

Abstract
A nonsense mutation adds a premature stop signal that hinders any further translation of a protein-coding gene, usually resulting in a null allele. To investigate the possible exceptions, we used the DMD gene as an ideal model. First, because dystrophin absence causes Duchenne muscular dystrophy (DMD), while its reduction causes Becker muscular dystrophy (BMD). Second, the DMD gene is X-linked and there is no second allele that can interfere in males. Third, databases are accumulating reports on many mutations and phenotypic data. Finally, because DMD mutations may have important therapeutic implications. For our study, we analyzed large databases (LOVD, HGMD and ClinVar) and literature and revised critically all data, together with data from our internal patients. We totally collected 2593 patients. Positioning these mutations along the dystrophin transcript, we observed a nonrandom distribution of BMD-associated mutations within selected exons and concluded that the position can be predictive of the phenotype. Nonsense mutations always cause DMD when occurring at any point in fifty-one exons. In the remaining exons, we found milder BMD cases due to early 5’ nonsense mutations, if reinitiation can occur, or due to late 3’ nonsense when the shortened product retains functionality. In the central part of the gene, all mutations in some in-frame exons, such as in exons 25, 31, 37 and 38 cause BMD, while mutations in exons 30, 32, 34 and 36 cause DMD. This may have important implication in predicting the natural history and the efficacy of therapeutic use of drug-stimulated translational readthrough of premature termination codons, also considering the action of internal natural rescuers. More in general, our survey confirm that a nonsense mutation should be not necessarily classified as a null allele and this should be considered in genetic counselling.
Introduction
A nonsense mutation is classically considered a loss-of-function change, with ribosomes that dissociate from mRNA and transcript degradation. Shortened protein products are usually quickly ubiquitinated and digested by the proteasome. All these mechanisms must be very efficient to prevent cell accumulation of toxic or ectopic protein garbage [1]. This suggests that the functional effect of a nonsense mutation may be considered equivalent to the full deletion of a gene (null or amorph allele). However, the difference between the two causes is striking: in the case of a nonsense mutation the cell retains almost all the genetic information, while in the case of deletion does not. We searched for exceptions by studying nonsense mutations of the DMD gene encoding a 427kDa- protein, named dystrophin. This is an ideal model. First, because DMD is X-linked and in hemizygous males there is no second allele that may complicate the genotype/phenotype correlation. Second, because in males the null alleles are fully penetrant in the form of Duchenne muscular dystrophy (DMD) and well distinct from the hypomorphic alleles that cause Becker muscular dystrophy (BMD). Third, because a huge number of different nonsense mutations and phenotypic data have been reported since 1992. Finally, because DMD nonsense mutations are the target for treatments based on readthrough strategies [2, 3]. Dystrophin defects disrupt the associated glycoprotein complex at the sarcolemma and several pathogenic cascades are thus activated [4]. They quickly lead to structural and functional disruption of the muscles and to a progressive muscle weakness. DMD is the most severe phenotype, in which the progressive muscle disruption cause an early loss of ambulation, skeletal alterations with respiratory and cardiac involvement, and sometimes cognitive impairment [5]. Conversely, BMD represents the milder phenotype with a slower progression of muscle weakness, tardive loss of ambulation, and variable cardiac and respiratory involvement [6]. DMD diagnosis cannot be questioned in teenagers, considering the dramatic phenotype in males, such as difficulty running, climbing stairs, getting up from the floor with a positive Gowers maneuver, creatine kinase values up to 100 times the normal maximum value, and the high accuracy of natural history data available. Even if phenotype variants have been reported, these are never strong enough to associate the dystrophin absence to a BMD phenotype. Another point regards clinical trials, because any phenotype variability in patients with nonsense mutations may reduce the statistical significance of any therapeutic improvement [7, 8].
The full mutational analysis of the DMD gene is considered part of the standard of care for DMD. The DMD gene, consisting of 79 exons generally separated by huge introns, is prone to intragenic deletions or duplications that when include exons cause DMD or BMD [9–11]. The first nonsense variants and other small defects were only identified six years after the DMD gene cloning [12, 13]. Unlike most disease genes, single nucleotide substitutions and small insertion/deletion of bases are a less frequent cause of disease [14, 15]. Random nonsense mutations were found in 10–15% of DMD cases [16]. This randomness of lethal X-linked mutations confirms the Haldane’s rule and offers a possibility of unbiased analysis [17]. In 1996, a pivotal study paved the way for a new therapeutic option for genetic disorders caused by nonsense alleles: gentamycin was shown to induce the readthrough of ribosome overcoming a single stop codon in the context of an open reading frame in the cystic fibrosis gene [18]. However, any possible therapeutic window was closed by severe side effects of gentamycin. A high-throughput screening of synthetic molecules resulted in the selection of a new compound, named PTC-124 from PTC Therapeutics (New Jersey, USA) that showed an important increase of protein production in cells and mdx mice, carrying a nonsense variant in exon 23 [2, 19, 20]. This drug, commercial name Ataluren (Translarna), can be administered orally and, compared with aminoglycosides, shows fewer side effects, in about 5% of treated subjects. These include vomiting, diarrhea, nausea (feeling sick), headache, stomachache and flatulence [21, 22]. Despite weak Phase II results, its use was approved in member states of the European Union, Iceland, Israel, Kazakhstan, Liechtenstein, Norway and the Republic of Korea, for the treatment (40 mg/kg/day) of ≥ 2 years DMD boys caused by nonsense mutation, or aged ≥ 5 years in Brazil and Chile [3, 23]. The possibility of readthrough-based treatments provided further impetus in searching for nonsense mutations in DMD boys as early as possible. Nowadays, next generation sequencing (NGS) protocols are being applied to fully sequence DNA in children with suspected muscular dystrophy [24–27].
