

Abstract
Recent approvals of TRK inhibitors have demonstrated the success of a tumor agnostic approach to oncogene-targeted therapy across cancers. Collective data from acquired resistance studies suggest that these mechanisms, which include both kinase domain mutations and bypass signaling via RTK-RAS-RAF-MAPK pathways, frequently recur regardless of tumor type, oncogene, and drug.
Precision oncology has advanced significantly in the last 20 years with an ever-growing list of targets and approved therapies. Oncogene targeted therapies that inhibit ABL, EGFR, ALK, and others via small molecule inhibitors comprise a large proportion of approved targeted therapies. Until recently, however, targeted therapies were developed and approved in a tumor type or histology-specific manner, likely due, at least in part, to two important factors. First, the earliest oncogene targets were all strongly associated with one tumor type (e.g., trastuzumab in HER2+ breast cancer, imatinib in BCR-ABL CML, erlotinib in EGFR mutant non-small cell lung cancer (NSCLC), and vemurafenib in BRAF V600E melanoma). Second, an early example of oncogene targeted therapy in colorectal cancer with BRAF V600E displayed poor tumor response rates compared to melanoma with BRAF V600E, leading to the (incorrect) notion that oncogene targeted therapies would perform very differently in different tumors harboring the same oncogene.
A paradigm shift occurred recently with the US FDA approval for TRK inhibitors, larotrectinib and entrectinib, which are not restricted to a tumor histology and only require the presence of a gene fusion involving NTRK1, NTRK2, or NTRK3, which encode the receptor tyrosine kinases TRKA, TRKB, and TRKC, respectively. These “tumor agnostic” approvals follow a clear biological rationale. Early preclinical work demonstrated that the small molecule TRKA/B/C inhibitor ARRY-470 (larotrectinib), could potently and selectively inhibit auto-activation of TRK fusion kinases, leading to inhibition of canonical cancer signaling pathways including MAPK and AKT (Vaishnavi et al., 2013). Using human-derived cancer cell lines from lung adenocarcinoma, colorectal cancer, and acute myeloid leukemia, all bearing a TRK fusion, larotrectinib was demonstrated to have a similar effect on target inhibition, downstream signaling, in vitro cellular proliferation, and in vivo tumor growth in xenograft mouse models (Doebele et al., 2015). Thus, it was not surprising that larotrectinib and entrectinib demonstrated robust anti-tumor activity across all tumor types bearing NTRK1/2/3 fusions (Drilon et al., 2018), leading to the first tumor agnostic oncogene targeted therapy approvals. Other small molecule tyrosine kinase inhibitors (TKIs) have also accumulated (mostly anecdotal) data to support tumor agnostic activity of oncogene targets including ALK, ROS1, RET fusions, and even BRAF V600E mutations. This approach has been labeled tumor agnostic because it disrupts the historical approach of histology-focused drug development, but a better term might be “biology-centric” to reflect that this strategy follows a clear cancer cell signaling program common to these oncogenes.
Despite the clear benefit for patient outcomes with oncogene-targeted therapies, drug resistance inevitably develops. Cocco et al. describe several drug resistance mechanisms in patients with gastrointestinal malignancies harboring NTRK1/2/3 fusions treated with TRK inhibitors (larotrectinib, entrectinib and LOXO-195) using patient-derived models and ctDNA obtained from patients at the time of disease progression (Cocco et al., 2019). Specifically, they describe kinase domain mutations (KDM) including the gatekeeper mutation (NTRK1 F589L) and the solvent front mutation (NTRK1 G595R). These mutations occurred in both colorectal and pancreatic cancers, demonstrating that drug resistance is also tumor agnostic or biology-centric. The development of these mutations was predicted from prior experience with TKIs in numerous disease and oncogene states. The exact frequencies and positions of the KDMs varies with the target and the structure of the drug, but resistance can be successfully overcome with rationally designed inhibitors that bind with high affinity to the oncogene targets harboring these mutations. Indeed, the ability to overcome NTRK1 KDM was demonstrated here using LOXO-195 (Cocco et al., 2019). TKI resistance therefore predictably proceeds through target- and drug-specific KDM, regardless of tumor type.
