

Abstract
To compare clinical characteristics and identify long-term outcomes of Chinese patients with systemic sclerosis (SSc) with and without muscle involvement.
We retrospectively investigated the medical records, laboratory results, and computed tomography images of 204 consecutive SSc patients. Kaplan–Meier analysis was performed to determine survival rates. Patients were allocated into groups with and without myopathy.
The prevalence of myopathy was 21.6%. The myopathy group was more likely to develop diffuse cutaneous involvement (90.9% vs 56%, P = .006), interstitial lung disease (90% vs 56%, P < .001), digestive system involvement (56.7% vs 29.3%, P = .001), pulmonary hypertension (29.5% vs 10.5%, P = .004), and pericardial effusion (25% vs. 10%, P = .019). Patients with myopathy had lower single-breath diffusing capacity of the lung for carbon oxide (46.5 ± 11.1 vs 57.1 ± 13.4, P < .001).Further, the myopathy group has similar results in interstitial lung disease associated higher resolution computed tomography score (186.8 ± 64.5 vs 152.3 ± 45.5, P = .037), Valentini score for disease activity (3.4 ± 0.9 vs 2.0 ± 0.9, P < .001) and modified Rodnan total skin score (19.4 ± 6.1 vs 15.1 ± 7.7, P = .002), compared with non-myopathy group. Kaplan–Meier survival analysis revealed decreased overall survival rate of the myopathy group (P = .028).
SSc Patients with myopathy had more severe clinical manifestations and higher disease activity compared with those without, which affected survival rates and indicated worse prognosis.
Keywords: myopathy, prognosis factors, systemic sclerosis
1. Introduction
Systemic sclerosis (SSc) is a disease of connective tissue with unknown pathogenesis, characterized by fibrosis and microvascular injury to the skin and internal organs.[1] SSc patients are classified into subtypes according to the classification system proposed by LeRoy and Medsger[2] as follows: limited cutaneous SSc (lcSSc), diffuse cutaneous SSc (dcSSc) and sine scleroderma. Most SSc patients have multiple systems involvement, including skin, joints, respiratory system, digestive system, and kidneys, which leads to low quality of life, disability, and possibly death.[3] The presence of antinuclear antibodies occurs with a prevalence ranging from 80% to 98%,[4] and Raynaud phenomenon (RP) occurs in approximately 90% to 95% of SSc patients.[5] SSc presents a large mortality than general population. Cumulative survival from diagnosis has been estimated at 74.9% at 5 years and 62.5% at 10 years. Pulmonary involvement represented the main cause of death.[6]
Although myopathy is not a rare symptom associated with SSc, it has not attracted sufficient attention. Approximately 1-third of SSc patients complain of muscle weakness,[7] among which 15% have objective muscle atrophy and 10% have elevated serum creatine kinase (CK).[8] The frequency of myopathy in SSc patients ranges from approximately 10% to 15% when systematically assessed, and muscle weakness occurs in as many as 90% of these patients.[8]
The clinical manifestation of skeletal muscle involvement, muscle magnetic-resonance imaging (MRI), and electromyography (EMG) of SSc patients are similar to those with polymyositis or dermatomyositis. For example, the Canadian Scleroderma Research Group found that myopathy was a poor prognostic feature of SSc patients, which affects survival rate, particularly in men with early dcSSc with positive anti-topoisomerase1 and anti-ribonucleoprotein autoantibodies, and interstitial lung disease (ILD).[9] The pathology and mechanisms of SSc-associated myopathy are complex and insufficiently characterized, in contrast to idiopathic inflammatory myopathy.[10] There is not a uniformly accepted case definition for myopathy in SSc or research on SSc-associated myopathy among patients in China.[11] Further, SSc associated myopathy is always neglected in clinical settings. The aim of the study was to delineate the clinical features and survival rates of 204 SSc patients with myopathy who were treated at our institution.
