

Abstract
Rare inherited anemias are a subset of anemias caused by a genetic defect along one of the several stages of erythropoiesis or in different cellular components that affect red blood cell integrity, and thus its lifespan. Due to their low prevalence, several complications on growth and development, and multi-organ system damage are not yet well defined. Moreover, during the last decade there has been a lack of proper understanding of the impact of rare anemias on maternal and fetal outcomes. In addition, there are no clear-cut guidelines outlining the pathophysiological trends and management options unique to this special population. Here, we present on behalf of the European Hematology Association, evidence- and consensus-based guidelines, established by an international group of experts in different fields, including hematologists, gynecologists, general practitioners, medical geneticists, and experts in rare inherited anemias from various European countries for standardized and appropriate choice of therapeutic interventions for the management of pregnancy in rare inherited anemias, including Diamond-Blackfan Anemia, Congenital Dyserythropoietic Anemias, Thalassemia, Sickle Cell Disease, Enzyme deficiency and Red cell membrane disorders.
Recommendations for pregnancy in rare anemias
Questions:
-
1.
Why specific guidelines for pregnancy in RIAs? What makes RIAs different from the most common conditions of anemia in pregnancy? When does an RIA deviate from the standard guidelines for anemia in pregnancy?
Rare inherited anemias (RIA) are a subset of anemias caused by a myriad of genetic defects in one of the stages of red blood cell (RBC) cellular component formation or erythropoiesis. The result of those defects is detrimental to the RBC integrity, and thus its lifespan or its production. Due to their rare prevalence, the full spectrum of disease complications and multi-organ system damage remains ill-defined. More importantly, less is known about RIA's impact on growth, development, and long-term morbidity. The significant medical advances made in the field of benign hematology have resulted in near normalization of RIA's patients’ longevity. Thus, the incidence of pregnancy among affected patients is steadily increasing. A better understanding of the impact of such rare anemias on maternal and fetal outcomes and evidence-based management guidelines is of paramount importance.
-
2.
When should a pregnancy in an inherited anemia be considered at risk?
Pregnancy in RIA should always be considered high risk regardless of underlying risk factors. All available data and case reports confirm the higher risk of complications seen throughout pregnancy in individuals with RIA. These complications vary depending on the type of anemia, its severity in addition to the predisposing pathophysiology of the disease.
-
3.
What should be addressed?
The approach that should be taken once a woman with RIA and her partner are planning a pregnancy should be multidisciplinary with collaboration among the hematologist, gynecologist and geneticist.-
a.The hematologist: has a role in optimizing the management of anemia, iron chelation, complications secondary to iron overload and anemia.
-
b.The gynecologist: has a role in addressing the risks of pregnancy on the mother along with the risk during delivery. He should monitor fetal complications such as growth restriction, fetal distress or hydrops fetalis.
-
a.
-
4.
Should the genetic and clinical counseling be given together?
The geneticist has a role prior and during pregnancy onset by explain risk of pregnancy and risks of having offspring with the disease mainly in particular genetic disorders. This consultation must include also the examination of the partner, searching for an inherited red cell defects that could modify the expected phenotype. Hence, genetic counselling should take place whenever inherited form of anemia is suspected based on the medical/family history of any of the partners.
-
5.
When to consider prenatal diagnosis?
Prenatal workup should be initiated whenever clinicians have a suspicion of an affected baby. This should be considered as early as possible once suspected. During pregnancy, close monitoring with regular fetal ultrasound can orient the clinicians on doing prenatal diagnosis.
Especially in the case of dominant condition or when the partner is also affected with an inherited form of anemia.
-
6.
Which tests are useful planning a pregnancy in RIA?
Maternal workup should be done prior to pregnancy to decrease risks of complications:- Evaluate iron status (using serum ferritin and transferrin saturation) and ensure adequate management of iron overload (if required liver and cardiac iron assessment by magnetic resonance imaging MRI)
- Assess cardiac status with echocardiography and manage accordingly
- Check viral status (Hepatitis B, Hepatitis C and HIV) and provide proper vaccination (Hepatitis B, Pneumococcus, seasonal influenza)
- Evaluate folate stores (as this population is at risk of folate deficiency) and offer preventive or curative supplementation
-
7.Which tests are useful during the pregnancy?
- Monitor hematological parameters, cardiac, hepatic, renal, endocrine (glucose tolerance) and thyroid functions
- Monthly Doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
- Splenectomized patient should be considered for aspirin therapy
- NTDT should be on a prophylactic dose of anticoagulation in peripartum
- Regular folic acid supplementation 800 to 1000 μg daily is recommended in all mothers to prevent superimposed megaloblastic anemia
- Delivery modality should be discussed between the patient and her gynecologist viewing the risks and complications that are possible to take place
- Optimize patient for vaginal vs cesarean section (C/S for high risk and poorly controlled patients)
-
8.
Special advice for the post-partum in RIAs?
Close monitoring should continue post-partum for at least first month. Anemia should be monitored, and iron chelation might need to be resumed earlier in cases with highly iron overloaded. Vitamin supplementation such as Calcium and Vitamin D is highly recommended.
-
9.
May a pregnancy help to suspect a diagnosis of RIA?
In rare situations, such in subclinical RIA women may develop severe anemias during pregnancy and become symptomatic putting her fetus at risk. Hence, in this case, clinicians should opt to manage these pregnancies same as the high-risk RIA pregnancies. In rare cases, pregnant women may have enhancement of their symptoms due to pregnancy which will cause delay in diagnosis and proper management of RIA.
-
10.
When should iron be considered in RIAs during pregnancy?
Iron supplementation is not recommended in RIA pregnancies, unless there is documentation of true iron deficiency through laboratory iron testing. On the other hand, many patients have iron overload due to multiple blood transfusions and require iron chelation.
-
11.
When should transfusion be considered in RIAs during pregnancy?
Maternal anemia, defined as Hb < 11.6 g/dL during the first trimester of pregnancy, or Hb < 9.7-9.5 g/dL during the second and third trimester, affects 7.5% to 8.6% of the population and is objectively proven to be an independent risk factor for maternal and fetal morbidity during pregnancy and deliver. Gestational anemia is objectively associated with higher rates of abnormal placentation, low birth weight, and preterm delivery. However, it is preferable that the decision of transfusion be based on maternal symptoms of anemia or risk of fetal distress, rather than hemoglobin level.
-
12.
Is there a role for erythropoietin (EPO) or other therapies?
The role of recombinant human EPO is controversial. Minimal data is available to show benefit with their use. However, some case reports have demonstrated its affectivity mainly in patients with NTDT in whom allo-immunization may complicate the availability of further safe transfusions.
-
13.
When to suspect an RIA in an asymptomatic patient with mild anemia?
RIA should be suspected whenever anemia is associated with clinical features that fulfill the specific characteristics of each disease: Diamond-Blackfan anemia, congenital dyserythropoietic anemias, thalassemia, sickle cell disease, pyruvate kinase deficiency, red cell membrane disorders.
Introduction
Rare inherited anemias (RIA) are a subset of anemias caused by a myriad of genetic defects affecting erythropoiesis stages or one red blood cell (RBC) component (Diamond-Blackfan anemia, congenital dyserythropoietic anemias, thalassemia, sickle cell disease, enzyme deficiencies, red cell membrane disorders).1 The result of those defects is detrimental to the RBC integrity, and thus its lifespan.2 Due to their rare prevalence, the full spectrum of disease complications and multi-organ system damage remains not well defined. More importantly, less is known about RIAs’ impact on growth, development, and long-term morbidity. The significant medical advances made in the field of benign hematology have resulted in near normalization of RIA patients’ longevity. Thus, the incidence of pregnancy among affected patients is steadily increasing.3,4 A better understanding of the impact of such rare anemias on maternal and fetal outcomes and evidence-based management guidelines is of paramount importance; however, high-quality evidence is lacking upon which to base guidelines. Nevertheless, recommendations are needed to help guide clinicians looking after these patients at this vulnerable time in their life.
Maternal anemia is defined as Hb <11 g/dL during the first trimester of pregnancy, as Hb <10.5 g/dL during the second trimester and as Hb <11 g/dL during the third trimester.5–7 It affects large groups of women worldwide and can range from 16.5% to 41.5% of pregnant women in High-income countries and Low-middle income countries, respectively.8 This number of maternal anemia represents 7.5% to 8.6% of the world population and is proven to be an independent risk factor for maternal and fetal morbidity during pregnancy and delivery.8–10 Gestational anemia has been shown to be associated with higher rates of abnormal placentation, low birth weight, and preterm delivery.11,12 In these recommendations, we analyzed the effects of RIAs on pregnancy outcomes based on available literature. This is an overview of the pathophysiology, followed by pregnancy-specific outcomes and management outlines for the most relevant forms of RIAs.
The aim of this paper was to develop European recommendations for the clinical management of pregnancies in patients with inherited anemias on behalf of the European Hematology Association (EHA). After a thorough review of the literature by experts in relevant fields, including hematologists, gynecologists, general practitioners, medical geneticists, and experts in inherited anemias from various European countries, a Delphi-like consensus process was used to produce statements and practical algorithms for the management of pregnancy in rare inherited anemias, including Diamond-Blackfan anemia, congenital dyserythropoietic anemias (CDAs), thalassemia, sickle cell disease, enzyme deficiency and red cell membrane disorders.