Our present survey on the positional effect of nonsense mutations may have important implication for therapeutic use of drug-stimulated translational readthrough of premature termination codons.
Methods
We collected the published unique nonsense variants in the dystrophin gene (DMD, NM_004006.2) from three main databases: Leiden Open Variation Database (LOVD) [28], Human Genome Variant Database (HGMD) [29], and ClinVar [30]. Data filtering was based on their classification as “Pathogenic” variants and considering their molecular consequence differently termed in the three databases (by using HGVS nomenclature in LOVD, “Term” in HGMD, and “nonsense” in ClinVar). We selected 702 nonsense variants in LOVD, 823 in HGMD and 236 in ClinVar. Removing the duplicates among the databases and integrating all the data, we obtained 849 unique nonsense mutations so far published (until April 2020). LOVD also provides a rough indication of the variant recurrence, as it allows researcher to resubmit a known variant found in additional patients [31]. Literature data were used to carefully correlate the specific phenotype to the nonsense variant observed in each patient. We reviewed these data together with our internal cohort of 1,102 patients that included already published cases [13, 15, 27] and further 128 cases. Genomic DNA was extracted from leucocyte according to the standard procedure [32]. We performed Multiplex Ligation-dependent Probe Amplification (MLPA), according to the manufacturer’s recommendations (MRC Holland) and/or Log-PCR, as previously described [33]. MLPA/LogPCR negative patients were analyzed for single nucleotide variants or small ins/del performing the NGS MotorPlex panel [26, 27] or by a panel focused on >5,200 genes responsible for Mendelian Disease (Sure Select Agilent Custom Constitutional Panel). We also used Human Splice Finder (HSF) [34], a bioinformatic tool able to predict possible effects of the mutations on canonical or cryptic splice sites and on specific exonic splicing enhancer/silencer sequences (ESE/ESS) [35]. ProteinPaint [36] was used to graphically represent the distribution of nonsense mutations along DMD gene. The Ethics Committee of Vanvitelli University approved the study with ID 5586/19 and 8635/19.
Results and discussion
To search for the most comprehensive number of annotated nonsense mutations in the DMD gene, we added to our internal cases all the variants retrieved from public databases (LOVD [28], HGMD [29] and ClinVar [30]) or from literature. The largest published study was carried out on 243 patients with nonsense mutations by Flanigan et al [37], but all recent papers were also considered [38, 39]. Since in some cases, nonsense variants reported in public databases did not have a clear clinical diagnosis, we critically reviewed the associated reports to be sure of the assigned phenotype. In our patient cohort, we had accurate information on 61 cases with nonsense mutations in the DMD gene, part of which was previously published (S1 Table) [13, 15, 40]. Altogether, we collected 2593 patients with 849 unique nonsense mutations (S2 Table). The reports were classified in five groups based on the phenotypic annotation of the patients: DMD, BMD, DMD/BMD, ND (Not Defined) and Other, as showed in Table 1.
Table 1. Summary of nonsense mutations and patients classified on the basis of reported phenotypes.
| Disease | Unique nonsense mutations | Number of patients |
|---|---|---|
| DMD | 579 | 2022 |
| BMD | 54 | 180 |
| DMD/BMD | 104 | 245 |
| ND | 88 | 103 |
| OTHER* | 25 | 43 |
| TOTAL | 849 | 2593 |
*This category includes heterozygous symptomatic carrier, hyperCK, or cardiomyopathic phenotypes.
To evaluate any positional effect of nonsense mutations, we first considered their distribution along the DMD gene in association with different phenotypes (Fig 1). Three main BMD-associated coding regions are evident: N-terminus, C- terminus and central part of the rod domain. BMD-associated nonsense mutations are listed in Table 2. Fig 2 describes the percentage of DMD/BMD frequency for each DMD exon with a blue color code for BMD cases and orange for DMD cases. From the analysis of this figure, we immediately observed a non-random distribution of milder cases in specific exons. We also found that mutations in adjacent exons, in the middle part of the gene, had completely different phenotypic consequences. For example, mutations in exons 30 and 32 were DMD-linked, while mutations in exons 29 and 31 were BMD-linked. We identified four exons with 15 unique nonsense mutations never associated with DMD. These were exons 2, 31, 72 and 73, for which we only found BMD patients or milder phenotypes. In addition, we highlighted other 21 exons with 236 nonsense mutations associated with BMD, DMD phenotype or undefined phenotypes at different frequencies (Table 3). Finally, nonsense mutation in the remaining 51 exons are associated with DMD in 100% of cases, as originally expected for “loss-of-function” mutations [41].
Fig 1. Graphical representation of the distribution of nonsense mutations in the DMD gene.

Nonsense mutations associated with DMD (orange) or BMD (blue) are reported. The ProteinPaint graph [36] highlights three main regions for BMD phenotypes, while the vast majority of nonsense mutations are associated with DMD. The number in the circle indicates that different nucleotide changes determine the same nonsense codon.