2. Materials and methods
2.1. Patients
We studied SSc patients referred to the Rheumatology Department of our hospital from December 2012 to December 2016. The diagnosis of SSc was made according to the 1980 or 2013 SSc diagnostic criteria proposed by the European League Against Rheumatism/American College of Rheumatology.[12] The Ethics Committee of our hospital approved the study.
2.2. Data collection
Clinical data and laboratory results, which were retrospectively obtained from patients’ medical records, included age, sex, autoantibody titers, SSc subsets, symptoms and signs, organ involvement as well as the modified Rodnan Total Skin Score (mRTSS). Survival status was confirmed by telephonic or hospital records.
2.3. SSc subsets
DcSSc was defined as rapid skin involvement of the trunk, face, proximal and distal extremities frequently associated with anti-topoisomerase antibodies.[13,14] Patients with dcSSc typically exhibited a progressive course of disease with an early onset of RP, often within 1 year after the onset of skin changes.
LcSSc was defined as thickening of the skin of the extremities distal to the knees and elbow joints, facial skin, and the occurrence of RP that typically appeared many years before skin involvement. These patients often produce anti-centromere antibodies.[13,14]
2.4. Definitions
SSc patients at diagnosis had symptoms of muscle involvement such as fatigue, muscle weakness, muscle pain, and at least 1 of the conditions as follows: serum CK over the normal value (145 U/L); inflammatory changes revealed by muscle biopsy, including necrosis, acute neurogenic atrophy, and fibrosis[15,16]; EMG showed low voltage, short duration potential during maximal contraction, fibrillation, or sharp waves[17]; muscle MRI[18] showed edema, atrophy, and hyperemia were defined as myopathy. Patients with polymyositis, dermatomyositis, rhabdomyolysis, drug-induced myopathy, necrotizing myopathy, infectious myopathy, toxic myopathy, or metabolic myopathy were excluded, because these diseases may increase serum CK. SSc patients were grouped according to the presence of myopathy.
2.5. Organ involvement
Patients underwent an extensive clinical evaluation that included demographics, disease duration (since the first non-RP symptom), disease subtypes, disease activity, clinical manifestations, and assessment of internal organ involvement.
Pulmonary hypertension (PH) was defined as the estimated systolic pulmonary artery pressure (≥45 mm Hg) according to electrocardiographic measurements of Doppler flow of the tricuspid regurgitant jet.[19] Pericardial effusion was also defined as electrocardiographic measurement.
ILD was diagnosed according to high-resolution computed tomography (HRCT) and pulmonary function tests.[20] The HRCT findings were scored on a scale of 1 to 6, according to the classification method proposed by Kazuya et al,[21,22] with the variables as follows:
-
(1)
normal attenuation,
-
(2)
ground glass attenuation without traction bronchiectasis or bronchiolectasis,
-
(3)
consolidation without traction bronchiectasis or bronchiolectasis,
-
(4)
ground glass attenuation with traction bronchiectasis or bronchiolectasis,
-
(5)
consolidation with traction bronchiectasis or bronchiolectasis,
-
(6)
honeycombing. An overall CT score was obtained by adding the 6 averaged scores (3 zones in each lung) assigned by a radiologist.
Gastrointestinal involvement was defined as gastrointestinal motility disturbance, dysphagia, nausea, malabsorption, oesophageal stenosis, gastro-oesophageal reflux, or intestinal pseudo-obstruction.[23]
Kidney involvement was defined as renal insufficiency caused by acute renal crisis (creatinine clearance age-related <80 mL/min).[24]
Disease activity was evaluated according to the Valentini score criteria for SSc.[25] Skin involvement was evaluated using the mRTSS, scale of 0 to 3, which assesses skin hardening and thickness determined by manual palpation of 17 body areas.[26]
2.6. Mortality
The in-hospital mortality was recorded with details of causes and dates of incidence. Details of home deaths were obtained via telephone interviews with each participant's relatives.
