

Abstract
The dynein adaptor Drosophila Bicaudal D (BicD) is auto-inhibited and activates dynein motility only after cargo is bound, but the underlying mechanism is elusive. In contrast, we show that the full-length BicD/F684I mutant activates dynein processivity even in the absence of cargo. Our X-ray structure of the C-terminal domain of the BicD/F684I mutant reveals a coiled-coil registry shift; in the N-terminal region, the two helices of the homodimer are aligned, whereas they are vertically shifted in the wild-type. One chain is partially disordered and this structural flexibility is confirmed by computations, which reveal that the mutant transitions back and forth between the two registries. We propose that a coiled-coil registry shift upon cargo binding activates BicD for dynein recruitment. Moreover, the human homolog BicD2/F743I exhibits diminished binding of cargo adaptor Nup358, implying that a coiled-coil registry shift may be a mechanism to modulate cargo selection for BicD2–dependent transport pathways.
Keywords: dynein, BicD2, coiled-coil, registry shift, auto-inhibition, Nup358, Rab6, nuclear positioning, intracellular transport, dynein adaptor
Graphical Abstract
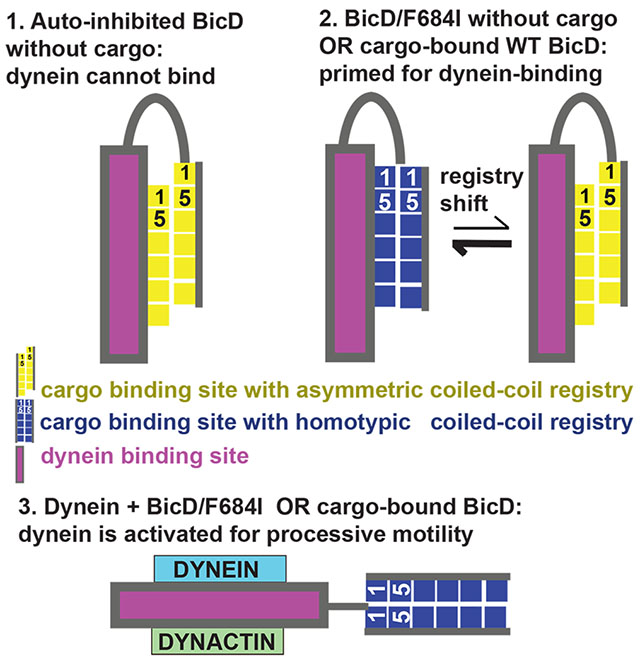
Introduction
Cytoplasmic dynein is the predominant minus-end directed microtubule motor and thus facilitates a vast number of cellular transport events.1 Dynein adaptor proteins, such as Drosophila Bicaudal D (Dm BicD)2 recognize cargo for dynein-dependent transport. Cargo-bound dynein adaptors are required to activate metazoan dynein for processive transport and are therefore an essential part of the dynein transport machinery.3–8 In the absence of cargo, BicD forms an auto-inhibited looped conformation, in which the C-terminal cargo binding region (CTD) binds to the N-terminal dynein/dynactin-binding site (coiled-coil domain 1, CC1), sterically preventing motor binding.2–15 The CTD is required for auto-inhibition, since a truncated BicD-CC1 construct without it activates dynein for processive transport in the absence of cargo.5,7 Auto-inhibition of full-length BicD is released upon cargo binding;2–15 however, the underlying molecular mechanism is elusive.
Notably, auto-inhibition is compromised in the classical Dm BicD mutant F684I, which recruits larger amounts of dynein from cell extracts compared to wild-type Dm BicD.9 The mutation causes dynein-dependent mRNA transport defects and subsequent anterior accumulation of the oskar mRNA pool,16–19 which result in a classical developmental phenotype that includes double-abdomen fly embryos.9,16,20,21 Therefore, the Dm BicD/F684I mutant could potentially serve as a tool to investigate the molecular mechanism of BicD-auto-inhibition.
Dm BicD facilitates the transport of mRNA and Golgi-derived vesicles and is recruited to these cargoes via protein cofactors that are termed cargo adaptors (whereas Dm BicD is a dynein adaptor). The most well characterized cargo adaptors for Dm BicD are the RNA-binding protein Egalitarian22 and Rab6GTP, which facilitate transport of mRNAs and Golgi-derived vesicles, respectively.15 The predominant cargo adaptors for the human homologs Bicaudal D2 (Hs BicD2) and Bicaudal D1 (Hs BicD1) are Rab6GTP,15 which engages in the transport of Golgi-derived and secretory vesicles and nuclear pore complex protein Nup358, which engages in transport of the cell nucleus.23 BicD2-dependent transport pathways are important for faithful chromosome segregation, neurotransmission at synapses, and essential for brain development.23–25 Mutations of BicD2 cause the neuromuscular disease spinal muscular atrophy.26–28 Cargo selection for BicD2-dependent transport is regulated by competition of cargo-adaptors,29 as well as the G2 phase specific kinase Cyclin-dependent kinase 1 (Cdk1),24 however, additional regulatory mechanisms remain to be identified in order to explain how BicD2 switches from selecting Rab6-positive vesicles for transport in G1/S phase to recruiting dynein to the cell nucleus via Nup358 in G2 phase.