Cocco et al., further describe several bypass signaling resistance mechanisms involving MET, ERBB2, KRAS, BRAF, and MAP2K1. It is arguable that these mechanisms also would have been predicted from prior studies. MET amplification was one of the earliest bypass signaling mechanisms described in EGFR mutant NSCLC, but has now been described for ALK in the post-crizotinib era. ERBB2 bypass signaling has been described for EGFR and ROS1 targeted therapies, whereas its family member, EGFR, has been described as a mediator of resistance for therapies targeting ALK, ROS1, RET and NTRK1 fusions, and HER3 via NRG1 for ALK fusions (Davies et al., 2013; McCoach et al., 2018; Vaishnavi et al., 2017). RAS-mediated resistance has been described for ALK (KRAS), ROS1 (NRAS), RET (NRAS), EGFR (KRAS), and other oncogene-driven cancers (Cargnelutti et al., 2015; Nelson-Taylor et al., 2017; Oxnard et al., 2018). BRAF-mediated resistance has been observed at resistance for ALK (unpublished data) and EGFR (Oxnard et al., 2018). MAP2K1 mutations have been observed in ALK+ NSCLC (Crystal et al., 2014). Bypass signaling is a common mechanism of drug resistance to small molecule kinase inhibitors and appears to occur through a common set of signaling nodes involved in the RTK-RAS-RAF-MAPK pathway. Thus, drug resistance can be thought of not only as tumor agnostic, but also as target agnostic, with cancer cells utilizing a relatively finite number of resistance pathways, regardless of the tumor type, the target or the drug.
Bypass signaling has been far harder to target than KDM, but deserves our collective attention. In this study, bypass signaling was observed in the majority of NTRK-resistance cases (75%), albeit in a relatively small number of cases. Although extensive testing for bypass signaling has not always been performed, it likely represents a significant proportion of cases not harboring KDM.
Several other features of bypass signaling are notable from this study and carry important clinical implications. First, tumors with bypass signaling do not respond to next generation targeted inhibitors given as monotherapy. This is expected given that bypass signaling by definition renders inhibition of the original oncogene futile and suggests that we need to employ broad next generation testing, not only targeted testing for KDM, prior to initiation of next generation inhibitors. Second, resistance via bypass signaling requires inhibition of not only the acquired bypass signaling track, but also the original oncogene, which has important clinical significance and makes clinical trials challenging.
NTRK1/2/3 fusions are rare oncogene targets (as are ROS1, ALK, RET and others), and resistance in this small study occurred through 5 different bypass signaling tracks in only 6 patients. It seems unlikely we can realistically hope to initiate and accrue to trials that may cover at best 10% of resistance in a rare oncogene population (e.g., TRK inhibitor plus MET inhibitor for MET-mediated resistance). One option is for physicians to prescribe off-label combinations, if available, or apply for compassionate use for non-approved, but promising agents. This allows for trial and error but will not help advance our understanding of efficacy and potential toxicity from new drug combinations. How then do we approach this growing problem?
If we are unable to run individual trials for rare oncogenes with rare resistance mechanisms, let us envision an agnostic trial platform for resistance in a similar way that we did for targeting oncogenes agnostic of tumor histology. Using the common MET bypass signaling as an example, a clinical trial adding a MET inhibitor to an existing EGFR, ALK, ROS1, TRK or other inhibitor at the time of MET mediated resistance may allow us to gather data from many more patients. This design can be similarly replicated with other common resistance pathways. We have at our disposal an ever-growing armamentarium of oncogene-targeted drugs, now is the time to start devising novel ways to test combinations for recurring, oncogene-agnostic bypass resistance mechanisms.
Figure 1.
