

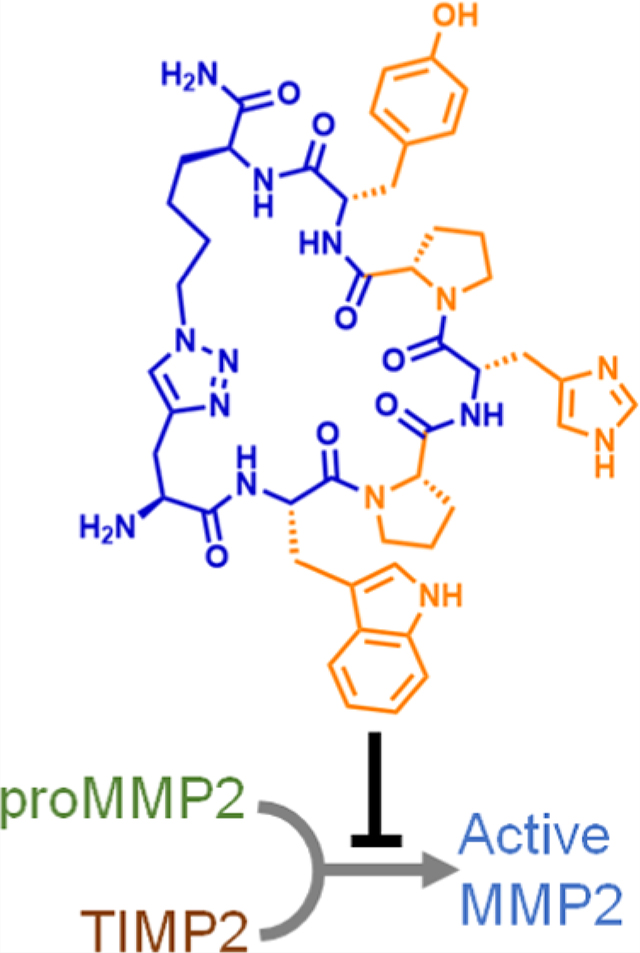
Abstract
We report on a cyclic peptide that inhibits matrix metalloproteinase-2 (MMP2) activation with a low-nM-level potency. This inhibitor specifically binds to the D570-A583 epitope on proMMP2 and interferes with the protein–protein interaction (PPI) between proMMP2 and tissue inhibitor of metalloproteinases-2 (TIMP2), thereby preventing the TIMP2-assisted proMMP2 activation process. We developed this cyclic peptide inhibitor through an epitope-targeted library screening process and validated its binding to proMMP2. Using a human melanoma cell line, we demonstrated the cyclic peptide’s ability to modulate cellular MMP2 activities and inhibit cell migration. These results provide the first successful example of targeting the PPI between proMMP2 and TIMP2, confirming the feasibility of an MMP2 inhibition strategy that has been sought after for 2 decades.
Graphical Abstract
INTRODUCTION
Matrix metalloproteinase-2 (MMP2) is a Zn-dependent endopeptidase with diverse biological functions, one of which is to facilitate cell migration by degrading the extracellular matrix and cell adhering molecules.1,2 Elevated MMP2 expression levels prevail in pathological processes such as tumor metastasis and atherosclerosis plaque rupture, where MMP2 activities often correlate with disease severity.2–4 Results from biomedical studies further support the hypothesis that elevated MMP2 activities promote the progression of those diseases. Consequently, MMP2 inhibitors have attracted significant interest but remain elusive despite decades of drug development efforts.5,6
Because MMP2 is an enzyme with a substrate-binding pocket, the most natural and popular strategy for MMP2 inhibition was to irreversibly halt the catalytic process using substrate analogues.7,8 More than 30 small-molecule inhibitors that came out of pharmaceutical pipelines shared this mechanism of action. They all showed promising efficacy in preclinical studies,9–11 but none of them demonstrated the desired outcomes in clinical trials. Subsequent analysis has linked those disappointing clinical results to the drugs’ off-target effects on other MMPs, which share significant structural homology with MMP2 but perform distinct physiological functions (Figure S1).12–16 These results highlight the necessity of improving target specificity during drug development, which has become the current consensus in the field of MMP inhibition.16,17 Nevertheless, the aforementioned structural homology makes this task difficult.
In light of the challenges associated with inhibiting MMP2 catalytic activity, targeting the MMP2 zymogen activation process has emerged as a promising strategy.17–20 As shown in Figure 1a, secreted proMMP2 must be cleaved by a membrane-tethered MMP to generate active MMP2.21 In most cases, this activation process requires the formation of a ternary protein complex that consists of proMMP2, tissue inhibitor of metalloproteinases-2 (TIMP2), and membrane-bound MMP14 (also known as MT1-MMP).22–24 Structural studies led to the hypothesis that inhibiting this protein complex could prevent the cleavage of proMMP2 and consequently decrease the amount of active MMP2 (Figure 1b).25
Figure 1.

Mechanism of inhibiting MMP2 activation by blocking the TIMP2–proMMP2 interaction. (a) Simplified illustration of the MMP2 activation process. The binding between TIMP2 and proMMP2 is critical to MMP2 activation mediated by another enzyme, MT1-MMP. (b) Blocking the PPI between TIMP2 and proMMP2 can prevent MMP2 activation. (c) Crystal structure of proMMP2 as adapted from PDB: 1GXD. The TIMP2-binding region on proMMP2 (D570-A583, highlighted in red) appears to be a structureless coil.
Indeed, previous studies have shown that anti-MT1-MMP antibodies could lower the production of active MMP2. In mice models, these antibodies demonstrated promising therapeutic effects.19 However, MT1-MMP’s biological functions are not limited to MMP2 activation. Therefore, global MT1-MMP inhibition methods, such as the use of antiMT1-MMP antibodies, still suffer from low specificity.20 The only exception is the murine 9E8 monoclonal antibody, which selectively inhibits MT1-MMP’s ability to activate MMP2.20 Nevertheless, developing therapeutics from this murine antibody requires challenging humanization processes. Indeed, 7 years after its discovery, no further progress has been reported. Currently, there is a pressing need for molecularly well-defined compounds (such as small molecules and short peptides) that target this protein complex and specifically inhibit MMP2 activation.
To this end, we propose to target the protein–protein interaction (PPI) between TIMP2 and proMMP2 instead. Because this PPI is unique to MMP2 activation, inhibitors acting on this PPI will be highly specific. Moreover, this PPI interface involves a critical epitope (D570-A583 on proMMP2) that shares little sequence homology with other MMPs and the rest of the human proteome (Figure S2).25 Therefore, a ligand that binds to this epitope promises less off-target effects. The technological difficulty here is that this epitope is structureless (Figure 1c) and it displays no binding pockets. Such a loop segment is a challenging target for traditional drug discovery strategies focusing on small molecules. As a result, almost 2 decades after the discovery of this PPI, a compound that can inhibit this PPI remains elusive.
Inspired by recent developments in cyclic peptide-based PPI inhibitors,26–34 we envisioned that we could address the unmet need using cyclic peptides. Herein, we report on a highly rigid cyclic peptide that specifically binds to the D570-A583 epitope of proMMP2 with a low nM affinity. We validated its ability to interfere with the TIMP2–proMMP2 interaction and tested its efficacy of modulating MMP2 activities using a human melanoma cell line.
RESULTS
Epitope-Targeted Library Screening Generated Hit Sequences
To identify compounds that can bind to the D570-A583 epitope, we performed a high-throughput screening using a one-bead–two-compound monocyclic/linear peptide library.26,35 We constructed this library through five iterations of split-and-pool processes, using 18 natural amino acids as building blocks (Figure 2a). We used Cu-catalyzed alkyne– azide cyclization and installed an extra N-terminal alkyne group (Figure S3).26,35 We removed the sequences that nonspecifically bound to a panel of dyes and proteins and screened this precleared library against a biotinylated D570-A583 epitope bearing an azide group (Figures S4 and S5). A strong binding between the synthetic epitope and a peptide sequence would promote a proximity-induced click reaction, allowing for hit identification through the biotin tag (Figure S5). The hit sequences were then determined by mass spectrometry, following established protocols.26,35 This screening generated 15 hit sequences, including four leucine/isoleucine isomers (Figure 2b).
Figure 2.

Validation of the hit sequences generated from library screening. (a) Generic structure of the cyclic peptide library. (b) Identified hits from the screening. (c) Förster resonance energy transfer (FRET) signals generated from the binding between 100 nM fluorescein-labeled epitope (Fluo-epitope) and varying concentrations of rhodamine B-modified ligand (Rhod-peptide). (d) Structure of cy(WPHPY) with and without the rhodamine B tag. (e) Competitive FRET results obtained using 100 nM Fluo-epitope, 100 nM Rhod-cy(WPHPY), and varying concentrations of cy(WPHPY) as the donor, acceptor, and competitor, respectively. The interaction between unlabeled cy(WPHPY) and Fluo-epitope displaced Rhod-cy(WPHPY), causing decreased FRET signals. (f) Fluorescence polarization (FP) assay results demonstrating the binding affinity between Rhod-cy(WPHPY) (100 nM) and the full-length recombinant proMMP2. (g) Competitive FP results demonstrating the binding between the unlabeled cy(WPHPY) and recombinant proMMP2 (100 nM). Here, the unlabeled cy(WPHPY) displaced Rhod-cy(WPHPY) (100 nM) and led to decreased FP values.