Cargo adaptors bind to the C-terminal domain (CTD) of all BicD homologs,2 and the structures of the Dm BicD-CTD9 as well as its homologs Hs BicD2-CTD30 and mouse (Ms) BicD1-CTD10 were determined, which all form homodimeric coiled-coils. Interestingly, Hs BicD2-CTD and Ms BicD1-CTD form a conformation with homotypic coiled-coil registry, in which the helices are aligned at equal height and the same residues from both chains engage in layers of knobs-into-holes interactions.10,30 In contrast, the Dm BicD-CTD has an asymmetric coiled-coil registry. In the N-terminal half of the CTD, the helices are vertically shifted by ~1 helical turn respective to each other (heterotypic coiled-coil registry), whereas in the C-terminal half, the chains are aligned in a homotypic coiled-coil registry.9 Coiled-coil registry shifts have so far only been reported for a few proteins, most notably for the stalk domain of dynein,30–39 but may potentially be an inherent property of many coiled-coil structures with important physiological functions. Thus, a clear demonstration of such dynamics in another functional context would be of broad interest. In the case of BicD2, a coiled-coil registry shift may relieve auto-inhibition. Notably, the BicD2 coiled-coil is parallel, whereas the dynein stalk has an anti-parallel coiled-coil,32 and thus it is expected that coiled-coil registry shifts in BicD2 and dynein are facilitated by distinct mechanisms.
We recently used molecular dynamics (MD) simulations to probe structural dynamics in the BicD2-CTD coiled-coil.30 These simulations support the idea that BicD2 can adopt both a homotypic coiled-coil registry and an asymmetric registry, as both states are similarly stable in simulations and defined by distinct conformations of F743 and F750, which stabilize either a homotypic or asymmetric coiled-coil registry.30 Notably, mutation of F743 to Ile (F684I in Dm) increases dynein recruitment in the Drosophila homolog compared to the wild type.9 In our MD simulations of the F743I mutant of Hs BicD2-CTD, a spontaneous coiled-coil registry shift from asymmetric to fully heterotypic coiled-coil registry was observed.30 We thus hypothesized that a coiled-coil registry shift upon cargo binding could relieve BicD-auto-inhibition and activate it for dynein recruitment, as has also been proposed earlier.9,10,30 In addition, in MD simulations of the R747C human disease mutant of Hs BicD2-CTD which causes spinal muscular atrophy, a spontaneous transient coiled-coil registry shift was observed that may be an underlying cause of the disease.30
Here we show that the F684I mutation in BicD allows cargo-independent activation of dynein motility. To investigate the structural basis for this activation, we determined the X-ray structure of the C-terminal cargo-binding domain (CTD) of Dm BicD-CTD/F684I, which has a homotypic coiled-coil registry in contrast to the wild-type. Furthermore, in the structure of the mutant, the region N-terminal of F684I is disordered for one chain. This structural flexibility is confirmed by MD simulations, in which the mutant transitions back and forth between homotypic and asymmetric registries on a time scale of tens of ns. Free energy calculations indicate conformations with homotypic and asymmetric registries to have similar stability. Our data suggest that the mutation promotes a registry shift and renders the coiled-coil flexible, likely resulting in the formation of multiple conformations.
Results
Full-length Dm BicD with the F684I mutation recruits dynein in the absence of cargo.
A single molecule TIRF (Total Internal Reflection Fluorescence) microscopy processivity assay was used to assess the functional properties of reconstituted dynein-dynactin-BicD (DDB) complexes. Complexes were reconstituted with either full-length Dm BicD (BicDWT), full-length Dm BicD/F684I (BicDF684I) or the truncated N-terminal fragment of Dm BicD (BicDCC1). Full-length WT Dm BicD is auto-inhibited and does not recruit dynein-dynactin, while the truncated N-terminal fragment fully activates dynein-dynactin for processive transport.5,7,11,12 Consistent with previous results,11 we did not observe processive directional movement for DDBWT on microtubule tracks, although some one-dimensional diffusive events of the Qdot-labeled dynein were observed (Fig. 1A). Because DDBWT showed no directional motion (Figure 1A, Movie S1), speed and run length were not measured. In contrast, DDBCC1 showed robust processivity, which was indistinguishable from that observed with DDBF684I (Figure 1A, Movie S2 and S3). The binding frequency of DDBCC1 to microtubule tracks is not statistically different from DDBF684I (p=0.17, one way ANOVA followed by Tukey’s post-hoc test) (Figure 1B). The speeds of both DDBCC1 and DDBF684I were fitted with a single Gaussian (Figure 1C). The DDBF684I complex moved at a speed of 0.42 ± 0.17 µm/s (n=72), which is not significantly different (p=0.9, Student’s t-test) from what was observed for DDBCC1 (0.40 ± 0.13 µm/s, n=57). Processive run-lengths were fitted with a standard exponential decay equation (y= Ae−bx), where A is the amplitude and 1/b is run length. The run length of DDBF684I (3.3 ± 0.08 µm, n=71) is not significantly different (p=0.6, Kolmogorov-Smirnov test) from that of DDBCC1 (2.9 ± 0.1 µm, n=57) (Figure 1D). Similar results were obtained using independent protein preparations of BicDWT, BicDF684I and dynein (Table S1).
