

Abstract
Spinal cord injury (SCI) is a serious central nervous system trauma that leads to loss of motor and sensory functions in the SCI patients. One of the cell death mechanisms is autophagy, which is ‘self-eating’ of the damaged and misfolded proteins and nucleic acids, damaged mitochondria, and other impaired organelles for recycling of cellular building blocks. Autophagy is different from all other cell death mechanisms in one important aspect that it gives the cells an opportunity to survive or demise depending on the circumstances. Autophagy is a therapeutic target for alleviation of pathogenesis in traumatic SCI. However, functions of autophagy in traumatic SCI remain controversial. Spatial and temporal patterns of activation of autophagy after traumatic SCI have been reported to be contradictory. Formation of autophagosomes following therapeutic activation or inhibition of autophagy flux is ambiguous in traumatic SCI studies. Both beneficial and harmful outcomes due to enhancement autophagy have been reported in traumatic SCI studies in preclinical models. Only further studies will make it clear whether therapeutic activation or inhibition of autophagy is beneficial in overall outcomes in preclinical models of traumatic SCI. Therapeutic enhancement of autophagy flux may digest the damaged components of the central nervous system cells for recycling and thereby facilitating functional recovery. Many studies demonstrated activation of autophagy flux and inhibition of apoptosis for neuroprotective effects in traumatic SCI. Therapeutic induction of autophagy in traumatic SCI promotes axonal regeneration, supporting another beneficial role of autophagy in traumatic SCI. In contrast, some other studies demonstrated that disruption of autophagy flux in traumatic SCI strongly correlated with neuronal death at remote location and impaired functional recovery. This article describes our current understanding of roles of autophagy in acute and chronic traumatic SCI, cross-talk between autophagy and apoptosis, therapeutic activation or inhibition of autophagy for promoting functional recovery, and future of autophagy in traumatic SCI.
Keywords: apoptosis, autophagy, autophagy modulation, neuroprotection, traumatic spinal cord injury
Introduction
Spinal cord injury (SCI), whether non-traumatic or traumatic, is always a serious neurological problem that significantly impairs locomotion, causes organ dysfunctions and infections, and reduces the life-span of its victim. Generally, mortality of patients with SCI is almost three times higher than normal people (Sabre et al., 2013). Broadly, SCI is classified into non-traumatic SCI and traumatic SCI based on the factors that cause their pathogenesis (Gedde et al., 2019). Non-traumatic injury to the spinal cord is the damage due to infection, lack of blood supply, compression caused by cancer, slow degeneration of spinal bones by osteoarthritis, vascular ischemia, multiple sclerosis, inflammatory disease, motoneuron disease, or radiation myelopathy (Grassner et al., 2016). However, most of the SCI patients in the United States are the victims of traumatic SCI caused by motor vehicle accidents, falls, gunshot wounds, stabbings, and sporting activities (Ge et al., 2018). Patients with acute traumatic SCI and chronic traumatic SCI suffer heavily from progressive pathogenesis and psychological problems (Farhadi et al., 2018; Mills et al., 2018). So, traumatic SCI is the intense focus of current research for understating of the cellular and molecular mechanisms of progressive pathogenesis to find out the appropriate therapeutic targets for prevention of neurodegeneration and improvement of neurological functions (Ahuja et al., 2017).
A mechanical or an accidental injury to the spinal cord is called the primary traumatic SCI that triggers a deleterious neurochemical cascade of secondary injury for pathogenesis and neurodegeneration leading to acute traumatic SCI. There are about 25 well documented secondary injury mechanisms that contribute to pathogenesis in acute traumatic SCI (Oyinbo, 2011). If acute traumatic SCI is not treated immediately or within a short period of time (a few hours), its progressive pathogenesis and neurodegeneration lead to chronic traumatic SCI. Chronic traumatic SCI at the thoracic, lumbar, or sacral spinal cord sections (T1 or below) causes paraplegia (paralysis from the waist down) while chronic traumatic SCI in the cervical spinal cord sections (C8 or above) causes quadriplegia or tetraplegia (paralysis from the shoulders down) (Nas et al., 2015). Unfortunately, there is still no effective treatment for chronic traumatic SCI. An interdisciplinary team led by a physiatrist and involvement of the patient’s family, physiotherapist, occupational therapist, dietician, psychologist, speech therapist, social worker, and other specialists is necessary for rehabilitation of patient with chronic traumatic SCI (Nas et al., 2015). Tetraplegic individuals with chronic traumatic SCI suffer the most from paralysis, organ dysfunction, and infection and the estimated life expectancy of these individuals, compared with general population, vary from 18.1 to 88.4% based on their ventilator dependency, type of injury, age, and gender (Savic et al., 2017).
The pathogenesis in traumatic SCI involves three main programmed cell death (PCD) mechanisms: apoptotic cell death (type I PCD), autophagic cell death (type II PCD), and necrotic cell death (type III PCD) (Hotchkiss et al., 2009; Booth et al., 2014). Apoptosis or apoptotic cell death (type I PCD) is a delayed cell death mechanism, which occurs due to mild to moderate extracellular or intracellular stimuli with activation of cysteine proteases (calpains and caspases), and it is preventable with appropriate therapeutic interventions in animal models of traumatic SCI (Ray et al., 2003, 2011). Apoptosis is usually a caspase-dependent cell death process. But autophagy and autophagic cell death are not synonymous. Autophagy is the name coined by the Belgian biochemist Christian de Duve in early 1960s (Klionsky, 2008). The discovery of the autophagy-related genes in yeast in the early 1990s in the laboratory of the Japanese autophagy investigator Yoshinori Ohsumi demonstrated the mechanisms of autophagy (Takeshige et al., 1992; Tsukada and Ohsumi, 1993) and led to his award of the 2016 Nobel Prize in Physiology or Medicine (Harnett et al., 2017). Autophagy (also called autophagocytosis), which is caspase-independent but cathepsin-dependent process, is still a contentious cell death mechanism that a cell under hypoxia or nutritional stress uses for lysosomal degradation of its damaged proteins, organelles, and other cellular materials for recycling and clearing. Currently, there are three well-known forms of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (Glick et al., 2010). Macroautophagy is simply known as autophagy; however, it should be clarified at this point that the remainder of this review article has circumstances where use of one specific term (macroautophagy or autophagy) is more appropriate than the other.
The execution of autophagy requires several sequential steps: (1) ‘nucleation’ starts with membranes of endoplasmic reticulum, Golgi, and mitochondria for its (2) ‘elongation’ to form (3) ‘phagophore’ for enclosing the cytosolic cargos (including long-lived and misfolded proteins, lipids, glycogen, ferritin, and damaged organelles) leading to formation of (4) ‘autophagosome’ that goes through (5) ‘maturation’ and then fusion with lysosome to form (6) ‘autolysosome’ where (7) ‘degradation’ of cargos by lysosomal hydrolases produces the products for their (8) ‘efflux’ into the cytosol (Figure 1). In macroautophagy, the autophagosome envelops the cargo and then fuses with lysosomes. In microautophagy, an invagination of the lysosomal membrane engulfs the cargo. In CMA, cognate proteins deliver substrates to the lysosomes. Each lysosome (0.2–0.5 µm) contains about 50 different degradative acid hydrolases (proteases, glycosidases, lipases, phosphatases, sulfatases, phospholipases, and nucleases), which are active inside the lysosome (pH 5.0) and inactive if released into the cytosol (pH 7.2). The acidic pH of lysosome is maintained because of transport of cytosolic protons across the lysosomal membrane by conformational changes in proton pumps (H+-ATPases located on lysosomal membrane) at the expense of hydrolysis of adenosine triphosphate (ATP) derived from mitochondria. Autophagy flux is the total dynamics of autophagy and thus it is the progression of cargo sequestration into autophagosomes, delivery to lysosomes, and degradation by lysosomal enzymes (Zhang et al., 2013). Autophagy flux is increased in mild mechanical injury (e.g., traumatic SCI) or metabolic stress (e.g., starvation) but decreased in suppression of the process at upstream steps (e.g., autophagosome formation) or downstream steps (e.g., autolysosome formation) (Wong and Holzbaur, 2015). The autophagic degradation and production of multiple metabolites can support diverse anabolic and biosynthetic pathways in the cells (Kaur and Debnath, 2015). When the metabolites or building blocks are back into the cytosol, they can be either recycled into metabolic and biosynthetic pathways or oxidized by the mitochondria to generate ATP for cell survival. However, autophagy may occur at high amounts due to high metabolic stress leading to autophagic cell death (type II PCD). The extent of autophagy and its role and contribution to cell death mechanism in traumatic SCI still remain controversial. Necrosis or necrotic cell death (type III PCD) is not preventable in traumatic SCI as it happens suddenly at the time of primary injury and probably due to extreme insults such as huge intracellular oxidative stress and cytosolic Ca2+ overload during secondary injury process.
Figure 1.

Sequential steps in induction of autophagy.