3. Statistical analysis
Descriptive statistics were used to summarize the baseline demographic and clinical characteristics of SSc patients. Statistical analysis was performed using SPSS, version 19.0, statistics for Windows. Survival analysis was performed using GraphPad Prism version 5.1. Continuous variables are presented as the mean ± standard deviation, and discrete variables are reported as the median (min-max). Continuous variables conforming to a Gaussian distribution were evaluated using a t test or the Wilcoxon rank-sum test. Discrete variables were evaluated using the chi-square test. Kaplan–Meier survival analysis was used to determine the survival rates of SSc patients with or without myopathy, and the P value was calculated using the log-rank test. The P values are 2-sided, α = 0.05.
4. Results
4.1. Patients’ characteristics
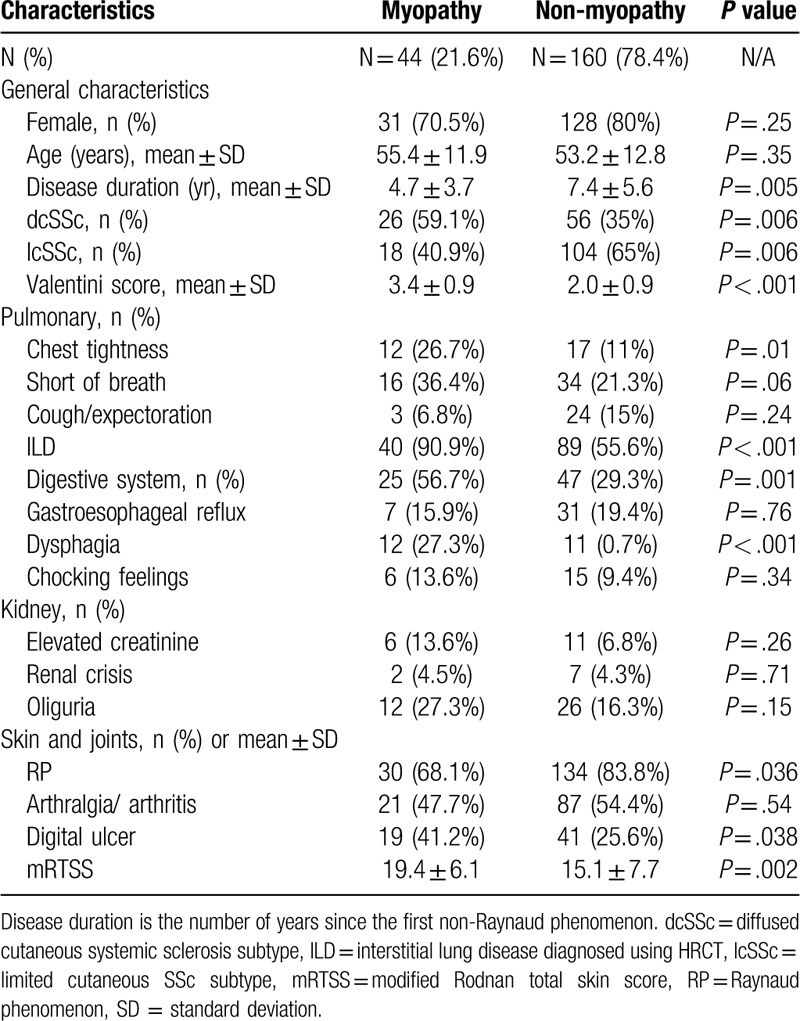
General characteristics and clinical features in SSc patients with and without myopathy were summarized in Table 1. In the 204 SSc patients, the mean age was 52.8 ± 12.9 years old with 3 (0.2-30) -year disease duration (since the first non-RP). There were 159 women (77.9%) and 45 men (22.1%), with a female/male ratio of 3.5:1. Of the 204 SSc patients, 44 (21.6%) developed myopathy. The 44 patients all had elevated CK, and 12 of them were diagnosed by EMG, 4 by MRI, 4 by biopsy, 24 by symptoms.