Table 2. Summary of reported nonsense mutations associated to BMD/mild phenotype.
| EXON | CDS | Protein | hg19 | Patients | Effect | Reference |
|---|---|---|---|---|---|---|
| 1 | c.8G>A | p.(Trp3*) | g.33229422C>T | 1 | - | [28] |
| 1 | c.9G>A | p.[Trp3*, Leu2_Met124del, Leu2_Met128del] | g.33229421C>T | 27 | exon 2-3-4-5 skipping | [38] |
| 1 | c.11G>A | p.(Trp4*) | g.33229419C>T | 6 | - | [42] |
| 2 | c.49C>T | p.(Gln17*) | g.33038300G>A | 1 | - | [39] |
| 2 | c.67A>T | p.(Lys23*) | g.33038282T>A | 1 | - | [28] |
| 2 | c.72G>A | p.(Trp24*) | g.33038277C>T | 1 | - | [43] |
| 3 | c.103C>T | p.(Gln35*) | g.32867928G>A | 2 | - | [44] |
| 3 | c.163G>T | p.[Glu55*, Phe32Metfs*13] | g.32867868C>A | 1 | frame-shift deletion of exons 3–7 | [45] |
| 5 | c.336G>A | p.(Trp112*) | g.32841433C>T | 3 | - | [42] |
| 21 | c.2704C>T | p.(Gln902*) | g.32503135G>A | 2 | - | [46] |
| 25 | c.3328G>T | p.[Glu1110*, Leu1093_Gln1144del] | g.32481660C>A | 1 | exon 25 skipping | [45] |
| 25 | c.3340A>T | p.(Lys1114*) | g.32481648T>A | 3 | - | [8, 9] |
| 25 | c.3352G>T | p.(Glu1118*) | g.32481636C>A | 2 | - | [47] |
| 25 | c.3358G>T | p.(Glu1120*) | g.32481630C>A | 1 | - | [48] |
| 25 | c.3409C>T | p.(Gln1137*) | g.32481579G>A | 3 | - | [49] |
| 25 | c.3413G>A | p.(Trp1138*) | g.32481575C>T | 4 | - | [50] |
| 26 | c.3515G>A | p.[Trp1172*, Val1145_Lys1201del] | g.32472867C>T | 1 | exon 26 skipping | [51, 52] |
| 27 | c.3631G>T | p.[Glu1211*; Arg1202_1262del; Arg1202_1357del] | g.32466728C>A | 1 | exons 27 or 27-28-29 skipping | [53, 54] |
| 27 | c.3700G>T | p.(Glu1234*) | g.32466659C>A | 1 | - | [9] |
| 28 | c.3843G>A | p.(Trp1281*) | g.32459375C>T | 2 | - | [55] |
| 28 | c.3850G>T | p.[Glu1284*, Glu1263_Asp1307del, Glu1263_Glu1357del] | g.32459368C>A | 2 | exon 28 or 28–29 skipping | [42] |
| 29 | c.3935T>A | p.[Leu1312*, Glu1263_Glu1357del, Ser1308_Glu1357del] | g.32456494A>T | 1 | exon 29 or 28–29 skipping | [28, 56] |
| 29 | c.3940C>T | p.[Arg1314*, Glu1263_Glu1357del, Ser1308_Glu1357del] | g.32456489G>A | 31 | exon 29 or 28–29 skipping | [51, 56] |
| 29 | c.4000G>T | p.[Gly1334*, Glu1263_Glu1357del, Ser1308_Glu1357del] | g.32456429C>A | 1 | exon 29 or 28–29 skipping | [28, 56] |
| 29 | c.4012G>T | p.(Glu1338*) | g.32456417C>A | 1 | - | [9] |
| 31 | c.4240C>T | p.(Gln1414*) | g.32408292G>A | 1 | - | [9] |
| 31 | c.4250T>A | p.[Leu1417*; Ile1413_Lys1449del] | g.32408282A>T | 4 | exon 31 skipping | [57] |
| 31 | c.4285A>T | p.(Lys1429*) | g.32408247T>A | 1 | - | [9] |
| 31 | c.4294C>T | p.[Gln1432*, Ile1413_Lys1449del] | g.32408238G>A | 3 | exon 31 skipping | [58] |
| 33 | c.4576G>T | p.(Gly1526*) | g.32404525C>A | 1 | - | [28] |
| 35 | c.4979G>A | p.(Trp1660*) | g.32383183C>T | 1 | - | [15] |
| 35 | c.4980G>A | p.(Trp1660*) | g.32383182C>T | 1 | - | [28] |
| 35 | c.5002G>T | p.(Glu1668*) | g.32383160C>A | 1 | - | [59] |
| 37 | c.5260G>T | p.(Glu1754*) | g.32380970C>A | 1 | - | [60] |
| 37 | c.5287C>T | p.[Arg1763*, Arg1719_Lys1775del] | g.32380943G>A | 22 | exon 37 skipping | [61] |
| 38 | c.5398G>T | p.(Glu1800*) | g.32366573C>A | 3 | - | [9] |
| 38 | c.5404C>T | p.(Gln1802*) | g.32366567G>A | 5 | - | [62] |
| 38 | c.5407C>T | p.[Gln1803*, Ala1776_Met1816del] | g.32366564G>A | 5 | exon 38 skipping | [46, 56] |
| 39 | c.5452G>T | p.(Glu1818*) | g.32364194C>A | 1 | - | [28] |
| 39 | c.5480T>A | p.[Leu1827*, Ala1776_Lys1862del, Asn1817_Lys1862del] | g.32364166A>T | 2 | exons 38–39 skipping | [28, 56] |
| 40 | c.5725G>T | p.(Glu1909*) | g.32361265C>A | 1 | - | [9] |
| 41 | c.5835G>T | p.(Glu1946*) | g.32360304C>A | 1 | - | [28] |
| 49 | c.7105G>T | p.[Glu2369*, Glu2367_Lys2400del] | g.31854930C>A | 5 | exon 49 skipping | [61] |
| 56 | c.8353A>T | p.(Lys2785*) | g.31525435T>A | 1 | - | [9] |
| 72 | c.10279C>T | p.[Gln3427*, Pro3422_Arg3443del] | g.31191705G>A | 7 | exon 72 skipping | [61] |
| 73 | c.10362T>A | p.(Tyr3454*) | g.31190497A>T | 1 | - | [28] |
| 74 | c.10412T>A | p.(Leu3471*) | g.31187701A>T | 2 | - | [57] |
| 74 | c.10429C>T | p.(Gln3477*) | g.31187684G>A | 1 | - | [28] |
| 74 | c.10543G>T | p.[Glu3515*, Ile3465_Arg3518delinsMet] | g.31187570C>A | 1 | exon 74 skipping | [52] |
| 75 | c.10759G>T | p.(Glu3587*) | g.31165430C>A | 1 | - | [28] |
| 75 | c.10792G>T | p.(Glu3598*) | g.31165397C>A | 1 | - | [46] |
| 76 | c.10873C>T | p.(Gln3625*) | g.31164456G>A | 1 | - | [54] |
| 76 | c.10888C>T | p.(Arg3630*) | g.31164441G>A | 1 | - | [9] |
| 76 | c.10910C>A | p.(Ser3637*) | g.31164419G>T | 4 | - | [61] |
Fig 2. Color representation of the distribution of phenotypes in relation to DMD exons.