Figure 1. Full-length Dm BicDF684I results in cargo-independent activation of dynein motility.
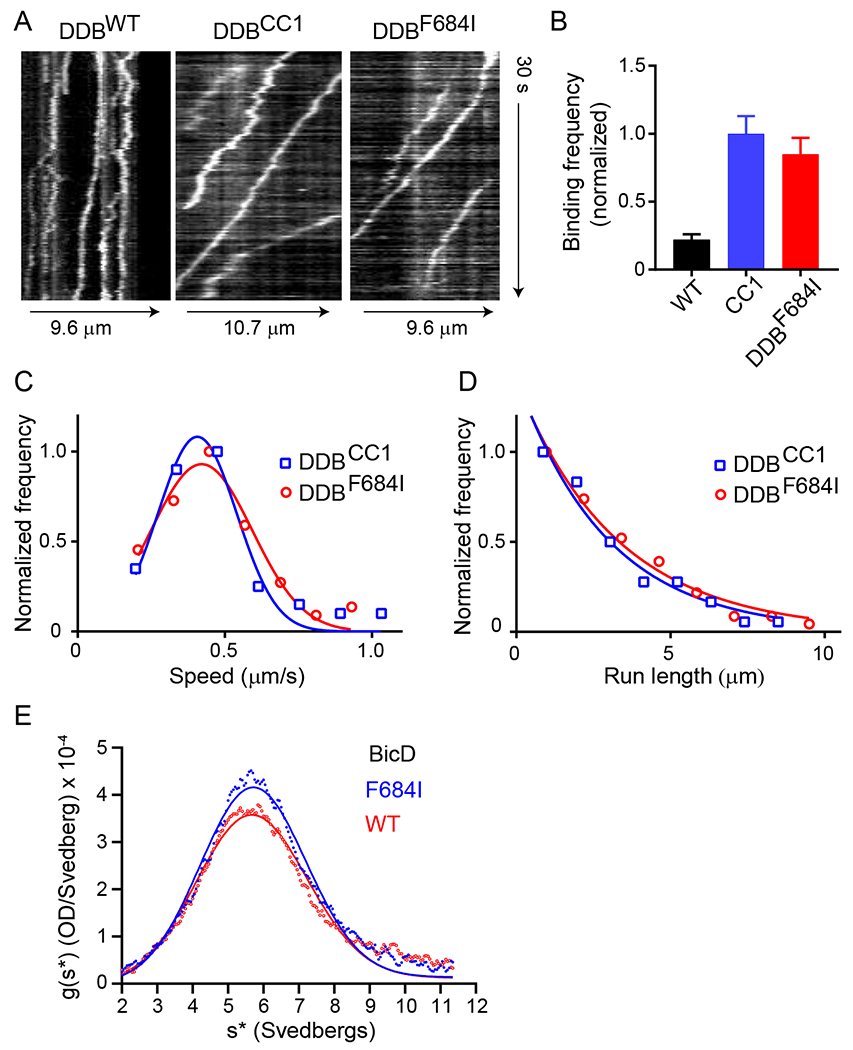
(A) Kymographs of the three DDB complexes. DDBWT is either static or diffusive, while the other two complexes show processive motion (sloped lines). (B) Normalized binding frequency of dynein-dynactin-BicD (DDB) with different Dm BicD constructs: full-length BicDWT (black), truncated BicDCC1 (blue), and full-length mutant BicDF684I (red). The binding frequency of DDBCC1 is normalized to one and reported as mean ± SE. Frequencies of DDBCC1 (n=33) and DDBF684I (n=35) are 4.5-fold (p<0.0001) and 3.8-fold (p<0.0001) higher, respectively, than DDBWT (n=24). Binding frequency is statistically insignificant between DDBCC1 and DDBF684I (p=0.17, one way ANOVA followed by a Tukey’s post-hoc test). Motion of DDBWT is mainly diffusive. (C) The speed of DDBCC1 (blue squares) and DDBF684I (red circles) are 0.40 ± 0.13 µm/s (n=57) and 0.42 ± 0.17 µm/s (n=72) (mean ± SD), respectively, which are not significantly different (p=0.9, Student’s t-test). (D) Run length of DDBCC1 (2.9 ± 0.1 µm, n=57) (blue squares) and DDBF684I (3.3 ± 0.08 µm, n=71) (red circles) (mean ± SE) are not significantly different (p=0.6, Kolmogorov-Smirnov test). Data from 2 independent experiments, using one protein preparation of each component, were pooled. See Movies S1–3. Table S1 tabulates similar data obtained with an independent protein preparation each of BicDWT, BicDF684I, and dynein. (E) Analytical ultracentrifugation of BicDWT and BicDF684I, with single species fits (solid lines) yielding sedimentation coefficients of 5.8S and 5.5S, respectively. See Table S2 for additional centrifugation data with an independent BicD preparation.68
Based on run frequency, speed and processive run length, we conclude that the full-length mutant Dm BicDF684I was fully capable of binding and activating dynein-dynactin for processive transport in the absence of cargo.