To validate the identified hits, we conjugated the identified cyclic peptide hits to rhodamine B (Rhod) and the epitope to fluorescein (Fluo) (Figures S6–S21). Binding between the cyclic peptide and the epitope would bring the two fluorophore tags into proximity, which would generate a Förster resonance energy transfer (FRET) signal. Therefore, we could evaluate binding affinities by titrating Fluo-epitope with Rhod-peptide and analyzing the FRET signals. As shown in Figure 2c, 7 of the 15 sequences bound to the epitope with low micromolar affinities (Figure S22). We chose Rhod-cy(WPHPY) for further studies because it showed the highest binding affinity (Figure 2d).
cy(WPHPY) Binds to proMMP2 with a Low nM Affinity
Because FRET-based titration experiments frequently suffer from artifacts such as microenvironment-induced quantum yield changes, we carried out more experiments to validate the binding between Rhod-cy(WPHPY) and the epitope. We found that the binding was insensitive to the titrant designation or the tag identity on the epitope (Figures S23 and S24). Conjugating cy(WPHPY) to a different fluorophore, however, destroyed the binding (Figure S25), indicating that N-terminal modifications to cy(WPHPY) could affect the binding significantly. Based on this result, we removed the N-terminal rhodamine B tag and tested its binding to the epitope using a competitive FRET approach (Figure S26). Here, cy(WPHPY) was used to titrate a mixture of Rhod-cy(WPHPY) (100 nM) and Fluo-epitope (100 nM). The cy(WPHPY)–epitope binding would disrupt the Rhodcy(WPHPY)–Fluo-epitope interaction and therefore lower the FRET signal. To our surprise, the result (Figure 2e) showed that the unconjugated cy(WPHPY) bound to the epitope with a much higher affinity (Kd = 1 nM) compared with Rhodcy(WPHPY).
To test if the observed cyclic peptide–epitope binding could translate to cyclic peptide–protein binding, we expressed and purified recombinant full-length proMMP2 and performed fluorescence polarization (FP) experiments to validate the binding. As shown in Figure 2f, Rhod-cy(WPHPY) was able to recognize recombinant proMMP2 with a high nM affinity (Kd = 280 nM), which was consistent with the epitope binding assay. This validated binding allowed us to implement a competitive FP assay to further test the binding between unmodified cy(WPHPY) and recombinant proMMP2. Here, we used unlabeled cy(WPHPY) to titrate a solution of Rhodcy(WPHPY) and recombinant proMMP2. Similar to the competitive FRET assay described above, cy(WPHPY) was expected to replace Rhod-cy(WPHPY) on proMMP2, causing decreased FP values. The result proved that the unmodified cy(WPHPY) peptide bound strongly to proMMP2 (Kd = 2.3 nM, Figure 2g), which also agreed with the epitope binding assay results.
Structure-Activity Relationship Analysis
We sought to elucidate the structure–activity relationship. The existence of two proline residues pointed to prominent backbone rigidity, which was expected to confer entropic advantages and contribute to enhanced binding affinities by providing privileged conformations.36 To evaluate the backbone rigidity, we performed molecular dynamics simulations of cy(WPHPY). We found that the two proline residues caused a unique kink in the backbone structure, which significantly limited the range of motion of the W, H, and Y residues (Figure 3a and Tables S1–S2, Figure S27). Substituting either of the two proline residues with alanine caused complete loss of binding to proMMP2, as demonstrated by the competitive FP results (Figure 3b,c). Also, alanine substitution at the W, H, and Y residues abolished the binding. These results indicated that both the identity and the orientation of the side-chain groups on cy(WPHPY) were critical to its binding affinity to proMMP2.
Figure 3.

Analysis of the structure–activity relationship. (a) Optimal conformations of cy(WPHPY) as calculated using molecular dynamics (MD) approaches. The blue wire is the most representative conformation generated from the conformation search. The orange wires illustrate the conformations that were evenly extracted every 2 ns from the MD simulation trajectory. (b) Alanine scanning variations of cy(WPHPY). (c) Competitive FP results obtained using alanine-substituted cy(WPHPY) variations. The competitive FP results of cy(WPHPY) were included as a reference (red line, peptide 1). (d) Partially scrambled epitope sequences in comparison with the original epitope. (e) FRET signals generated from the binding between 100 nM Fluo-epitope variations and varying concentrations of Rhod-cy(WPHPY). The result from the original Fluo-epitope was included as a reference. C5 was not able to bind to Rhod-cy(WPHPY), while M5 and N5 retained the binding affinity. (f) Competitive FRET assay results demonstrating the lack of binding affinity between the unlabeled cy(WPHPY) and the partially scrambled epitopes (M5 and N5). Unlabeled cy(WPHPY) could not displace Rhod-cy(WPHPY) from the epitope; therefore, the FRET intensities did not change. The result from the original Fluo-epitope was included as a reference.
To map the critical interaction sites on the epitope, we synthesized partially scrambled epitopes and evaluated their binding affinities to the cy(WPHPY) peptide using the aforementioned FRET approach (Figures 3d and S28–S31). The results showed that altering the C terminal of the epitope caused a loss of binding (Figure 3e), while scrambling the middle and N-terminal segments retained the binding to Rhodcy(WPHPY). Interestingly, we also found that preserving the C terminal alone could not maintain the binding to Rhodcy(WPHPY) (Figure S32). These results suggested that Rhodcy(WPHPY) interacted with multiple segments on the epitope, and this interaction was slightly more dependent on the C terminal. We further evaluated the binding between the unlabeled cy(WPHPY) and the scrambled epitopes, using the competitive FRET method. Even though M5 and N5 were able to bind to Rhod-cy(WPHPY), they were not able to recognize the unlabeled cy(WPHPY) (Figure 3f). Since the original and partially scrambled epitopes all lack secondary structures (Figures S28–S31), these results hinted that the whole epitope participated in the binding, perhaps by adopting a favored conformation that was induced by the cyclic peptide ligand. In addition, we found that the cy(WPHPY)–epitope interaction caused an increase of Trp fluorescence (Figures S33 and S34), while the alanine-substituted peptide, cy(WAHPY), did not. Because Trp fluorescence is sensitive to local environments, this increase was likely due to the epitope conformation change.37
cy(WPHPY) Inhibits the TIMP2–MMP2 Interaction through Binding to the MMP2 Hemopexin Domain
The TIMP2-binding region on the hemopexin domain remains intact during MMP2 activation. Therefore, we expected that cy(WPHPY) could bind to both pro- and active MMP2. To validate if cy(WPHPY) could interfere with the TIMP2– MMP2 protein–protein interaction, we performed a competitive binding test based on enzyme-linked immunosorbent assay (ELISA). WM115 cells (a human metastatic melanoma cell line) were cultured in serum-free media, which were used as a source of MMP2.38 As shown in Figure 4a, surface-immobilized recombinant TIMP2 was able to pull down MMP2 (pro- and active) from culture media, and cy(WPHPY) addition led to decreased MMP2 levels in a concentration-dependent manner. These results agreed with our hypothesis that cy(WPHPY) could inhibit the PPI between TIMP2 and MMP2.
Figure 4.

cy(WPHPY) inhibited the TIMP2–proMMP2 interaction and prevented proMMP2 activation. (a) Principle and results of the ELISA-based competitive binding assay. Surface-immobilized recombinant TIMP2 can pull down MMP2 from the solution, while cy(WPHPY) binds to MMP2 and blocks this interaction. Phosphate-buffered saline (PBS) was used in place of cy(WPHPY) in the control sample. (b–f) Schematic illustration and results of the in vitro bioactivity assay evaluating the function of cy(WPHPY). (b) Media from WM115 cell culture were collected and incubated with varying concentrations of cy(WPHPY). The corresponding MMP2 activities were analyzed using an MMP2-specific fluorogenic substrate. The assay buffer was used in place of cy(WPHPY) in the control sample. (c) WM115 culture media were incubated with TIMP2 and cy(WPHPY), and the corresponding MMP2 activities were evaluated. For the control samples, the assay buffer was used in place of cy(WPHPY) and TIMP2. (d–f) WM115 cells were cultured with varying concentrations of cy(WPHPY), and the corresponding media were collected for subsequent analyses. Serum-free media without cy(WPHPY) were used for the control samples. (d) Gelatin zymography and western blotting results demonstrate that cy(WPHPY) inhibited the MMP2 activity in the WM115 cell culture. (e, f) Enzymatic activity assays show that cy(WPHPY) was highly specific to MMP2 activities and did not affect MMP9. (g, h) Confocal microscopy images of WM115 cells showing membrane-bound MMP2 immunofluorescence. The scale bars are 10 μm. Media without cy(WPHPY) were used in the control sample. (i) Comparison of the single-cell MMP2 immunofluorescence signals extracted from the confocal images. A total of 30 single cells were analyzed for each image. The boxes denote the middle two quartiles, and the horizontal blue bars represent median values. The whiskers show the standard deviation values.
