 40 female athymic nude mice (n = 10 per group) at 5–6 weeks of age (Harlan Laboratories, Indianapolis, IN, USA). Each tumor type consisted of 20 mice clustered into a saline control group (n = 10) and virus group (n = 10) .When tumors reached 5 mm3 in diameter, the mice received a single intratumoral dose of 1 × 107 TCID50 units of VSV-FH or saline (negative control) in 30 μL of volume on day 0. Three mice in the virus-treated groups (Hep3B or BxPC-3) were euthanized at day 3 to study short-term effects of VSV-FH regardless of health status. Body weights, tumor volumes, and clinical observations were recorded at least three times per week until the end of the study. Mice were euthanized when tumor size was larger than 2,000 mm3 or when the end of the study occurred (59 days post-treatment). Tissues and blood were collected at necropsy. No detectable amount of live virus was recovered from tumor samples collected from day 3; however, VSV-FH was detected in 70% of all tumor samples (Hep3B and BxPC-3).
Blood Tests
The CD46 IFN-αR WT/WT mice were bled via the venous retro-orbital plexus of the eye at days 2 and 21 and by heart stick at day 45 after treatment with VSV-FH or saline solution. Blood was collected in BD Microtainer tubes with ethylenediaminetetraacetic acid or lithium heparin (Becton Dickinson, Franklin Lakes, NJ, USA) for CBCs and blood chemistry tests, respectively, or in BD Microtainer Serum Separator Tubes (SSTs) (Becton Dickinson) for serum analysis. CBC analysis was performed in an Abaxis Piccolo Xpress chemistry analyzer (Abaxis, Union City, CA, USA), and blood chemistry analysis was done in a VetScan HM5 Hematology Analyzer (Abaxis).
Immune Response Analysis
For antibody production analysis, serum was obtained from whole blood collected in BD Microtainer tubes. The presence of anti-MV or anti-VSV antibodies was determined by enzyme-linked immunosorbent assay (ELISA). Briefly, ELISA plates were coated with VSV or MV. After a blocking step, plates were incubated with serum from treated mice. Plates were then incubated with biotinylated goat anti-mouse (SC-2072; Santa Cruz Biotechnology, Dallas, Texas, USA), followed by incubation with streptavidin peroxidase conjugate (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA). Signal was developed using a tetramethylbenzidine substrate.
Statistical Analysis
All values were expressed as the mean ± SD, and the results were analyzed by 1-way analysis of variance followed by the Tukey test for multiple comparisons and the Kaplan-Meier method for survival, using statistical software in GraphPad Prism, version 7.03 (GraphPad Software, La Jolla, CA, USA). A p value less than 0.05 was considered to be significant.
Author Contributions
B.M.N. performed experiments, analyzed and interpreted data, and wrote the manuscript. C.A.B. contributed to study design and data analysis. M.B.S., N.J.J., and M.J.B. contributed to study concept and design, acquisition of data, interpretation of data, and critical revision of the manuscript. A.T.B., O.B., D.G.D., Y.Z., M.A., and J.M.B. contributed to drafting and critical revision of the manuscript. K.W.P., S.J.R., L.R.R., and M.J.B. contributed to study concept and design, interpretation of data, and critical revision of the manuscript. M.J.B. and K.W.P. conceived and directed the study. All authors approved the final submitted version of the manuscript.
Conflicts of Interest
Study design, contents, and conclusions are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. M.J.B. has received grants to the institution from Senhwa Biosciences, Adaptimmune, Agios Pharmaceuticals, Halozyme Pharmaceuticals, Celgene Pharmaceuticals, EMD Serono, Toray Medical, Dicerna Pharmaceuticals, Taiho Pharmaceutical, Sun BioPharma, Isis Pharmaceuticals, RedHill Biopharma, Boston Biomedical, Basilea, Incyte, Mirna Therapeutics, MedImmune, Bioline, SillaJen, ARIAD Pharmaceuticals, PUMA Biotechnology, Novartis Pharmaceuticals, QED Pharmaceuticals, and Pieris Pharmaceuticals; consultancy from ADC Therapeutics, Exelixis, Inspyr Therapeutics, G1 Therapeutics, Immunovative Therapies, OncBioMune Pharmaceuticals, Western Oncolytics, and Lynx Group; and travel support from AstraZeneca. D.G.D. received consultant fees from Bayer, Simcere Pharmaceuticals, and Bristol-Myers Squibb and research grants from Bayer, Exelixis, and Bristol-Myers Squibb. The remaining authors declare no competing interests.