Table 1.
Comparison of patients’ clinical characteristics.
4.2. Comparisons of clinical characteristics
Patients with dcSSc and shorter disease duration were more likely to develop myopathy compared with those with lcSSc. The frequencies of ILD (90.0% vs 55.6%, P < .001), digestive system involvement (56.7% vs 29.3%, P = .001), and digital ulcers (41.2 vs 25.6%, P = .038) were significantly higher in patients with myopathy compared with those without. Further, the myopathy group had a higher mRTSS and higher disease activity-related Valetini score (19.4 ± 6.1 vs 15.1 ± 7.7, P = .002 and 3.4 ± 0.9 vs 2.0 ± 0.9, P < .001, respectively). However, there were no significant differences between groups in the frequencies of arthritis, or kidney involvement.
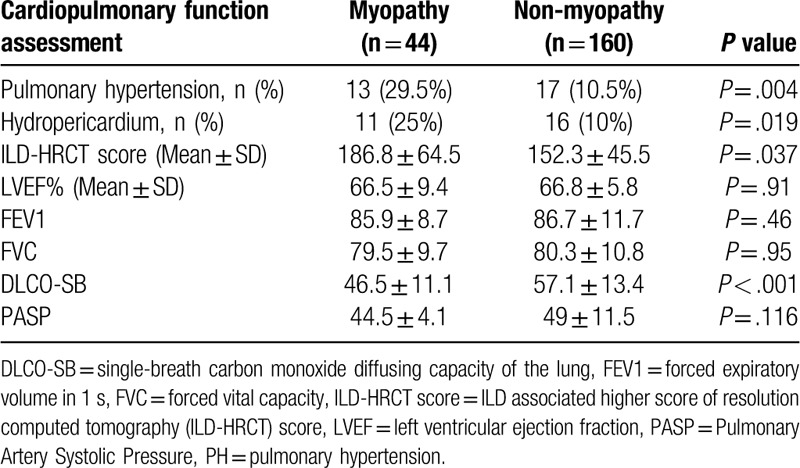
The variables of cardiopulmonary function associated with visceral organ involvement were assessed and are summarized in Table 2. The myopathy group had a higher incidence of PH, pericardial effusion, higher ILD-HRCT scores, and lower single breath diffusing lung capacity for carbon monoxide. However, there were no differences between left ventricular ejection fraction%, FEV1, or FVC between groups.
Table 2.
Assessment of cardiopulmonary function.
4.3. Laboratory results
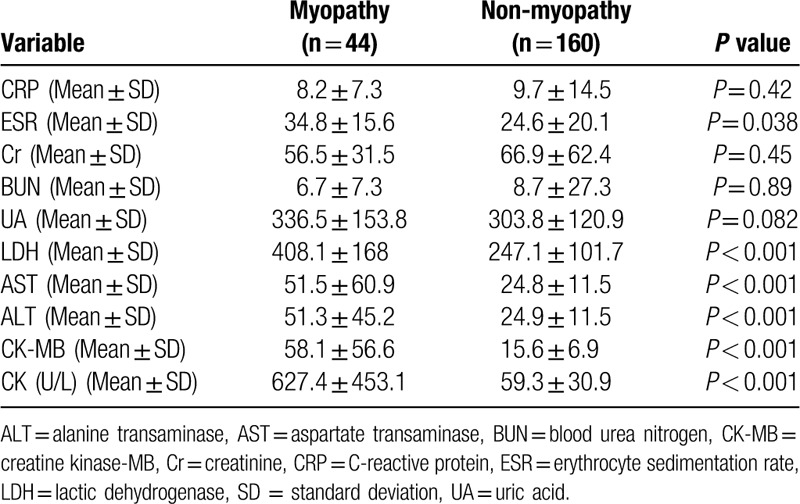
Table 3 shows the laboratory test data of the 2 subgroups. The CK values in the 2 subsets were 627.4 ± 453.1 U/L and 59.3 ± 30.9 U/L, respectively. The erythrocyte sedimentation rate, lactic dehydrogenase, alanine transaminas, and aspartate transaminase were higher in the myopathy group (Table 3), and there was no significant difference in the titers of autoantibodies.