In addition to the hemopexin domain shared by pro- and active MMP2, the catalytic domain of active MMP2 is also a TIMP2 target. This alternative PPI is the basis of TIMP2mediated MMP2 inhibition. Therefore, we must rule out the possibility that cy(WPHPY) was interfering with the TIMP2–MMP2 (catalytic domain) interaction.23,39–41 To address this concern, we first mixed the WM115 culture media with different concentrations of cy(WPHPY) and then used an MMP2-specific fluorogenic substrate to evaluate the corresponding MMP2 activity (Figure S35). Because cy(WPHPY) did not interact with the fluorescent substrate (Figure S36), our result proved that cy(WPHPY) did not affect MMP2 activity (Figure 4b). Nevertheless, because the size of cy(WPHPY) was small, even if it bound to the catalytic domain, it might not block the substrate’s access to the catalytic center. To further validate our hypothesis, we introduced recombinant TIMP2 into the same experiment. Here, we found that cy(WPHPY) was not able to rescue the MMP2 activity from TIMP2 inhibition (Figure 4c), which proved that the PPI of TIMP2–MMP2 at the MMP2 catalytic domain was not affected. Taken together, these results supported our hypothesis that cy(WPHPY) specifically acted on the TIMP2–MMP2 interaction through the MMP2 hemopexin domain.
cy(WPHPY) Specifically Inhibits proMMP2 Activation In Vitro
We then moved on to test our hypothesis that disturbing the TIMP2–MMP2 interaction through the hemopexin domain will prevent proMMP2 activation. We treated WM115 cells with different concentrations of cy-(WPHPY) and analyzed the MMP2 content in the corresponding culture media. We first used gelatin zymography to evaluate how cy(WPHPY) affected MMP2 activity. As shown in Figure 4d, cy(WPHPY) treatment caused decreased MMP2 activity in the culture media, and the effect was dependent on the cy(WPHPY) concentration. To rule out the possibility that the observed decrease was due to cy(WPHPY) inhibiting MMP2 catalytic activity, we performed western blotting to quantify the amount of active MMP2 in the media. As expected, we observed a similar trend (Figure 4d). Based on the band intensity on the gel, we estimated an IC50 value of ∼20 nM (Figure S37), which was consistent with the Kd value obtained from binding assays (Figure 2g). These results supported our proposed mechanism of action that cy(WPHPY) inhibited proMMP2 activation.
Because our strategy targeted the unique D570-A583 epitope, which shares little homology with other MMPs, the inhibition was expected to be highly specific. To test this hypothesis, we used enzyme-specific fluorogenic substrates to analyze the activities of MMP2 and its most similar gelatinase relative, MMP9, in the culture media (Figures S35, S38, and S39). As shown in Figure 4e,f, cy(WPHPY) specifically acted on MMP2 and did not affect MMP9. In addition, we found that the linear sequence, WPHPY, showed no inhibition effect on MMP2 in the same experiment (Figure S40).
According to our proposed mechanism of action, cy(WPHPY) was expected to prevent the cell membrane association of proMMP2. To visualize such an effect, we performed confocal microscopy experiments. We treated WM115 cells with cy(WPHPY) for 15 h and subsequently fixed the cells using paraformaldehyde. We then stained MMP2 using anti-MMP2 antibodies and evaluated the corresponding immunofluorescence. Because no permeabilizer was used, the majority of the fluorescence signal came from surface-bound MMP2. We found that cy(WPHPY) treatment led to decreased MMP2 signals (Figure 4g,h), which proved that fewer copies of MMP2 were associated with the cell surface. By extracting single-cell immunofluorescence data from the confocal images, we also validated that the observed MMP2 signal difference was statistically significant (Figure 4i).
cy(WPHPY) Inhibits Cell Migration
To validate if the inhibitory effect of cy(WPHPY) can translate into decreased cell mobility, we performed a wound-healing scratch assay. We used a micropipette tip to create artificial wounds on the cell culture and incubated the cells with cy(WPHPY) for 24 h (Figure 5a). Because these cells were pretreated with a mitotic inhibitor (mitomycin C), cells that appeared in the wound were due to cell migration. As shown in Figure 5b, cy(WPHPY) treatment led to decreased cell numbers in the wound region, demonstrating its ability to inhibit cell mobility.
Figure 5.

cy(WPHPY) inhibited cell migration. (a) Schematic illustration of the scratch wound-healing experiment. (b) Results of the scratch wound-healing experiment demonstrate that cy(WPHPY) treatments led to fewer cells migrating to the scratch wound. (c) Schematic illustration of the transwell cell invasion assay. Active MMP2 was required for the cells to digest the Matrigel and invade through the porous membrane. Before the final imaging stage, cells were fixed and stained with crystal violet. (d) Results of the transwell assay demonstrate that cy(WPHPY) significantly inhibited cell invasion. Images were taken after 24 h of incubation.
To further validate our findings in a more quantitative manner, we carried out a transwell cell invasion assay using a two-chamber apparatus.42 In the inner chamber, we cultured WM115 cells on Matrigel in serum-free media. A porous membrane separated this inner chamber from the outer chamber, which contained full media. To access the full media, cells must secret MMP2 to digest the Matrigel and migrate to the other side of the membrane (Figure 5c). We found that cy(WPHPY) treatment led to fewer cells on the full media side of the membrane, indicating that it inhibited cell migration (Figure 5d). This effect was consistent with the observation from the wound-healing experiment. In addition, the inhibition ability matched the Kd and IC50 values from previous assays (Figures 2g and S37), supporting our proposed mechanism of action.
DISCUSSION
Inhibiting MMP2 activities has profound therapeutic implications. Unfortunately, a clinically viable drug remains elusive, despite more than 3 decades of research efforts. Results from the failed clinical trials have underscored the need of developing highly specific MMP2 inhibitors. However, this is substantially challenging because of the prevalent structural similarities among the catalytic domain of different MMPs.16 Consequently, targeting the enzymatic activity of MMP2 has become a less-favored approach.17 Evidently, research interests have shifted toward alternative strategies, such as inhibiting the zymogen activation process involving the proMMP2/TIMP2/ MT1-MMP ternary complex.17–20 Pioneering work has focused on membrane-tethered MT1-MMP, as represented by the development of DX2400 and 9E8 antibodies.19,20 These studies have demonstrated the feasibility of achieving MMP2 inhibition by preventing proMMP2 activation. Nevertheless, therapeutic development based on these antibody leads faces significant obstacles, such as the lack of functional specificity of DX2400 and the murine origin of 9E8 mAb.20
On the other hand, targeting the interaction between proMMP2 and TIMP2 remains underexplored. The structural basis for this PPI involves two epitopes on proMMP2, the C-terminal fourth blade of the hemopexin domain and the loop between the third and fourth hemopexin blades.25 Because the fourth hemopexin blade exhibits very high sequence homology to many other proteins (Figure S41), it is not a suitable target for drug development. In contrast, the second epitope has a unique amino acid sequence. Nevertheless, this second epitope is structureless, which makes it a challenging target for developing high-affinity binders. Although studies on inhibiting a related PPI, i.e., between proMMP9 and TIMP1, have been successful,43,44 it is largely because that many parts of the proMMP9 hemopexin domain are structurally distinct and easier to target.45 Therefore, it was unclear whether proMMP2 could be targeted similarly. To this end, our results provide the first demonstration of blocking the proMMP2–TIMP2 interaction to inhibit proMMP2 activation, proving the feasibility of this long-sought strategy.
In this study, removing the rhodamine tag improved the binding affinity by 100-fold. Such a result may seem astonishing if one puts peptides and proteins in the same therapeutics category. However, the cyclic peptide presented here has a molecular weight less than 1 kDa, which makes it more similar to a small molecule than large biologics. Considering that the rhodamine tag size is more than half of the cyclic peptide size, it is expected that this tag would significantly affect the binding affinity. Besides, because a structureless loop was targeted, it was expected that this binding would be sensitive to structure variations. The alanine scanning and the epitope randomization results (Figure 3) further demonstrated this point. On the other hand, it was to our surprise that the epitope-based competitive FRET results matched extremely well with the recombinant protein-based competitive FP data. Although the target epitope resides at the peripheral region of the protein and its structureless nature is conserved in the full-length protein, neighboring epitopes and the protein’s tertiary structure could influence the peptide–epitope interaction in an unforeseeable manner. Therefore, it is always crucial to validate the binding using full-length proteins. Nevertheless, this extra effort does not diminish the feasibility or application of the epitope-targeted screening approach, which has been demonstrated in many studies.26,35,46
Our results also shed light on the structural biology of the TIMP2–MMP2 interaction. Instead of facilitating MMP2 activation, a more well-known role of TIMP2 is inhibiting active MMP2. The detailed mechanism of this inhibition is elusive, as the structure of the active MMP2–TIMP2 complex has not been solved. The current hypothesis is that the Nterminal domain of TIMP2 can bind to the catalytic domain of MMP2, thereby blocking the substrate entrance to the catalytic center.25,47 Moreover, an auxiliary binding between TIMP2 and the MMP2 hemopexin domain is believed to stabilize the interaction further.47 However, it is unclear if this interaction involves the same hemopexin epitope as the one implicated in proMMP2 activation. Because the hemopexin domain on proMMP2 is conserved through its activation process, the cy(WPHPY) peptide is expected to bind to both pro- and active MMP2. Our results (Figure 4c) demonstrated that binding of cy(WPHPY) to active MMP2 did not interfere with the inhibitory TIMP2–MMP2 interaction. Based on these data, we believe that the inhibitory PPI between TIMP2 and MMP2 does not involve the loop between the third and fourth hemopexin blades on MMP2.