Acknowledgments
We thank the personnel of the Viral Toxicology Pharmacology Core at Mayo Clinic in Rochester, Minnesota, for their assistance during the characterization of the toxicologic, pharmacologic, and efficacy effects of VSV-FH in animal models. This work was supported by the National Cancer Institute (NCI) grant K01CA234324-01 and the Mayo Clinic Cancer Center Kathryn H. and Roger Penske Career Development Award to B.M.N., NCI grants 1DP2CA195764-01 and 1R01FD004781-01A1 to M.J.B., and NCI grant 3R01CA175795-02S1 to K.W.P.
References
- 1.Felt S.A., Grdzelishvili V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J. Gen. Virol. 2017;98:2895–2911. doi: 10.1099/jgv.0.000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velazquez-Salinas L., Naik S., Pauszek S.J., Peng K.W., Russell S.J., Rodriguez L.L. Oncolytic Recombinant Vesicular Stomatitis Virus (VSV) Is Nonpathogenic and Nontransmissible in Pigs, a Natural Host of VSV. Hum. Gene Ther. Clin. Dev. 2017;28:108–115. doi: 10.1089/humc.2017.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L., Steele M.B., Jenks N., Grell J., Suksanpaisan L., Naik S., Federspiel M.J., Lacy M.Q., Russell S.J., Peng K.W. Safety Studies in Tumor and Non-Tumor-Bearing Mice in Support of Clinical Trials Using Oncolytic VSV-IFNβ-NIS. Hum. Gene Ther. Clin. Dev. 2016;27:111–122. doi: 10.1089/humc.2016.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdullahi S., Jäkel M., Behrend S.J., Steiger K., Topping G., Krabbe T., Colombo A., Sandig V., Schiergens T.S., Thasler W.E. A Novel Chimeric Oncolytic Virus Vector for Improved Safety and Efficacy as a Platform for the Treatment of Hepatocellular Carcinoma. J. Virol. 2018;92:e01386-18. doi: 10.1128/JVI.01386-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banadyga L., Stein D.R., Qiu X., Safronetz D. Pre-clinical development of a vaccine against Lassa fever. Can. Commun. Dis. Rep. 2018;44:139–147. doi: 10.14745/ccdr.v44i06a04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emanuel J., Callison J., Dowd K.A., Pierson T.C., Feldmann H., Marzi A. A VSV-based Zika virus vaccine protects mice from lethal challenge. Sci. Rep. 2018;8:11043. doi: 10.1038/s41598-018-29401-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapadia S.U., Rose J.K., Lamirande E., Vogel L., Subbarao K., Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174–182. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimpel J., Urbiola C., Koske I., Tober R., Banki Z., Wollmann G., von Laer D. The Oncolytic Virus VSV-GP Is Effective against Malignant Melanoma. Viruses. 2018;10:E108. doi: 10.3390/v10030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu R., Wang J., Shao Y., Wang X., Zhang H., Shuai L., Ge J., Wen Z., Bu Z. A recombinant VSV-vectored MERS-CoV vaccine induces neutralizing antibody and T cell responses in rhesus monkeys after single dose immunization. Antiviral Res. 2018;150:30–38. doi: 10.1016/j.antiviral.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melzer M.K., Zeitlinger L., Mall S., Steiger K., Schmid R.M., Ebert O., Krackhardt A., Altomonte J. Enhanced Safety and Efficacy of Oncolytic VSV Therapy by Combination with T Cell Receptor Transgenic T Cells as Carriers. Mol. Ther. Oncolytics. 2018;12:26–40. doi: 10.1016/j.omto.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naik S., Galyon G.D., Jenks N.J., Steele M.B., Miller A.C., Allstadt S.D., Suksanpaisan L., Peng K.W., Federspiel M.J., Russell S.J., LeBlanc A.K. Comparative Oncology Evaluation of Intravenous Recombinant Oncolytic Vesicular Stomatitis Virus Therapy in Spontaneous Canine Cancer. Mol. Cancer Ther. 2018;17:316–326. doi: 10.1158/1535-7163.MCT-17-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suder E., Furuyama W., Feldmann H., Marzi A., de Wit E. The vesicular stomatitis virus-based Ebola virus vaccine: From concept to clinical trials. Hum. Vaccin. Immunother. 2018;14:2107–2113. doi: 10.1080/21645515.2018.1473698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muik A., Stubbert L.J., Jahedi R.Z., Geiβ Y., Kimpel J., Dold C., Tober R., Volk A., Klein S., Dietrich U. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014;74:3567–3578. doi: 10.1158/0008-5472.CAN-13-3306. [DOI] [PubMed] [Google Scholar]