To rule out that the F684I mutation caused aggregation of BicD that could alter dynein recruitment, we performed analytical ultracentrifugation on the two BicD constructs. Both BicDF684I and BicDWT showed similar sedimentation profiles, yielding sedimentation coefficients of 5.8S and 5.5S, respectively (Fig. 1E and Table S2). Moreover, the similar sedimentation coefficients surprisingly imply that both the WT and mutant BicD constructs form the auto-inhibited, looped conformation in the absence of motor or cargo, despite the ability of BicDF684I to recruit dynein in the absence of cargo.
Crystal structure of the Dm BicD-CTD/F684I mutant provides mechanistic insights into cargo-independent activation.
To gain structural insights into the molecular mechanism of activation of BicD, we determined the structure of the C-terminal domain of the Drosophila melanogaster BicD/F684I mutant (Dm BicD-CTD/F684I, residues 656-745). Crystals were obtained in the space group P31 2 1. The structure was determined by molecular replacement in the PHENIX suite,40 using coordinates from the wild-type structure9 that were truncated N-terminal of residue 692 as the search model. The structure was refined to 2.35Å resolution, with an Rfree of 25.99% and an Rwork of 25.06% (Table 1). In the structure, Dm BicD-CTD/F684I forms a homodimeric coiled-coil. In contrast to the structure of the wild type, in the structure of the F684I mutant, a ~20 residue N-terminal region upstream of the mutated residue I684 is not resolved in the electron density map for one chain of the dimer (Figure 2). However, the same region is well defined in the electron density map for the second chain. Consequently, the model contains residues 666-740 and 684-741 for the two chains, respectively. The homo-dimer has an interface area of 1427 Å2. An analysis of the crystal contacts reveals six interfaces with symmetry-related dimers with interface areas of 376-205 Å2, which are small compared to the homo-dimer interface (Figure S1).
Table 1.
Crystallographic statistics
| Data collection statistics | |
| Space group | P 31 2 1 |
| Unit cell parameters a, b, c (Å) | 60.0, 60.0, 142.6 |
| Unit cell parameters α, β, γ | 90°, 90°, 120° |
| Wavelength (Å) | 0.9791 |
| Resolution range (Å) | 19.47-2.35 (2.434-2.35) |
| Total Reflections | 125766 (12111) |
| Unique Reflections | 12955 (1263) |
| Redundancy | 9.7 (9.6) |
| Completeness (%) | 99.39 (99.92) |
| I/σ(I) | 27.98 (1.26) |
| Wilson B-factor (Å2) | 72.37 |
| Rmerge | 0.0443 (1.621) |
| Rmeas | 0.04684 (1.713) |
| Rpim | 0.01498 (0.549) |
| CC1/2 | 1 (0.573) |
| CC* | 1 (0.854) |
| Refinement statistics | |
| Rfree (%) | 25.99 (38.38) |
| Rwork (%) | 25.06 (36.04) |
| Reflections (work/test sets) | 12948/640 |
| R.m.s.d. bonds (Å)/angles | 0.005/0.77 |
| Average B-factor (Å2) | 96.46 |
| MolProbity validation | |
| Ramachandran plot favored | 100% |
| Ramachandran plot allowed outliers | 0% |
| Ramachandran plot outliers | 0% |
| Rotamer outliers | 0.85% |
| Clash score | 1.83 |
The statistics for the high-resolution shell are shown in parenthesis.
Figure 2. Dm BicD-CTD/F684I assumes a conformation with homotypic coiled-coil registry.