Table 3.
Laboratory data.
4.4. Survival analysis
The follow-up duration ranged from 1 to 48 months. The mean follow-up time was 17.5 months and 13 patients died in total. 4 patients died of PH, 5 patients died of malignant tumor, 2 patients died of heart failure, 1 patient died of ILD, and 1 patient died of unknow cause. In myopathy group, 3 died of PH, 1 died of ILD, and 1 died of tumor. In non-myopathy group, 1 died of PH, 4 died of tumor, 2 died of heart failure, and 1 died of unknow cause.
We analyzed the survival rates at baseline. Disease duration was recorded as the number of years since the first non-RP. Kaplan–Meier analysis revealed that compared with patients without myopathy, those with myopathy had lower 3-year (97.7% vs 98.8%), 5-year (95.1% vs 97.8%), and 10-year (81.4% vs 94.1% survival rates; P = 0.028), calculated using the log-rank test (Fig. 1).
Figure 1.
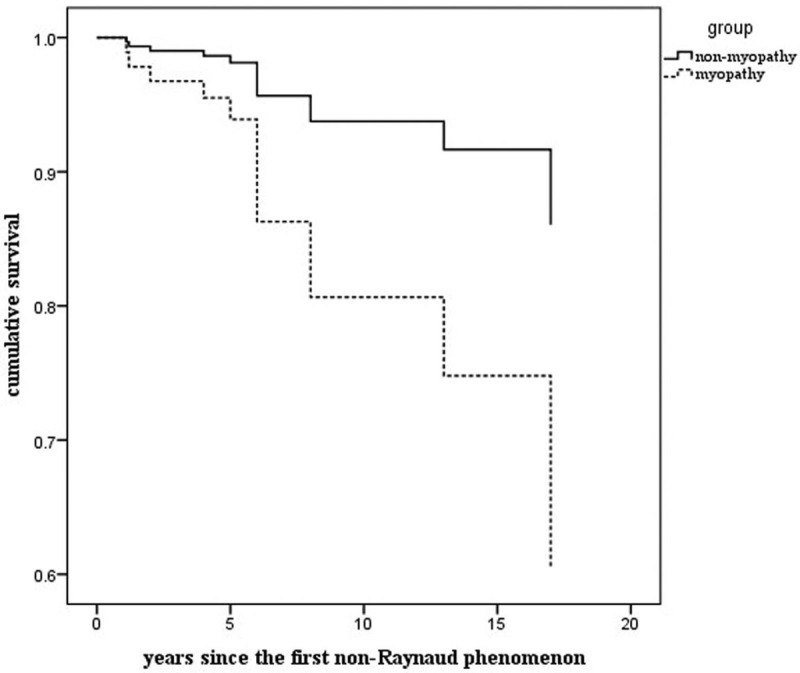
Survival curves of patients with systemic sclerosis with and without myopathy: Disease duration is the number of years since the first non-Raynaud phenomenon. SSc patients with myopathy had lower survival rates. Kaplan–Meier analysis revealed that the 3-year, 5-year, and 10-year survival rates for SSc patients with myopathy were 97.7%, 95.1%, and 81.4% respectively, compared with those of patients without myopathy, which were 98.8%, 97.8%, and 94.1%, respectively, (P = .028)(the black arrow).