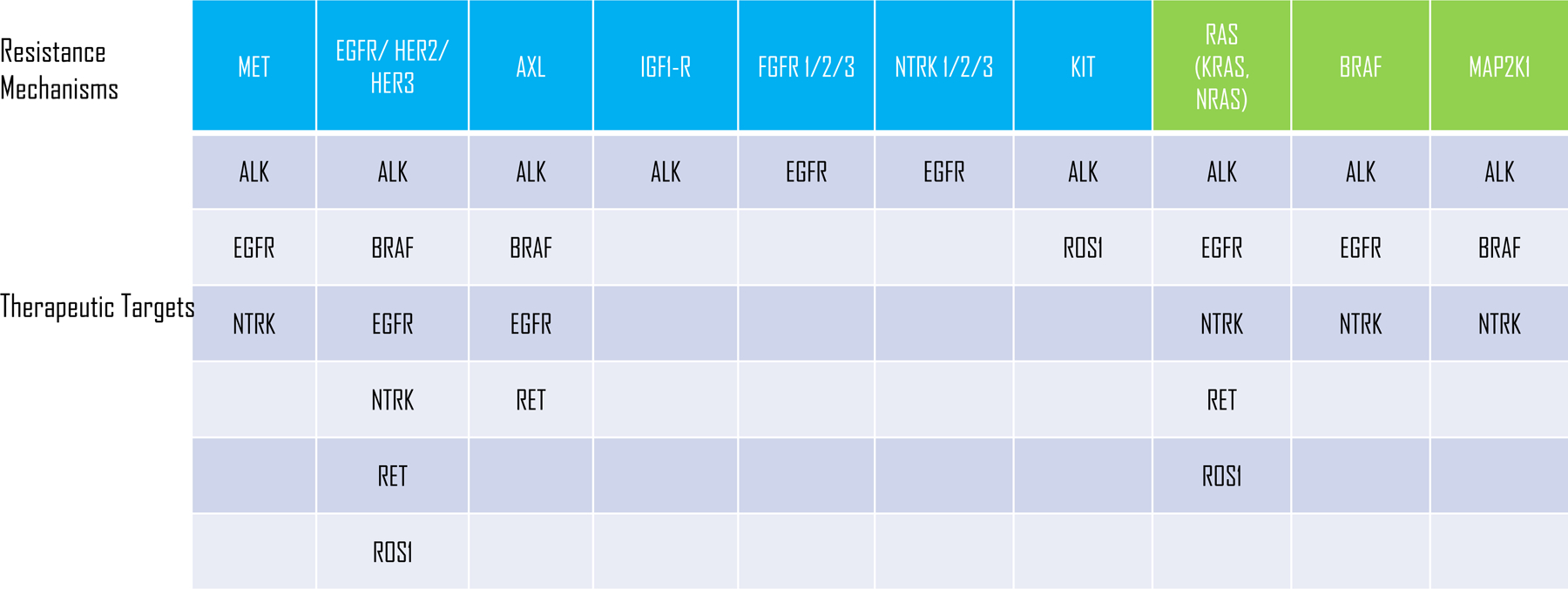
Oncogene target agnostic bypass resistance mechanisms. Resistance to small molecule inhibitors targeting the indicated oncogenes can be mediated by bypass mechanisms involving receptor tyrosine kinases (blue boxes) and RAS/RAF/MAPK pathway (green boxes).
Acknowledgements
RCD has received funding from NIH/NCI 5R01CA193935 and 5P50CA058187.
Footnotes
Declaration of Interests
RCD is a founder, shareholder and member of the scientific advisory board of Rain Therapeutics and has received licensing fees for a patent from Rain Therapeutics. RCD has received licensing fees for a patent from Abbott Molecular. RCD has received licensing fees for biologic materials from Genentech, Foundation Medicine, Black Diamond, Pearl River, Ariad Pharmaceuticals, Chugai, Blueprint Medicines, Loxo Oncology and Ignyta. RCD has served on ad hoc advisory boards for Loxo, Ignyta, Genentech/Roche, AstraZeneca, Takeda/Millenium and Bayer.
References
- Cargnelutti M, Corso S, Pergolizzi M, Mevellec L, Aisner DL, Dziadziuszko R, Varella-Garcia M, Comoglio PM, Doebele RC, Vialard J, and Giordano S (2015). Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget 6, 5182–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. (2014). Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, Vaishnavi A, Bunn PA Jr., Heasley LE, Tan AC, et al. (2013). Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One 8, e82236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, Jimeno A, Varella-Garcia M, Aisner DL, Li Y, et al. (2015). An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo AS, et al. (2018). Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, Estrada-Bernal A, Davies KD, Merrick DT, Bunn PA Jr., et al. (2018). Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin Cancer Res 24, 3334–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Taylor SK, Le AT, Yoo M, Schubert L, Mishall KM, Doak A, Varella-Garcia M, Tan AC, and Doebele RC (2017). Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated By Reactivation of RAS/MAPK Signaling. Mol Cancer Ther 16, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, et al. (2018). Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol 4, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, et al. (2013). Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med 19, 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, et al. (2017). EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer Res 77, 3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]