- 14.Naik S., Nace R., Barber G.N., Russell S.J. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-β. Cancer Gene Ther. 2012;19:443–450. doi: 10.1038/cgt.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson J.E., Nasar F., Coleman J.W., Price R.E., Javadian A., Draper K., Lee M., Reilly P.A., Clarke D.K., Hendry R.M., Udem S.A. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quiroz E., Moreno N., Peralta P.H., Tesh R.B. A human case of encephalitis associated with vesicular stomatitis virus (Indiana serotype) infection. Am. J. Trop. Med. Hyg. 1988;39:312–314. doi: 10.4269/ajtmh.1988.39.312. [DOI] [PubMed] [Google Scholar]
- 17.Jenks N., Myers R., Greiner S.M., Thompson J., Mader E.K., Greenslade A., Griesmann G.E., Federspiel M.J., Rakela J., Borad M.J. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum. Gene Ther. 2010;21:451–462. doi: 10.1089/hum.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Pol A.N., Dalton K.P., Rose J.K. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 2002;76:1309–1327. doi: 10.1128/JVI.76.3.1309-1327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tesfay M.Z., Ammayappan A., Federspiel M.J., Barber G.N., Stojdl D., Peng K.W., Russell S.J. Vesiculovirus neutralization by natural IgM and complement. J. Virol. 2014;88:6148–6157. doi: 10.1128/JVI.00074-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ammayappan A., Peng K.W., Russell S.J. Characteristics of oncolytic vesicular stomatitis virus displaying tumor-targeting ligands. J. Virol. 2013;87:13543–13555. doi: 10.1128/JVI.02240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu L., Huang T.G., Meseck M., Altomonte J., Ebert O., Shinozaki K., García-Sastre A., Fallon J., Mandeli J., Woo S.L. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 2008;19:635–647. doi: 10.1089/hum.2007.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayala-Breton C., Barber G.N., Russell S.J., Peng K.W. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 2012;23:484–491. doi: 10.1089/hum.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wollmann G., Drokhlyansky E., Davis J.N., Cepko C., van den Pol A.N. Lassa-vesicular stomatitis chimeric virus safely destroys brain tumors. J. Virol. 2015;89:6711–6724. doi: 10.1128/JVI.00709-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanauer J.D.S., Rengstl B., Kleinlützum D., Reul J., Pfeiffer A., Friedel T., Schneider I.C., Newrzela S., Hansmann M.L., Buchholz C.J., Muik A. CD30-targeted oncolytic viruses as novel therapeutic approach against classical Hodgkin lymphoma. Oncotarget. 2018;9:12971–12981. doi: 10.18632/oncotarget.24191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayala Breton C., Wikan N., Abbuhl A., Smith D.R., Russell S.J., Peng K.W. Oncolytic potency of HER-2 retargeted VSV-FH hybrid viruses: the role of receptor ligand affinity. Mol. Ther. Oncolytics. 2015;2:15012. doi: 10.1038/mto.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayala-Breton C., Russell L.O., Russell S.J., Peng K.W. Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J. Virol. 2014;88:8332–8339. doi: 10.1128/JVI.03823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noyce R.S., Richardson C.D. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol. 2012;20:429–439. doi: 10.1016/j.tim.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Noyce R.S., Bondre D.G., Ha M.N., Lin L.T., Sisson G., Tsao M.S., Richardson C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011;7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence D.M., Patterson C.E., Gales T.L., D’Orazio J.L., Vaughn M.M., Rall G.F. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J. Virol. 2000;74:1908–1918. doi: 10.1128/jvi.74.4.1908-1918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cathomen T., Mrkic B., Spehner D., Drillien R., Naef R., Pavlovic J., Aguzzi A., Billeter M.A., Cattaneo R. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 1998;17:3899–3908. doi: 10.1093/emboj/17.14.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson B.D., Nakamura T., Russell S.J., Peng K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 32.Shang Y., Chai N., Gu Y., Ding L., Yang Y., Zhou J., Ren G., Hao X., Fan D., Wu K., Nie Y. Systematic immunohistochemical analysis of the expression of CD46, CD55, and CD59 in colon cancer. Arch. Pathol. Lab. Med. 2014;138:910–919. doi: 10.5858/arpa.2013-0064-OA. [DOI] [PubMed] [Google Scholar]