Methods
Our recommendations are based on a systematic literature search. All articles on pregnancy and inherited anemias were identified by a PubMed, Online Mendelian Inheritance in Man, and Textbook search, including all the additional relevant references cited in the articles found. The key search terms “inherited anemias” and “chronic anemias” were used alone and in combination with the term “pregnancy”. The examined period was from June 2019 to January 2020. We used the same search method as in our previously published systematic review.13 All reviewed articles and abstracts were restricted to those published in the English language.
The steering committee and expert panel included 20 members who have been selected for their recognized expertise in research and clinical practice in RIA, with a wide geographical representation in order to provide an international perspective. EHA verified the absence of relevant competing conflict of interests of the authors and supervised the project in regard to compliance to, and consistency with, the methodology adopted by its Committee on Guidelines. EHA provided the organizational and logistic assistance and funding for the necessary face-to-face meetings or teleconferences. With this background, a series of questions regarding pregnancy in inherited anemias were formulated and the relative recommendations were produced. An agreement was obtained using a Delphi-like approach. Multiple rounds of questionnaires have been generated by a steering committee and a final questionnaire has been sent to a panel of experts. The responses by this expert panel have been aggregated and shared with the group after each round. The experts adjusted their answers in subsequent rounds; thus, the final response has been reached through consensus.
Practical recommendations for all rare anemias
The pregnancy in women affected by rare anemias must be planned ahead of time with close monitoring by an experienced multidisciplinary team. The immune system of pregnant women with rare anemias is often compromised due to the elevated estrogen levels of pregnancy, the variable state of iron overload, and the absence of a spleen in some cases.14 Therefore, all women with a rare anemia should be screened for the human immunodeficiency virus (HIV), Hepatitis B, Hepatitis C, and rubella. Of note, genetic counseling should be offered to all RIAs women, preferably before any pregnancies. Therapy should be recommended for HIV positive or Hepatitis C positive women before pregnancy.15 Treatment of the hepatitis C virus with either interferon or ribavirin should be discontinued six months prior to pregnancy. Patients who have been splenectomized should be on penicillin prophylaxis and be up to date with all of their vaccines, in particular the 5-yearly pneumococcal booster.16 Moreover, endocrinopathies including hypogonadism which is secondary to iron overload should be expected whenever women with RIA present for pre-pregnancy workup. Lastly, genetic counseling with both parents should be offered along with antenatal testing when medical or family history is suggestive. Table 1 summarizes the practical recommendations that should be taken into consideration before conception, during pregnancy and delivery, and postpartum.
Table 1.
Summary of the Practical Recommendations for all Rare Anemias Throughout Pregnancy.

Diamond-Blackfan anemia
Diamond-Blackfan anemia (DBA) is a rare form of congenital pure red-cell aplasia caused by a genetic disorder in ribosome synthesis that causes pro-apoptotic hematopoiesis leading to bone marrow failure. Central to the pathophysiology of DBA is erythropoiesis failure, which is due to an intrinsic progenitor defect leading to insensitivity to erythropoietin and to other erythroid factors.17 Phenotypically, the disease manifests as a variable combination of severe anemia, congenital malformations, growth retardation, and cancer predisposition.18,19
The most commonly seen mutations are in the gene encoding the Ribosomal Protein S19 (RPS19).20,21 The mode of inheritance is heterogeneous and mostly sporadic, in around 75% of the time. However, familial DBA exist and can be as autosomal dominant with incomplete penetrance or rarely as X-linked transmission. Approximately 55% to 75% of affected patients have a de novo variant.20 The common presentation of DBA includes macrocytic normochromic anemia and reticulocytopenia with no other affected cell lineages. Usually, it presents early in life. Bone marrow inspection shows normal cellularity with a paucity of erythroid precursors.22
Although corticosteroid therapy is the first-line treatment in DBA, their mode of action is still unknown. More than 80% of DBA patients will respond initially to steroid treatment. However, some will become refractory to the first-line treatment and will require chronic transfusion therapy.23 Hematopoietic stem cell transplantation has recently proven to be a curative treatment for anemia in DBA patients.24 Forty percent of affected individuals are corticosteroids dependent, 40% transfusion-dependent, and 20% experience remission.25,26
Pregnancy-specific complications
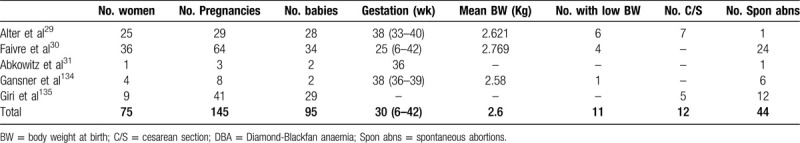
Advances in the treatment of DBA have improved the demographics of affected individuals. DBA is no longer limited to children but has shifted to becoming a chronic disorder with an increasing incidence among adults.27 Women are now able to reach childbearing age with limited morbidity and higher quality of life, as well as a positive reproductive potential. On the other hand, maternal, fetal, and neonatal complications are numerous.28 Due to the sparsity of available data, we still lack a clear description of risk factors and outcomes. Several case reports of DBA in pregnancy were summarized in one review article.29 Twenty-five women affected by DBA, with a total of 29 pregnancies were included. Overall, 40% of the pregnancies required multiple transfusions, and 30% required steroids. All steroid-treated patients were also transfused. They reported an increased risk of cesarean section (C/S) (25%), low birth weight (21%), and premature delivery of offspring with and without DBA.29
Another large prospective study of 64 pregnancies in 24 DBA-affected women reported adverse outcomes in 66% of pregnancies. Complications included worsening of anemia, increased transfusion requirements, resistance to corticosteroid, higher rates of intrauterine growth retardation, fetal malformation, preterm delivery, fetal death in utero, and miscarriage.30 Around 41% of the liveborn offspring were affected by DBA. Congenital malformations were noted in 12.5% of all pregnancies and were independent of the DBA status of the child.30 It is essential to stress that the direct causes of these complications are not clear—whether from DBA itself, anemia, iron overload, or even steroid use.
An interesting case report describes three pregnancy outcomes of a woman affected by DBA.31 The patient's first pregnancy at the age of 33 ended with a stillbirth at week 30. An interesting observation made by the author is that her hematocrit (Hct) spontaneously increased after week 25 of gestation. During the patient's second pregnancy, she received transfusions during the first trimester to keep her Hct > 25%. After week 19 of gestation, her Hct spontaneously increased. She delivered a healthy newborn at 36 weeks gestational age. Postpartum, her hematocrit level remained elevated during the 23 months of breastfeeding. Her third pregnancy was identical to the second one with spontaneous remission during pregnancy and breastfeeding, followed by relapse.32 The authors hypothesized that the leading cause of this peripartum remission was the high level of prolactin. Previous studies and case reports have shown the role of prolactin in erythropoiesis. Supraphysiologic prolactin levels during pregnancy and breastfeeding may be the reason behind improved outcomes in DBA. Additionally, patients treated with Metoclopramide, a prolactin agonist, experienced significant improvement in their anemia.31 The pregnancy outcomes of women affected by DBA are summarized in Table 2.
Table 2.
Summary of the Pregnancy Outcomes of the Largest Published Case Series of DBA.
Management during pregnancy
Preconception consultations with maternal-fetal medicine, medical geneticist, and hematology specialists are crucial for DBA-affected women planning a pregnancy. The risk of transmitting DBA to the offspring should be discussed. There are no clear guidelines on the use of iron chelators in DBA patients and in general, management of iron chelation therapy during pregnancy remains controversial.33 However, recommendations from other rare anemias, such as thalassemia, can be taken into account. Hence, ideally iron chelation should be optimized well ahead of conception since the use of most chelators is not recommended during pregnancy. In cases of severe iron overload, however, iron chelation could be considered during the last trimester with a preference for deferoxamine—based on the data available in pregnant women with thalassemia.34 Anemia should also be regularly corrected for optimal fetal oxygenation, although there is no consensus on how low hemoglobin levels can be tolerated. Moreover, blood transfusion should be considered once hemoglobin level drops below 10 g/dL26; although the decision to transfuse should be driven by the clinical presentation of the patient and fetal growth status rather than maternal Hb per se. Aspirin 80 to 100 mg daily has shown to be beneficial in some cases since many of the observed complications have similar presentations to placental vasculopathy.30 Finally, utero-placental Doppler ultrasound should be used to continuously monitor and screen for any congenital abnormalities in the fetus or possible placental vascular disorder.