The extracellular stress signals such as nutrient deficiency, hypoxia, and injury trigger intracellular activation of adenosine 5' monophosphate-activated protein kinase (AMPK) to initiate formation of the Unc-51 like kinase (ULK) complex that in turn leads to generation of the vacuolar protein sorting 34 (VPS34) complex for bringing about the isolation membrane, which is the first step in induction of autophagy, followed by seven subsequent steps. Autolysosome, in addition to macroautophagy, may perform microautophagy and chaperone-mediated autophagy (CMA) and lead to the next step for lysosomal digestion of all cargos. Efflux, the final step of autophagy, releases the metabolites that are new building blocks (amino acids, fatty acids, nucleotides, sugars, etc.) into the cytosol for recycling. Recycling of amino acids and fatty acids via the Krebs cycle, also known as the tricarboxylic acid (TCA) cycle, in the mitochondria generates plenty of adenosine triphosphate (ATP) molecules to fuel biosynthesis of cellular macromolecules such as proteins (using amino acids), lipids (using fatty acids) and genetic materials (DNA and RNA molecules using nucleotides and sugars) to support and sustain cell survival.
Some earlier studies have suggested that an increase in expression of Beclin-1, which is a Bcl-2-interacting protein and promoter of autophagy, activates autophagy as a novel cell death mechanism to contribute to neural tissue damage at the lesion site in traumatic SCI in mice (Kanno et al., 2009a, b). More recent investigations indicate that rapamycin, which is an inhibitor of the mechanistic target of rapamycin (mTOR) signaling, can promote autophagy and reduce secondary damage, exert neuroprotective effects, promote neuroregeneration, and improve locomotor function in traumatic SCI in mice (Kanno et al., 2012; Sekiguchi et al., 2012) as well as in rats (Wang et al., 2014). Autophagy uses lysosome for degradation of the damaged mitochondria and dysfunctional components in the cell to block apoptosis (type I PCD) and thus produces cellular building blocks, which can be used for cell survival and proliferation (Dombi et al., 2018). Autophagic elimination of the damaged mitochondria is a salvage mechanism that a cell uses to defy apoptosis. However, autophagy is a double-edged sword as it contributes to cell proliferation and participates in promotion of cell death (Das et al., 2018). There is a well-recognized interplay between autophagy and apoptosis depending on the context and stimuli (Mariño et al., 2014). Induction of appropriate amount of autophagy can clean up the damaged organelles to potentiate the neuroprotective and recovery processes in traumatic SCI. On the other hand, inhibition of excessive autophagy appears to be another therapeutic option in treating traumatic SCI.
Activation and Amount of Autophagy in Neural Cells in Traumatic Spinal Cord Injury
Activation and amount of autophagy appear to have highly complicated roles in altering the course of pathogenesis in traumatic SCI. The functions of autophagy in traumatic SCI are still controversial and have not yet been completely clarified (Lipinski et al., 2015). Macroautophagy, simply called autophagy as mentioned earlier, is an important mechanism under stress for bulk cytosolic degradation of the damaged organelles, toxic agents, long-lived and unwanted proteins, fats, carbohydrates, and nucleic acids for efficient turnover, recycling, and cellular homeostasis through an autophagosomal–lysosomal pathway. The mechanism of autophagy has three main phases: formation of autophagosome, fusion of autophagosome with lysosome, and lysosomal degradation in the autolysosome (Glick et al., 2010). There are several well-defined biomarkers such as Beclin-1 (which is encoded by BECN1 gene, an ortholog of the yeast autophagy-related 6 gene or Atg6 gene) (Shravage et al., 2013), microtubule-associated protein 1 light chain 3B (hereafter called LC3B, which is encoded by MAP1LC3B gene, an ortholog of the yeast Atg8 gene) (Tanida et al., 2014), and p62/SQSTM1 (also called p62/sequestosome1, which is encoded by the SQSTM1 gene, an ortholog of the yeast Atg19 gene) (Kraft et al., 2010; Mao et al., 2013) that can be readily used to monitor autophagy in traumatic SCI in humans (Figure 1). Autophagy flux may be either increased or decreased depending on the location and severity of traumatic SCI (Lipinski et al., 2015). Activation of autophagy in simple terms involves formation of autophagosomes to sequester cytoplasmic components such as damaged organelles and toxic protein aggregates, fusion of autophagosomes with lysosomes, and then degradation of cargo by lysosomal enzymes. The processing of cargo through the autophagy flux is usually a cytoprotective function. In course of pathogenesis in traumatic SCI, autophagy flux may be inhibited causing accumulation of dysfunctional autophagosomes and induction of cell death (Guo et al., 2018).
Increases in biomarkers of autophagy (Beclin-1 and LC3B II) and accumulation of autophagosomes have been observed in acute phase of secondary injury, which is initiated within hours after primary traumatic central nervous system (CNS) injuries in mice (Lai et al., 2008; Kanno et al., 2011) as well as in human autopsy samples (Sakai et al., 2014). The animal and human studies also documented that increases in biomarkers of autophagy were sustained for weeks and months after the primary traumatic injury (Chen et al., 2014; Sakai et al., 2014). The prolonged activation of autophagy may reflect its potential functions in the acute phase of secondary injury for neuroprotection as well as in the chronic phase of secondary injury for neurodegeneration and/or neuroregeneration. On the contrary, disruption of autophagy flux after traumatic SCI has recently been reported to be associated with endoplasmic reticulum (ER) stress and induction of motoneuron apoptosis (Liu et al., 2015). Motoneurons with the disrupted autophagy did co-express the ER-stress-associated initiator caspase-12 and the cleaved executioner caspase-3, suggesting that traumatic SCI caused lysosomal dysfunction and contributed to autophagy disruption and ER stress for induction of motoneuron apoptosis (Liu et al., 2015). There are no reasonable clarifications yet to account for the differences in autophagy flux in different traumatic SCI studies. Preclinical animal models of traumatic SCI have been categorized into mild, moderate, and severe injury models (Verma et al., 2019). It can be speculated that different injury severity continuum in different injury models may differentially modulate activation of autophagy and autophagy flux (Lipinski et al., 2015). It is plausible that a relatively mild primary injury may cause upstream activation of autophagy and maintenance of autophagy flux to provide neuroprotection (Lipinski et al., 2015), a relatively moderate primary injury may inhibit autophagy flux leading to neurodegeneration unless autophagy flux is stimulated by therapeutic interventions (Zhou et al., 2016; Zhang et al., 2017), and a relatively severe primary injury may cause hyper activation of autophagy flux leading to autophagic neuronal death during secondary injury process in traumatic SCI (Hao et al., 2013; Lipinski et al., 2015). Inhibition of autophagy flux in the acute phase and hyper activation of autophagy flux in the chronic phase may be accounted for progressive neurodegeneration in moderate traumatic SCI. Generation of excessive reactive oxygen species due to mitochondrial dysfunction, activation of calpain and cleavage of the chaperone heat shock protein 70 (HSP70), and activity of the channel forming pro-apoptotic Bax protein are potential mediators for breakdown of lysosomal membrane permeability leading to lysosomal dysfunction (Azbill et al., 1997; Yamashima and Oikawa, 2009; Ray et al., 2011), while ER stress may cause destabilization of autophagosomal membrane (Liu et al., 2015) leading to inhibition of autophagy flux and neurological deficits in the acute phase of moderate traumatic SCI in animal model (Figure 2). High levels of reactive oxygen species can cause oxidative damage to the lysosomal membrane and inhibit autophagy (Sarkar et al., 2011). Increase in Bax can induce channel formation on lysosomes and prevent non-functional lysosomes from participation in autophagy (Bové et al., 2014). Calpain is readily activated due to intracellular Ca2+-overload in acute traumatic SCI (Ray et al., 2003) and calpain mediated cleavage and inhibition of HSP70 can cause lysosomal rupture and thereby put a break on autophagy flux (Yamashima and Oikawa, 2009), leading to increases in neuronal and oligodendrocyte death and neurological dysfunction in traumatic SCI.
Figure 2.
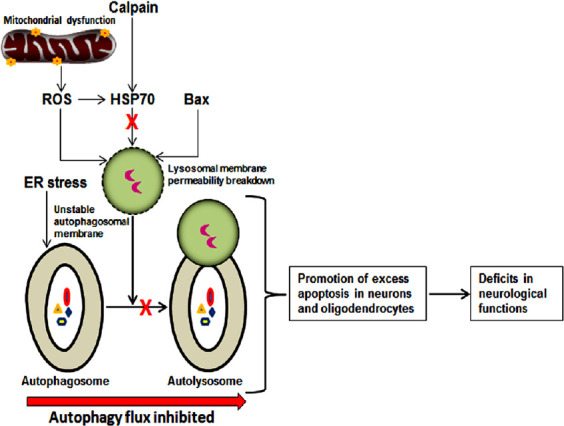
Plausible mechanism of inhibition of autophagy flux in secondary injury process during acute phase of moderate traumatic SCI and the outcomes.
Several factors such as excessive generation of ROS due to mitochondrial dysfunction, cleavage of HSP70 by calpain activity, and channel-forming Bax are likely to contribute to the damage in lysosomal membrane leading to lysosomal dysfunction and ER stress may cause destabilization of autophagosomal membrane leading to failure in formation of functional autolysosome and thus inhibition of autophagy flux. Inhibition of autophagy flux promotes excessive apoptosis in neurons and oligodendrocytes resulting in neurological dysfunctions. ER: Endoplasmic reticulum; HSP70: heat shock protein 70; ROS: reactive oxygen species; SCI: spinal cord injury.