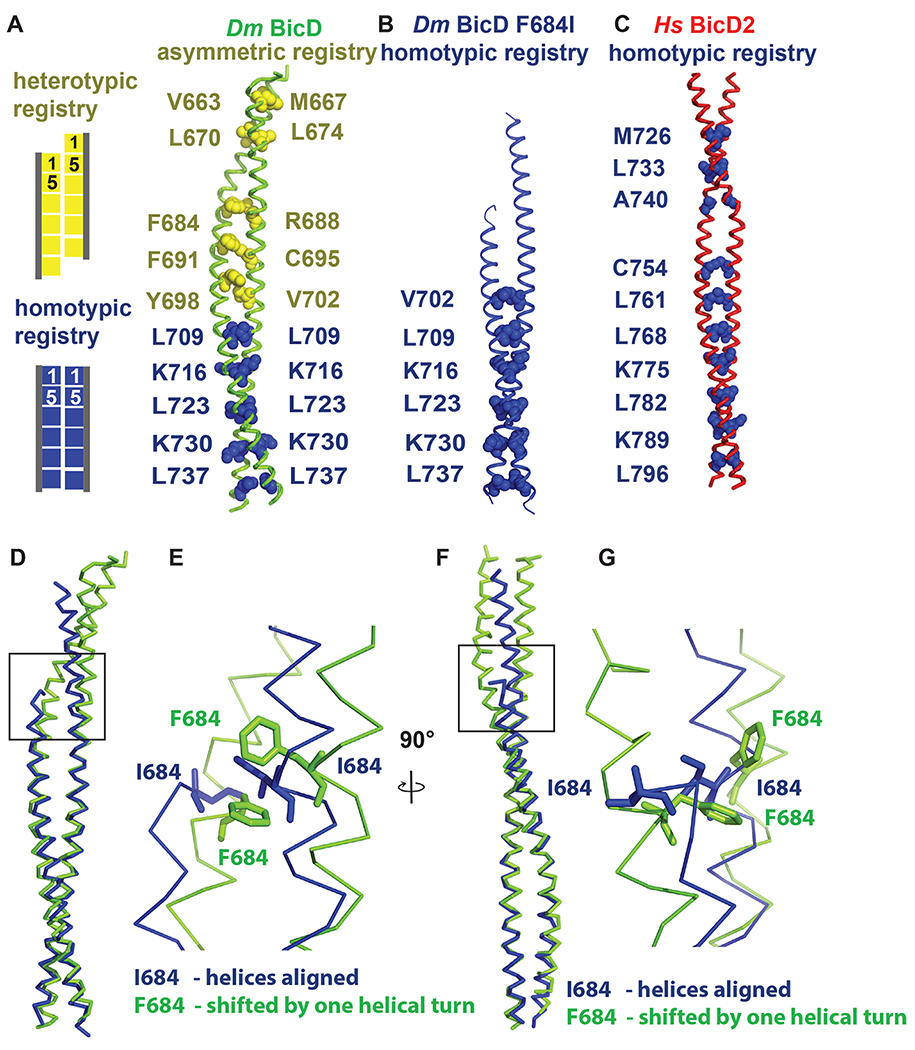
(A) The structure of Dm BicD-CTD wild type (PDB ID 4BL6) 9 which has an asymmetric coiled-coil registry is shown in cartoon representation next to a schematic illustrating coiled-coil registries (left panel). Knob residues in the “a” position of the heptad repeat are shown in spheres representation (heterotypic registry yellow, homotypic registry dark blue). (B) Structure of Dm BicD-CTD/F684I, which has a homotypic registry. (C) Structure of Hs BicD2-CTD (PDB ID 6OFP) 30, which has a homotypic registry. (D-G) Least squares superimposed structures of the Dm BicD-CTD wild type (green) and the F684I mutant (dark blue) are shown as C-α traces, and are rotated by 90° in (D, F). (E, G) Close-up of the boxed area in (D, F). Residues F684 and I684 are shown in stick representation. Note that in the structure of the mutant, the I684 residues from both chains of the dimer are aligned at the same height, consistent with a homotypic registry, while in the wild-type structure, the F684 residues from both monomers are vertically shifted by one helical turn respective to each other, consistent with a heterotypic registry. See Figures S1 and S2.
Coiled-coils such as Dm BicD-CTD are characterized by heptad repeats ‘abcdefg’ in the sequence, where residues at ‘a’ and ‘d’ positions are predominantly hydrophobic. These residues form characteristic “knobs-into-holes interactions”, where a knob from one chain (either an ‘a’ or ‘d’ position residue) fits into a hole formed of four residues on the opposite chain.41,42 Notably, structures of distinct BicD homologs with distinct coiled-coil registries have been determined. Hs BicD2-CTD has a homotypic coiled-coil registry with characteristic layers of knobs-into-holes interactions, which are formed by the same knob residues from both chains, and therefore the helices are aligned at equal height. Wild-type Dm BicD-CTD (Figure 2A) however has an asymmetric coiled-coil registry. The N-terminal half of the coiled-coil has a heterotypic registry, in which residue i from one chain is paired up with residue i+4 from the second chain to form layers of knobs-into-holes interactions, resulting in a vertical displacement of the helices by õne helical turn. In the C-terminal half, the helices are aligned to form a coiled-coil with homotypic registry.
To determine the coiled-coil registry, we assigned the heptad register of the structure of Dm BicD-CTD/F684I (Figure 2, Figure S2). As observed for the wild-type Dm BicD-CTD (Figure 2A) and Hs BicD2-CTD (Figure 2C), the C-terminal half of the mutant structure has a homotypic coiled-coil registry (Figure 2B, see Table 2 for residue numbering in Hs and Dm homologs). Notably, one additional layer of knobs-into-holes interactions with homotypic coiled-coil registry was identified in the F684I mutant compared to the Dm BicD wild type. This layer is formed by knob residue V702 from both chains (Figure 2, Figure S2). In contrast to the F684I mutant, in the Dm BicD wild type, V702 forms a layer of knobs-into-holes interactions with residue Y698 and therefore part of the region that has a heterotypic coiled-coil registry. Wild-type Dm BicD-CTD also has additional layers of knob-into-holes interactions with heterotypic registry in its N-terminal region, resulting in a vertical displacement of the helices by approximately one helical turn. However, no additional layers of knobs-into-holes interactions N-terminal of V702 were identified in the structure of Dm BicD-CTD/F684I, because a ~20 residue region upstream of residue I684 is not resolved for one chain (Figure 2).