Congenital dyserythropoietic anemias
Congenital dyserythropoietic anemias (CDAs) are a heterogeneous subset of hypo-productive anemias where the defect lies in abnormal late-stage differentiation and proliferation of the erythroid lineage.35 There are currently 6 types of CDA, which are distinguished by a combination of bone marrow morphology, laboratory findings, characteristic physical presentations, and well-identified causative genes. Common hallmarks of the diseases are chronic hypo-regenerative anemia, relative reticulocytopenia, a hemolytic component, and iron overload.36 Cholelithiasis, splenomegaly, and secondary hemochromatosis are complications found in the most common types—I and II.35 CDAs are rare inherited disorders, with 0.04 cases/million in North Europe and 2.49 cases/million in Mediterranean countries. Allelic heterogeneity is found in the different types of CDA, and the founder effect explains the appearance of specific mutations within different geographical groups worldwide.37,38
The following criteria should generally be met to diagnose a suspected case of congenital dyserythropoietic anemia: 1. evidence of anemia, jaundice, and splenomegaly; 2. evidence of ineffective erythropoiesis; 3. occurrence of typical morphological features of erythroblasts at bone marrow (BM) examination; and 4. exclusion of other congenital anemias that fulfill criteria 1 and 2, such as thalassemia syndromes and other inherited bone marrow failure syndromes.39 Up to 20% of CDA patients can present with congenital anomalies, particularly syndactyly in hands or feet, absence of nails or supernumerary toes, and pigeon chest deformity, among others.40,41 Proper use of genetic confirmation, along with sensitive laboratory tools and BM morphology findings, can aid in the differential diagnosis.38
Depending on the severity of the case, some patients may be born with hydrops fetalis or require transfusions for survival while others may remain asymptomatic even in adulthood, requiring no intervention.38 Regular follow up with CBC, iron balance parameters, and spleen/gallbladder evaluation is the typical management, with RBC transfusions used as required. Splenectomy has been helpful in transfusion dependent CDA II cases only. Response to IFN-α therapy, with high variability, has been documented with CDA I patients. Finally, BM transplantation has been efficacious in CDA, mostly in type I and II leading to a curative state.38,42,43
Pregnancy-specific complications
Knowledge about maternal and fetal pregnancy outcomes in CDA is limited to case reports and a short review of a Bedouin tribe with CDA type I.44 Historical data of CDA II, previously coined HEMPAS (Hereditary Erythroblastic Multinuclearity associated with a Positive Acidified Serum test) before proper genetic classification, described 7 cases of affected pregnant women whose deliveries were only complicated by the worsening of their anemias. They required blood transfusion when their condition became severe, but there were no adverse fetal outcomes.45,46 The most extensive pregnancy outcome data comes from 28 spontaneous pregnancies in six Bedouin women with CDA I.12,47 Sixty-four percent of these pregnancies were complicated: there was one first-trimester spontaneous abortion and one stillbirth; 42% had lower birth weights and 36% had cesarean sections.48 A more recent case of a 26-year-old primigravida woman with CDA I was reported, whereby she suffered from pre-eclampsia and required C/S.47 When a North Swedish family with CDA III was followed, common symptoms of weakness, fatigue, and headache were reported to be worsening during pregnancy. Five patients (20%) received blood transfusions, 4 of them in late pregnancy or after delivery.49 Scarce data exist on the incidence of pregnancy in CDA women. Some references consider that women with CDA who achieved a complete pregnancy with minimal complications might have milder disease at baseline or they were more carefully followed throughout the course.48 Hence, it is important to have proper counseling, close monitoring, and adequate management during pregnancy.
Management during pregnancy
Blood transfusion was used once during pregnancy in 4 cases of the Bedouin registry, and all women required blood transfusions at delivery, despite being transfusion independent before pregnancy.50 Another previously asymptomatic 32-year-old female with CDA I suffered from severe anemia in her third trimester, and required RBC transfusions for 6 consecutive months, along with a successful IFN-α treatment for 4 months before her Hb stabilized again.51
Careful follow up of total Hb levels at least once a month during pregnancy was significantly associated with better fetal outcomes (SGA rate) in CDA I pregnancies. Expert recommendations are to transfuse the mothers once symptomatic or based on Hb levels if below 8 g/dL or in the case of a rapid fall by 2 g/dL, to perform monthly ultrasonography follow-ups of fetal growth starting from week 20 of gestational age, and close fetal monitoring in the third trimester.48,50 High dose folic acid supplementation is recommended (800–1000 μg daily), and if significant iron overload is suspected, endocrine and cardiac status should be checked. The high platelet counts associated with splenectomy may justify the use of low doses of aspirin to minimize thromboembolic risk.47
On the other hand, regarding iron overload management, we suggest applying the guidelines for iron chelation of thalassemic patients which has been used with successful results in CDA cases of iron overload.36
Thalassemia
Thalassemia patients can be classified as transfusion dependent thalassemia (TDT) or non-transfusion dependent thalassemia (NTDT) based on their blood transfusion requirement. Patients with TDT commonly present with severe anemia in their early childhood and they require regular lifelong blood transfusions for survival. Patients with β-thalassaemia major and severe forms of hemoglobin E/β-thalassaemia are classified as having TDT.52 Patients with NTDT usually present with mild/moderate anemia during their late childhood or even in adulthood and may necessitates intermittent or occasional transfusions in certain clinical settings and for the prevention or management of certain disease manifestations. Patients with β-thalassaemia intermedia, hemoglobin H disease, and mild-to-moderate forms of hemoglobin E/β-thalassaemia often fall under the classification of NTDT.53 Key milestones have been achieved in optimizing the management of thalassemia with transfusion and iron chelation therapy, in addition to new non-invasive methods for early detection of iron overload, which have prolonged the survival of patients and improved their overall quality of life. As a result, pregnancy, which was almost impossible, in recent decades has proven to be not only possible but also safe, with remarkable improvements in maternal and fetal survival.
Transfusion dependent thalassemia (TDT)
Pregnancy-specific complications
Although conception is possible in TDT patients who are well-transfused and well-chelated and undergo normal puberty and menstrual function, the majority of patients aged >25 years of age are sub-fertile mainly due to hypogonadotropic hypogonadism as a consequence of transfusional hemosiderosis and endocrinopathies.54 To note, endocrinopathies including diabetes mellitus, hypothyroidism, hypoparathyroidism, and hypogonadism are common in TDT especially in patients with elevated serum ferritin levels >2500 μg/l or spelenctomized.55 Women who fail to achieve a spontaneous pregnancy might benefit from assisted reproductive techniques (ART). A planned pregnancy is essential both in spontaneous, or ART conceptions since pregnancies in TDT-affected women are high risk for both the mother and the baby.56
Management during pregnancy
In patients with TDT, certain precautions before and during pregnancy should be considered. Patients are subject to cardiac impairment, liver dysfunction, and chronic viral infections. A multidisciplinary assessment preconception is essential to support a good pregnancy outcome. The evaluation of cardiac function is important given that cardiac complications remain the leading cause of death in transfusion-dependent patients.57 During pregnancy, the cardiac load is increased by at least 25% to 30% as a result of an increase in both heart rate and stroke volume. Cardiac stress, in addition to an increased iron load, can lead to cardiac failure. It is therefore essential for all TDT patients to undergo a pre-conceptional, complete cardiological evaluation including echocardiography testing for left ventricular ejection fraction and other functional parameters, and a 24-hour Holter monitoring to check for any rhythm disorders.56 Moreover, a pre-pregnancy cardiac T2∗ of the heart of greater than or at least equal to 20 ms should ideally be achieved. Patients with evidence of significant cardiac iron overload (T2∗ MRI ≤ 10 ms), or those with cardiac dysfunction, should be discouraged from planning a pregnancy at that time pending optimization to their normal values.58
As for liver dysfunction before pregnancy, all TDT women should have their liver iron load assessed preferentially by MRI or in cases of potential liver disease by liver biopsy. An abdominal ultrasound for liver and spleen volume as well as a gallbladder evaluation should be obtained. Thalassemia patients have an increased risk of gallstones: in those patients with evidence of sludge or stones, cholecystectomy should be considered before conception.59 In terms of management of these patients, for women who were transfusion-dependent prior to pregnancy, regular transfusions should continue with a pre-transfusion hemoglobin goal of ≥ 10 g/dL. The use of iron chelation therapy during pregnancy has not been addressed in clinical studies and remains controversial. The current standard of practice is to discontinue chelation therapy as soon as pregnancy is established. Reproductive studies in non-iron-loaded rats and rabbits have indicated that deferiprone (DFP) is teratogenic and embryotoxic.60,61 Although successful pregnancies following unintentional treatment with DFP or deferasirox (DFX) have been reported in the literature,61–63 the use of oral iron chelators is contraindicated in pregnant women, because of the lack of controlled data.
Women of childbearing age on oral chelators who are planning a pregnancy should be counseled and advised to switch to deferoxamine (DFO). However, discontinuation of iron chelation with DFO should be recommended once a pregnancy has been achieved.34 Nevertheless, it may be reasonable to consider restarting chelation therapy with DFO during the third trimester in patients in patients with severe iron overload in the heart and liver, when the potential benefits outweigh the potential risks to the fetus.34,64 After delivery, DFO can be recommenced because concentrations are very low in breast milk and because it is not absorbed when taken orally.