Activation of autophagy is an important event and promotion or inhibition of autophagy can be a promising therapeutic strategy for prevention of pathogenesis in traumatic SCI. Autophagy can be considered a pro-survival mechanism as it restricts neural cells death and promotes neuroprotection in traumatic SCI (Lipinski et al., 2015). Among the neural cells, neurons are most likely to activate autophagy to eliminate toxic proteins and damaged mitochondria (mitophagy) and inhibit apoptosis for promoting neuroprotection in traumatic SCI (Yu et al., 2013; Tang et al., 2014). Other neural cell types such as astrocytes, oligodendrocytes, and microglia also upregulate autophagy in traumatic SCI but functions of autophagy in these glial cells in traumatic SCI still remain unresolved (Kanno et al., 2009a; Tang et al., 2014; Liu et al., 2015). Spatial and temporal patterns of activation of autophagy, autophagy flux, and overall functions of autophagy after traumatic SCI have been found to be contradictory in various studies (Zhou et al., 2017). Formation of autophagosomes following therapeutic activation or inhibition of autophagy flux is ambiguous in many traumatic SCI studies. Both beneficial and harmful outcomes due to enhancement of autophagy have been reported in traumatic SCI studies in preclinical models. Only further studies will make it clear whether therapeutic activation or inhibition of autophagy is beneficial in overall outcomes in preclinical models of traumatic SCI. Initiation of autophagosome biogenesis requires activity of class III phosphatidylinositol 3-kinase (PI3K), lysosomal biogenesis is dependent on activity of the transcription factor EB (TFEB), and both class III PI3K and TFEB are negatively regulated by mTOR; inhibition of mTOR by rapamycin strongly stimulates class III PI3K and TFEB activities leading to hyperactivation of autophagy flux (Lipinski et al., 2015). Too little activation or inhibition of autophagy flux in acute phase of moderate traumatic SCI may be associated with too little clearance of the damaged cells and cellular components. Probably, too much activation of autophagy flux due to intracellular inhibition of mTOR activity following injury (Kanno et al., 2012) causes autophagic cell death and further enhances neurodegeneration and locomotor dysfunction in the chronic phase of moderate traumatic SCI in animal model (Figure 3).
Figure 3.

Plausible mechanism of enhancement of autophagy flux in secondary injury process during chronic phase of moderate traumatic SCI and the outcomes.
Class III PI3K and TFEB (with no negative regulation by mTOR) enhance biogenesis of autophagosome and lysosome, respectively, causing hyper activation of autophagy flux and autophagic cell death in neurons and oligodendrocytes leading to loss of locomotor functions. mTOR: Mechanistic target of rapamycin; PI3K: phosphatidylinositol 3-kinase; SCI: spinal cord injury; TFEB: transcription factor EB.
Our current concepts about how autophagy works, as a neuroprotective mechanism or as a neurodegenerative mechanism, in traumatic SCI are still evolving. Although it has initially been heralded as an alternative form of cell death, autophagy is now known to be not only a homeostatic mechanism in healthy cells but also a cytoprotective process in response to metabolic challenges in the CNS diseases and injuries (Nixon and Yang, 2012). Autophagy acts as a stress response and quality control mechanism in human diseases and injuries (Murrow and Debnath, 2013). In traumatic SCI, impairment of autophagy pathway at various steps can cause accumulation of pathogenic proteins and damaged organelles, compromising or abolishing the pro-survival and anti-apoptotic effects of autophagy on neurons and glial cells. Neurodegenerative disorders are associated with alterations in extents of autophagy and apoptosis for progressive pathogenesis, which poses enormous challenges and also provides therapeutic opportunities (Ghavami et al., 2014). In the CNS, autophagy fights neurodegeneration and plays an important role in self-digestion by mass clearance of the damaged proteins, organelles, and cells to maintain the normal neurological functions (Mizushima et al., 2008). Two important intracellular systems for degradation of proteins are the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system, which are connected and collaborators for neuroprotection (Nedelsky et al., 2008). The UPS is a highly useful catabolic mechanism for small amounts of specific regulatory proteins, which are short-lived or damaged for fine-tuning their steady state levels in the cytosol and nucleus in mammalian cells; while autophagy is an essential and massive degradative mechanism for not only the damaged long-lived proteins but also for the non-functional nucleic acids, useless carbohydrates, and the damaged cell itself for significant clearing and producing cellular building blocks (e.g., amino acids, nucleic acids, sugars) for recycling. As autophagy is needed for neuroprotection, impairment or absence of basal level of autophagy in the CNS may give rise to neurodegeneration and severely compromise the normal neurological functions (Radad et al., 2015). Therefore, it is important that activation of autophagy at the appropriate level needs to be maintained for clearance of the damaged cells and cellular components to reduce pathogenesis and promote neuroprotection in traumatic SCI. Recent findings show that impairment of autophagy in neurodegenerative disorders prompts activation of an alternative Golgi-mediated degradation pathway for expulsion of toxic proteins, nuclear breakdown, and neuronal cell death (Barthet and Ryan, 2018).
Interplay between Autophagy and Apoptosis in Traumatic Spinal Cord Injury
Interplay between autophagy and apoptosis has been extensively studied (Fimia and Piacentini, 2010). Depending on circumstance, activation of autophagy is known to help either cell survival or cell death. Switching from cell survival to cell death, autophagy can significantly control the pathological process in traumatic SCI. Autophagy is sometimes a harbinger of apoptosis, which unambiguously causes degeneration of neural cells in traumatic SCI. However, complete spatial and temporal patterns of contradictory or cooperative roles of autophagy and apoptosis in traumatic SCI still remain unclear (Zhou et al., 2017). Basal level of autophagy in normal conditions maintains cellular homeostasis, but an increase in activation of autophagy is a stress response to make the cell survive in an adverse condition such as traumatic SCI. One of the main tasks of autophagy is recycling of the damaged proteins, aggregates, carbohydrates, nucleic acids, lipids, and organelles in an attempt to clean up the mess in the cell to offer it a fighting chance for using new building blocks to replenish cellular components for survival under stress and short supply of nutrients (Mizushima et al., 2008; Yang and Klionsky, 2010). This process thus provides the cell with a protection from nutrient deprivation and an opportunity to adjust with the situation of scarcity. However, autophagy may cause excessive self-degradation leading to autophagic cell death. Defective or uncontrolled autophagy may cause cell death either by selective degradation of indispensable cellular components such as catalase and mitochondria or by non-selective degradation of different cellular components to the extent that makes the cell unable to survive any more (Yu et al., 2006; Lartigue et al., 2009). There exists another possibility that autophagy may stop producing enough amino acids and other cellular building blocks for cell survival, causing the cell to trigger induction of apoptosis. Indeed, there have been reports suggesting that under certain circumstances autophagy itself is a cell death mechanism and activity of autophagy stops to promote apoptotic cell death (Kroemer and Levine, 2008). Rapidly emerging evidence strongly suggests several direct molecular connections between autophagy and apoptosis (Gump and Thorburn, 2011). Currently, we know multiple direct and indirect interactions that strongly suggest mechanistic communication and collaboration between the autophagy and apoptosis regulatory proteins to determine cell fate (Thorburn, 2008; Eisenberg-Lerner et al., 2009). Thus, molecular connections and interactions between autophagy and apoptosis may determine occurrence of either neuroprotection or neurodegeneration in traumatic SCI. Beclin-1 and p62/SQSTM1 are two autophagy regulatory proteins, which are at the core of interactions between autophagy and apoptosis. Activities of both Beclin-1 and p62/SQSTM1 after a mild traumatic SCI may promote autophagy and slow down or inhibit apoptosis for prevention of neurodegeneration (Figure 4).
Figure 4.
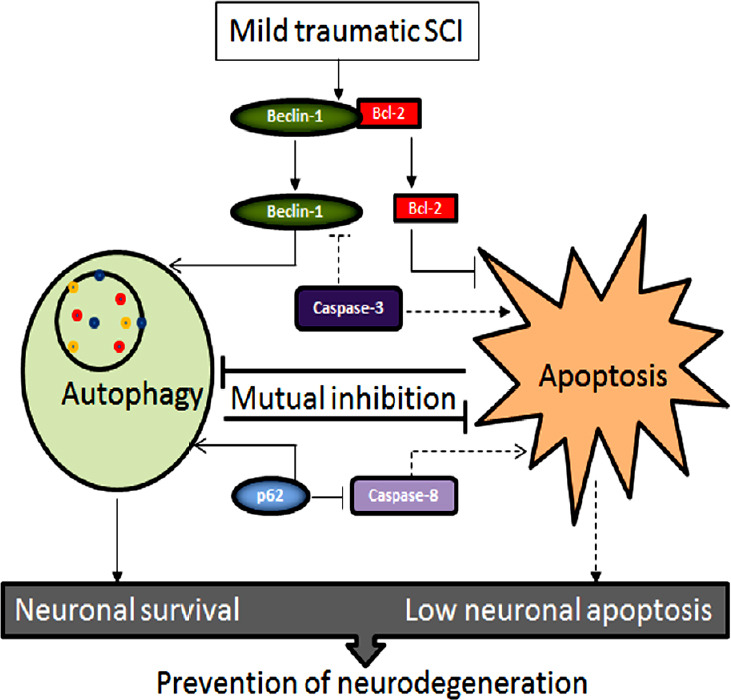
The cross-talk between autophagy and apoptosis for prevention of neurodegeneration in mild traumatic SCI.