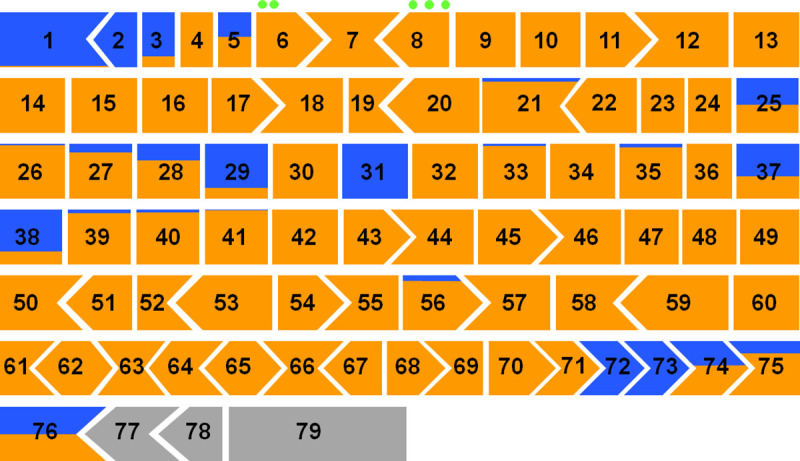
Table 3. Patients with nonsense mutations in the same exons but with discordant phenotypes.
| DMD EXON | FRAME | TOT | DMD | BMD | DMD/BMD | Not Defined | Other | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nonsense mutations | Patients | Nonsense mutations | Patients | Nonsense mutations | Patients | Nonsense mutations | Patients | Nonsense mutations | Patients | Nonsense mutations | Patients | ||
| 1 | - | 4 | 35 | 1 | 1 | 3 | 34 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | out | 4 | 5 | 0 | 0 | 3 | 3 | 0 | 0 | 1 | 2 | 0 | 0 |
| 3 | in | 5 | 16 | 2 | 12 | 2 | 3 | 1 | 1 | 0 | 0 | 0 | 0 |
| 5 | in | 7 | 18 | 3 | 4 | 1 | 3 | 3 | 11 | 0 | 0 | 0 | 0 |
| 21 | out | 18 | 44 | 12 | 37 | 1 | 2 | 1 | 1 | 3 | 3 | 1 | 1 |
| 25 | in | 18 | 58 | 7 | 15 | 6 | 14 | 4 | 27 | 1 | 2 | 0 | 0 |
| 26 | in | 21 | 48 | 13 | 37 | 1 | 1 | 1 | 1 | 5 | 8 | 1 | 1 |
| 27 | in | 13 | 19 | 9 | 14 | 2 | 2 | 0 | 0 | 1 | 1 | 1 | 2 |
| 28 | in | 12 | 24 | 5 | 10 | 2 | 4 | 3 | 8 | 0 | 0 | 2 | 2 |
| 29 | in | 12 | 57 | 4 | 8 | 4 | 34 | 1 | 10 | 3 | 5 | 0 | 0 |
| 31 | in | 7 | 12 | 0 | 0 | 4 | 9 | 2 | 2 | 1 | 1 | 0 | 0 |
| 33 | in | 14 | 32 | 11 | 27 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 3 |
| 35 | in | 15 | 55 | 8 | 48 | 3 | 3 | 2 | 2 | 2 | 2 | 0 | 0 |
| 37 | in | 10 | 39 | 8 | 16 | 2 | 23 | 0 | 0 | 0 | 0 | 0 | 0 |
| 38 | in | 9 | 34 | 2 | 4 | 3 | 13 | 4 | 17 | 0 | 0 | 0 | 0 |
| 39 | in | 16 | 65 | 8 | 49 | 2 | 3 | 1 | 5 | 3 | 3 | 2 | 5 |
| 40 | in | 16 | 34 | 9 | 21 | 1 | 1 | 3 | 6 | 2 | 2 | 1 | 4 |
| 41 | in | 20 | 76 | 10 | 58 | 1 | 1 | 4 | 11 | 4 | 4 | 1 | 2 |
| 49 | in | 5 | 10 | 1 | 1 | 1 | 5 | 1 | 1 | 2 | 3 | 0 | 0 |
| 56 | out | 8 | 11 | 6 | 9 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| 72 | in | 2 | 10 | 0 | 0 | 1 | 7 | 1 | 3 | 0 | 0 | 0 | 0 |
| 73 | in | 2 | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| 74 | in | 10 | 18 | 4 | 5 | 3 | 4 | 2 | 8 | 1 | 1 | 0 | 0 |
| 75 | out | 8 | 14 | 3 | 7 | 2 | 2 | 1 | 3 | 1 | 1 | 1 | 1 |
| 76 | out | 6 | 13 | 2 | 6 | 3 | 6 | 1 | 1 | 0 | 0 | 0 | 0 |
For each exon, the fraction of blue color is proportional to the percentage of independent BMD cases with nonsense mutations, while the fraction of orange color is proportional to the percentage of DMD cases; exons without nonsense mutations are in gray. For each exon, the shape of box extremities represents the phase, where in-frame junctions are indicated by vertical lines.An arrow shape represents an exon starting (or ending) at the 2nd or 3rd nucleotide of a codon. Methionines in exons 6 and 8 are reported with a green circle.