Non-transfusion dependent thalassemia (NTDT)
Pregnancy-specific complications
Pregnancy has been reported to be safe in most women with NTDT under multidisciplinary management. Pregnancies in this patient population have mostly been achieved without routine assisted reproductive technologies.63,65 The physiologic anemia of pregnancy becomes exaggerated in NTDT patients, and the increased oxygen demand by the fetus makes blood transfusion a tempting option. The major factors of pregnancy-related adversities in NTDT females are iron overload and increased thrombotic risk. In NTDT, where the levels of hepcidin are abnormally low, excess iron is absorbed into the system leading to preferential portal and hepatocyte iron loading and free iron in the bloodstream resulting in organ damage and pregnancy complications.66 Venous thromboembolism is the most serious complication seen in this patient population; especially in women with additional prothrombotic risk factors such as splenectomy, age and low Hb level.67,68 An incidence greater than 30% of venous thromboembolic events has been reported in NTDT patients, reflecting a hypercoagulable state that is driven by several mechanisms including endothelial damage secondary to the iron-mediated free radical formation.69 Other studies, however, show that pregnancy in NTDT patients was not shown to increase the risk of thromboembolic complications compared to non-pregnant NTDT women.68
Management during pregnancy
Blood transfusion therapy is a common consideration in pregnant NTDT women due to the worsening of anemia during gestation. There are no clear cutoffs on when to transfuse, and the only determining factor is the clinical status of the patient. Data from non-thalassemia cohorts suggest that hemoglobin levels ≥ 10 g/dL during gestation are recommended for optimal fetal growth and development.12 However, an Italian case series showed that targeting a 10 g/dL cutoff level proved beneficial only in 78% of the pregnant NTDT patients.63 As such, the introduction of blood transfusions for pregnant patients with NTDT should rely on hemoglobin level, maternal general health, and cardiac status along with fetal growth status. Furthermore, pregnant women with NTDT who were previously never- or minimally-transfused, should be considered at high risk of alloimmunization if blood transfusions are to be administered during pregnancy.70 If blood transfusion is necessary, then an extended genotype and antibody screening should be performed at first. A recent study describes 4 patients who developed alloimmune hemolytic anemia after transfusion that necessitated postpartum splenectomy in two of them.71 Hence, splenectomy should be considered for NTDT patients with hypersplenism or splenomegaly before conception or postpartum.
Thalassemia, in particular for the patients who are non-transfused and splenectomized, is a hypercoagulable state that involves interactions of disrupted thalassemic red blood cell membrane surfaces, platelets, and endothelium.72,73 Therefore, pregnant women with NTDT should receive a prophylactic dose of low-molecular-weight heparin (LMWH) peripartum.70 Patients with a history of recurrent abortions or who are at increased risk of thromboembolic events may be prescribed LMWH throughout pregnancy. Splenectomized patients should also be considered for acetyl salicylic acid (ASA) therapy if they are not prescribed LMWH.70
Sickle cell disease
Sickle cell disease (SCD) is a worldwide distributed hereditary red cell disorder that affects approximately 100,000 individuals in the United States and between 20,000 and 25,000 individuals in Europe, mainly through immigrants from endemic areas such as Sub-Saharan Africa.74–76 Estimates of the number of affected new-born in 2010 are of approximately 312,302 subjects, 75.5% of which were born in Africa.77 SCD is caused by a point mutation in the β-globin gene resulting in the synthesis of pathological hemoglobin S (HbS) with subsequent generation of rigid, dense and dehydrated red cells.78–80 The main clinical manifestations of SCD are chronic hemolytic anemia and acute vaso-occlusive crisis, leading to an impaired blood flow with ischemic/reperfusion injury.78,81–83 A complex perturbation of hemostasis has been reported in SCD both under a steady state and during acute events. Clinical manifestations of the pro-thrombotic state of SCD patients include venous thromboembolism, in situ thrombosis and stroke, (most in splenectomized patients or with functional asplenia).78
Hydroxyurea or hydroxycarbamide (HU) is still today the key therapeutic tool for SCD that is approved by both the Food and Drug Administration (FDA) and European Medicines Agency (EMA). In addition, different blood transfusion regimens, such as transfusion on demand or chronic transfusion, automatized/manual red cell exchange, may be used in the clinical management of both acute and chronic sickle cell-related complications. The main objective of the transfusion approach is to rapidly reduce/maintain low percentage of HbS in the peripheral circulation. In SCD, transfusion related iron-overload organ damage is generally underestimated, mainly due to difficulties in collecting information on patient transfusion history. Indeed, an adult patient with SCD might be treated with red cell transfusion for acute vaso-occlusive crises (VOCs) or even placed on chronic transfusion program for the management of chronic sickle cell pain although the benefit is controversial.84
Pregnancy-specific complications
In the last decade, progress in the clinical management of SCD has improved the survival of patients with SCD.85 Thus, a larger number of SCD women have reached the reproductive age, thereby requiring a multidisciplinary care team to support their pregnancy.4,86–89 Indeed, poor outcomes for pregnancy in SCD women with high maternal and fetal mortality and morbidity have been reported.88,90 Three main categories of complications have been identified: (i) the severe clinical manifestations related to SCD (ie, recurrent acute VOC, acute chest syndrome); (ii) the medical complication of pregnancy (ie, urinary infection, pyelonephritis); and (iii) the obstetrical complications (pre-eclampsia, intrauterine growth retardation-IUGR).87–89,91 In SCD, prediction of the course of pregnancy is often very difficult to establish due to the variability in the severity of SCD phenotype. Thus, a proper assessment of chronic disease complications in a multidisciplinary setting (ie, screening pulmonary hypertension, iron overload, hypertension and/or proteinuria, for nephropathy and/or hepatic dysfunction, proliferative retinopathy) should be made and the presence of alloantibodies should be carried out ideally prior to conception.92 Due to the recurrent blood transfusions, SCD patients are at higher risk of alloimmunization.93 Almost half of the patients will have alloantibodies which will increase the risk of hemolytic disease in the fetus. Hence, any SCD women with high frequency of transfusions should be tested for RBC alloantibodies and managed accordingly.94 This will allow to differentiate women with SCD with a mild phenotype from those with a more severe phenotype (identify chronic organ damages), who require a more intense therapeutic approach and rigorous follow-up.
Management during pregnancy
SCD women should have a strict follow up at the reference center as well as at the obstetric-sickle clinics at least every 2 to 3 weeks and preferably weekly starting from 32 weeks of gestation. During pregnancy, follow-up of chronic disease complications if they exist as well as obstetric complications are the basis of care. Careful monitoring of fetal growth by serial ultrasonography combined with determination of arterial impedance (umbilical arterial Doppler) is recommended for the risk of intra-uterine growth restriction (IUGR). Hydroxyurea and iron chelation therapy should be discontinued at least 3 months before conception or as soon as the pregnancy test is positive if conception has inadvertently occurred while on the medication. Regular monitoring should include an urinalysis and blood pressure measurement to identify and treat the urinary tract infections or pre-eclampsia as early as possible.87–89,91 Although there is no evidence about the role of ASA therapy in pregnant SCD women, it has been shown that ASA can reduce the risk of pre-eclampsia in women with high risk pregnancies.95 Thus, 80 to 100 mg daily dose of oral ASA may be appropriate after week 12 of gestation, unless major contra-indications are present. However, no evidence-based consensus on this issue has been published. Although the risk of venous thromboembolism (VTE) in SCD women increases fivefold during pregnancy, and studies of placental pathology have indicated the presence of ischemic-reperfusion damage and local thrombosis,96 there is no consensus on the use of anticoagulant treatment. However, SCD women with a history of VTE, at risk for the occurrence of VTE or who are hospitalized for acute medical complications should receive anticoagulation prophylaxis with LMWH during pregnancy and for a period of time after delivery.4,89,97
The use of prophylactic RBC transfusion during pregnancy to reduce sickle cell-related clinical complications and ameliorate maternal/fetal outcomes is supported by a randomized study.98 A recent meta-analysis demonstrated a positive association between prophylactic transfusion and reduction in maternal mortality and neonatal complications in SCD.86 A further randomized controlled study comparing prophylactic transfusions with standard of care is currently under way: https://clinicaltrials.gov/ct2/show/NCT03975894. In addition, a few retrospective studies have also shown the beneficial effects of prophylactic erythrocytapheresis on maternal/fetal outcomes in SCD women.99–101 Data on prophylactic transfusion during pregnancy is controversial. As pregnancy outcome is not clearly correlated with routinely prophylactic transfusion, it is not recommended in SCD women with an uncomplicated pregnancy. A recent dual center retrospective cross sectional study on pregnancies in SCD women with history of severe sickle cell-related clinical manifestations treated with early prophylactic erythrocytapheresis (10.7 +/− 5.2 weeks of gestation) and anticoagulation prophylaxis with enoxaparin showed improvement of maternal outcomes and newborn weights.96 A prophylactic transfusion regimen should be initiated as early as possible in women with severe sickle cell-related clinical manifestations or pre-eclampsia/eclampsia.102
The timing and mode of delivery should be according to obstetric indications and/or the severity of SCD clinical manifestations. SCD women with a mild phenotype might go to spontaneous labor and normal vaginal delivery. Oxygen support should be provided during labor (to maintain oxygen saturation ≥ 94%), the patient should be kept warm, and hydration should be adequate to prevent life-threatening complication such as ACS.87,103 In the case of cesarean delivery, preoperative transfusion therapy to reach a hemoglobin level of 10 g/dL and an HbS <40% is strongly recommended. Thromboprophylaxis with LMWH is recommended in these patients pre- and post a cesarean section.
Enzyme deficiencies
Pyruvate kinase deficiency (PKD) is the most frequent enzyme abnormality of the glycolytic pathway (estimated prevalence is 1–5 per 100,000 in the Caucasian population; higher frequencies in the Pennsylvania Amish and the Gypsy communities from a founder effect).104 Clinical manifestations reflect the symptoms and complications of lifelong chronic hemolysis, including anemia, jaundice, gallstones, splenomegaly, and iron overload. The severity of symptoms is highly variable, ranging from fully compensated hemolysis to life-threatening anemia requiring neonatal exchange transfusions and/or later regular transfusions. PKD is inherited in an autosomal recessive fashion and is caused by homozygous or compound heterozygous variants in the PKLR gene located on chromosome 1q21. To date, more than 300 mutations have been reported, and genotype-phenotype correlation studies have shown that patients with severe hemolytic anemia more commonly have deleterious mutations or missense pathogenic variants that affect the active site or stability of the PK protein.