Beclin-1 (pro-autophagic protein) and Bcl-2 (anti-apoptotic protein) after separation from each other cause induction of autophagy and inhibition of apoptosis, respectively. Also, p62 participates to promote autophagy and inhibit apoptosis. The end result of the cross-talk between the molecular components of autophagy and apoptosis is prevention of neurodegeneration. Dash lines in this diagram indicate the paths of petty operational. SCI: Spinal cord injury.
Beclin-1, which controls initiation of autophagy, is a novel BH3-only protein that can directly interact with the anti-apoptotic Bcl-2 (Sinha and Levin, 2008) and Bcl-xL (Maiuri et al., 2007) proteins. Autophagy is not activated if Beclin-1 is bound to its inhibitor Bcl-2 or Bcl-xL. However, autophagy is initiated with Beclin-1 following its phosphorylation on BH3 domain by the death-associated protein kinase (a Ca2+/calmodulin-dependent Ser/Thr kinase) and release from Bcl-2 (Levin-Salomon et al., 2014) or Bcl-xL (Zalckvar et al., 2009). Beclin-1 may also be on the loose for initiating autophagy following Bcl-2 phosphorylation by c-Jun N-terminal protein kinase 1 (Wei et al., 2008). On the other hand, overexpression of Bcl-2 or Bcl-xL is known to block activation of autophagy (Levine et al., 2008). Also, cleavage of Beclin-1 by caspase-3 produces a truncated protein that does not activate autophagy, effectively causing inhibition of autophagy to promote apoptosis (Zhu et al., 2010). Thus, various components of the apoptosis machinery regulate Beclin-1 to either promote or inhibit autophagy. The above examples of mutual regulation of autophagy and apoptosis make it clear that when autophagy is activated, apoptosis is blocked; when autophagy is inhibited, apoptosis is promoted.
p62/SQSTM1 is an important player in selective autophagy for degradation of many aggregate-prone proteins and unwanted organelles in the cells (Komatsu and Ichimura, 2010). Although autophagy is widely known for its ability for non-selective degradation, many recent studies show that like the UPS, selective autophagy is also essential and linked to the UPS for regulation of diverse cellular processes and induction of cell death (Liu et al., 2016; Nam et al., 2017; Fan et al., 2018). In selective autophagy, cytoplasmic components are selected and tagged before being sequestered into the autophagosomes using the selective autophagy receptors such as p62/SQSTM1 (Lamark et al., 2017). p62/SQSTM1 is known to interact directly with the apoptotic pathway protein caspase-8 (Moscat and Diaz-Meco, 2009). p62/SQSTM1 is important for cost-effective activation of caspase-8 (Jin et al., 2009) but caspase-8 also targets and cleaves p62/SQSTM1 during death receptor activation (Norman et al., 2010). Interestingly, another study showed that p62/SQSTM1 degraded caspase-8 (Hou et al., 2010). These studies suggest the presence of models where autophagy and apoptosis are in intricate balancing acts showing that autophagy regulates the amount of apoptosis whereas apoptosis regulates degradation of p62/SQSTM1 and caspase-8 (Figure 4).
Autophagy may regulate apoptosis via active degradation of pro-apoptotic proteins and organelles (e.g., caspase-8, mitochondria), but the implication of these autophagic degradations for changing the amount of apoptotic cell death in traumatic SCI is currently unknown. Selective autophagic degradation of mitochondria is mitophagy that targets only a few and not all the mitochondria in a cell (Youle and Narendra, 2011). Mitophagy may selectively eliminate the damaged or excessive mitochondria to maintain mitochondrial quality and homeostasis after a traumatic SCI. It is still unclear whether the remaining healthy mitochondria in the CNS cells experience any apoptotic stimulus to cause release of cytochrome c into the cytosol to promote formation of apoptosome and induction of apoptosis in traumatic SCI. There is a possibility that mitophagy mediated reduction in the number of mitochondria may change the threshold of apoptotic stimulus, which is necessary to induce apoptosis. It thus seems that autophagy may control the threshold of apoptotic stimulus in the CNS cells for determining activation or inhibition of apoptosis. Although we have evidence of interplay and communication between autophagy and apoptosis, we still lack a clear molecular mechanism to explain how autophagy can inhibit or activate apoptosis in traumatic SCI.
The protein networks that control the initiation and the execution phases of autophagy and apoptosis are highly interconnected (Kang et al., 2011; Wu et al., 2014). The cross-talk between autophagy and apoptosis is generally known to exist in traumatic SCI (Gordy and He, 2012). Activation of autophagy following traumatic SCI is an attempt to enable the CNS cells to cope with stress and protect them from apoptosis. Interestingly, depending on circumstances and cell signaling mechanisms, autophagy may often end with apoptosis and apoptosis may begin with autophagy (Booth et al., 2014). It is reasonable to think that mutual inhibitory cross-talk between autophagy and apoptosis prevents simultaneous activation of both cell survival and cell death pathways in acute traumatic SCI. However, activation of excessive autophagy may contribute to cell death depending on the context (Cooper, 2018). In course of progressive and aggressive pathogenesis in severe traumatic SCI, increase in autophagy flux may promote cell execution through excessive self-digestion (autophagic cell death) or/and via activation of apoptosis and other cell death mechanisms (Figure 5).
Figure 5.
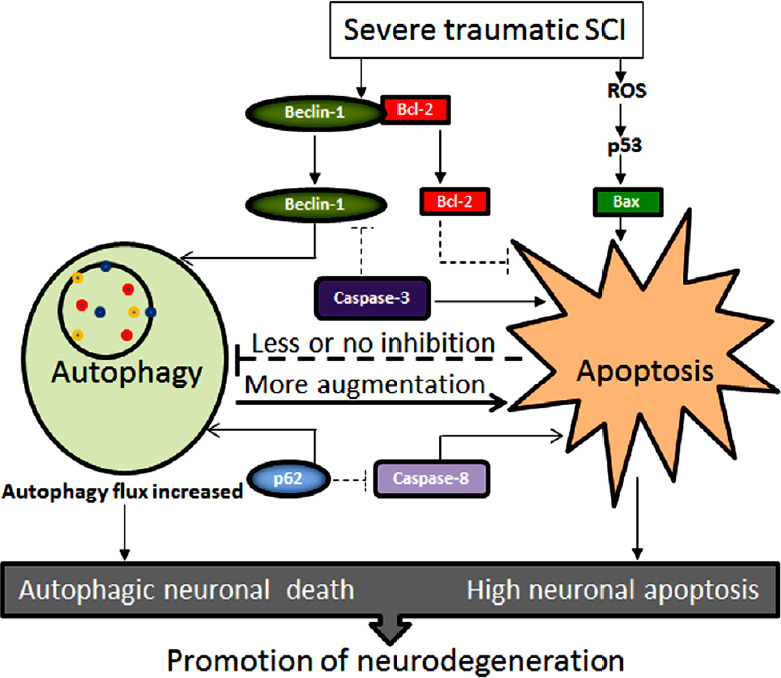
The cross-talk between autophagy and apoptosis for enhancement of neurodegeneration in severe traumatic SCI.
Severe traumatic SCI not only triggers separation of Beclin-1 and Bcl-2 but also produces excessive ROS for p53 signaling for generation of free Bax. Beclin-1 and p62 enhance autophagy flux and thus cause autophagic cell death while presence of pro-apoptotic factors (e.g., Bax, caspase-8, caspase-3) enhance induction of apoptosis. There is minimal or no mutual inhibition of two cell death mechanisms. Autophagic cell death may even augment induction of apoptosis and other cell death mechanisms for increasing neurodegeneration. Dash lines in this diagram indicate the paths of trivial operational. ROS: Reactive oxygen species; SCI: spinal cord injury.
The general concept is that activation of appropriate amount of autophagy during acute phase of secondary injury is helpful for clearing of the damaged cellular components and damaged cells to limit inflammation and toxic triggers that otherwise cause neurodegeneration in traumatic SCI. This concept begets therapeutic activation and maintenance of appropriate amount of autophagy during acute phase of secondary injury to promote neuroprotection in traumatic SCI. Too much hypoxia or nutritional stress may trigger too much autophagy or lead to activation of apoptosis and other cell death pathways in traumatic SCI. In this circumstance, therapeutic inhibition of high amount of autophagy may be warranted to block apoptosis as well.