N-terminus
The distribution of nonsense mutations along the dystrophin molecule (Fig 2) is also quite surprising. Although it may be expected that the effect of a mutation at the beginning of the nascent polypeptide chain can be recovered from a re-initiation phenomenon [63], it is not clear how this can occur much further downstream. After the first start codon, the following methionines are two in exon 6 (position 124 and 128) [38] and three in exon 8 (230, 253 and 272) [64]. This could explain why nonsense mutations in exon 1, 2, 3 and 5 may be also associated with non-DMD phenotypes, but not why exon 4 mutations appear to be 100% DMD-linked.
C-terminus
At the 3’ end, premature stop codons are understandably associated with milder phenotypes, because major part of the proteins has already been produced and therefore the truncated products may be partially functional. This prediction is supported by nonsense mutations of exons 72–76, also considering that most 3’ DMD exons are alternatively spliced [58, 65, 66]. No nonsense mutation in exons 77, 78 and 79 has been so far described in DMD/BMD patients. Recently, the last gnomAD v.3 reports a nonsense variant at position p.Arg3681* in exon 78, found in six African individuals (5 females and 1 male), and reported as variant of uncertain significance [67, 68].
This could suggest that nonsense mutations at the last 3’ end of the gene are not deleterious for the dystrophin function.
Internal rod (in-frame exons)
A nonsense mutation in the middle of an open reading frame (ORF) generally undergoes nonsense mediated (mRNA) decay, a translation-coupled mechanism that eliminates mRNAs containing premature translation-termination codons [69]. Thus, even if it is possible a therapeutic induction of translation readthrough, the mRNA is degraded and therefore the expected phenotype should be severe. It is overly complex to measure the percentage of reduction of transcripts from muscle tissue in relation to the position of each nonsense mutation, but it seems clear that in many cases the phenomenon could be not stringent. Indeed, alternatively spliced isoforms could be actively selected by this mechanism, enriching the mRNA fraction with an ORF compared to those with stop codons.
On the other hand, if a portion of mRNA skips the exon with a mutation, a smaller protein could still be produced on the condition that the skipped exon is in-frame. Previous works hypothesized that mutations in in-frame exons might cause milder phenotypes via spontaneous exon skipping of the mutated exon, which may weaken the mutation consequence [14, 37]. This favorable precondition is the rule for most central dystrophin exons: all of them between 23 and 42 are in-frame. Apart from exon 29 that is alternatively spliced in normal muscle, all these other exons appear to be required [70]. Interestingly, consecutive exons may have divergent phenotypic associations. The skipping could restore the transcript and several reports have demonstrated that specific nonsense mutation can convert exonic splicing enhancer sequences (ESE) to silencer elements (ESS) [37, 71–73]. However, the situation is very strange for some exons such as 25, 31, 37 and 38 where many different nonsense mutations all lead to a mild phenotype (Table 2) [8, 45, 46, 56, 58, 61]. What is the explanation? Are these four exons easily skippable and thus are lost wherever they are mutated?
In addition, there is also the possibility of a multiple exon skipping. Nonsense mutations in the exon 27 cause the skipping of the exons 27–29 [53, 54]. Finally, it has been described that nonsense mutations in the exon 28 and 29 induce the skipping of single involved exon or the skipping of double exons (exons 28–29); moreover, mutations in the exon 39 cause 38–39 exons skipping [28, 42, 51, 56].
Internal rod (out-of-frame exons)
The explanation remains obscure for a few cases in out-of-frame exons. To provide an hypothesis, we checked two BMD-associated nonsense mutations in exons 21 and 56. The splice-site predictor software HSF [34] indicates that c.8353A>T, p.Lys2785*, in the exon 56, could cause the creation of two new splice acceptor sites. Only one allows to maintain the protein frame, thus explaining the BMD phenotype (Fig 3). By contrast, investigating the consequence of the variation c.2704C>T, p.Gln902*, in the exon 21, no splicing alteration was predicted. It is possible that self-correcting exon skipping may involve more than one exon and in this case prediction of phenotype effect based on small mutations location is not possible [74]. Therefore, to explain the reported association with BMD phenotype, it could be speculated about a potential coupled skipping of the exons 21–22, which could restore the protein frame (Fig 2).
Fig 3. Prediction of a cryptic acceptor splice site in DMD exon56.

By analyzing the c.8353A>T (p.Lys2785*) variant using the splice-site predictor software HSF, the nucleotide change is predicted to activate a cryptic acceptor splice site able to partially rescue reading-frame of exon 56, retaining the last 12 amino acids. A second weaker cryptic acceptor splice site is underscored, no rescuing reading-frame.