Current management of PKD is based on red cell transfusions, chelation, and splenectomy. More than 80% of individuals with PKD have been transfused at least once in their lifetime. Splenectomy, which is performed in about 50% of patients with PKD, ameliorates anemia in the majority of patients (median rise in hemoglobin of 1.6 g/dL) and decreases the transfusion burden. However, it carries the risk of post-splenectomy sepsis due to encapsulated organisms, thus is typically delayed until 5 years of age if possible. Splenectomy also carries the risk of thrombosis (11% in the PKD series, all splenectomized). Moreover, about 10% to 15% of splenectomized cases still remain transfusion-dependent, and surgery does not abolish hemolysis, jaundice and the risk of gallstones. Finally, iron overload occurs in about half of PKD patients, and correlates with disease severity and transfusion requirement, but is also observed in milder cases. Therefore, timing and risk of post-splenectomy sepsis and thrombosis must be weighed against the risks of iron loading in patients treated with regular transfusions.104
Regarding the other enzyme defects, there is no specific information available. For this reason, we recommend the general approaches suggested for PK deficiency.
Pregnancy-specific complications
Data on pregnancy in women with PKD are scarce, mainly based on case reports105,106 and the recently published large series on the PKD natural history.105–107 Older series report that pregnancy had been associated with a good maternal and fetal outcome. However, more accurate data come from the 79 post-menarchal females among the 254 patients enrolled in the PKD Natural History study, of whom 39 (about 50%) had at least one pregnancy (total of 115) prior to enrollment. Pregnancy outcomes included 82 live births, of whom 70 full term and 12 preterm. Therefore, preterm births occur in about 10%, and miscarriages in 18% of pregnancies (7 were elective termination), with rates not different from the healthy female population.
Pregnancy is recognized as a hemolytic trigger,104–106,108,109 requiring transfusion support, with frequencies comparable to that of infectious episodes. The great majority of females enrolled in the PKD Natural History study have been transfused during pregnancy. In fact, 75% were transfused during pregnancy and 38% after delivery, as compared with only 6% prior to pregnancy.
Management during pregnancy
The recommendations for pregnancy management include transfusions to support a higher hemoglobin nadir in the mother and close monitoring of fetal growth by a multidisciplinary approach (hematologist and expert obstetrician).110 However, there are no specific recommendations for having a hemoglobin threshold for transfusion, other than mother wellness and fetal growth. Some obstetricians recommend monthly ultrasounds to assess fetal growth after 20 weeks and biophysical profiles by at least 32 weeks.106 Folic acid supplementation should be given generously prior to conception and during pregnancy (800–1000 μg daily). Iron supplementation is not recommended, unless there is a documentation of true iron deficiency by laboratory iron testing. Prior to conception, screening for hepatitis B and C and HIV in previously transfused PKD patients, in addition to an echocardiogram to evaluate cardiac function, and correction of iron overload are also advisable.
Finally, fertility in women with PKD appears to be like that of the general population, and the majority of pregnancies (98%) did not require female assisted reproduction. Of note, 94% of patients reached a normal puberty, and only a small fraction of them had a delayed one and/or required hormone administration.
Red cell membrane disorders
Red cell membrane disorders encompass a vast group of hemolytic anemias that greatly differ by clinical, morphological, laboratory and molecular features. The hereditary forms can be broadly classified into 2 types: (1) erythrocyte disorders caused by altered membrane structural organization, such as hereditary spherocytosis (HS), and (2) erythrocyte disorders caused by altered membrane transport function, such as dehydrated hereditary stomatocytosis (DHS).111 Red cell membrane disorders can be associated with a wide range of genetical alterations depending on their subtype. While some can have recessive inheritance pattern such as mutation in SPTA1 gene seen in HS, others are dominant like SPTB, SLC4A1, ANK1 genes.112 The primary clinical manifestations are hemolytic anemia, jaundice, reticulocytosis, splenomegaly, and cholelithiasis.111,113 HS is the most common non-immune hemolytic anemia with a prevalence of 1:2000–5000 in the Caucasian population.113 HS can present a broad spectrum of clinical severity, ranging from asymptomatic to severe transfusion-dependent forms, even within the same family. The intra-familial heterogeneity can be ascribed to the co-inheritance of genetic variants involved in erythrocyte defects themselves or in other disorders.114 Dehydrated hereditary stomatocytosis (DHS) is the most common form of hereditary stomatocytosis. Patients have a well-compensated hemolytic anemia, with a high reticulocyte count, a tendency to macrocytosis and mild jaundice. Secondary hemochromatosis is the most harmful complication in DHS patients, particularly in those with mutations in the PIEZO1 gene.115–117
The first-line treatment is based on supportive care. For example, HS neonatal patients may need phototherapy or exsanguinous-transfusions.118 Some newborns may necessitate transfusions. In these patients, recombinant erythropoietin can reduce transfusion requirements.119 Transfusions may be administered as needed in adulthood, mostly during aplastic or hemolytic crises.113 Splenectomy could be suggested in several cases of red cell membrane defects, resulting in an increased life span of erythrocytes. Currently, total splenectomy is recommended in adult patients with severe forms of HS; otherwise, in patients with mild or moderate anemia, splenectomy is so far not recommended.13 Conversely, splenectomy is contraindicated in DHS due to the increased risk of thromboembolic complications, probably related to the increased number of circulating stomatocytes.113,115
Pregnancy-specific complications
There is very minimal data in the literature on pregnancy complicated by red blood membrane defects. However, some reports describe the effect of pregnancy on the course of HS.120,121 In most investigated women, pregnancy per se did not cause any problems. Nevertheless, an increase in spherocytes, osmotic fragility, and auto-hemolysis during pregnancy were reported.120 Approximately 40% of patients during pregnancy developed new or worsening anemia, with one third as a consequence of a hemolytic crisis. In a few cases, pregnancy led to megaloblastic anemia secondary to folate deficiency. In splenectomized patients the incidence of complaints was reduced.121 Anemia requiring transfusion during pregnancy occurred in 30.7% of the births121
Regarding the fetal outcome, fetal losses in women with HS during the first trimester have been reported.121 Several cases of nonimmune hydrops fetalis associated with recessive HS have been also described. In particular, these cases are due to near-complete or complete α-spectrin deficiency, caused by homozygous or compound heterozygous nonsense variants in the SPTA1 gene that can cause fatal hydrops fetalis in the third trimester of pregnancy or perinatally.122,123 Similarly, recurrent prenatal ascites and hydrops secondary to DHS have been also described.124–127 A recent study showed perinatal edema secondary to DHS in 17% of the cases, mostly associated with PIEZO1 mutations.126 The main forms of perinatal edema secondary to DHS are hydrops, pleural effusion, and ascites.126 Of note, in DHS the occurrence of perinatal edema is very variable even between patients carrying identical PIEZO1 mutations. For example, there are several cases of patients with the R2456H mutation in PIEZO1 without perinatal edema;128 while incompletely penetrant perinatal edema was noted in two other families carrying the same mutation.129
Management during pregnancy
In patients with increased folate demands, such as pregnant women, folic acid supplementation is recommended.113 In particular, 800 to 1000 μg of folic acid daily is necessary for pregnant women with chronic hemolytic anemias such as EPB42-related HS.130 Monitoring for the exacerbation of anemia with a CBC and reticulocyte count is recommended in pregnant women with HS, as both hemolytic crisis and persistent anemia have been reported during pregnancy, especially in women who have not undergone splenectomy.121 All the patients must follow the regular follow up for pregnant women with repeated ultrasound to monitor the fetus growth.
The cases of severe fetal anemia are generally managed with in utero erythrocyte transfusions,123,125 while the cases of severe perinatal edema may require in utero punctures or after-birth fluid drainage to avoid severe complications and fatal outcomes.126,131 Symptomatic treatment of the perinatal edema is still being debated to prevent pulmonary hypoplasia or Prune Belly syndrome since spontaneous resolution may occur in the first months of life. However, in published cases, ascites and amniotic fluid were drained as a precaution.129
Conclusions
In conclusion, routine obstetrical care for all patients with rare anemias during pregnancy before and after delivery is strongly recommended. Proper counseling prior to pregnancy should take into account a multidisciplinary global assessment of the disease and individualization of each patient. The importance of collaboration among different specialists taking care of the pregnant patient should be emphasized. An international registry tracking all pregnancies in women with rare anemias reported in the literature is needed and could be of use in outlining pathophysiological trends and management recommendations unique to this special population.
Box 1. Summary of the Practical Recommendations for DBA.
Monitor Hb level and transfuse if symptomatic or fetal growth distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Genetic counseling on risk of fetal attaint with DBA
Acetyl salicylic acid 80 to 100 mg once daily during pregnancy
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Box 2. Summary of the Practical Recommendations for CDA.
Monitor Hb level and transfuse if symptomatic or fetal growth distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Acetyl salicylic acid 80 to 100 mg once daily during pregnancy
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Box 3. Summary of the Practical Recommendations for TDT.
Monitor Hb level and maintain the pre-transfusion Hb no less than 9.5g/dL
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Assess cardiac status with echocardiography (preferably every trimester) and manage accordingly
Consider DFO towards the end of the second trimester in patients with severe iron overload in the heart and/or liver
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Splenectomized patient should be considered for acetyl salicylic acid 80 to 100 mg once daily during pregnancy
Box 4. Summary of the Practical Recommendations for NTDT.