5. Discussion
The present study was specifically designed to analyze the clinical features and survival rates of SSc patients with and without myopathy. We found that myopathy was not rare in SSc patients (prevalence = 21.6%), using the definition of myopathy described above, which was higher compared with those of other studies (10%–17%).[10]
SSc patients with myopathy have more severe skin involvement such as sclerosis and pigmentation changes.[27] Kahan et al reported that 302 Japanese patients with dcSSc experienced myopathy more frequently,[17] and the most common clinical manifestations included muscle atrophy, muscle weakness, and muscle pain, which typically involved the proximal limb muscles such as those of the shoulder muscles, gluteus, and tendon sheath.[28] Ranque et al conducted a descriptive and prognostic study of SSc associated myopathies, based on 35 muscle biopsies,[29] that described the pathological muscle features of SSc patients. This study found that the main myopathological characteristics are mononuclear inflammation (63%), muscle atrophy (60%), necrosis (59%), regeneration (44%), fibrosis (24%), or microangiopathy (27%); and in the 4.4-year follow-up, 90% of SSc patients with myopathy responded well to glucocorticoids compared with 38% of those without myopathy.[29]
Comprehensive analysis may reveal that inflammation and microvasculitis play important roles in SSc patients with myopathy, which may involve a significantly different mechanism of pathogenesis compared with that of SSc patients without myopathy. Early detection of muscle involvement in SSc patients is critical for designing effective therapies of SSc-associated myopathy, which is associated with poor prognosis.
We demonstrate here that SSc patients with myopathy were more likely to have PH, ILD, and pericardial effusion, which are risk factors of higher mortality. Further, SSc patients with myopathy were more likely to develop cardiac involvement, a prognostic indicator of poor outcomes. For example, SSc patients with myopathy are relatively prone to suffer heart complications, including cardiomyopathy, heart valve disease, left ventricular dysfunction, changes in heart capacity, myocardial perfusion abnormalities as well as cardiac inflammation and fibrosis.[30] Here we found that the frequencies of ILD, PH, and cardiac involvement were significantly higher in SSc patients with myopathy. These manifestations lead to serious adverse consequence. Therefore, early screening for myopathy and timely aggressive intervention are crucial for improving a patients’ life quality and prognosis.[31]
Digestive system involvement, mainly esophageal motility dysfunction, was common in SSc patients (approximately 20%–95%), and the incidence is associated with myopathy, disease subtype, disease duration, and autoantibodies.[32] Digestive system involvement can be considered as a risk factor that decreases the 5-year survival rate.[33] In the present study, patients with myopathy experienced significantly higher frequencies of gastrointestinal symptoms such as choking feelings and dysphagia.
Previous reports show that myopathy predicts poor outcomes,[9] which is consistent with our present findings that the survival rates of patients with myopathy decreased more rapidly compared with those without, and that myopathy had significant adverse effects on patients’ long-term outcomes.
Several limitations should be considered in this study. First, this is a retrospective cross-sectional study, SSc related myopathy was defined at first visit/registration within our database but not at diagnosis, there is a lack of information on the timing of onset of SSc myopathy. Second, relatively small sample size and short follow-up time, there is a likely to be a survival bias. Finally, the causes of death were collected by organ system involvement or other reasons not related to SSc, so we can not determine precisely what proportion, if any, relates to sudden death.
6. Conclusion
Despite the limitations, our results show that SSc patients with myopathy usually suffered from more severe clinical manifestations, such as cardiac, pulmonary, and esophageal involvement, particularly ILD and PH. Myopathy was associated with higher disease activity, which affected the survival rate, leading to worse prognosis. Further studies are required to determine the differences between SSc patients with or without myopathy.
Author contributions
Acquisition of data: Zhou, Nie, Chen, Du, Xue.
Analysis and interpretation of data: Zhou, Chen, Du, Xue.
Manuscript preparation: Zhou, Du, Sutikno, Zhang, Sun, Xue.
Statistical analysis: Zhou, Chen, Du, Xue.
Study conception & design: Zhou, Du, Xue.