Conclusions
By positioning all reported nonsense mutations along the dystrophin transcript, we observed a skewed concentration of BMD within selected exons. Previous data from large cohort of patients, [37, 61] and the present survey show that a milder than expected phenotype can be produced by the spontaneous elimination of a nonsense mutation from dystrophin mRNA in some central exons. The reported exceptions further confirm that natural mechanisms for rescue do exist. The observation suggests that exon skipping in the specific exons identified in this work could be a biologically more favored therapeutic approach than recovering deletions. Antisense oligonucleotides (AON) or new molecules, designed to induce the jump of specific exons are desirable. While on another fifty exons on the effects of the readthrough strategies can be more easily monitored.
Our graphical output may be of practical use both in genetic counselling and in recruitment of patients for translational readthrough of premature termination codons. From a more general point of view, our data confirm that multiple mechanisms can partially rescue nonsense mutations that should be not necessarily classified as null variants. This should be considered for the interpretation of NGS results.
Supporting information
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Francesco Musacchia for NGS analyses [75], Alessandra Varavallo for bioinformatic support and Anna Cuomo for the Sanger sequence validation process.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This survey was partially supported by unrestricted grant from PTC Therapeutics Italy (www.ptcbio.it), Fondazione Telethon (Italy) (GSP15001). This project has also received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 779257 (Solve‐RD): “SOLVE RD”. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kervestin S, Jacobson A (2012) NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712. 10.1038/nrm3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aurino S, Nigro V (2006) Readthrough strategies for stop codons in Duchenne muscular dystrophy. Acta Myol 25: 5–12. [PubMed] [Google Scholar]
- 3.Mullard A (2014) EMA reconsiders 'read-through' drug against Duchenne muscular dystrophy following appeal. Nat Biotechnol 32: 706 10.1038/nbt0814-706 [DOI] [PubMed] [Google Scholar]
- 4.Nigro V, Piluso G (2015) Spectrum of muscular dystrophies associated with sarcolemmal-protein genetic defects. Biochim Biophys Acta 1852: 585–593. 10.1016/j.bbadis.2014.07.023 [DOI] [PubMed] [Google Scholar]
- 5.Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, et al. (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17: 251–267. 10.1016/S1474-4422(18)30024-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrews JG, Lamb MM, Conway K, Street N, Westfield C, et al. (2018) Diagnostic Accuracy of Phenotype Classification in Duchenne and Becker Muscular Dystrophy Using Medical Record Data1. J Neuromuscul Dis 5: 481–495. 10.3233/JND-180306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anthony K, Cirak S, Torelli S, Tasca G, Feng L, et al. (2011) Dystrophin quantification and clinical correlations in Becker muscular dystrophy: implications for clinical trials. Brain 134: 3547–3559. 10.1093/brain/awr291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santos R, Goncalves A, Oliveira J, Vieira E, Vieira JP, et al. (2014) New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet 59: 454–464. 10.1038/jhg.2014.54 [DOI] [PubMed] [Google Scholar]
- 9.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, et al. (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30: 1657–1666. 10.1002/humu.21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, et al. (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 30: 934–945. 10.1002/humu.20976 [DOI] [PubMed] [Google Scholar]
- 11.Flanigan KM (2014) Duchenne and Becker muscular dystrophies. Neurol Clin 32: 671–688, viii. 10.1016/j.ncl.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 12.Roberts RG, Bobrow M, Bentley DR (1992) Point mutations in the dystrophin gene. Proc Natl Acad Sci U S A 89: 2331–2335. 10.1073/pnas.89.6.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nigro V, Politano L, Nigro G, Romano SC, Molinari AM, et al. (1992) Detection of a nonsense mutation in the dystrophin gene by multiple SSCP. Hum Mol Genet 1: 517–520. 10.1093/hmg/1.7.517 [DOI] [PubMed] [Google Scholar]
- 14.Neri M, Rossi R, Trabanelli C, Mauro A, Selvatici R, et al. (2020) The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front Genet 11: 131 10.3389/fgene.2020.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torella A, Trimarco A, Blanco Fdel V, Cuomo A, Aurino S, et al. (2010) One hundred twenty-one dystrophin point mutations detected from stored DNA samples by combinatorial denaturing high-performance liquid chromatography. J Mol Diagn 12: 65–73. 10.2353/jmoldx.2010.090074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, et al. (2011) Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther 19: 830–840. 10.1038/mt.2011.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bucher K, Ionasescu V, Hanson J (1980) Frequency of new mutants among boys with Duchenne muscular dystrophy. Am J Med Genet 7: 27–34. 10.1002/ajmg.1320070107 [DOI] [PubMed] [Google Scholar]
- 18.Howard M, Frizzell RA, Bedwell DM (1996) Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 2: 467–469. 10.1038/nm0496-467 [DOI] [PubMed] [Google Scholar]
- 19.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, et al. (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447: 87–91. 10.1038/nature05756 [DOI] [PubMed] [Google Scholar]
- 20.Nelson SF, Crosbie RH, Miceli MC, Spencer MJ (2009) Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol 22: 532–538. 10.1097/WCO.0b013e32832fd487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agency EM (2020, May 07) EMA.