Table 2.
Numbering of homologous key residues in Dm BicD and Hs BicD2
| Dm BicD | Hs BicD2 | |
|---|---|---|
| Conversion for residue # | Residue i | Residue i+59 |
| N-terminal dynein/dynactin binding site | 21–380 11 | 1-400 5 |
| C-terminal cargo-binding domain (CTD) | 656-745 9 | 715-804 30 |
| Dm BicD/F684I mutant | F684I | F743I |
| Key aromatic residues | F684, F691, Y698 | F743, F750, Y757 |
| Nup358 interface residues* | L687, R688, M690, R694 | L746, R747, M749, R753 |
A least-squares superimposition of the structures of the Dm BicD-CTD/F684I mutant and the wild type revealed that in the mutant, both I684 residues are aligned at equal height, as observed for the homologous residue F743 in Hs BicD2 with homotypic registry (Figure 2D–G). However, in the wild-type Dm BicD structure, which has an asymmetric coiled-coil registry, residues F684 from both chains are vertically shifted by approximately one helical turn with respect to each other (Figure 2D–G). F684 from one chain lines up with R688 from the second chain to form a layer of knobs-into-holes interactions (heterotypic registry) (Figure 2A).
To conclude, several pieces of data suggest that the structure of the Dm BicD2-CTD/F684I mutant has a homotypic coiled-coil registry: One additional layer of knobs-into-holes interactions with homotypic coiled-coil registry is formed by residues V702 in the structure of the F684I mutant compared to the wild type. Furthermore, residues I684 are aligned in the structure of the mutant at equal height (homotypic registry), whereas in the wild-type structure, residues F684 are vertically shifted respectively to each other by approximately one helical turn (heterotypic registry). However, since a ~20 residue area of one monomer in the F684I mutant is not resolved, no additional knobs-into-holes interactions were identified. Therefore, the coiled-coil registry N-terminal of I684 is unknown.
Distinct conformations of F684 and F691 stabilize distinct coiled-coil registries.
Another key residue that is important to stabilize either a homotypic or asymmetric coiled-coil registry is F691 (Figure 3). In the Dm BicD-CTD wild-type structure with the asymmetric coiled-coil registry, phenylalanine side chains of F691 from both chains interact in a face-to-edge aromatic interaction, which leads to vertical displacement of the chains by ~1 helical turn (Figure 3B). However, in the BicD homologs with homotypic registry (Ms BicD1, Hs BicD2) the homologous phenylalanine residues interact with face-to-face aromatic interaction, which allows the chains to be aligned in the homotypic registry (Figure 3D, E). In the Dm BicD F684I mutant, the phenylalanine side chains of F691 from both chains interact face-to-face (Figure 3A, C, F), which suggests a homotypic registry. A least-squares superimposition of the structures of the Dm BicD wild type and the F684I mutant confirms that the F691 side chains form a face-to-face aromatic interaction in the structure of the mutant and a face-to-edge aromatic interaction in the wild-type structure with the asymmetric registry (Figure 3C). These data suggest that in the structure of the Dm BicD F684I mutant, the F691 residues assume a conformation that is found in Hs BicD2 with homotypic coiled-coil registry.
Figure 3. Conformation of key aromatic residues in Dm BicD-CTD/F684I.
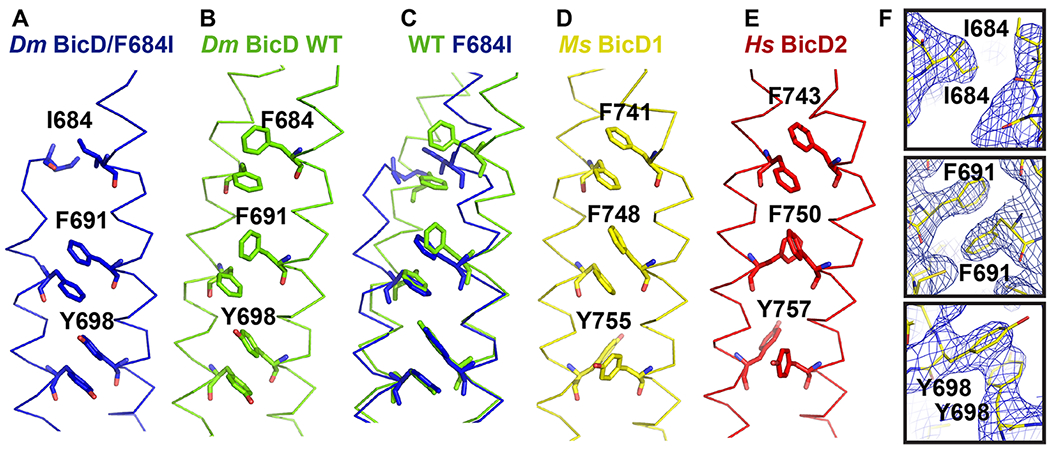
(A) The Cα-trace of the structure of Dm BicD-CTD/F684I is shown (blue). Residues I684, F691, Y698 are labeled and shown in stick representation. (B) Structure of the wild-type Dm BicD-CTD (green, PDB ID 4BL6).9 (C) Least squares superimposed structures of Dm BicD-CTD/F684I and the wild type. (D, E) Structures of the Dm BicD homologs (D) Ms BicD1-CTD (yellow, PDB ID 4YTD)10 and (E) Hs BicD2-CTD (red, PDB ID 6OFP).30 (F) Structure of the Dm BicD-CTD/F684I mutant in stick representation overlaid with the 2FO-FC electron density map (blue mesh). Close-ups of residues I684, F691 and Y698 are shown in three panels. Note that residues F691 from both chains are oriented face-to-face, as observed in the structures with the homotypic registries.