Promotion of Autophagy for Neuroprotection and Functional Recovery in Traumatic Spinal Cord Injury
Many animal model studies suggest that promotion of autophagy is very essential for protecting the CNS cells and promoting the recovery of locomotor function in traumatic SCI (Table 1). Loss of locomotor function occurs due to demyelination of axons in acute (0–1 week), subacute (1–4 weeks), and chronic (1–6 months) traumatic SCI in adult Sprague-Dawley rats (James et al., 2011). There is an innate need in the CNS to activate enough autophagy in acute phase of secondary injury following a mild traumatic SCI to promote cell survival. But autophagy flux in acute phase of secondary injury following a severe traumatic SCI may fail due to lack of appropriate autophagy stimuli, accumulation of excessive reactive oxygen species, increased level of pro-apoptotic Bax protein, and decreased level of HSP70 (Lipinski et al., 2015). It is unfortunate that autophagy flux fades in the acute phase of secondary injury due to aggressive and progressive nature of pathogenesis in severe traumatic SCI lesion and its penumbra.
Table 1.
Induction of autophagy for neuroprotection and functional recovery in traumatic SCI
| Inducer of autophagy | Type of SCI model | Mechanisms and outcomes | Reference(s) |
|---|---|---|---|
| Rapamycin | Acute traumatic SCI in mice and rats | Inhibition of mTOR signaling promoted autophagy with upregulation of Beclin-1 and LC3B II, reduced neuronal apoptosis, and significantly increased locomotor function. | Sekiguchi et al., 2012; Wang et al., 2014 |
| Rapamycin | Acute traumatic SCI in mice | Activation of autophagy flux (degradation of p62/SQSTM1 and p70S6K), decreases in macrophage/neutrophil infiltration and microglia activation, inflammation, astrocyte proliferation, and increase in p-Akt promoted neuronal survival and axonogenesis. | Goldshmit et al., 2015 |
| VEGF165 | Acute and subacute traumatic SCI in rats | Increases in the autophagy biomarkers Beclin-1 and LC3B II and decreases in inflammatory factors (IL-1β, TNF-α, and IL-10) and loss of motoneurons promoted locomotor function. | Wang et al., 2015 |
| Retinoic acid | Subacute traumatic SCI in rats | Activation of autophagy flux with increase in LC3B II and decrease in p62, reduction in BSCB permeability and loss of tight junction molecules (P120, β-catenin, occludin, and claudin5) improved functional recovery. | Zhou et al., 2016 |
| Metformin | Acute and subacute traumatic SCI in rats | Stimulation of autophagy flux with increases in Beclin-1 and LC3B II and decreases in p62 and inhibition of apoptosis promoted functional recovery. | Zhang et al., 2017 |
| Atorvastatin | Acute, subacute, and chronic traumatic SCI in rats | Activation of autophagy (increases in Beclin-1 and LC3B II) and inhibition of apoptosis (deceases in caspase-9 and caspase-3) promoted recovery of neurological function. | Gao et al., 2016 |
| Resveratrol | Acute and subacute traumatic SCI in rats | Activation of AMPK/SIRT1 signaling pathway promoted autophagy (increases in Beclin-1 and LC3B II), inhibited apoptosis (decreases in Bax and active caspase-9 and caspase-3), and provided functional neuroprotection. | Zhao et al., 2017 |
| Curcumin | Acute and subacute traumatic SCI in rats | Increases in autophagy (inhibition of Akt/mTOR signaling pathway), spinal cord integrity, and remyelination, and prevention of apoptosis and inflammation promoted functional recovery. | Li et al., 2019a |
| FG-4592 or dimethyloxalylglycine | Acute and subacute traumatic SCI in rats | Activation of autophagy via HIF-1α/BNIP3 signaling pathway promoted neuroprotection, and axonal regeneration and improved functional recovery. | Wu et al., 2016; Li et al., 2019b |
AMPK: Adenosine 5′ monophosphate-activated protein kinase; BNIP3: Bcl-2 and adenovirus E1B 19-kDa interacting protein 3; BSCB: blood-spinal cord barrier; HIF-1α: hypoxia inducible factor-1α; IL: interleukin; LC3B II: microtubule-associated protein 1 light chain 3B; mTOR: mechanistic target of rapamycin; SCI: spinal cord injury; SIRT1: Sirtuin 1; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor.
Fortunately, there are well documented therapeutic strategies to sustain an autophagy flux in acute traumatic SCI to clear the damaged cellular components, organelles, and cells and to promote neuroprotection and functional recovery. Rapamycin, an inhibitor of the Ser/Thr kinase mTOR signaling that negatively regulates autophagy, has recently been shown to promote autophagy with upregulation of Beclin-1 and LC3B II, reduce neuronal loss due to apoptosis, and cause significant increase in locomotor function in acute traumatic SCI in rats (Wang et al., 2014) and mice (Sekiguchi et al., 2012). A single injection of rapamycin can eliminate p62/SQSTM1 indicating induction of an active autophagy flux, inhibit the mTORC1 downstream effector p70S6K, reduce macrophage/neutrophil infiltration into the lesion site, prevent microglia activation and inflammation, block astrocyte proliferation, increase level of p-Akt (active Akt), and promote neuronal survival and axonogenesis at the lesion site following traumatic SCI in mice (Goldshmit et al., 2015). VEGF promotes angiogenesis and re-vascularization in traumatic SCI. VEGF is also thought to be a potent neurotrophic factor for survival of spinal cord neurons. A recent study showed that injection of VEGF165 directly into the lesion epicenter in young male Wistar rats after traumatic SCI increased the Basso, Beattie, and Bresnahan (BBB) scores indicating improvement of locomotor function, reduced the loss of motoneurons, decreased the inflammatory factors such as interleukin-1β (IL-1β), tumor necrosis factor alpha, and IL-10, and increased the expression of the autophagy biomarkers such as Beclin-1 and LC3B II (Wang et al., 2015). The results from this study indicated that VEGF165 provided neuroprotection and functional recovery in traumatic SCI in rats though inhibition of inflammation and activation of autophagy (Wang et al., 2015). More recent studies also show that therapeutic inhibition of inflammation is associated with activation of autophagy for neuroprotection and improvement of neurological outcome in traumatic SCI (Wang et al., 2016; Yang et al., 2017; Meng et al., 2018).
Traumatic SCI disrupts the blood-spinal cord barrier, leading to infiltration of blood cells, inflammatory responses, and neuronal cell death for progression of secondary injury. An important role of retinoic acid, which is the active metabolite of vitamin A, is the generation of the blood-brain barrier during human and mouse development. A recent investigation showed that retinoic acid reduced blood-spinal cord barrier permeability, decreased the loss of tight junction molecules such as p120-catenin, β-catenin, occludin, and claudin5, improved functional recovery, and increased the expression of LC3B II and decreased the expression of p62 indicating the activation of autophagy flux after traumatic SCI in rats (Zhou et al., 2016). Metformin is a potent anti-hyperglycemic drug that causes glucose starvation due to an acute decrease in hepatic glucose production, mostly via mild and transient inhibition of the mitochondrial respiratory-chain complex 1 leading to activation of the cellular metabolic sensor adenosine 5′ monophosphate-activated protein kinase (AMPK), lowering of cAMP, and thus reducing the expression of gluconeogenic enzymes (Viollet et al., 2012; Rena et al., 2017). Metformin treatment of traumatic SCI in rats improved functional recovery, which was paralleled by a decrease in apoptosis, increases in formation of autophagosomes and expression of the autophagy biomarkers such as Beclin-1 and LC3B II, and attenuation of accumulation of the autophagy substrate protein p62 and ubiquitinated proteins, suggesting a stimulation of the autophagy flux (Zhang et al., 2017). These investigators also detected activation of AMPK and inhibition of its downstream mTOR signaling due to metformin treatment in vivo and in vitro (Zhang et al., 2017). Atorvastatin is a lipid-lowering drug that also provides neuroprotective effects, although the precise mechanisms of its action for neuroprotection remain mostly unclarified (Patel and Pisklakov, 2012). Atorvastatin activated autophagy (increased Beclin-1 and LC3B II), inhibited apoptosis (deceased caspase-9 and caspase-3 expression and TUNEL positive cells), and thereby promoted recovery of neurological function (significantly increased BBB scores) in traumatic SCI in rats (Gao et al., 2016). Resveratrol, which is a polyphenolic compound, is known to possess neuroprotective effects (Rege et al., 2014). Activation of AMPK directly phosphorylates its downstream target Sirtuin 1 (SIRT1) for inactivation of SIRT1 deacetylase activity, which essentially regulates cell energy metabolism, cell stress, cell protein homeostasis, and cell fate (Wątroba et al., 2017). A recent study determined the neuroprotective effects of resveratrol in traumatic SCI and the potential relationship among AMPK/SIRT1 signaling pathway, autophagy, and apoptosis (Zhao et al., 2017). Resveratrol treatment significantly increased BBB scores and reduced the loss of motoneurons and lesion size by promoting expression of p-AMPK, SIRT1, Beclin-1, LC3B II, and Bcl-2, while inhibiting expression of p62, Bax, and active caspase-9 and caspase-3. These results revealed that resveratrol provided functional neuroprotection in traumatic SCI in vivo via AMPK/SIRT1 signaling pathway promoting autophagy and inhibiting apoptosis (Zhao et al., 2017).