- 22.Ebrahimi-Fakhari D, Dillmann U, Flotats-Bastardas M, Poryo M, Abdul-Khaliq H, et al. (2018) Off-Label Use of Ataluren in Four Non-ambulatory Patients With Nonsense Mutation Duchenne Muscular Dystrophy: Effects on Cardiac and Pulmonary Function and Muscle Strength. Front Pediatr 6: 316 10.3389/fped.2018.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messina S, Vita GL (2018) Clinical management of Duchenne muscular dystrophy: the state of the art. Neurol Sci 39: 1837–1845. 10.1007/s10072-018-3555-3 [DOI] [PubMed] [Google Scholar]
- 24.Nigro V, Piluso G (2012) Next generation sequencing (NGS) strategies for the genetic testing of myopathies. Acta Myol 31: 196–200. [PMC free article] [PubMed] [Google Scholar]
- 25.Nigro V, Savarese M (2016) Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr Opin Neurol 29: 621–627. 10.1097/WCO.0000000000000371 [DOI] [PubMed] [Google Scholar]
- 26.Savarese M, Di Fruscio G, Mutarelli M, Torella A, Magri F, et al. (2014) MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun 2: 100 10.1186/s40478-014-0100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savarese M, Di Fruscio G, Torella A, Fiorillo C, Magri F, et al. (2016) The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology 87: 71–76. 10.1212/WNL.0000000000002800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leiden Open Variation Database (2020, April 14) LOVD.
- 29.Human Genome Variant Database (2020, April 14) HGMD.
- 30.NCBI (2020, April 14) ClinVar.
- 31.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34: 135–144. 10.1002/mus.20586 [DOI] [PubMed] [Google Scholar]
- 32.Ausubel F.M. BR, Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl k. (1988) Current Protocols in Molecular Biology: Wiley & Sons. [Google Scholar]
- 33.Trimarco A, Torella A, Piluso G, Maria Ventriglia V, Politano L, et al. (2008) Log-PCR: a new tool for immediate and cost-effective diagnosis of up to 85% of dystrophin gene mutations. Clin Chem 54: 973–981. 10.1373/clinchem.2007.097881 [DOI] [PubMed] [Google Scholar]
- 34.Human Splicing Finder (2020, April 14) HSF.
- 35.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, et al. (2009) Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37: e67 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.St. Jude Children's Research Hospital (2020, March 17) ProteinPaint.
- 37.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Howard MT, et al. (2011) Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat 32: 299–308. 10.1002/humu.21426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurvich OL, Maiti B, Weiss RB, Aggarwal G, Howard MT, et al. (2009) DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum Mutat 30: 633–640. 10.1002/humu.20913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witting N, Duno M, Vissing J (2013) Becker muscular dystrophy with widespread muscle hypertrophy and a non-sense mutation of exon 2. Neuromuscul Disord 23: 25–28. 10.1016/j.nmd.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 40.Nigro V, Nigro G, Esposito MG, Comi LI, Molinari AM, et al. (1994) Novel small mutations along the DMD/BMD gene associated with different phenotypes. Hum Mol Genet 3: 1907–1908. 10.1093/hmg/3.10.1907 [DOI] [PubMed] [Google Scholar]
- 41.Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, Baena M, Verdura E, et al. (2015) DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS One 10: e0135189 10.1371/journal.pone.0135189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma P, Zhang S, Zhang H, Fang S, Dong Y, et al. (2018) Comprehensive genetic characteristics of dystrophinopathies in China. Orphanet J Rare Dis 13: 109 10.1186/s13023-018-0853-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oitani Y, Ishiyama A, Kosuga M, Iwasawa K, Ogata A, et al. (2018) Interpretation of acid alpha-glucosidase activity in creatine kinase elevation: A case of Becker muscular dystrophy. Brain Dev 40: 837–840. 10.1016/j.braindev.2018.05.001 [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Planells J, Torres-Puente M, Vilchez JJ, Perez-Alonso M (2009) Novel human pathological mutations. Gene symbol: DMD. Disease: muscular dystrophy, Duchenne. Hum Genet 126: 338. [PubMed] [Google Scholar]
- 45.Sedlackova J, Vondracek P, Hermanova M, Zamecnik J, Hruba Z, et al. (2009) Point mutations in Czech DMD/BMD patients and their phenotypic outcome. Neuromuscul Disord 19: 749–753. 10.1016/j.nmd.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 46.Okubo M, Goto K, Komaki H, Nakamura H, Mori-Yoshimura M, et al. (2017) Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J Rare Dis 12: 149 10.1186/s13023-017-0703-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lo IF, Lai KK, Tong TM, Lam ST (2006) A different spectrum of DMD gene mutations in local Chinese patients with Duchenne/Becker muscular dystrophy. Chin Med J (Engl) 119: 1079–1087. [PubMed] [Google Scholar]
- 48.Tomar S, Moorthy V, Sethi R, Chai J, Low PS, et al. (2019) Mutational spectrum of dystrophinopathies in Singapore: Insights for genetic diagnosis and precision therapy. Am J Med Genet C Semin Med Genet 181: 230–244. 10.1002/ajmg.c.31704 [DOI] [PubMed] [Google Scholar]
- 49.Bonnal RJ, Severgnini M, Castaldi A, Bordoni R, Iacono M, et al. (2010) Reliable resequencing of the human dystrophin locus by universal long polymerase chain reaction and massive pyrosequencing. Anal Biochem 406: 176–184. 10.1016/j.ab.2010.07.022 [DOI] [PubMed] [Google Scholar]