The disordered region is present in the crystal, and α-helical.
Because a ~20 residue region of one chain is not resolved in the crystal structure, we dissolved crystals of Dm BicD-CTD/F684I and analyzed them by SDS-PAGE, to assess whether the crystals contained the intact protein (Figure 4A). A comparison of the SDS-PAGE of the dissolved crystals, the purified Dm BicD-CTD/F684I protein and the wild-type protein suggests that indeed the intact Dm BicD-CTD/F684I protein is present in crystal, suggesting that the unresolved N-terminal region is disordered.
Figure 4. The intact Dm BicD-CTD/F684I is present in the crystal and it is fully folded.
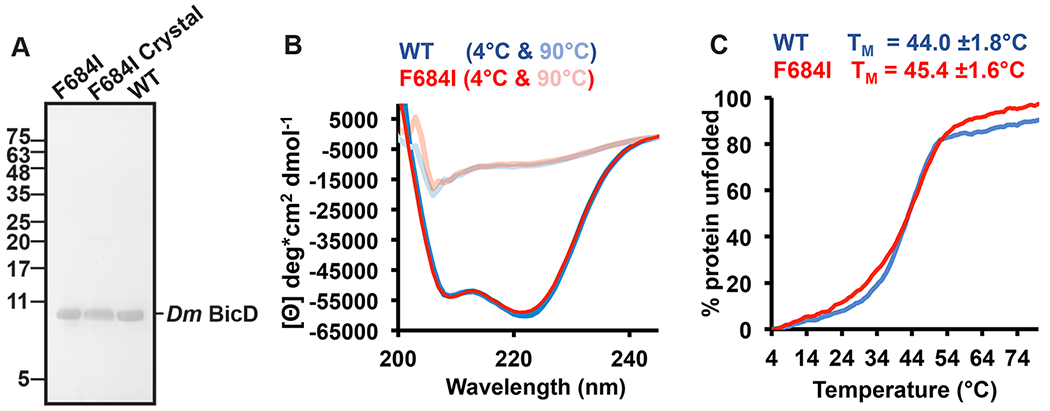
(A) SDS-PAGE analysis of purified Dm BicD-CTD/F684I (left lane), Dm BicD-CTD/F684I crystals (middle lane), and purified wild-type protein (right lane). The position of molecular weight standards (kDa) is indicated on the left. Note that a faint dimer band (21.8 kDa) is visible in all samples. For the crystal sample, 20 crystals were washed three times with reservoir buffer before being dissolved in gel filtration buffer. Two crystal samples were analyzed, with similar results. (B) CD wavelength scans of Dm BicD-CTD WT (blue) and Dm BicD-CTD/F684I (red) at 4°C (native) and 90°C (random coil). The mean residue molar ellipticity [Θ] versus the wavelength is shown. Experiments were repeated three times, representative scans are shown. (C) Thermal unfolding curves of wild type (blue) and F684I (red) were recorded by CD spectroscopy at 222 nm. Molar ellipticity [Θ] versus temperature is plotted. 0% and 100% protein unfolded represent the values of [Θ]min and [Θ]max, respectively. Representative experiments are shown; melting temperatures TM of Dm BicD-CTD WT and F684I were averaged from three experiments. See Figures S3, S4 and Table S3.
In order to assess whether the disordered portion of the helix is α-helical (rather than misfolded), we probed the secondary structure content of Dm BicD-CTD/F684I by circular dichroism (CD) wavelength scans. The CD wavelength spectra of the Dm BicD-CTD/F684I mutant and the wild-type both have minima at 208nm and 222nm that are characteristic for α-helical proteins. Notably, the spectra of the F684I mutant and the wild-type overlay perfectly, suggesting that both structures have a very similar α-helical content. Our calculations of the experimental error and the expected signal (below) suggest that the CD spectra of the mutant would be significantly different from the wild-type, if the ~20 residue disordered region in the structure was unfolded rather than α-helical.