Aside from the TIMP2/MT1-MMP-mediated process, other mechanisms of proMMP2 activation also exist. For instance, in human astrocytic tumor cases, prominent correlations exist among the MT2-MMP expression level, the MMP2 activity, and the tumor grade.48 These observations are consistent with the in vitro result that MT2-MMP can activate proMMP2 without help from MT1-MMP.49 Other types of TIMPs can also bind to proMMP2 to facilitate its activation.47 Therefore, our method of directly targeting proMMP2, rather than TIMP2 or MT1-MMP, promises more generalization potential. The uncertainty here is whether our targeted epitope on the hemopexin domain is also involved in these alternative MMP2 activation routes. As a natural extension to this work, we are currently testing the efficacy of cy(WPHPY) in MT1MMP- and TIMP2-deficient models. The result will provide structural insights into how proMMP2 interacts with other types of MT-MMPs and TIMPs.
The work presented here, together with recent reports from other groups and us, highlights the feasibility of using cyclic peptides to target protein–protein interactions. The two proline residues in cy(WPHPY) provided substantial backbone rigidity, which was proven critical to the binding. This finding is consistent with previous reports that increased structural rigidity in cyclic peptides translates into higher binding affinities to disordered protein structures. In light of these results, we are currently implementing medicinal chemistry optimization strategies on cy(WPHPY), with an emphasis on preserving the structural rigidity and the orientation of sidechain functional groups. We believe that such efforts promise more potent and specific inhibitors against MMP2 activation and ultimately a clinically viable strategy for MMP2 inhibition.
CONCLUSIONS
In conclusion, we have identified a cyclic peptide, cy(WPHPY), which inhibits MMP2 activation with a low-nM-level potency and high specificity. We have demonstrated that cy(WPHPY) functions by binding to the D570-A583 epitope on proMMP2, thus impeding the protein–protein interaction between TIMP2 and proMMP2. Using a human melanoma cell line, we have also confirmed that cy(WPHPY) can effectively inhibit cell invasion and migration (IC50 ∼ 20 nM), which underscores its potential clinical applications.
EXPERIMENTAL SECTION
Materials
TentaGel S-NH2 resin (loading capacity 0.28 mmol/g) was purchased from Rapp Polymere GmbH and Rink amide MBHA resin (loading capacity 0.678 mmol/g) from Aapptec (Louisville, KY). All of the Fmoc-protected amino acids were purchased from Anaspec (Fremont, CA) except Fmoc-L-propargylglycine (Pra) and FmocLys(N3)-OH (Az4), which were purchased from Chempep (Wellington, FL) and Chem-Impex (Wood Dale, IL), respectively. The coupling reagent 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 99.6%) was obtained from Chem-Impex (Wood Dale, IL). Diisopropylethylamine (DIEA, 99.5%) was purchased from ACROS (Germany). Phenyl isothiocyanate (PhNCS) and triisopropylsilane (TIPS) were obtained from TCI (Portland, OR). Piperidine was purchased from Alfa Aesar (Ward Hill, MA). Cyanogen bromide (CNBr) and 5(6)-carboxyfluorescein were obtained from ACROS (Pittsburg, PA). Rhodamine B (Rhod), cuprous iodide (CuI), and α-cyano-4-hydroxycinnamic acid (CHCA) were obtained from Sigma-Aldrich (St. Louis, MO). 5-Bromo-4chloro-3-indolyl-phosphate and nitro blue tetrazolium (BCIP/NBT) were acquired from Promega (Madison, WI). Tris base, sodium phosphate dibasic anhydrous (Na2HPO4, 99.6%), sodium phosphate monobasic monohydrate (NaH2PO4, 99.4%), sodium chloride (NaCl), ascorbic acid, Tween 20, sodium dodecyl sulfate (SDS), bovine serum albumin (BSA), acetonitrile (CH3CN), diethyl ether (Et2O), chloroform (CHCl3), ethyl acetate (EA), N,N′-dimethylformamide (DMF), and dichloromethane (DCM) were purchased from Thermo Fisher Scientific (Waltham, MA). Recombinant TIMP2 (125639) was purchased from Abcam (Cambridge, MA).
Preparative Reversed-Phase (RP) High-Performance Liquid Chromatography (HPLC)
Preparative HPLC was performed on a Thermo Ultimate 3000BX HPLC instrument, using a Phenomenex C18 reversed-phase preparative column (Kinetex 5 μm EVO, 250 × 21.2 mm2). Nonlinear gradients of 0–100% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) were employed, and the gradient parameters were adjusted for each product to achieve desired separation efficiencies. A multiwavelength UV–vis detector was used to monitor the absorbance at 215, 280, 480, and 560 nm.
Analytical HPLC
The purity of the peptide was analyzed on a Thermo Ultimate 3000SD HPLC instrument, using a Phenomenex C18 reversed-phase analytical column (Kinetex 2.6 μm EVO, 250 × 4.6 mm2). A gradient of 0–100% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) was employed with a flow rate of 1 mL/min. A UV–vis detector was used to monitor the absorbance at 280 or 560 nm. The purity of all cyclic peptides used for binding assays and biological activity assays was >95%.
Mass Spectrometry
The MS and MS/MS spectra were obtained using a SCIEX 5800 matrix-assisted laser desorption ionization timeof-flight (MALDI-TOF) mass spectrometer.
Solid-Phase Peptide Synthesis
The peptides were synthesized following the standard Fmoc SPPS coupling process. Unless otherwise noted, Rink Amide MBHA resin was used for the synthesis. To couple amino acids to the resin, the Fmoc group on the resin was first removed by 20% piperidine/DMF solution (10 min, three times). Fmoc-AA-OH (3 equiv), DIEA (5 equiv), and HBTU (2.8 equiv) were mixed in DMF for 10 min, and the solution was then introduced to the deprotected resin. The mixture was gently agitated at room temperature for 1 h, followed by draining and washing (DMF, methanol, and DCM, three times each). To label the peptides with fluorophores, poly(ethylene glycol) (PEG), or biotin, the corresponding dye-COOH, Fmoc-PEG-OH, and biotin were coupled at the N-terminal using the SPPS procedure described above.
For constructing cyclic peptides, Fmoc-propargylglycine-OH (Pra) and Fmoc-azidolysine-OH (Az4) were inserted at the N and C terminals, respectively. A Cu-catalyzed click reaction was used for cyclization. Specifically, resins were incubated in 20% piperidine/ DMF with CuI (2.5 equiv) and L-ascorbic acid (5 equiv) at room temperature overnight. After cyclization, the beads were washed with sodium diethyldithiocarbamate (5% w/v) and DIEA (5% v/v) in DMF to remove the copper catalyst.
To cleave peptides off from the resin, a cleavage solution composed of TFA/TIPS/ddH2O (95:2.5:2.5) was used. The crude peptides were purified by preparative RP-HPLC, and the product purity and identity were confirmed by analytical RP-HPLC and mass spectrometry.
Rhod-cy(WPHPY), C80H96N17O11, [M + H]+ calculated 1471.76, found 1471.12. Rhod-cy(EWWFR), C86H104N19O12, [M + H]+ calculated 1595.90, found 1595.67. Rhod-cy(THNWP), C74H93N18O12, [M + H]+ calculated 1426.67, found 1426.65. Rhodcy(IDYSP), C71H93N14O14, [M + H]+ calculated 1366.61, found 1366.62. Rhod-cy(VEYSP), C71H93N14O14, [M + H]+ calculated 1366.61, found 1366.71. Rhod-cy(RNWIR), C77H107N22O11, [M + H]+ calculated 1516.85, found 1516.69. Rhod-cy(IHWRW), C84H106N21O10, [M + H]+ calculated 1569.91, found 1569.71. Rhod-cy(RNWLR), C77H107N22O11, [M + H]+ calculated 1516.85, found 1516.93. Rhod-cy(WRLKG), C75H104N19O10, [M + H]+ calculated 1431.78, found 1431.84. Rhod-cy(WRIKG), C75H104N19O10, [M + H]+ calculated 1431.78, found 1431.85. Rhod-cy(QTNWF), C75H104N19O10, [M + H]+ calculated 1467.72, found 1467.80. Rhod-cy(WFQDS), C76H93N16O14, [M + H]+ calculated 1454.68, found 1454.80. Rhod-cy(LDYSP), C71H93N14O14, [M + H]+ calculated 1366.61, found 1365.71. Rhodcy(LHWRW), C84H106N21O10, [M + H]+ calculated 1569.91, found 1569.90. Rhod-cy(WWPAR), C80H100N19O10, [M + H]+ calculated 1487.80, found 1487.74. cy (WPHPY), C47H58N14O8, [M + H]+ calculated 947.07, found 947.46. cy(WPHPA), C41H54N14O7, [M + H]+ calculated 854.97, found 854.56. cy(WPHAY), C45H56N14O8, [M + H]+ calculated 921.03, found 920.89. cy(WPAPY), C44H56N12O8, [M + H]+ calculated 880.43, found 880.52. cy(WAHPY), C45H56N14O8, [M + H]+ calculated 920.44, found 920.56. cy(APHPY), C39H53N13O8, [M + H]+ calculated 831.94, found 831.78. Biotin-PEG5-epitope-Az4, C100H153N25O28S, [M + H]+ calculated 2185.53, found 2186.24. Fluo-epitope, C92H114N18O25, [M + H]+ calculated 1872.03, found 1872.72. Fluo-C5, C92H114N18O25, [M + H]+ calculated 1872.03, found 1872.69. Fluo-M5, C92H114N18O25, [M + H]+ calculated 1872.03, found 1872.72. Fluo-N5, C92H114N18O25, [M + H]+ calculated 1872.03, found 1872.71.