Promotion of autophagy in traumatic SCI provides an opportunity for degradation and recycling of the intracellular contents for neuronal survival in an environment of trophic factor deficiency. Therefore, pharmacological activation of autophagy in traumatic SCI continues to be a promising avenue for neuroprotection (Zhang et al., 2018; Thellung et al., 2019). A very recent study reported that treatment of traumatic SCI in rats with curcumin caused enhancement of autophagy through inhibition of the Akt/mTOR signaling pathway and promoted functional recovery due to prevention of neuronal apoptosis, improvement of spinal cord integrity, occurrence of remyelination, and suppression of inflammatory response (Li et al., 2019a). After traumatic SCI, the expression of hypoxia inducible factor-1α (HIF-1α) is known to be increased to provide a neuroprotective effect but this endogenous expression of HIF-1α is not high enough for promoting functional recovery. Two recent studies showed that sustained stabilization of expression of HIF-1α by the prolylhydroxylase inhibitor FG-4592 or dimethyloxalylglycine activated autophagy via the HIF-1α/BNIP3 signaling pathway, promoted neuroprotection, and increased axonal regeneration to improve functional recovery in traumatic SCI in animals (Wu et al., 2016; Li et al., 2019b). Another very recent investigation examined role of the tectonic family member 2 (TCTN2) long non-coding RNA (lncRNA) in apoptosis and autophagy in a rat model of traumatic SCI (Ren et al., 2019). The results from this investigation showed that TCTN2 lncRNA was decreased while microRNA-216b (miR-216b) was increased in the spinal cord tissues but overexpression of TCTN2 lncRNA blocked neuronal apoptosis by inducing autophagy through the miR-216b/Beclin-1 pathway, improving neurological function in traumatic SCI in rats.
Inhibition of Autophagy for Neuroprotection and Functional Recovery in Traumatic Spinal Cord Injury
When autophagy potentiates neurodegeneration in traumatic SCI, therapeutic inhibition of autophagy is arguably an attractive alternative treatment strategy for neuroprotection and functional recovery. There have been several studies in experimental animal models of different traumatic SCI showing that inhibition of autophagy is beneficial (Table 2) and supposed to be explored in clinical settings as well.
Table 2.
Inhibition of autophagy for neuroprotection and functional recovery in traumatic SCI
| Inhibitor of autophagy | Type of SCI model | Mechanisms and outcomes | Reference |
|---|---|---|---|
| VPA | Chronic traumatic SCI in rats | Decrease in autophagic cell death in ventral horn motoneurons and prevention of myelin sheath damage promoted recovery of motor function. | Hao et al., 2013 |
| E2 | Acute and subacute traumatic SCI in rats | Inhibition of excessive autophagy (decreases in expression of Beclin-1 and LC3B II) decreased loss of motoneurons leading to neuroprotective effects of E2 and improvement of locomotor function. | Lin et al., 2016 |
| 3-MA | Acute hemisection SCI in rats | Inhibitor of autophagy (blockage of formation of autophagosomes via inhibition of PI3K) significantly promoted survival of rubrospinal neurons at remote regions and improved spontaneous functional recovery. | Bisicchia et al., 2017 |
| Ginsenoside Rb1 | Acute and subacute traumatic SCI in rats | Inhibition of autophagy (decreases in expression of Beclin-1 and LC3B II) reduced loss of motoneurons and promoted functional recovery. | Wang et al., 2018 |
3-MA: 3-Methyladenine; E2: 17β-Estradiol; LC3B II: microtubule-associated protein 1 light chain 3B; PI3K: class III phosphatidylinositol 3-kinase; SCI: spinal cord injury; VPA: valproic acid.
It should be noted that autophagy flux is different in different traumatic SCI models. Autophagy flux is increased in hemisection SCI model (Cloud et al., 2012; Tang et al., 2014) and moderate compression SCI model (a crushing injury via compression with a vascular clip, 15 g force for 1 minute, to the exposed spinal cord) (Zhou et al., 2015). But autophagy flux is blocked in severe contusion SCI model (produced by dropping a 10 g weight from a height of 25 mm onto an impounder placed on the exposed spinal cord without disrupting the dura) (Liu et al., 2015) and severe compression SCI model (30 g force for 1 minute) (Zhang et al., 2017). Autophagy agonists (e.g., rapamycin) and antagonists (e.g., valproic acid or VPA, 17β-estradiol or E2) (Hao et al., 2013; Lin et al., 2016) have already been used to target autophagy for augmentation and attenuation, respectively, of autophagy flux in experimental animal models of traumatic SCI.
Some investigators argue that autophagy plays an important role in neurodegeneration and pathogenesis in various CNS diseases, cerebral ischemia, traumatic brain injury, and traumatic SCI as well. So, therapeutic intervention for inhibition of autophagy has been explored for prevention of neural tissue damage and improvement of neurological functions in different animal models of traumatic SCI. A study detected significantly increase in LC3B II level at 2 hours and then its decline at 1 week after contusion SCI in rats, co-localization of LC3B II positive cells with neuronal nuclei but not with glial fibrillary acidic protein indicating existence of autophagic cell death mostly in neurons soon after contusion SCI, and capability of methylprednisolone (a synthetic glucocorticoid hormone) in decreasing LC3B II expression at 2 hours after contusion SCI (Chen et al., 2012). Thus, this study suggested that autophagic cell death might play a significant role in neuronal death after traumatic SCI. A subsequent study also speculated that autophagy could result in cell death and play a key role in the progression of pathogenesis in contusion SCI (Hao et al., 2013). This study reported that both the mRNA and protein levels of Beclin-1 and LC3B II were significantly increased at 1, 2, and 6 hours after contusion SCI and peaked at 2 hours. VPA (a histone deacetylase inhibitor), a known neuroprotective agent in certain experimental animal models, markedly decreased the biomarkers of autophagy at 2 hours post-injury in the animals (Hao et al., 2013). Besides, post-injury treatment with VPA improved the BBB scores, increased the number of ventral horn motoneurons, and reduced myelin sheath damage at 42 days after the traumatic SCI. Together, the results from this study demonstrated that autophagic cell death occurred in motoneurons following traumatic SCI; and VPA reduced autophagic cell death and promoted recovery of motor function (Hao et al., 2013). A more recent investigation employed E2, also a well-known neuroprotective agent in different CNS diseases, for treatment showing improvement in locomotor function and decrease in loss of motoneurons due to decreases in expression of Beclin-1 and LC3B II leading to the conclusion that neuroprotective effects of E2 in traumatic SCI were partly related to inhibition of excessive autophagy (Lin et al., 2016).
Neuronal death at the primary injury site and at the remote regions, which are functionally connected to the primary injury site, is a major contributor to neurological deficits following traumatic SCI. A recent study examined the function and effects of modulation of autophagy on the fate of axotomized rubrospinal neurons in a rat model of hemisection SCI at the cervical level (Bisicchia et al., 2017). Induction of hemisection SCI caused an accumulation of LC3B II positive autophagosomes (APs) in the axotomized neurons at 1 and 5 days after injury and this accumulation was not due to an increase in initiation of autophagy but due to decrease in clearance of APs, as demonstrated by an accrual of p62; and disruption of autophagy flux strongly correlated with neuronal death at remote regions and worse functional recovery. 3-Methyladenine is an inhibitor of autophagy as it blocks formation of APs via inhibition of the class III PI3K. Inhibition of formation of APs by 3-methyladenine significantly attenuated neurodegeneration at remote regions and improved spontaneous functional recovery, indicating the detrimental effects of autophagy in causing remote damage after hemisection SCI (Bisicchia et al., 2017). A more recent study used Ginsenoside Rb1, one of the major active components from Panax Ginseng CA Meyer, as an inhibitor of autophagy for treatment of traumatic SCI in rats and showed that Ginsenoside Rb1 inhibited autophagy and reduced loss of motoneurons and thereby helped functional recovery in traumatic SCI in animals (Wang et al., 2018).
Conclusion and Future Direction
Roles of autophagy in traumatic SCI remain controversial and further studies are warranted to define consequences of activation of autophagy in spatial and temporal terms. Both school of thoughts, activation of autophagy and inhibition of autophagy, are gathering supports from studies in animal models of traumatic SCI. So far, there have been more reports of activation of autophagy than inhibition of autophagy for functional neuroprotection in preclinical animal models of traumatic SCI. All in all, it appears that successful therapeutic strategy for activation or inhibition of autophagy in experimental animal models of different traumatic SCI may depend on the type of primary injury, location of primary injury, and acute or choric phase of secondary injury. Further studies need to be conducted to confirm these proclamations.
Footnotes
Conflicts of interest: The author declares no conflicts of interest.
Financial support: This work was supported in part by the Investigator Initiated Research grant (SCIRF-2015-I-0) from the South Carolina Spinal Cord Injury Research Fund (SCIRF, Columbia, SC, US), an incentive award from the Soy Health Research Program (SHRP, United Soybean Board, Chesterfield, MO, US), and also the R01 grants (CA91460 and NS057811) from the National Institutes of Health (Bethesda, MD, US).