Du and Xue have full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Abbreviations: CK = creatine kinase, dcSSc = diffuse cutaneous SSc, DLCO-SB = single breath diffusing lung capacity for carbon monoxide, EMG = electromyography, HRCT = high-resolution computed tomography, ILD = interstitial lung disease, LcSSc = limited cutaneous SSc, LVEF = left ventricular ejection fraction, MRI = magnetic-resonance imaging, mRTSS = modified Rodnan total skin score, PH = pulmonary hypertension, RP = Raynaud phenomenon, SSc = systemic sclerosis.
How to cite this article: Zhou M, Jiang L, Nie L, Chen T, Zhang T, Sun W, Sutikno J, Du Y, Xue J. Myopathy is a Risk Factor for Poor Prognosis of Patients with Systemic Sclerosis: a retrospective cohort study. Medicine. 2020;99:33(e21734).
YD and JX have equally contributed to this work.
The corresponding authors have the right to grant on behalf of all authors and do grant on behalf of all authors. Supported in part by National Natural Science Foundation of China (No. 81501388), Medical Science and Technology Project of Zhejiang Province (No. 2018KY422), Public Technology Applied Research Project of Zhejiang Province (No. 2015C33177), Medical Science and Technology Plan Project of Zhejiang Province (No. 2017KY381), and the General Research Program of Zhejiang Provincial Department of Health (No. 2018240038)
This study was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine (Hangzhou, China). All the patients provided written informed consent.
The work described has not been submitted elsewhere for publication, in whole or in part, and all the authors listed have approved the manuscript that is enclosed.
The authors have no conflicts of interest to disclose.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Tamby MC, Chanseaud Y, Guillevin L, et al. New insights into the pathogenesis of systemic sclerosis. Autoimmun Rev 2003;2:152–7. [DOI] [PubMed] [Google Scholar]
- [2].van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Domsic RT, Nihtyanova SI, Wisniewski SR, et al. Derivation and validation of a prediction rule for two-year mortality in early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol 2014;66:1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Villalta D, Imbastaro T, Giovanni SD, et al. Diagnostic accuracy and predictive value of extended autoantibody profile in systemic sclerosis. Autoimmun Rev 2012;12:114. [DOI] [PubMed] [Google Scholar]
- [5].Cappelli L, Wigley FM. Management of Raynaud phenomenon and digital ulcers in scleroderma. Rheum Dis Clin North Am 2015;41:419–38. [DOI] [PubMed] [Google Scholar]
- [6].Rubio-Rivas M, Royo C, Simeon CP, et al. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum 2014;44:208–19. [DOI] [PubMed] [Google Scholar]
- [7].Clements PJ, Furst DE, Wong WK, et al. High-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis: analysis of a two-year, double-blind, randomized, controlled clinical trial. Arthritis Rheum 1999;42:1194–203. [DOI] [PubMed] [Google Scholar]
- [8].Walker UA, Tyndall A, Czirják L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis 2007;66:754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jung M, Baron M, Hudson M, et al. Myopathy is a poor prognostic feature in systemic sclerosis. results from the canadian scleroderma research group (CSRG) cohort. Scand J Rheumatol 2014;43:217–20. [DOI] [PubMed] [Google Scholar]
- [10].Ranque B, Authier FJ, Berezne A, et al. Systemic sclerosis-associated myopathy. Ann N Y Acad Sci 2007;1108:268–82. [DOI] [PubMed] [Google Scholar]
- [11].Walker UA, Clements PJ, Allanore Y, et al. Muscle involvement in systemic sclerosis: points to consider in clinical trials. Rheumatology (Oxford) 2017;56:v38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Masi AT. ARA Preliminary criteria for the classification of systemic sclerosis (scleroderma): Special article. Arthritis Rheumatol 1980;23:581–90. [DOI] [PubMed] [Google Scholar]