One-Bead–Two-Compound (OBTC) Library Construction
The one-bead–two-compound peptide library was constructed using the split-and-pool strategy on TentaGel S-NH2 resins. Methionine was first coupled to the resin, followed by glycine. Then, Az4 was coupled at 80% loading capacity. Eighteen natural amino acids (Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Trp, Tyr, and Val, all L-stereoisomers) were used as building blocks for the subsequent five randomized positions, and a Pra residue was attached at the N-terminal. The library was cyclized, as described above. Afterward, an additional Pra was attached to the N-terminal. The protecting groups were removed as described above. The deprotected resins were then washed with DMF, methanol, and DCM and kept under Argon for further use.
OBTC Library Screening
The precleared library (see the Supporting Information) was incubated with 10 μM biotinylated epitope (biotin-PEG5-D-P-G-F-P-K-L-I-A-D-A-W-N-A-Az4) in 0.1% BSA/TBST at room temperature for 6 h. The library was then washed with 0.1% BSA/TBST, TBST, and TBS solution and incubated with 7.5 M guanidine-HCl (pH 2.0) at room temperature for 2 h. Afterward, the beads were washed with water and incubated overnight in 1% BSA/TBST at 4 °C. The library was then incubated with streptavidin-AP (1:10 000 dilution) and its substrate BCIP/NBT, giving rise to dark purple hit beads. After incubation, the resins were washed with HCl (pH 2.0) to quench the reaction and then with 0.1% BSA/TBST (5 min × 3) at 4 °C. The colored hits were then picked out; washed with 7.5 M guanidine-HCl (pH 2.0), water, DMF, and DCM; and dried under vacuum.
Sequencing of the Hits
Edman Degradation
The first round of Edman degradation was performed to remove the click handle that was conjugated to the epitope.50 The hit beads were transferred into a glass vial with 50 μL of 2.5% PhNCS in pyridine/H2O (1:1) solution and flushed with Ar for about 15 s. The vial was then placed in a water bath at 50 °C for 30 min, and the solution was removed. The beads were then washed with ethyl acetate (three times) and DCM (once) and air-dried. Subsequently, 100 μL of TFA solution was added to the vial, flushed with Ar, and left for incubation in the water bath for 10 min at 50 °C. Afterward, the solution was removed, and the beads were washed three times with ethyl acetate and then with DCM. These procedures were repeated for another round of Edman degradation. Later the beads were incubated with a 20% TFA/water solution and heated at 60 °C for 15 min. After 15 min, the solution was removed, and the beads were washed with ethyl acetate and DCM and then air-dried.
Cleavage
After the Edman degradation, individual beads were transferred into a microcentrifuge tube containing 10 μL of water and 10 μL of a 0.50 M CNBr/0.2 M HCI solution. The tubes were microwaved for 1 min and dried using a centrifugal vacuum chamber at 45 °C.51
Sequencing by MALDI-TOF MS/MS
First, 0.55 μL of a 4 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) solution (50:50 ACN/ water with 0.1% TFA) was added in each microcentrifuge tube. The mixture was spotted on a 384-spot MALDI sample plate and air-dried.
The MS/MS spectra were obtained on a SCIEX 5800 mass spectrometer and analyzed using mMass to solve for the sequences.52
FRET Binding Assay
The fluorescein-labeled epitope was incubated with varying concentrations of the rhodamine-tagged cyclic peptides. The final concentration of the Fluo-epitope was kept at 100 nM in all samples. Then, 65 μL of the mixed solution was placed in a Greiner black 384-well low binding plate and incubated for 30 min. The FRET intensities were measured using a Synergy H1 microplate reader. For the competitive FRET assay, the concentrations of the epitope and the rhodamine-tagged cyclic peptide were kept at 100 nM in all samples, and the concentration of the unlabeled cyclic peptide (the competitor) was varied. Furthermore, 65 μL of the mixtures was added into a Greiner black 384-well low binding plate and incubated at room temperature for 30 min. The FRET intensities were measured using a Synergy H1 microplate reader. All of the FRET experiments were performed using individually prepared triplicate samples. The values from the triplicate were averaged and analyzed. Every experiment was independently repeated at least three times on different days, and a consistent trend was observed. Representative experiment results are presented in the paper.
proMMP2 Expression and Purification
The cDNA fragment encoding full-length human proMMP2 was inserted into an in-house expression vector in which it was N-terminally fused to a hexahistidine (His6)-MBP tag and a tobacco etch virus (TEV) protease cleavage site. The resulting plasmid was transformed into BL21(DE3) RIL cells (Novagen Inc.). After the cell density reached an optical density at 600 nm (OD 600) of ∼0.8, the protein expression was induced by 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and the cells continued to grow at 18 °C overnight. The cells were harvested and lysed in a buffer containing 25 mM Tris-HCl (pH 7.5), 25 mM imidazole, 1 M NaCl, 5% glycerol, and 1 mM PMSF. The proMMP2 fusion protein was first purified using a nickel column and eluted with a buffer containing 25 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5% glycerol, and 300 mM imidazole. Subsequently, the eluted protein sample was supplemented with 5 mM dithiothreitol (DTT) and incubated with TEV protease overnight. The tag-free MMP2 protein was further purified by ion-exchange chromatography on a Q HP column (GE Healthcare) and size-exclusion chromatography on a Hiload 16/600 Superdex 200 pg column (GE Healthcare) preequilibrated with a buffer containing 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 5% glycerol, and 5 mM DTT. The final protein sample was stored at −80 °C in a freezer before use.
FP Binding Assay
The rhodamine-tagged ligand Rhod-cy(WPHPY) was mixed with varying concentrations of the recombinant proMMP2 protein in PBS. The final concentration of Rhodcy(WPHPY) was kept at 100 nM in all samples. Then, 65 μL of the mixture was added in a Greiner black 384-well low binding plate and incubated at room temperature for 30 min. The fluorescence polarization values were measured with a Synergy H1 microplate reader. For the competitive polarization assay, the concentrations of the recombinant protein and Rhod-cy(WPHPY) were kept at 100 nM in all samples. The concentration of the unlabeled ligand cy(WPHPY) was varied. The mixtures were added into a Greiner black 384-well plate, and the fluorescence polarization values were measured with a Synergy H1 microplate reader. All of the FP experiments were performed in triplicates using individually prepared samples, and the results from the triplicates were averaged and analyzed. Each experiment was repeated at least three times on different days, and consistent trends were observed. Representative results are presented in the paper.
Conformational Search and Molecular Dynamics Simulation
To study the properties of cy(WPHPY) in silico, conformational search and molecular dynamics simulation were performed. The standard amino acid part of the peptide (WPHPY) was parameterized with the protein.ff14SB force field.53 The organic linkage that connects the amino acid was built by Avogadro v1.2.0 and then processed by Antechamber to calculate the AM1-BCC charge and assign the gaff2 force field parameters.54–56 After the amino acids and the small organic fragment were parameterized, they were assembled using xleap from AmberTools18.57 The topology and coordinate files for the hybrid peptide were saved as prmtop and inpcrd files, which were converted into counterpart top and crd files, respectively, using the script provided in the VM2 package.58 The whole peptide was treated as a flexible set to enable a thorough conformational search by using the VM2 package.
For the molecular dynamics simulation, the selected conformation of the peptide was immersed in a rectangular box of TIP3P water molecules with a margin distance of 12.0 Å.59 The solvated box was further neutralized with a Cl− counter ion using the tleap program. The water molecules and the whole system then went through a 3000step and 5000-step minimization to correct any inconsistencies, respectively. Next, we relaxed the system by slowly heating it up during an equilibrium course of 1 ns at 50, 100, 150, 200, 250, and 298 K. By using the GPU accelerated pmemd.cuda code in Amber18, 20 ns simulations were performed at 298 K in an NPT (isothermal–isobaric) ensemble.60 Particle mesh Ewald (PME) was employed to calculate the long-range electrostatic interactions, and the nonbonded energy cutoff was set to 10.0 Å.61
Cell Culture
The human metastatic melanoma cell line, WM115, was a gift from Prof. Yinsheng Wang (UC Riverside). It was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Corning) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 100 U/mL penicillin/streptomycin (Sigma). Cells were cultured under 5% CO2 in a 37 °C incubator. A trypsin-EDTA solution (0.05%, Sigma) was used for passaging once the cells reached 80–90% confluency.