Monitor Hb level and transfuse if symptomatic or fetal growth distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Assess cardiac status with echocardiography and manage accordingly
Discontinue use of hydroxyurea
Prophylactic dose of LMWH should be considered in peripartum period
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Splenectomized patient should be considered for acetyl salicylic acid 80 to 100 mg once daily during pregnancy unless prophylactic use of LMWH is prescribed
Box 5. Summary of the Practical Recommendations for SCD.
Monitor Hb level and transfuse if symptomatic or fetal growth distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Discontinue use of hydroxyurea
Regular urinalysis and blood pressure measurement at every clinic visit at least monthly
During Labor: Oxygen support (to maintain oxygen saturation ≥ 94%), the patient should be kept warm, and proper intravenous hydration. HbS <40% is strongly recommended in case of cesarean section
Acetyl salicylic acid 80 to 100 mg daily during pregnancy after week 12 of gestational age
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Peripartum thromboprophylaxis with LMWH is recommended in case of cesarean section or vaginal delivery
Box 6. Summary of the Practical Recommendations for Enzyme Deficiency.
Monitor Hb level and transfuse if symptomatic or fetal distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Acetyl salicylic acid 80 to 100 mg once daily during pregnancy
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Box 7. Summary of the Practical Recommendations for Red Cell Membrane Disorders.
Monitor Hb level and transfuse if symptomatic or fetal growth distress (consider transfusion if Hb < 8 to 10g/dL)
Monthly utero-placental doppler ultrasound starting week 20 of gestational age to monitor growth restriction, fetal distress or hydrops fetalis
Genetic counseling on risk of fetal attaint with red cell membrane disorder
Evaluate iron overload status (Ferritin, Transferrin Saturation) and ensure adequate management of iron overload
Acetyl salicylic acid 80 to 100 mg once daily during pregnancy
Regular folic acid supplementation of 800–1000 μg daily to prevent superimposed megaloblastic anemia
Footnotes
Citation: Taher AT, Iolascon A, Matar CF, Bou-Fakhredin R, de Franceschi L, Cappellini MD, Barcellini W, Russo R, Andolfo I, Tyan P, Gulbis B, Aydinok Y, Anagnou NP, Bencaiova GA, Tamary H, Martinez PA, Forni G, Vindigni R. Recommendations for Pregnancy in Rare inherited Anemias. HemaSphere, 2020;4:4(e446). http://dx.doi.org/10.1097/HS9.0000000000000446
The authors have no conflicts of interest to disclose.
References
- 1.Dokal I, Vulliamy T. Inherited bone marrow failure syndromes. Haematologica. 2010;95:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chirnomas SD, Kupfer GM. The inherited bone marrow failure syndromes. Pediatr Clin North Am. 2013;60:1291–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malhotra M, Sharma JB, Batra S, et al. Maternal and perinatal outcome in varying degrees of anemia. Int J Gynaecol Obstet. 2002;79:93–100. [DOI] [PubMed] [Google Scholar]
- 4.Boulet SL, Okoroh EM, Azonobi I, et al. Sickle cell disease in pregnancy: maternal complications in a Medicaid-enrolled population. Matern Child Health J. 2013;17:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbassi-Ghanavati M, Greer LG, Cunningham FG. Pregnancy and laboratory studies: a reference table for clinicians. Obstet Gynecol. 2009;114:1326–1331. [DOI] [PubMed] [Google Scholar]
- 6.American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 95: Anemia in Pregnancy. Obstetr Gynecol. 2008;112:201–207. [DOI] [PubMed] [Google Scholar]
- 7.Pavord S, Myers B, Robinson S, et al. UK guidelines on the management of iron deficiency in pregnancy. Br J Haematol. 2012;156:588–600. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization The global prevalence of anaemia in 2011. Geneva: World Health Organization; 2015. [Google Scholar]
- 9.Bencaiova G, Burkhardt T, Breymann C. Anemia--prevalence and risk factors in pregnancy. Eur J Intern Med. 2012;23:529–533. [DOI] [PubMed] [Google Scholar]
- 10.Recommendations to prevent and control iron deficiency in the United States Centers for Disease Control and Prevention. MMWR Recomm Rep. 1998;47(RR-3):1–29. [PubMed] [Google Scholar]
- 11.Geelhoed D, Agadzi F, Visser L, et al. Maternal and fetal outcome after severe anemia in pregnancy in rural Ghana. Acta Obstet Gynecol Scand. 2006;85:49–55. [DOI] [PubMed] [Google Scholar]
- 12.Levy A, Fraser D, Katz M, et al. Maternal anemia during pregnancy is an independent risk factor for low birthweight and preterm delivery. Eur J Obstet Gynecol Reprod Biol. 2005;122:182–186. [DOI] [PubMed] [Google Scholar]
- 13.Iolascon A, Andolfo I, Barcellini W, et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margaret P Adam, Editor-in-Chief; Senior Editors: Holly H Ardinger, Roberta A Pagon, and Stephanie E Wallace. Molecular Genetics: Lora JH Bean and Karen Stephens. Anne Amemiya, Genetic Counseling. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993-2020.
- 15.Petrakos G, Andriopoulos P, Tsironi M. Pregnancy in women with thalassemia: challenges and solutions. Int J Womens Health. 2016;8:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies JM, Lewis MP, Wimperis J, et al. Review of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen: prepared on behalf of the British Committee for Standards in Haematology by a working party of the Haemato-Oncology task force. Br J Haematol. 2011;155:308–317. [DOI] [PubMed] [Google Scholar]
- 17.Tsai PH, Steven Arkin, Lipton Jeffrey M. An intrinsic progenitor defect in Diamond-Blackfan anaemia. Br J Haematol. 1989;73:112–120. [DOI] [PubMed] [Google Scholar]
- 18.Gripp KW, et al. Bilateral microtia and cleft palate in cousins with Diamond-Blackfan anemia. Am J Med Genetics. 2001;101:268–274. [DOI] [PubMed] [Google Scholar]
- 19.Lipton JM. Diamond blackfan anemia: new paradigms for a “not so pure” inherited red cell aplasia. Semin Hematol. 2006;43: [DOI] [PubMed] [Google Scholar]
- 20.Orfali KA, Yaw Ohene-Abuakwa, Ball Sarah E. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol. 2004;125:243–252. [DOI] [PubMed] [Google Scholar]
- 21.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. [DOI] [PubMed] [Google Scholar]
- 22.Vlachos A, Lionel Blanc, Jeffrey M, et al. Diamond Blackfan anemia: a model for the translational approach to understanding human disease. Expert Rev Hematol. 2014;7:359–372. [DOI] [PubMed] [Google Scholar]
- 23.Raghavachar A. Pure red cell aplasia: review of treatment and proposal for a treatment strategy. Ann Hematol. 1990;61:47–51. [DOI] [PubMed] [Google Scholar]
- 24.Vlachos A, Federman N, Reyes-Haley C, et al. Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant. 2001;27:381–386. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Warszawski J, Bader-Meunier B, et al. Diamond-blackfan anemia and growth status: the French registry. J Pediatr. 2005;147:669–673. [DOI] [PubMed] [Google Scholar]
- 26.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116:3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rijhsinghani A, Wiechert RJ. Diamond-blackfan anemia in pregnancy. Obstet Gynecol. 1994;83(5 Pt 2):827–829. [PubMed] [Google Scholar]
- 29.Alter BP, Kumar M, Lockhart LL, et al. Pregnancy in bone marrow failure syndromes: Diamond-Blackfan anaemia and Shwachman-Diamond syndrome. Br J Haematol. 1999;107:49–54. [DOI] [PubMed] [Google Scholar]
- 30.Faivre L, et al. High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica. 2006;91:530–533. [PubMed] [Google Scholar]
- 31.Abkowitz JL, Schaison G, Boulad F, et al. Response of Diamond-Blackfan anemia to metoclopramide: evidence for a role for prolactin in erythropoiesis. Blood. 2002;100:2687–2691. [DOI] [PubMed] [Google Scholar]
- 32.Faivre L, Meerpohl J, Da Costa L, et al. High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica. 2006;91:530–533. [PubMed] [Google Scholar]
- 33.Diamantidis MD, Neokleous N, Agapidou A, et al. Iron chelation therapy of transfusion-dependent beta-thalassemia during pregnancy in the era of novel drugs: is deferasirox toxic? Int J Hematol. 2016;103:537–544. [DOI] [PubMed] [Google Scholar]
- 34.Tsironi M, Karagiorga M, Aessopos A. Iron overload, cardiac and other factors affecting pregnancy in thalassemia major. Hemoglobin. 2010;34:240–250. [DOI] [PubMed] [Google Scholar]
- 35.Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica. 2012;97:1786–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gambale A, Iolascon A, Andolfo I, et al. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev Hematol. 2016;9:283–296. [DOI] [PubMed] [Google Scholar]
- 37.Heimpel H, Matuschek A, Ahmed M, et al. Frequency of congenital dyserythropoietic anemias in Europe. Eur J Haematol. 2010;85:20–25. [PubMed] [Google Scholar]
- 38.Russo R, Gambale A, Langella C, et al. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: definition of clinical and molecular spectrum and identification of new diagnostic scores. Am J Haematol. 2014;89:E169–E175. [DOI] [PubMed] [Google Scholar]
- 39.H. Heimpel, A. Iolascon. Congenital dyserythropoietic anaemias. Chapter 7. ESH Handbook on Disorders of Iron Metabolism (2009).