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: N. Scott Litofsky, University of Missouri School of Medicine, USA.
Funding: This work was supported in part by the Investigator Initiated Research grant (SCIRF-2015-I-0) from the South Carolina Spinal Cord Injury Research Fund (SCIRF, Columbia, SC, US), an incentive award from the Soy Health Research Program (SHRP, United Soybean Board, Chesterfield, MO, US), and also the R01 grants (CA91460 and NS057811) from the National Institutes of Health (Bethesda, MD, US).
P-Reviewer: Litofsky NS; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Ahuja CS, Wilson JR, Nori S, Kotter MRN, Druschel C, Curt A, Fehlings MG. Traumatic spinal cord injury. Nat Rev Dis Primers. 2017;3:17018. doi: 10.1038/nrdp.2017.18. [DOI] [PubMed] [Google Scholar]
- 2.Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- 3.Barthet VJA, Ryan KM. Autophagy in neurodegeneration: can’t digest it, spit it out! Trends Cell Biol. 2018;28:171–173. doi: 10.1016/j.tcb.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Bisicchia E, Latini L, Cavallucci V, Sasso V, Nicolin V, Molinari M, D’Amelio M, Viscomi MT. Autophagy inhibition favors survival of rubrospinal neurons after spinal cord hemisection. Mol Neurobiol. 2017;54:4896–4907. doi: 10.1007/s12035-016-0031-z. [DOI] [PubMed] [Google Scholar]
- 5.Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26:549–555. doi: 10.1016/j.cellsig.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bové J, Martínez-Vicente M, Dehay B, Perier C, Recasens A, Bombrun A, Antonsson B, Vila M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy. 2014;10:889–900. doi: 10.4161/auto.28286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen HC, Fong TH, Lee AW, Chiu WT. Autophagy is activated in injured neurons and inhibited by methylprednisolone after experimental spinal cord injury. Spine (Phila Pa 1976) 2012;37:470–475. doi: 10.1097/BRS.0b013e318221e859. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Fu Q, Shen B, Huang X, Wang K, He P, Li F, Zhang F, Shen H. Enhanced p62 expression triggers concomitant autophagy and apoptosis in a rat chronic spinal cord compression model. Mol Med Rep. 2014;9:2091–2096. doi: 10.3892/mmr.2014.2124. [DOI] [PubMed] [Google Scholar]
- 9.Cloud BA, Ball BG, Chen BK, Knight AM, Hakim JS, Ortiz AM, Windebank AJ. Hemisection spinal cord injury in rat: the value of intraoperative somatosensory evoked potential monitoring. J Neurosci Methods. 2012;211:179–184. doi: 10.1016/j.jneumeth.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper KF. Till death do us part: The marriage of autophagy and apoptosis. Oxid Med Cell Longev. 2018;2018:4701275. doi: 10.1155/2018/4701275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018;37:749–766. doi: 10.1007/s10555-018-9727-z. [DOI] [PubMed] [Google Scholar]
- 12.Dombi E, Mortiboys H, Poulton J. Modulating mitophagy in mitochondrial disease. Curr Med Chem. 2018;25:5597–5612. doi: 10.2174/0929867324666170616101741. [DOI] [PubMed] [Google Scholar]
- 13.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 14.Fan L, Yin S, Zhang E, Hu H. Role of p62 in the regulation of cell death induction. Apoptosis. 2018;23:187–193. doi: 10.1007/s10495-018-1445-z. [DOI] [PubMed] [Google Scholar]
- 15.Farhadi HF, Kukreja S, Minnema A, Vatti L, Gopinath M, Prevedello L, Chen C, Xiang H, Schwab JM. Impact of admission imaging findings on neurological outcomes in acute cervical traumatic spinal cord injury. J Neurotrauma. 2018;35:1398–1406. doi: 10.1089/neu.2017.5510. [DOI] [PubMed] [Google Scholar]
- 16.Fimia GM, Piacentini M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci. 2010;67:1581–1588. doi: 10.1007/s00018-010-0284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao S, Zhang ZM, Shen ZL, Gao K, Chang L, Guo Y, Li Z, Wang W, Wang AM. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen Res. 2016;11:977–982. doi: 10.4103/1673-5374.184498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge L, Arul K, Ikpeze T, Baldwin A, Nickels JL, Mesfin A. Traumatic and nontraumatic spinal cord injuries. World Neurosurg. 2018;111:e142–148. doi: 10.1016/j.wneu.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Gedde MH, Lilleberg HS, Aßmus J, Gilhus NE, Rekand T. Traumatic vs. non-traumatic spinal cord injury: A comparison of primary rehabilitation outcomes and complications during hospitalization. J Spinal Cord Med. 2019;3:1–7. doi: 10.1080/10790268.2019.1598698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Łos MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldshmit Y, Kanner S, Zacs M, Frisca F, Pinto AR, Currie PD, Pinkas-Kramarski R. Rapamycin increases neuronal survival, reduces inflammation and astrocyte proliferation after spinal cord injury. Mol Cell Neurosci. 2015;68:82–91. doi: 10.1016/j.mcn.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead. Protein Cell. 2012;3:17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grassner L, Marschallinger J, Dünser MW, Novak HF, Zerbs A, Aigner L, Trinka E, Sellner J. Nontraumatic spinal cord injury at the neurological intensive care unit: spectrum, causes of admission and predictors of mortality. Ther Adv Neurol Disord. 2016;9:85–94. doi: 10.1177/1756285615621687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection. Trends Cell Biol. 2011;21:387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018;28:3–13. doi: 10.1111/bpa.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hao HH, Wang L, Guo ZJ, Bai L, Zhang RP, Shuang WB, Jia YJ, Wang J, Li XY, Liu Q. Valproic acid reduces autophagy and promotes functional recovery after spinal cord injury in rats. Neurosci Bull. 2013;29:484–492. doi: 10.1007/s12264-013-1355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harnett MM, Pineda MA, Latré de Laté P, Eason RJ, Besteiro S, Harnett W, Langsley G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed J. 2017;40:9–22. doi: 10.1016/j.bj.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James ND, Bartus K, Grist J, Bennett DL, McMahon SB, Bradbury EJ. Conduction failure following spinal cord injury: functional and anatomical changes from acute to chronic stages. J Neurosci. 2011;31:18543–18555. doi: 10.1523/JNEUROSCI.4306-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanno H, Ozawa H, Sekiguchi A, Itoi E. Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol Dis. 2009a;33:143–148. doi: 10.1016/j.nbd.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Kanno H, Ozawa H, Sekiguchi A, Itoi E. The role of autophagy in spinal cord injury. Autophagy. 2009b;5:390–392. doi: 10.4161/auto.5.3.7724. [DOI] [PubMed] [Google Scholar]
- 36.Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine (Phila Pa 1976) 2011;36:E1427–E1434. doi: 10.1097/BRS.0b013e3182028c3a. [DOI] [PubMed] [Google Scholar]
- 37.Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Tateda S, Yahata K, Itoi E. The role of mTOR signaling pathway in spinal cord injury. Cell Cycle. 2012;11:3175–3179. doi: 10.4161/cc.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 39.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 40.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 41.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 42.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH, Watkins SC, Clark RS. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- 44.Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
- 45.Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell. 2009;20:4871–4884. doi: 10.1091/mbc.E09-07-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine B, Sinha SC, Kroemer G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levin-Salomon V, Bialik S, Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014;19:346–356. doi: 10.1007/s10495-013-0918-3. [DOI] [PubMed] [Google Scholar]
- 48.Li W, Yao S, Li H, Meng Z, Sun X. Curcumin promotes functional recovery and inhibits neuronal apoptosis after spinal cord injury through the modulation of autophagy. J Spinal Cord Med. 2019a;4:1–9. doi: 10.1080/10790268.2019.1616147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Han W, Wu Y, Zhou K, Zheng Z, Wang H, Xie L, Li R, Xu K, Liu Y, Wang X, Xiao J. Stabilization of hypoxia inducible factor-1α by dimethyloxalylglycine promotes recovery from acute spinal cord injury by inhibiting neural apoptosis and enhancing axon regeneration. J Neurotrauma. 2019b;36:3394–3409. doi: 10.1089/neu.2018.6364. [DOI] [PubMed] [Google Scholar]
- 50.Lin CW, Chen B, Huang KL, Dai YS, Teng HL. Inhibition of autophagy by estradiol promotes locomotor recovery after spinal cord injury in rats. Neurosci Bull. 2016;32:137–144. doi: 10.1007/s12264-016-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipinski MM, Wu J, Faden AI, Sarkar C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal. 2015;23:565–577. doi: 10.1089/ars.2015.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski MM, Wu J. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6:e1582. doi: 10.1038/cddis.2014.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. 2013;26:9–18. doi: 10.1016/j.devcel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meng HY, Shao DC, Li H, Huang XD, Yang G, Xu B, Niu HY. Resveratrol improves neurological outcome and neuroinflammation following spinal cord injury through enhancing autophagy involving the AMPK/mTOR pathway. Mol Med Rep. 2018;18:2237–2244. doi: 10.3892/mmr.2018.9194. [DOI] [PubMed] [Google Scholar]