Enzyme-Linked Immunosorbent Assay (ELISA)
A 96-well Nicoated plate was incubated with His-tagged recombinant TIMP2 protein (9 pmol/well) for an hour at room temperature. Afterward, the protein solution was removed, and the plate was washed three times with PBST (phosphate buffer saline, 0.05% Tween 20). To minimize any nonspecific binding, the plate was blocked with His6 peptide (9 pmol/well) for an hour at room temperature and washed with PBST three times.
WM115 cells were cultured to 80% confluency and then incubated in serum-free media for 24 h. The culture media were collected and concentrated using centrifugal membrane filters (10 kDa cutoff, Amicon). The concentrated media were aliquoted at 200 μL each. PBS solutions of cy(WPHPY) with varying concentrations of cy(WPHPY) (20 μL) were mixed with the aliquots. For the positive control, PBS was mixed with the aliquot. The mixtures were added into the above-mentioned 96-well plate. The plate was incubated on an orbital shaker at room temperature for an hour. Then, the wells were washed with PBST, and 100 μL of MMP2 primary antibody solution (Rabbit mAb, #40994, Cell Signaling Technology, 1:200 in 5% BSA/PBST) was added into each well. The plate was incubated at room temperature for an hour and then washed with PBST three times. Furthermore, 100 μL of secondary antibody (Goat Anti-Rabbit IgG, HRP-linked, ab97051, Abcam, 1:10 000 in 5% BSA/PBST) was added into each well, and the plate was incubated for another hour at room temperature. To generate luminescence readouts, the wells were washed with PBST three times, and then a luminol substrate was added into the wells (SuperSignal West Pico PLUS Chemiluminescent Substrate, Thermo). The luminescence was quantified using a SYNERGY H1 microplate reader (Biotek) after 5 min of additional incubation. For each cy(WPHPY) concentration, three individually prepared replicates were used, and the luminescence values were averaged and reported. To ensure reproducibility, this ELISA experiment was independently repeated three times with a different plate each time, and consistent results were observed.
Fluorogenic Substrate Assay
WM115 cells were cultured in full media to 80% confluency. Afterward, the media were replaced by serum-free media, and the cells were cultured for another 24 h. The media were then collected and concentrated with centrifugal membrane filters (10 kDa cutoff, Amicon). Once concentrated, the total protein content was quantified using a bicinchoninic acid (BCA) assay kit (Thermo) and normalized. In a 96-well microplate, 10 μL (1 μg/μL protein content) of the media was mixed with 90 μL of a tenascin C (TNC) assay buffer (50 mM Tris base, 0.15 M NaCl, 10 mM CaCl2, pH 7.5) and 10 μL of cy(WPHPY) (varying concentrations in PBS). For the positive control sample, 10 μL of the TNC assay buffer was added in place of cy(WPHPY). The plate was left for 5 min at 37 °C for equilibration, and 100 μL of MMP-specific substrates (Sigma, 3 μM) was added in the mix.62 The plate was then incubated at 37 °C for 20 min. The fluorescence was measured using a SYNERGY H1 microplate reader with an excitation wavelength of 325 nm and an emission wavelength of 393 nm.
For the experiment shown in Figure 4c, the same procedure was followed as that described above. The difference was that an additional 10 μL of the recombinant TIMP2 (1 μg/μL in PBS) was added to all of the mixtures.
For the experiment shown in Figure 4e,f, WM115 cells were cultured in full media to 80% confluency. Afterward, the media were changed to serum-free media containing varying concentrations of cy(WPHPY), and the cells were cultured for 24 h. The control sample contained no cy(WPHPY). The media were then collected, concentrated, and quantified as described above. In a 96-well microplate, 10 μL (1 μg/μL protein content) of the media was mixed with 90 μL of the TNC assay buffer and 100 μL of MMPspecific substrates (Sigma, 3 μM). The plate was then incubated at 37 °C for 20 min, and the fluorescence was measured as described above.
In every fluorogenic substrate experiment, three individually prepared replicates were used for each cy(WPHPY) concentration (including control), and the averaged values from the replicates were reported. To ensure reproducibility, all experiments were independently repeated three times, using fresh substrate each time. Consistent trends were observed.
Gelatin Zymography
In a 6-well plate, 600 k cells were seeded in full DMEM medium containing 10% FBS and 1% PS. After 8 h, each well was washed twice with serum-free media. Then, 2 mL of serum-free media containing different concentrations of cy(WPHPY) was added into the wells. For the positive control, no cy(WPHPY) was added. The plate was kept in the 5% CO2 incubator at 37 °C for 24 h. Afterward, the media were collected and concentrated using centrifugal membrane filters (10 kDa cutoff, Amicon). The protein content in each of the samples was quantified using a BCA assay kit (Thermo), and the solutions were normalized accordingly to reach a concentration of 10 μg/mL in each sample. A 7.5% acrylamide gel was prepared using 2 mL of gelatin (4 mg/mL in water) and a 1 mm electrophoresis plate. Then, 10 μL of the sample (10 μg/mL protein content) was added into each well, and a standard protocol was followed for running the gel.63 After the run, the gel was washed (2 × 30 min) with washing buffer (pH 7.5, 2.5% Triton X-100, 50 mM Tris-HCl, 5 mM CaCl2, 1 μM ZnCl2) at room temperature and then kept in the incubation buffer (1% Triton X-100, 50 mM Tris-HCl, 5 mM CaCl2, 1 μM ZnCl2) for 10 min at 37 °C. Later the buffer was replaced with a new incubation buffer and was kept in the incubator at 37 °C for 20 h. After incubation, the gel was washed using a washing buffer and was stained using a 0.3% Coomassie blue water solution (containing 4% methanol and 1% acetic acid) for 30 min at room temperature. Afterward, the gel was washed with H2O to remove the excess coloring. Then, the gel was incubated in the destain solution (4% methanol and 1% acetic acid in H2O) until the bands on the gel became clear. Following this process, we carried out three independent experiments. All three experiments showed similar results. A representative result is presented in Figure 4d.
Western Blot
WM115 cells were treated with different concentrations of the ligand in serum-free media for 24 h. For the positive control, PBS was added instead of the ligand. After preparing the samples, western blot was carried out following standard protocols.64 The antibodies (#13132, Cell Signaling Technology; ab97051, Abcam) were used at 1:1000 and 1:10 000 dilutions, as recommended by the manufacturers. The resulting membrane was developed using SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Scientific), and the image was taken using an Odyssey Fc imaging system (LI-COR Biosciences, Lincoln, NE). We followed this process to run three independent western blots, with freshly collected medium samples from new batches of cells each time. All three experiments showed similar results, and a representative one is presented in Figure 4d.
Immunofluorescence and Confocal Imaging
WM115 cells were cultured in 6-well plates in full media until reaching the desired confluency. For cy(WPHPY)-treated cells, cy(WPHPY) was added to reach a final concentration of 10 nM, and the cells were incubated for 15 h. The cells were fixed using 4% paraformaldehyde at room temperature for 15 min. Note that no membrane permeabilizer (such as Triton X-100) was used here because we wanted to assess membrane-associated MMP2. Subsequently, fixed cells were washed with PBS (three times) and blocked in a blocking buffer (PBS + 5% normal goat serum) for 1 h at room temperature. Afterward, cells were incubated with a 1:500 dilution of anti-MMP2 (#40994, Cell Signaling Technology) in the blocking buffer overnight at 4 °C and washed with PBS (three times). Then, cells were incubated with a 1:2000 dilution of Alexa Fluor 594-conjugated goat anti-rabbit IgG (#8889, Cell Signaling Technology) in the blocking buffer for 2 h at room temperature and washed thoroughly with PBS.
To image the cells, a Zeiss 880 inverted confocal laser scanning microscope (Carl Zeiss, Germany) was used. Image acquisition and analyses were carried out using the manufacturer’s software (ZEN, Carl Zeiss). Quantification of fluorescence intensity in single cells was performed using Fiji software. Thirty single cells were analyzed for each image.
The experiment was repeated three times, with different WM115 cells each time. Consistent results were observed, and the representative images are presented in Figure 4.
Scratch Wound-Healing Assay
The scratch assay was performed in 6-well plates. To coat the wells with gelatin, 2 mL of a 0.1% gelatin/H2O solution was added into each well, and the plates were incubated at 37 °C for 2 h. Afterward, the gelatin solution was removed, and the wells were washed with PBS three times. WM115 cells were seeded into the wells and cultured in media containing 0.3% FBS. When the cells reached 90% confluency, 3 μg/mL mitomycin C (Sigma-Aldrich) was added and incubated for 3 h to stop cell proliferation. A 10 μL pipet tip was used to create scratch wounds in a straight line. The wells were then washed three times with serum-free media. Subsequently, serum-free media containing different concentrations of cy(WPHPY) were added in the wells. For a positive control, no cy(WPHPY) was added. The cell images were immediately taken (0 h). During the imaging process, once a spot was selected, the area was marked so that the same spot could be located and imaged after 24 h. The cells were incubated at 37 °C for 24 h, and the images were retaken at the marked locations. This process was followed to carry out three independent scratch assays, all of which showed similar results. The representative result is presented in Figure 5b.