- 40.Iolascon A, Russo R, Delaunay J. Congenital dyserythropoietic anemias. Curr Opin Hematol. 2011;18:146–151. [DOI] [PubMed] [Google Scholar]
- 41.El-Sheikh AA, Hashem H, Holman C, et al. Congenital dyserythropoietic anemia type I presenting as persistent pulmonary hypertension with pigeon chest deformity. Pediatr Blood Cancer. 2014;61:1460–1462. [DOI] [PubMed] [Google Scholar]
- 42.Ayas M, al-Jefri A, Baothman A, et al. Transfusion-dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplant. 2002;29:681–682. [DOI] [PubMed] [Google Scholar]
- 43.Miano M, Eikema D-J, Aljurf M, et al. Stem cell transplantation for congenital dyserythropoietic anemia: an analysis from the European Society for Blood and Marrow Transplantation. Haematologica. 2019;104:e335–e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamary H, Shalev H, Luria D, et al. Clinical features and studies of erythropoiesis in Israeli Bedouins with congenital dyserythropoietic anemia type I. Blood. 1996;87:1763–1770. [PubMed] [Google Scholar]
- 45.Crookston JH, Crookston MC, Burnie KL, et al. Hereditary erythroblastic multinuclearity associated with a positive acidified-serum test: a type of congenital dyserythropoietic anaemia. Br J Haematol. 1969;17:11–26. [DOI] [PubMed] [Google Scholar]
- 46.Verwilghen RL, Lewis SM, Dacie JV, et al. Hempas: congenital dyserythropoietic anaemia (type II). Q J Med. 1973;42:257–278. [PubMed] [Google Scholar]
- 47.Ion R, Hayman R, Hyde J. Congenital dyserythropoietic anaemia and pregnancy. Arch Dis Childhood-Fetal Neonatal Ed. 2012;97(Suppl 1):A64–A70. [Google Scholar]
- 48.Shalev H, Perez Avraham G, Hershkovitz R, et al. Pregnancy outcome in congenital dyserythropoietic anemia type I. Eur J Haematol. 2008;81:317–321. [DOI] [PubMed] [Google Scholar]
- 49.Sandstrom H, Wahlin A. Congenital dyserythropoietic anemia type III. Haematologica. 2000;85:753–757. [PubMed] [Google Scholar]
- 50.Heimpel H, Schwarz K, Ebnother M, et al. Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on long-term observation. Blood. 2006;107:334–340. [DOI] [PubMed] [Google Scholar]
- 51.Salihoglu A, Elverdi T, Eskazan AE, et al. Congenital Dyserythropoietic Anemia Type 1: Report of One Patient and Analysis of Previously Reported Patients Treated with Interferon Alpha. Indian J Hematol Blood Transfus. 2016;32(Suppl 1):272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cappellini MD, Cohen A, Porter J, et al. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 3rd edition. Nicosia, Cyprus: Thalassaemia International Federation. Cyprus. 2014 [PubMed]
- 53.Sleiman J, Tarhini A, Bou-Fakhredin R, et al. Non-transfusion-dependent thalassemia: an update on complications and management. Int J Mol Sci. 2018;19:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skordis N, Christou S, Koliou M, et al. Fertility in female patients with thalassemia. J Pediatr Endocrinol Metab. 1998;11(Suppl 3):935–943. [PubMed] [Google Scholar]
- 55.Belhoul KM, Bakir ML, Saned M-S, et al. Serum ferritin levels and endocrinopathy in medically treated patients with β thalassemia major. Ann Hematol. 2012;91:1107–1114. [DOI] [PubMed] [Google Scholar]
- 56.Origa R, Comitini F. Pregnancy in Thalassemia. Mediterr J Hematol Infect Dis. 2019;11:e2019019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paul A, Thomson VS, Refat M, Al-Rawahi B, et al. Cardiac involvement in beta-thalassemia: current treatment strategies. Postgrad Med. 2019;131:261–267. [DOI] [PubMed] [Google Scholar]
- 58.Carlberg KT, Singer ST, Vichinsky EP. Fertility and pregnancy in women with transfusion-dependent thalassemia. Hematol Oncol Clin North Am. 2018;32:297–315. [DOI] [PubMed] [Google Scholar]
- 59.Cassinerio E, Baldini IM, Alameddine RS, et al. Pregnancy in patients with thalassemia major: a cohort study and conclusions for an adequate care management approach. Ann Hematol. 2017;96:1015–1021. [DOI] [PubMed] [Google Scholar]
- 60.Galanello R, Campus S. Deferiprone chelation therapy for thalassemia major. Acta Haematol. 2009;122:155–164. [DOI] [PubMed] [Google Scholar]
- 61.Berdoukas V, Bentley P, Frost H, et al. Toxicity of oral iron chelator L1. Lancet. 1993;341:1088. [DOI] [PubMed] [Google Scholar]
- 62.Anastasi S, Lisi R, Abbate G, et al. Absence of teratogenicity of deferasirox treatment during pregnancy in a thalassaemic patient. Pediatr Endocrinol Rev. 2011;8(Suppl 2):345–347. [PubMed] [Google Scholar]
- 63.Origa R, Piga A, Quarta G, et al. Pregnancy and beta-thalassemia: an Italian multicenter experience. Haematologica. 2010;95:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsironi M, Ladis V, Margellis Z, et al. Impairment of cardiac function in a successful full-term pregnancy in a homozygous beta-thalassemia major: does chelation have a positive role? Eur J Obstet Gynecol Reprod Biol. 2005;120:117–118. [DOI] [PubMed] [Google Scholar]
- 65.Voskaridou E, Balassopoulou A, Boutou E, et al. Pregnancy in beta-thalassemia intermedia: 20-year experience of a Greek thalassemia center. Eur J Haematol. 2014;93:492–499. [DOI] [PubMed] [Google Scholar]
- 66.Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391:155–167. [DOI] [PubMed] [Google Scholar]
- 67.Roumi JE, Moukhadder HM, Graziadei G, et al. Pregnancy in β-thalassemia intermedia at two tertiary care centers in Lebanon and Italy: a follow-up report on fetal and maternal outcomes. Am J Hematol. 2017;92:E96–E99. [DOI] [PubMed] [Google Scholar]
- 68.Nassar AH, Usta IM, Rechdan JB, et al. Pregnancy in patients with beta-thalassemia intermedia: outcome of mothers and newborns. Am J Hematol. 2006;81:499–502. [DOI] [PubMed] [Google Scholar]
- 69.Cappellini MD, Robbiolo L, Bottasso BM, et al. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111:467–473. [DOI] [PubMed] [Google Scholar]
- 70.Taher A, Vichinsky E, Musallam K, et al. Guidelines for the management of non-transfusion dependent thalassaemia (NTDT). Nicosia (Cyprus): Thalassaemia International Federation. 2013. [PubMed] [Google Scholar]
- 71.Roumi JE, Moukhadder HM, Graziadei G, et al. Pregnancy in beta-thalassemia intermedia at two tertiary care centers in Lebanon and Italy: A follow-up report on fetal and maternal outcomes. Am J Hematol. 2017;92:E96–E99. [DOI] [PubMed] [Google Scholar]
- 72.Lao TT. Obstetric care for women with thalassemia. Best Pract Res Clin Obstet Gynaecol. 2017;39:89–100. [DOI] [PubMed] [Google Scholar]
- 73.Leung TY, Lao TT. Thalassaemia in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2012;26:37–51. [DOI] [PubMed] [Google Scholar]
- 74.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murray CJ, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. [DOI] [PubMed] [Google Scholar]
- 76.Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- 77.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Franceschi L, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost. 2011;37:226–236. [DOI] [PubMed] [Google Scholar]
- 79.Matte A, Zorzi F, Mazzi F, et al. New therapeutic options for the treatment of sickle cell disease. Mediterr J Hematol Infect Dis. 2019;11:e2019002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Franceschi L, Corrocher R. Established and experimental treatments for sickle cell disease. Haematologica. 2004;89:348–356. [PubMed] [Google Scholar]
- 81.Ballas SK, Smith ED. Red blood cell changes during the evolution of the sickle cell painful crisis. Blood. 1992;79:2154–2163. [PubMed] [Google Scholar]
- 82.Vinchi F, De Franceschi L, Ghigo A, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127:1317–1329. [DOI] [PubMed] [Google Scholar]
- 83.Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. 2009;9:271–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lottenberg R, Hassell KL. An evidence-based approach to the treatment of adults with sickle cell disease. Hematology Am Soc Hematol Educ Program. 2005;58–65. [DOI] [PubMed] [Google Scholar]
- 85.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512–S521. [DOI] [PubMed] [Google Scholar]
- 86.Malinowski AK, Shehata N, D'Souza R, et al. Prophylactic transfusion for pregnant women with sickle cell disease: a systematic review and meta-analysis. Blood. 2015;126:2424–2435. [DOI] [PubMed] [Google Scholar]
- 87.Villers MS, Jamison MG, De Castro LM, et al. Morbidity associated with sickle cell disease in pregnancy. Am J Obstet Gynecol. 2008;199:125.e1-5. [DOI] [PubMed] [Google Scholar]
- 88.Oteng-Ntim E, Meeks D, Seed PT, et al. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: systematic review and meta-analysis. Blood. 2015;125:3316–3325. [DOI] [PubMed] [Google Scholar]