- 58.Mills PB, Vakil AP, Phillips C, Kei L, Kwon BK. Intra-rater and inter-rater reliability of the Penn Spasm Frequency Scale in people with chronic traumatic spinal cord injury. Spinal Cord. 2018;56:569–574. doi: 10.1038/s41393-018-0063-5. [DOI] [PubMed] [Google Scholar]
- 59.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nam T, Han JH, Devkota S, Lee HW. emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol Cells. 2017;40:897–905. doi: 10.14348/molcells.2017.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nas K, Yazmalar L, Şah V, Aydın A, Öneş K. Rehabilitation of spinal cord injuries. World J Orthop. 2015;6:8–16. doi: 10.5312/wjo.v6.i1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691–699. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012;4:a008839. doi: 10.1101/cshperspect.a008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 67.Oyinbo CA. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars) 2011;71:281–299. doi: 10.55782/ane-2011-1848. [DOI] [PubMed] [Google Scholar]
- 68.Patel A, Pisklakov SV. Statins as potentially neuroprotective agents: A Review. J Anesth Clin Res. 2012;3:251. [Google Scholar]
- 69.Radad K, Moldzio R, Al-Shraim M, Kranner B, Krewenka C, Rausch WD. Recent advances in autophagy-based neuroprotection. Expert Rev Neurother. 2015;15:195–205. doi: 10.1586/14737175.2015.1002087. [DOI] [PubMed] [Google Scholar]
- 70.Ray SK, Hogan EL, Banik NL. Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Rev. 2003;42:169–185. doi: 10.1016/s0165-0173(03)00152-8. [DOI] [PubMed] [Google Scholar]
- 71.Ray SK, Samantaray S, Smith JA, Matzelle DD, Das A, Banik NL. Inhibition of cysteine proteases in acute and chronic spinal cord injury. Neurotherapeutics. 2011;8:180–186. doi: 10.1007/s13311-011-0037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rege SD, Geetha T, Griffin GD, Broderick TL, Babu JR. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front Aging Neurosci. 2014;6:218. doi: 10.3389/fnagi.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ren XD, Wan CX, Niu YL. Overexpression of lncRNA TCTN2 protects neurons from apoptosis by enhancing cell autophagy in spinal cord injury. FEBS Open Bio. 2019;9:1223–1231. doi: 10.1002/2211-5463.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabre L, Rekand T, Asser T, Kõrv J. Mortality and causes of death after traumatic spinal cord injury in Estonia. J Spinal Cord Med. 2013;36:687–694. doi: 10.1179/2045772313Y.0000000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakai K, Fukuda T, Iwadate K. Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: association with time between the injury and death. Am J Forensic Med Pathol. 2014;35:38–44. doi: 10.1097/PAF.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 77.Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O’Kane CJ, Rubinsztein DC. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Savic G, DeVivo MJ, Frankel HL, Jamous MA, Soni BM, Charlifue S. Long-term survival after traumatic spinal cord injury: a 70-year British study. Spinal Cord. 2017;55:651–658. doi: 10.1038/sc.2017.23. [DOI] [PubMed] [Google Scholar]
- 79.Sekiguchi A, Kanno H, Ozawa H, Yamaya S, Itoi E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J Neurotrauma. 2012;29:946–956. doi: 10.1089/neu.2011.1919. [DOI] [PubMed] [Google Scholar]
- 80.Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH. Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development. 2013;140:1321–1329. doi: 10.1242/dev.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 27 Suppl. 2008;1:S137–S148. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang P, Hou H, Zhang L, Lan X, Mao Z, Liu D, He C, Du H, Zhang L. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Mol Neurobiol. 2014;49:276–287. doi: 10.1007/s12035-013-8518-3. [DOI] [PubMed] [Google Scholar]
- 84.Tanida I, Ueno T, Kominami E. In vitro assays of lipidation of mammalian Atg8 homologs. Curr Protoc Cell Biol. 2014;64:11. doi: 10.1002/0471143030.cb1120s64. [DOI] [PubMed] [Google Scholar]
- 85.Thellung S, Corsaro A, Nizzari M, Barbieri F, Florio T. Autophagy activator drugs: a new opportunity in neuroprotection from misfolded protein toxicity. Int J Mol Sci. 2019;20:E901. doi: 10.3390/ijms20040901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13:1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 88.Verma R, Virdi JK, Singh N, Jaggi AS. Animals models of spinal cord contusion injury. Korean J Pain. 2019;32:12–21. doi: 10.3344/kjp.2019.32.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang C, Liu C, Gao K, Zhao H, Zhou Z, Shen Z, Guo Y, Li Z, Yao T, Mei X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem Biophys Res Commun. 2016;477:534–540. doi: 10.1016/j.bbrc.2016.05.148. [DOI] [PubMed] [Google Scholar]
- 91.Wang H, Wang Y, Li D, Liu Z, Zhao Z, Han D, Yuan Y, Bi J, Mei X. VEGF inhibits the inflammation in spinal cord injury through activation of autophagy. Biochem Biophys Res Commun. 2015;464:453–458. doi: 10.1016/j.bbrc.2015.06.146. [DOI] [PubMed] [Google Scholar]
- 92.Wang P, Lin C, Wu S, Huang K, Wang Y, Bao X, Zhang F, Huang Z, Teng H. Inhibition of autophagy is involved in the protective effects of Ginsenoside Rb1 on spinal cord injury. Cell Mol Neurobiol. 2018;38:679–690. doi: 10.1007/s10571-017-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang ZY, Liu WG, Muharram A, Wu ZY, Lin JH. Neuroprotective effects of autophagy induced by rapamycin in rat acute spinal cord injury model. Neuroimmunomodulation. 2014;21:257–267. doi: 10.1159/000357382. [DOI] [PubMed] [Google Scholar]
- 94.Wątroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res Rev. 2017;40:11–19. doi: 10.1016/j.arr.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 95.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wong YC, Holzbaur EL. Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci. 2015;128:1259–1267. doi: 10.1242/jcs.161216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, Wu Q, Zhang J, Hong Y. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci. 2014;10:1072–1083. doi: 10.7150/ijbs.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu K, Zhou K, Wang Y, Zhou Y, Tian N, Wu Y, Chen D, Zhang D, Wang X, Xu H, Zhang X. Stabilization of HIF-1α by FG-4592 promotes functional recovery and neural protection in experimental spinal cord injury. Brain Res. 2016;1632:19–26. doi: 10.1016/j.brainres.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 99.Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 100.Yang Y, Guo C, Liao B, Cao J, Liang C, He X. BAMBI inhibits inflammation through the activation of autophagy in experimental spinal cord injury. Int J Mol Med. 2017;39:423–429. doi: 10.3892/ijmm.2016.2838. [DOI] [PubMed] [Google Scholar]
- 101.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu D, Li M, Ni B, Kong J, Zhang Z. Induction of neuronal mitophagy in acute spinal cord injury in rats. Neurotox Res. 2013;24:512–522. doi: 10.1007/s12640-013-9397-0. [DOI] [PubMed] [Google Scholar]
- 104.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang D, Wang F, Zhai X, Li XH, He XJ. Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy. Neural Regen Res. 2018;13:2191–2199. doi: 10.4103/1673-5374.241473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang D, Xuan J, Zheng BB, Zhou YL, Lin Y, Wu YS, Zhou YF, Huang YX, Wang Q, Shen LY, Mao C, Wu Y, Wang XY, Tian NF, Xu HZ, Zhang XL. Metformin improves functional recovery after spinal cord injury via autophagy flux stimulation. Mol Neurobiol. 2017;54:3327–3341. doi: 10.1007/s12035-016-9895-1. [DOI] [PubMed] [Google Scholar]
- 108.Zhang XJ, Chen S, Huang KX, Le WD. Why should autophagic flux be assessed. Acta Pharmacol Sin. 2013;34:595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao H, Chen S, Gao K, Zhou Z, Wang C, Shen Z, Guo Y, Li Z, Wan Z, Liu C, Mei X. Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience. 2017;348:241–251. doi: 10.1016/j.neuroscience.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 110.Zhou K, Sansur CA, Xu H, Jia X. The temporal pattern, flux, and function of autophagy in spinal cord injury. Int J Mol Sci. 2017;18:466. doi: 10.3390/ijms18020466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou KL, Zhou YF, Wu K, Tian NF, Wu YS, Wang YL, Chen DH, Zhou B, Wang XY, Xu HZ, Zhang XL. Stimulation of autophagy promotes functional recovery in diabetic rats with spinal cord injury. Sci Rep. 2015;5:17130. doi: 10.1038/srep17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou Y, Zheng B, Ye L, Zhang H, Zhu S, Zheng X, Xia Q, He Z, Wang Q, Xiao J, Xu H. Retinoic acid prevents disruption of blood-spinal cord barrier by inducing autophagic flux after spinal cord injury. Neurochem Res. 2016;41:813–825. doi: 10.1007/s11064-015-1756-1. [DOI] [PubMed] [Google Scholar]
- 113.Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang X, Jin H, Xu H, Chen Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 2010;1:468–477. doi: 10.1007/s13238-010-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]