Cell Migration and Invasion Assay
The 24-well plates with inserts were purchased from Corning (8.0 μm, pore size; 6.5 mm, diameter; and 0.33 cm2, growth area) and the ECM gel from SigmaAldrich. The cell culture insert was coated with 100 μL of the ECM gel (2 mg/mL) and left in the 5% CO2 incubator at 37 °C for 30 min. The excess gel solution was removed carefully, and the insert was washed gently with serum-free media twice. Then, 100 μL of the WM115 cell suspension (200 k cells in serum-free media) was added into the gel-coated insert. Full medium with 10% FBS was added to the lower chamber, and the insert was placed into the wells. Furthermore, 100 μL of the cy(WPHPY) solution in serum-free media was added into the insert chamber. For a positive control, serum-free medium with no ligand was added to the chamber. The assembly was kept in the cell incubator for 24 h. The insert was then removed from the well and washed three times with PBS. The cells were fixed with a 4% paraformaldehyde solution in PBS (10 min at room temperature). After the removal of the paraformaldehyde solution, the insert was washed three times with PBS. The insert was then incubated in methanol for 20 min at room temperature and then washed three times with PBS. After that, a 0.2% crystal violet/ methanol solution was added, and the insert was incubated for 15 min at room temperature. The staining solution was removed, and the insert was washed three times with PBS. Cells above the membrane (noninvasive cells) were scraped off, and the cells beneath the membrane were imaged using an inverted microscope. Three independent experiments were performed to ensure reproducibility, and individually prepared triplicate samples were used in each experiment. All of the experiments showed similar results, and a representative result is shown in Figure 5d.
Data and Statistical Analysis
Results from the triplicate measurements are reported as average ± standard deviation. The binding assay (FRET and FP) results were processed using OriginPro 2019 software. A Hill function was used for fitting the binding curves and calculating the Kd. For the single-cell immunofluorescence analysis, the p-value was calculated using a Mann–Whitney test.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the following agencies for financial support: US National Institutes of Health Grants 1R21EB025393 (M.X.) and 1R35GM119721 (J.S.) and US National Science Foundation Grant MCB-1932984 (C.-E.A.C).
ABBREVIATIONS
- TIPS
triisopropylsilane
- TFA
trifluoroacetic acid
- PhNCS
phenyl isothiocyanate
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- DIEA
diisopropylethylamine
- Az4
Fmoc-azidolysine-OH
- RP-HPLC
reverse-phase high-pressure liquid chromatography
- Rhod
rhodamine B
- CNBr
cyanogen bromide
- FBS
fetal bovine serum
- PBS
phosphate buffer saline
- TBS
Tris-buffered saline
- TBST
Tris-buffered saline, 0.1% Tween 20
- AP
alkaline phosphatase
- DMEM
Dulbecco’s modified Eagle’s medium
- FP
fluorescence polarization
- MS/MS
tandem mass spectrometry
- MMP2
matrix metalloproteinase-2
- TIMP2
tissue inhibitor of metalloproteinases 2
- MT1-MMP
membrane type I-matrix metalloproteinase
- PPI
protein–protein interaction
- IC50
half-maximal inhibitory concentration
- His6 peptide
hexahistidine peptide
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00180.
Characterization of the cyclic peptide ligands and epitopes, validation of binding affinities, molecular dynamics calculations, and other related experimental results (PDF)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00180
Contributor Information
Priyanka Sarkar, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Zhonghan Li, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Wendan Ren, Department of Biochemistry, University of California, Riverside, Riverside, California 92521, United States.
Siwen Wang, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Shiqun Shao, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Jianan Sun, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Xiaodong Ren, Medical College, Guizhou University, Guiyang, Guizhou 550025, China.
Nicole G. Perkins, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States
Zhili Guo, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Chia-En A. Chang, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
Jikui Song, Department of Biochemistry, University of California, Riverside, Riverside, California 92521, United States.
Min Xue, Department of Chemistry, University of California, Riverside, Riverside, California 92521, United States.
REFERENCES
- (1).Visse R; Nagase H Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839. [DOI] [PubMed] [Google Scholar]
- (2).Page-McCaw A; Ewald AJ; Werb Z Matrix Metalloproteinases and the Regulation of Tissue Remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Li YY; McTiernan CF; Feldman AM Interplay of Matrix Metalloproteinases, Tissue Inhibitors of Metalloproteinases and Their Regulators in Cardiac Matrix Remodeling. Cardiovasc. Res. 2000, 46, 214–224. [DOI] [PubMed] [Google Scholar]
- (4).Curran S; Murray GI Matrix Metalloproteinases: Molecular Aspects of Their Roles in Tumour Invasion and Metastasis. Eur. J. Cancer 2000, 36, 1621–1630. [DOI] [PubMed] [Google Scholar]
- (5).Vartak DG; Gemeinhart RA Matrix Metalloproteases: Underutilized Targets for Drug Delivery. J. Drug Targeting 2007, 15, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Henriet P; Emonard H Matrix Metalloproteinase-2: Not (Just) a “Hero” of the Past. Biochimie 2019, 166, 223–232. [DOI] [PubMed] [Google Scholar]
- (7).Hopkins ALG; Colin R The Druggable Genome. Nat. Rev. Drug Discovery 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- (8).Pavlaki M; Zucker S Matrix Metalloproteinase Inhibitors (MMPIs): The Beginning of Phase I or the Termination of Phase III Clinical Trials. Cancer Metastasis Rev. 2003, 22, 177–203. [DOI] [PubMed] [Google Scholar]
- (9).Vandenbroucke RE; Libert C Is There New Hope for Therapeutic Matrix Metalloproteinase Inhibition? Nat. Rev. Drug Discovery 2014, 13, 904–927. [DOI] [PubMed] [Google Scholar]
- (10).Dormán G; Cseh S; Hajdu I; Barna L; Konya D; Kupai K.; Kovacs L; Ferdinandy P. Matrix Metalloproteinase Inhibitors: A Critical Appraisal of Design Principles and Proposed Therapeutic Utility. Drugs 2010, 70, 949–964. [DOI] [PubMed] [Google Scholar]
- (11).Shi ZG; Li JP; Shi LL; Li X An Updated Patent Therapeutic Agents Targeting MMPs. Recent Pat. Anticancer Drug Discovery 2012, 7, 74–101. [DOI] [PubMed] [Google Scholar]
- (12).Fingleton B Matrix Metalloproteinase Inhibitors for Cancer Therapy: The Current Situation and Future Prospects. Expert Opin. Ther. Targets 2003, 7, 385–397. [DOI] [PubMed] [Google Scholar]
- (13).Coussens LM; Fingleton B; Matrisian LM Matrix Metalloproteinase Inhibitors and Cancer: Trials and Tribulations. Science 2002, 295, 2387–2392. [DOI] [PubMed] [Google Scholar]
- (14).Overall CM; Kleifeld O Tumour Microenvironment Opinion: Validating Matrix Metalloproteinases as Drug Targets and Anti-Targets for Cancer Therapy. Nat. Rev. Cancer 2006, 6, 227–239. [DOI] [PubMed] [Google Scholar]
- (15).Cathcart J; Pulkoski-Gross A; Cao J Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fields GB Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cathcart J; Pulkoski-Gross A; Cao J Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yosef G; Arkadash V; Papo N Targeting the MMP-14/ MMP-2/Integrin αvβ3 Axis with Multispecific N-TIMP2–Based Antagonists for Cancer Therapy. J. Biol. Chem. 2018, 293, 13310–13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Devy L; Huang L; Naa L; Yanamandra N; Pieters H; Frans N; Chang E; Tao Q; Vanhove M; Lejeune A; van Gool R; Sexton DJ; Kuang G; Rank D; Hogan S; Pazmany C; Ma YL; Schoonbroodt S; Nixon AE; Ladner RC; Hoet R; Henderikx P; TenHoor C; Rabbani SA; Valentino ML; Wood CR; Dransfield DT Selective Inhibition of Matrix Metalloproteinase-14 Blocks Tumor Growth, Invasion, and Angiogenesis. Cancer Res. 2009, 69, 1517–1526. [DOI] [PubMed] [Google Scholar]
- (20).Shiryaev SA; Remacle AG; Golubkov VS; Ingvarsen S; Porse A; Behrendt N; Cieplak P; Strongin AY A Monoclonal Antibody Interferes with TIMP-2 Binding and Incapacitates the MMP-2-Activating Function of Multifunctional, Pro-Tumorigenic MMP-14/MT1–MMP. Oncogenesis 2013, 2, No. e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Itoh Y; Takamura A; Ito N; Maru Y; Sato H; Suenaga N; Aoki T; Seiki M Homophilic Complex Formation of MT1-MMP Facilitates ProMMP-2 Activation on the Cell Surface and Promotes Tumor Cell Invasion. EMBO J. 2001, 20, 4782–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Deryugina EI; Quigley JP Matrix Metalloproteinases and Tumor Metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [DOI] [PubMed] [Google Scholar]