- 89.Oteng-Ntim E, Ayensah B, Knight M, et al. Pregnancy outcome in patients with sickle cell disease in the UK--a national cohort study comparing sickle cell anaemia (HbSS) with HbSC disease. Br J Haematol. 2015;169:129–137. [DOI] [PubMed] [Google Scholar]
- 90.Meeks D, Robinson SE, Macleod D, et al. Birth weights in sickle cell disease pregnancies: a cohort study. PLoS One. 2016;11:e0165238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lesage N, Deneux Tharaux C, Saucedo M, et al. Maternal mortality among women with sickle-cell disease in France, 1996-2009. Eur J Obstet Gynecol Reprod Biol. 2015;194:183–188. [DOI] [PubMed] [Google Scholar]
- 92.Aroke D, Momo Kadia B, Njim T. Iron stores in pregnant women with sickle cell disease: a protocol for a systematic review and meta-analysis. BMJ Open. 2019;9:e026497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aygun B, Padmanabhan S, Paley C, et al. Clinical significance of RBC alloantibodies and autoantibodies in sickle cell patients who received transfusions. Transfusion. 2002;42:37–43. [DOI] [PubMed] [Google Scholar]
- 94.Mitra A, Patel K, Gittens-Williams L. Sickle cell disease in pregnancy. Topics Obstetr Gynecol. 2019;39:1–7. [Google Scholar]
- 95.Brunelli E, Seidenari A, Germano C, et al. External validation of a simple risk score based on the ASPRE trial algorithm for preterm preeclampsia considering maternal characteristics in nulliparous pregnant women: a multicentric retrospective cohort study. BJOG. 2020 April 10. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 96.Vianello A, Vencato E, Cantini M, et al. Improvement of maternal and fetal outcomes in women with sickle cell disease treated with early prophylactic erythrocytapheresis. Transfusion. 2018;58:2192–2201. [DOI] [PubMed] [Google Scholar]
- 97.Porter B, Key NS, Jauk VC, et al. Impact of sickle hemoglobinopathies on pregnancy-related venous thromboembolism. Am J Perinatol. 2014;31:805–809. [DOI] [PubMed] [Google Scholar]
- 98.Koshy M, Burd L, Wallace D, et al. Prophylactic red-cell transfusions in pregnant patients with sickle cell disease. A randomized cooperative study. N Engl J Med. 1988;319:1447–1452. [DOI] [PubMed] [Google Scholar]
- 99.Gilli SC, De Paula EV, Biscaro FP, et al. Third-trimester erythrocytapheresis in pregnant patients with sickle cell disease. Int J Gynaecol Obstet. 2007;96:8–11. [DOI] [PubMed] [Google Scholar]
- 100.Ngo C, Kayem G, Habibi A, et al. Pregnancy in sickle cell disease: maternal and fetal outcomes in a population receiving prophylactic partial exchange transfusions. Eur J Obstet Gynecol Reprod Biol. 2010;152:138–142. [DOI] [PubMed] [Google Scholar]
- 101.Asma S, Kozanoglu I, Tarim E, et al. Prophylactic red blood cell exchange may be beneficial in the management of sickle cell disease in pregnancy. Transfusion. 2015;55:36–44. [DOI] [PubMed] [Google Scholar]
- 102.Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4:327–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Asare EV, Olayemi E, Boafor T, et al. Third trimester and early postpartum period of pregnancy have the greatest risk for ACS in women with SCD. Am J Hematol. 2019;94:E328–E331. [DOI] [PubMed] [Google Scholar]
- 104.Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131:2183–2192. [DOI] [PubMed] [Google Scholar]
- 105.Fanning J, Hinkle RS. Pyruvate kinase deficiency hemolytic anemia: two successful pregnancy outcomes. Am J Obstet Gynecol. 1985;153:313–314. [DOI] [PubMed] [Google Scholar]
- 106.Wax JR, Pinette MG, Cartin A, et al. Pyruvate kinase deficiency complicating pregnancy. Obstet Gynecol. 2007;109(2 Pt2):553–555. [DOI] [PubMed] [Google Scholar]
- 107.Grace RF, Bianchi P, van Beers EJ, et al. The clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131:2183–2192. [DOI] [PubMed] [Google Scholar]
- 108.Amankwah KS, Dick BW, Dodge S. Hemolytic anemia and pyruvate kinase deficiency in pregnancy. Obstet Gynecol. 1980;55(3 Suppl):42S–S44. [DOI] [PubMed] [Google Scholar]
- 109.Rider NL, Strauss KA, Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old-order Amish cohort: longitudinal risk and disease management. Am J Hematol. 2011;86:827–834. [DOI] [PubMed] [Google Scholar]
- 110.Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184:721–734. [DOI] [PubMed] [Google Scholar]
- 111.Iolascon A, Andolfo I, Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol. 2019;187:13–24. [DOI] [PubMed] [Google Scholar]
- 112.Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21:1–20. [DOI] [PubMed] [Google Scholar]
- 113.Andolfo I, Russo R, Gambale A, et al. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101:1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Russo R, Andolfo I, Manna F, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93:672–682. [DOI] [PubMed] [Google Scholar]
- 115.Andolfo I, Russo R, Gambale A, et al. Hereditary stomatocytosis: An underdiagnosed condition. Am J Hematol. 2018;93:107–121. [DOI] [PubMed] [Google Scholar]
- 116.Andolfo I, Russo R, Rosato BE, et al. Genotype-phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am J Hematol. 2018;93:1509–1517. [DOI] [PubMed] [Google Scholar]
- 117.Andolfo I, Rosato BE, Manna F, et al. Gain-of-function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am J Hematol. 2020;95:188–197. [DOI] [PubMed] [Google Scholar]
- 118.Da Costa L, Galimand J, Fenneteau O, et al. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27:167–178. [DOI] [PubMed] [Google Scholar]
- 119.Tchernia G, Delhommeau F, Perrotta S, et al. Recombinant erythropoietin therapy as an alternative to blood transfusions in infants with hereditary spherocytosis. Hematol J. 2000;1:146–152. [DOI] [PubMed] [Google Scholar]
- 120.Ho-Yen DO. Hereditary spherocytosis presenting in pregnancy. Acta Haematol. 1984;72:29–33. [DOI] [PubMed] [Google Scholar]
- 121.Pajor A, Lehoczky D, Szakacs Z. Pregnancy and hereditary spherocytosis. Report of 8 patients and a review. Arch Gynecol Obstet. 1993;253:37–42. [DOI] [PubMed] [Google Scholar]
- 122.Chonat S, Risinger M, Sakthivel H, et al. The Spectrum of SPTA1-Associated Hereditary Spherocytosis. Front Physiol. 2019;10:815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hannah DM, Tressler TB, Taboada CD. Nonimmune hydrops fetalis due to autosomal recessive hereditary spherocytosis. Case Rep Womens Health. 2017;16:4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jenner B, Brockelsby J, Thomas W. Dehydrated hereditary stomatocytosis causing fetal hydrops and perinatal ascites. Br J Haematol. 2018;182:620. [DOI] [PubMed] [Google Scholar]
- 125.Ogburn PL, Jr, Ramin KD, Danilenko-Dixon D, et al. In utero erythrocyte transfusion for fetal xerocytosis associated with severe anemia and non-immune hydrops fetalis. Am J Obstet Gynecol. 2001;185:238–239. [DOI] [PubMed] [Google Scholar]
- 126.Picard V, Guitton C, Thuret I, et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients. Haematologica. 2019;104:1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poret H, Simon EG, Herve P, et al. Dehydrated hereditary stomatocytosis and recurrent prenatal ascites. J Obstet Gynaecol. 2013;33:527. [DOI] [PubMed] [Google Scholar]
- 128.Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121:3925–3935. [DOI] [PubMed] [Google Scholar]
- 129.Beneteau C, Thierry G, Blesson S, et al. Recurrent mutation in the PIEZO1 gene in two families of hereditary xerocytosis with fetal hydrops. Clin Genet. 2014;85:293–295. [DOI] [PubMed] [Google Scholar]
- 130.Kalfa TA, Connor JA, Begtrup AH. EPB42-related hereditary spherocytosis. In: Adam MP, Ardinger HH, Pagon RA, et al (eds). GeneReviews® [Internet]. Seattle, Washington: University of Washington; 1993. [PubMed]
- 131.Le Vaillant C, Riteau AS, Eveillard M, et al. Dehydrated hereditary stomatocytosis: prenatal management of ascites and pleural effusions. Taiwan J Obstet Gynecol. 2018;57:323–324. [DOI] [PubMed] [Google Scholar]
- 132.Rogers BB, Bloom SL, Buchanan GR. Autosomal dominantly inherited Diamond-Blackfan anemia resulting in nonimmune hydrops. Obstet Gynecol. 1997;89(5 Pt 2):805–807. [DOI] [PubMed] [Google Scholar]
- 133.Kotera A, Miyazaki N, Hashimoto M, et al. Anesthetic management of the cesarean section in a patient with aplastic anemia. Masui. 2010;59:776–779. [PubMed] [Google Scholar]
- 134.Gansner JM, Achebe MM, Gray KJ, et al. Pregnancy outcomes in inherited bone marrow failure syndromes. Blood. 2017;130:1671–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Giri N, Stratton P, Savage SA, et al. Pregnancies in patients with inherited bone marrow failure syndromes in the NCI cohort. Blood. 2017;130:1674–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]