- [13].Jiucun W, Shervin A, Gang G, et al. Clinical and serological features of systemic sclerosis in a Chinese cohort. Clin Rheumatol 2013;32:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390:1685–99. [DOI] [PubMed] [Google Scholar]
- [15].Paik JJ, Wigley FM, Lloyd TE, et al. Spectrum of muscle histopathologic findings in forty-two scleroderma patients with weakness. Arthritis Care Res (Hoboken) 2015;67:1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Paik JJ, Wigley FM, Shah AA, et al. Association of fibrosing myopathy in systemic sclerosis and higher mortality. Arthritis Care Res (Hoboken) 2017;69:1764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mimura Y, Ihn H, Jinnin M, et al. Clinical and laboratory features of scleroderma patients developing skeletal myopathy. Clin Rheumatol 2005;24:99–102. [DOI] [PubMed] [Google Scholar]
- [18].Paik JJ, Mammen AL, Wigley FM, et al. Myopathy in scleroderma, its identification, prevalence, and treatment: lessons learned from cohort studies. Curr Opin Rheumatol 2014;26:124–30. [DOI] [PubMed] [Google Scholar]
- [19].Stone AC, Machan JT, Jeffery M, et al. Echocardiographic evidence of pulmonary hypertension is associated with increased 1-year mortality in patients admitted with chronic obstructive pulmonary disease. Lung 2011;189:207–12. [DOI] [PubMed] [Google Scholar]
- [20].Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest 2013;143:814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zou J, Guo Q, Chi J, et al. HRCT score and serum ferritin level are factors associated to the 1-year mortality of acute interstitial lung disease in clinically amyopathic dermatomyositis patients. Clin Rheumatol 2015;34:707–14. [DOI] [PubMed] [Google Scholar]
- [22].Ichikado K, Suga M, Muranaka H, et al. Prediction of prognosis for acute respiratory distress syndrome with thin-section CT: validation in 44 cases. Radiology 2006;238:321–9. [DOI] [PubMed] [Google Scholar]
- [23].Gyger G, Baron M. Systemic sclerosis: gastrointestinal disease and its management. Rheum Dis Clin North Am 2015;41:459–73. [DOI] [PubMed] [Google Scholar]
- [24].Galluccio F, Mã1/4Ller-Ladner U, Furst DE, et al. Points to consider in renal involvement in systemic sclerosis. Rheumatology 2017;56:v49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Della Rossa A, Valentini G, Bombardieri S, et al. European multicentre study to define disease activity criteria for systemic sclerosis. I. Clinical and epidemiological features of 290 patients from 19 centres. Ann Rheum Dis 2001;60:585–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol 1995;22:1281–5. [PubMed] [Google Scholar]
- [27].Jinnin M. Mechanisms of skin fibrosis in systemic sclerosis. J Dermatol 2010;37:11–25. [DOI] [PubMed] [Google Scholar]
- [28].Randone SB, Guiducci S, Cerinic MM. Musculoskeletal involvement in systemic sclerosis. Best Pract Res Clin Rheumatol 2008;22:339–50. [DOI] [PubMed] [Google Scholar]
- [29].Ranque B, Authier FJ, Le-Guern V, et al. A descriptive and prognostic study of systemic sclerosis-associated myopathies. Ann Rheum Dis 2009;68:1474–7. [DOI] [PubMed] [Google Scholar]
- [30].Meune C, Vignaux O, Kahan A, et al. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010;103:46–52. [DOI] [PubMed] [Google Scholar]
- [31].Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010;69:1809–15. [DOI] [PubMed] [Google Scholar]
- [32].Kahan A, Menkés CJ. Gastrointestinal involvement in systemic sclerosis. Clin Dermatol 1994;12:259. [DOI] [PubMed] [Google Scholar]
- [33].Domsic RT, Nihtyanova SI, Wisniewski SR, et al. Derivation and external validation of a prediction rule for five-year mortality in patients with early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol 2016;68:993–1003. [DOI] [PubMed] [Google Scholar]