- (23).Nagase H; Visse R; Murphy G Structure and Function of Matrix Metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [DOI] [PubMed] [Google Scholar]
- (24).Zucker S; Drews M; Conner C; Foda HD; DeClerck YA; Langley KE; Bahou WF; Docherty AJP; Cao J Tissue Inhibitor of Metalloproteinase-2 (TIMP-2) Binds to the Catalytic Domain of the Cell Surface Receptor, Membrane Type 1-Matrix Metalloproteinase 1 (MT1-MMP). J. Biol. Chem. 1998, 273, 1216–1222. [DOI] [PubMed] [Google Scholar]
- (25).Morgunova E; Tuuttila A; Bergmann U; Tryggvason K Structural Insight into the Complex Formation of Latent Matrix Metalloproteinase 2 with Tissue Inhibitor of Metalloproteinase 2. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Das S; Nag A; Liang J; Bunck DN; Umeda A; Farrow B; Coppock MB; Sarkes DA; Finch AS; Agnew HD; Pitram S;Lai B; Yu MB; Museth AK; Deyle KM; Lepe B; RodriguezRivera FP; McCarthy A; Alvarez-Villalonga B; Chen A; Heath J; Stratis-Cullum DN; Heath JR A General Synthetic Approach for Designing Epitope Targeted Macrocyclic Peptide Ligands. Angew. Chem., Int. Ed. 2015, 54, 13219–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dougherty PG; Qian Z; Pei D Macrocycles as Protein–Protein Interaction Inhibitors. Biochem. J. 2017, 474, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Heinis C; Winter G Encoded Libraries of Chemically Modified Peptides. Curr. Opin. Chem. Biol. 2015, 26, 89–98. [DOI] [PubMed] [Google Scholar]
- (29).Moellering RE; Cornejo M; Davis TN; Del Bianco C; Aster JC; Blacklow SC; Kung AL; Gilliland DG; Verdine GL; Bradner JE Direct Inhibition of the Notch Transcription Factor Complex. Nature 2009, 462, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Qian Z; Dougherty PG; Pei D Targeting Intracellular Protein–Protein Interactions with Cell-Permeable Cyclic Peptides. Curr. Opin. Chem. Biol. 2017, 38, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tavassoli A; Lu Q; Gam J; Pan H; Benkovic SJ; Cohen SN Inhibition of HIV Budding by a Genetically Selected Cyclic Peptide Targeting the Gag–Tsg101 Interaction. ACS Chem. Biol. 2008, 3, 757–764. [DOI] [PubMed] [Google Scholar]
- (32).Cardote TAF; Ciulli A Cyclic and Macrocyclic Peptides as Chemical Tools to Recognise Protein Surfaces and Probe Protein– Protein Interactions. ChemMedChem 2016, 11, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Boehm M; Beaumont K; Jones R; Kalgutkar AS; Zhang L; Atkinson K; Bai G; Brown JA; Eng H; Goetz GH; Holder BR; Khunte B; Lazzaro S; Limberakis C; Ryu S; Shapiro MJ; Tylaska L; Yan J; Turner R; Leung SSF; Ramaseshan M; Price DA; Liras S; Jacobson MP; Earp DJ; Lokey RS; Mathiowetz AM; Menhaji-Klotz E Discovery of Potent and Orally Bioavailable Macrocyclic Peptide–Peptoid Hybrid CXCR7 Modulators. J. Med. Chem. 2017, 60, 9653–9663. [DOI] [PubMed] [Google Scholar]
- (34).Naylor MR; Bockus AT; Blanco M-J; Lokey RS Cyclic Peptide Natural Products Chart the Frontier of Oral Bioavailability in the Pursuit of Undruggable Targets. Curr. Opin. Chem. Biol. 2017, 38, 141–147. [DOI] [PubMed] [Google Scholar]
- (35).Shao S; Li Z; Cheng H; Wang S; Perkins NG; Sarkar P; Wei W; Xue M A Chemical Approach for Profiling Intracellular AKT Signaling Dynamics from Single Cells. J. Am. Chem. Soc. 2018, 140, 13586–13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Joo SH Cyclic Peptides as Therapeutic Agents and Biochemical Tools . Biomol. Ther. 2012, 20, No. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen Y; Barkley MD Toward Understanding Tryptophan Fluorescence in Proteins. Biochemistry 1998, 37, 9976–9982. [DOI] [PubMed] [Google Scholar]
- (38).Ciolczyk-Wierzbicka D; Laidler P The Inhibition of Invasion of Human Melanoma Cells through N-Cadherin Knock-Down . Med. Oncol. 2018, 35, No. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Olson MW; Gervasi DC; Mobashery S; Fridman R Kinetic Analysis of the Binding of Human Matrix Metalloproteinase-2 and –9 to Tissue Inhibitor of Metalloproteinase (TIMP)-1 and TIMP2. J. Biol. Chem. 1997, 272, 29975–29983. [DOI] [PubMed] [Google Scholar]
- (40).Nguyen Q; Willenbrock F; Cockett MI; O’Shea M; Docherty AJ; Murphy G Different Domain Interactions Are Involved in the Binding of Tissue Inhibitors of Metalloproteinases to Stromelysin-1 and Gelatinase A. Biochemistry 1994, 33, 2089–2095. [DOI] [PubMed] [Google Scholar]
- (41).Stetler-Stevenson WG; Krutzsch HC; Liotta LA Tissue Inhibitor of Metalloproteinase (TIMP-2). A New Member of the Metalloproteinase Inhibitor Family. J. Biol. Chem. 1989, 264, 17374–17378. [PubMed] [Google Scholar]
- (42).Lechapt-Zalcman E; Pruliere-Escabasse V; Advenier D; Galiacy S; Charriere-Bertrand C; Coste A; Harf A; d’Ortho MP; Escudier E Transforming Growth Factor-Beta1 Increases Airway Wound Repair via MMP-2 Upregulation: A New Pathway for Epithelial Wound Repair? AJP Lung Cell. Mol. Physiol. 2006, 290, L1277–1282. [DOI] [PubMed] [Google Scholar]
- (43).Dufour A; Sampson NS; Li J; Kuscu C; Rizzo RC; DeLeon JL; Zhi J; Jaber N; Liu E; Zucker S; Cao J SmallMolecule Anticancer Compounds Selectively Target the Hemopexin Domain of Matrix Metalloproteinase-9. Cancer Res. 2011, 71, 4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Alford VM; Kamath A; Ren X; Kumar K; Gan Q; Awwa M; Tong M; Seeliger MA; Cao J; Ojima I; Sampson NS Targeting the Hemopexin-Like Domain of Latent Matrix Metalloproteinase-9 (ProMMP-9) with a Small Molecule Inhibitor Prevents the Formation of Focal Adhesion Junctions. ACS Chem. Biol. 2017, 12, 2788–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cha H; Kopetzki E; Huber R; Lanzendö M.; Brandstetter H. Structural Basis of the Adaptive Molecular Recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079. [DOI] [PubMed] [Google Scholar]
- (46).Agnew HD; Coppock MB; Idso MN; Lai BT; Liang J; McCarthy-Torrens AM; Warren CM; Heath JR ProteinCatalyzed Capture Agents . Chem. Rev. 2019, 119, 9950–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Brew K; Nagase H The Tissue Inhibitors of Metalloproteinases (TIMPs): An Ancient Family with Structural and Functional Diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nakada M; Nakamura H; Ikeda E; Fujimoto N; Yamashita J; Sato H; Seiki M; Okada Y Expression and Tissue Localization of Membrane-Type 1, 2, and 3 Matrix Metalloproteinases in Human Astrocytic Tumors. Am. J. Pathol. 1999, 154, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Morrison CJ; Butler GS; Bigg HF; Roberts CR; Soloway PD; Overall CM Cellular Activation of MMP-2 (Gelatinase a) by MT2-MMP Occurs via a TIMP-2-Independent Pathway. J. Biol. Chem. 2001, 276, 47402–47410. [DOI] [PubMed] [Google Scholar]
- (50).Klemm P Manual Edman Degradation of Proteins and Peptides . Methods Mol. Biol. 1984, 1, 243–254. [DOI] [PubMed] [Google Scholar]
- (51).Lee SS; Lim J; Cha J; Tan S; Heath JR Rapid Microwave-Assisted Cnbr Cleavage of Bead-Bound Peptides. J. Comb. Chem. 2008, 10, 807–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Niedermeyer TH; Strohalm M mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS One 2012, 7, No. e44913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminf. 2012, 4, No. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jakalian A; Jack DB; Bayly CI Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (56).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (57).Case D; Babin V; Berryman J; Betz R; Cai Q; Cerutti D; Cheatham T III; Darden T; Duke R; Gohlke H The ff14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- (58).Chang CE; Gilson MK Tork: Conformational Analysis Method for Molecules and Complexes. J. Comput. Chem. 2003, 24, 1987–1998. [DOI] [PubMed] [Google Scholar]
- (59).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- (60).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Darden T; York D; Pedersen L Particle Mesh Ewald: An N· Log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (62).Troeberg L; Nagase H Monitoring Metalloproteinase Activity Using Synthetic Fluorogenic Substrates . Curr. Protoc. Protein Sci. 2004, 33, 21.16.21–21.16.29. [DOI] [PubMed] [Google Scholar]
- (63).Toth M; Fridman R Assessment of Gelatinases (MMP-2 and MMP-9 by Gelatin Zymography. Methods Mol. Med. 2001, 57, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Mahmood T; Yang PC Western Blot: Technique, Theory, and Trouble Shooting. North Am. J. Med. Sci. 2012, 4, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.