- 33.Sherbenou D.W., Aftab B.T., Su Y., Behrens C.R., Wiita A., Logan A.C., Acosta-Alvear D., Hann B.C., Walter P., Shuman M.A. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Invest. 2016;126:4640–4653. doi: 10.1172/JCI85856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee C.N., Heidbrink J.L., McKinnon K., Bushman V., Olsen H., FitzHugh W., Li A., Van Orden K., He T., Ruben S.M., Moore P.A. RNA interference characterization of proteins discovered by proteomic analysis of pancreatic cancer reveals function in cell growth and survival. Pancreas. 2012;41:84–94. doi: 10.1097/MPA.0b013e3182236385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lok A., Descamps G., Tessoulin B., Chiron D., Eveillard M., Godon C., Le Bris Y., Vabret A., Bellanger C., Maillet L. p53 regulates CD46 expression and measles virus infection in myeloma cells. Blood Adv. 2018;2:3492–3505. doi: 10.1182/bloodadvances.2018025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z., Zhang C., Cui J., Song Q., Wang L., Kang J., Li P., Hu X., Song H., Yang J., Sun Y. Bioinformatic analysis of the membrane cofactor protein CD46 and microRNA expression in hepatocellular carcinoma. Oncol. Rep. 2014;31:557–564. doi: 10.3892/or.2013.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravindranath N.M., Shuler C. Cell-surface density of complement restriction factors (CD46, CD55, and CD59): oral squamous cell carcinoma versus other solid tumors. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2007;103:231–239. doi: 10.1016/j.tripleo.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 38.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 39.Razumilava N., Gores G.J. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGuigan A., Kelly P., Turkington R.C., Jones C., Coleman H.G., McCain R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018;24:4846–4861. doi: 10.3748/wjg.v24.i43.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rimassa L., Pressiani T., Merle P. Systemic Treatment Options in Hepatocellular Carcinoma. Liver Cancer. 2019;8:427–446. doi: 10.1159/000499765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayala-Breton C., Suksanpaisan L., Mader E.K., Russell S.J., Peng K.W. Amalgamating oncolytic viruses to enhance their safety, consolidate their killing mechanisms, and accelerate their spread. Mol. Ther. 2013;21:1930–1937. doi: 10.1038/mt.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng K.W., Donovan K.A., Schneider U., Cattaneo R., Lust J.A., Russell S.J. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101:2557–2562. doi: 10.1182/blood-2002-07-2195. [DOI] [PubMed] [Google Scholar]
- 44.Myers R.M., Greiner S.M., Harvey M.E., Griesmann G., Kuffel M.J., Buhrow S.A., Reid J.M., Federspiel M., Ames M.M., Dingli D. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin. Pharmacol. Ther. 2007;82:700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roscic-Mrkic B., Schwendener R.A., Odermatt B., Zuniga A., Pavlovic J., Billeter M.A., Cattaneo R. Roles of macrophages in measles virus infection of genetically modified mice. J. Virol. 2001;75:3343–3351. doi: 10.1128/JVI.75.7.3343-3351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weaver E.A. Vaccines within vaccines: the use of adenovirus types 4 and 7 as influenza vaccine vectors. Hum. Vaccin. Immunother. 2014;10:544–556. doi: 10.4161/hv.27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel M.R., Jacobson B.A., Ji Y., Drees J., Tang S., Xiong K., Wang H., Prigge J.E., Dash A.S., Kratzke A.K. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget. 2015;6:33165–33177. doi: 10.18632/oncotarget.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaustad J.V., Simonsen T.G., Wegner C.S., Rofstad E.K. Vascularization, Oxygenation, and the Effect of Sunitinib Treatment in Pancreatic Ductal Adenocarcinoma Xenografts. Front. Oncol. 2019;9:845. doi: 10.3389/fonc.2019.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murakami T., Hiroshima Y., Matsuyama R., Homma Y., Hoffman R.M., Endo I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019;3:130–137. doi: 10.1002/ags3.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S., Toyokawa H., Yamao J., Satoi S., Yanagimoto H., Yamamoto T., Hirooka S., Yamaki S., Inoue K., Matsui Y., Kwon A.H. Antitumor effect of angiotensin II type 1 receptor blocker losartan for orthotopic rat pancreatic adenocarcinoma. Pancreas. 2014;43:886–890. doi: 10.1097/MPA.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 51.Guedan S., Rojas J.J., Gros A., Mercade E., Cascallo M., Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010;18:1275–1283. doi: 10.1038/mt.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]