- 50.Prior TW, Bridgeman SJ (2005) Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn 7: 317–326. 10.1016/S1525-1578(10)60560-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo R, Zhu G, Zhu H, Ma R, Peng Y, et al. (2015) DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J Hum Genet 60: 435–442. 10.1038/jhg.2015.43 [DOI] [PubMed] [Google Scholar]
- 52.Hofstra RM, Mulder IM, Vossen R, de Koning-Gans PA, Kraak M, et al. (2004) DGGE-based whole-gene mutation scanning of the dystrophin gene in Duchenne and Becker muscular dystrophy patients. Hum Mutat 23: 57–66. 10.1002/humu.10283 [DOI] [PubMed] [Google Scholar]
- 53.Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y, et al. (1997) Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest 100: 2204–2210. 10.1172/JCI119757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, et al. (2010) Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 55: 379–388. 10.1038/jhg.2010.49 [DOI] [PubMed] [Google Scholar]
- 55.de Almeida PAD, Machado-Costa MC, Manzoli GN, Ferreira LS, Rodrigues MCS, et al. (2017) Genetic profile of Brazilian patients with dystrophinopathies. Clin Genet 92: 199–203. 10.1111/cge.12975 [DOI] [PubMed] [Google Scholar]
- 56.Le Guedard-Mereuze S, Vache C, Molinari N, Vaudaine J, Claustres M, et al. (2009) Sequence contexts that determine the pathogenicity of base substitutions at position +3 of donor splice-sites. Hum Mutat 30: 1329–1339. 10.1002/humu.21070 [DOI] [PubMed] [Google Scholar]
- 57.Tuffery-Giraud S, Saquet C, Thorel D, Disset A, Rivier F, et al. (2005) Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. Eur J Hum Genet 13: 1254–1260. 10.1038/sj.ejhg.5201478 [DOI] [PubMed] [Google Scholar]
- 58.Okubo M, Noguchi S, Hayashi S, Nakamura H, Komaki H, et al. (2020) Exon skipping induced by nonsense/frameshift mutations in DMD gene results in Becker muscular dystrophy. Hum Genet 139: 247–255. 10.1007/s00439-019-02107-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Gao M, Zhang K, Jin R, Lv Y, et al. (2019) Molecular Genetics Analysis of 70 Chinese Families With Muscular Dystrophy Using Multiplex Ligation-Dependent Probe Amplification and Next-Generation Sequencing. Front Pharmacol 10: 814 10.3389/fphar.2019.00814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamed S, Sutherland-Smith A, Gorospe J, Kendrick-Jones J, Hoffman E (2005) DNA sequence analysis for structure/function and mutation studies in Becker muscular dystrophy. Clin Genet 68: 69–79. 10.1111/j.1399-0004.2005.00455.x [DOI] [PubMed] [Google Scholar]
- 61.Deburgrave N, Daoud F, Llense S, Barbot JC, Recan D, et al. (2007) Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat 28: 183–195. 10.1002/humu.20422 [DOI] [PubMed] [Google Scholar]
- 62.Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J (2005) MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics 6: 29–35. 10.1007/s10048-004-0204-1 [DOI] [PubMed] [Google Scholar]
- 63.Malhotra SB, Hart KA, Klamut HJ, Thomas NS, Bodrug SE, et al. (1988) Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science 242: 755–759. 10.1126/science.3055295 [DOI] [PubMed] [Google Scholar]
- 64.Winnard AV, Mendell JR, Prior TW, Florence J, Burghes AH (1995) Frameshift deletions of exons 3–7 and revertant fibers in Duchenne muscular dystrophy: mechanisms of dystrophin production. Am J Hum Genet 56: 158–166. [PMC free article] [PubMed] [Google Scholar]
- 65.Lidov HG, Kunkel LM (1997) Dp140: alternatively spliced isoforms in brain and kidney. Genomics 45: 132–139. 10.1006/geno.1997.4905 [DOI] [PubMed] [Google Scholar]
- 66.Austin RC, Howard PL, D'Souza VN, Klamut HJ, Ray PN (1995) Cloning and characterization of alternatively spliced isoforms of Dp71. Hum Mol Genet 4: 1475–1483. 10.1093/hmg/4.9.1475 [DOI] [PubMed] [Google Scholar]
- 67.gnomAD (2020, April 14) DMD/gnomAD.
- 68.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brogna S, Wen J (2009) Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 16: 107–113. 10.1038/nsmb.1550 [DOI] [PubMed] [Google Scholar]
- 70.Bouge AL, Murauer E, Beyne E, Miro J, Varilh J, et al. (2017) Targeted RNA-Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci Rep 7: 39094 10.1038/srep39094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Y, Deng H, Chen X, Li H, Yang C, et al. (2019) Skipping of an exon with a nonsense mutation in the DMD gene is induced by the conversion of a splicing enhancer to a splicing silencer. Hum Genet 138: 771–785. 10.1007/s00439-019-02036-2 [DOI] [PubMed] [Google Scholar]
- 72.Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, Verdura E, Lazaro K, et al. (2013) Interplay between DMD point mutations and splicing signals in Dystrophinopathy phenotypes. PLoS One 8: e59916 10.1371/journal.pone.0059916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miro J, Bourgeois CF, Claustres M, Koenig M, Tuffery-Giraud S (2018) Identification of Splicing Factors Involved in DMD Exon Skipping Events Using an In Vitro RNA Binding Assay. Methods Mol Biol 1687: 157–169. 10.1007/978-1-4939-7374-3_11 [DOI] [PubMed] [Google Scholar]
- 74.Aartsma-Rus A, Ginjaar IB, Bushby K (2016) The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 53: 145–151. 10.1136/jmedgenet-2015-103387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Musacchia F, Ciolfi A, Mutarelli M, Bruselles A, Castello R, et al. (2018) VarGenius executes cohort-level DNA-seq variant calling and annotation and allows to manage the resulting data through a PostgreSQL database. BMC Bioinformatics 19: 477 10.1186/s12859-018-2532-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.