We calculated the experimental error by determining the standard deviation of the molar ellipticity at 208 and 222 nm from ten experiments for which the samples were independently prepared (including the determination of the protein concentration). The experimental error was 3.5% at 208 nm and 5.2% at 222 nm. The error from the CD instrument itself is even lower (the error of the ellipticity at 200 nm is <0.04 mdeg, Jasco). The wild-type Dm BicD-CTD has an α-helical content of 83%, which would decrease by 18% to 65% in the mutant, if the disordered region was unfolded. To determine the expected signal for such structural change, we recorded CD melting curves of the mutant and wild type Dm BicD-CTD (Figure 4C, Figure S3). Protein unfolding was monitored by CD spectroscopy at 222 nm. The protein is 50% unfolded at the apparent melting temperature, and fully denatured at the plateau. In Figure S3, a graph of the molar elllipticity plotted versus % unfolding is shown. Based on these data, an 18% reduction in the α-helical content would result in an 18% change of molar ellipticity at 222 nm (Δ[Θ]= 9,142 deg*cm2 dmol−1) (Figure S3). This expected signal is 3.5 times higher than the experimental error of 5.2% (the standard deviation of [Θ] at 222 nm is 2,930 deg*cm2 dmol−1).
To conclude, CD spectroscopy is sensitive enough to distinguish whether the ~20 amino acid residue region that is disordered in the crystal structure of the F684I mutant is α-helical or unfolded, and the CD spectra of the wild-type and the F684I mutant are very similar, confirming that this region is α-helical (Figure 4B).
In order to assess whether differences in the crystallization conditions of the Dm BicD-CTD/F684I mutant and the wild-type protein contributed to the observed structural differences, we also recorded CD wavelength spectra in modified crystallization buffers (Figure S4). These spectra confirmed that the compounds of the crystallization buffers and a temperature of 20°C do not affect the α-helical content of either the mutant or the wild-type protein and therefore do not cause the observed structural disorder in the mutant.
Furthermore, we compared the dimer interface of the structure of the F684I mutant with the wild type (Table S3). Since the N-terminal region of one of two chains is disordered in the mutant, the interface area is smaller (1427 Å2) compared to the wild type (1764 Å2), and the dimer interface of the F684I mutant contains eighteen fewer interacting residues as well as one less hydrogen bond and one less salt bridge compared to the wild type (Table S3). It is unknown if the disordered region engages in interactions that stabilize the dimer, however, in the absence of additional interactions one would expect that the F684I mutant would be less stable than the wild type.
Thus, we probed thermodynamic stability of the F684I mutant and the wild type by recording circular dichroism spectroscopy melting curves (Figure 4C). Protein unfolding was monitored by CD spectroscopy at 222 nm. The apparent melting temperature TM of Dm BicD-CTD/F684I was 45.4±1.6°C, which is similar to the TM of the wild-type protein (44.0±1.8°C) (Figure 4C). Based on the melting temperatures, the Dm BicD-CTD/F684I mutant has comparable thermodynamic stability as the wild type, despite the N-terminal disordered portion of one helix. Therefore, it is conceivable that the disordered portion still interacts with the other chain, as it would explain the observed similar thermodynamic stability.
To conclude, the ~20 residue disordered region is present in the crystal and folded, and since the thermodynamic stability of the mutant is comparable to the wild type, this region is likely to still interact with the other chain. These data suggest that the region N-terminal of I684 is flexible in the mutant in one chain and possibly assuming multiple conformations.
MD simulations suggest that the N-terminal region of the mutant can switch between homotypic and heterotypic registries.
In the X-ray structure, the F684I mutant of Dm BicD-CTD assumes a conformation with a homotypic coiled-coil registry, and the region N-terminal of I684 is disordered for one of two chains. In order to gain insight into the disorder in the N-terminal region of Dm BicD-CTD/F684I, we used MD simulations to assess if this region would sample multiple conformations. For these simulations of Dm BicD-CTD/F684I, the structure of the homolog Hs BicD2-CTD was chosen as a starting point, since it has a fully resolved homotypic coiled-coil registry (unlike Dm BicD WT), and the protein sequence was mutated to Dm BicD-CTD/F684I. Of the three independent simulations carried out with different initial atomic velocities, two occasionally showed local heterotypic registry-like conformations in the region around I684. These conformations were however not stable, indicating barriers along key coordinates for the registry shift that could not be fully overcome in these simulations. In the third simulation, the region N-terminal of residue Y698 of the Dm BicD-CTD/F684I coiled-coil switched back and forth between a homotypic and heterotypic registry, while the region C-terminal of Y698 retained a homotypic registry, in line with the various crystal structures. After an initial 10 ns equilibration with restraints on the backbone heavy atoms and an additional unrestrained 50 ns simulation, the overall coiled-coil registry of Dm BicD-CTD/F684I started switching after ~30 ns from homotypic to asymmetric, with the shift complete at ~45 ns, and reverted back to homotypic after ~105 ns (Figure 5A–C). This suggests that the disorder in the region N-terminal of residue I684 of one of the chains, as seen in the crystal structure, is likely caused by the ability of this region to easily switch between the homotypic and heterotypic registries.
Figure 5. MD simulations suggest that the Dm BicD-CTD/F684I mutant switches between homotypic and asymmetric registries.
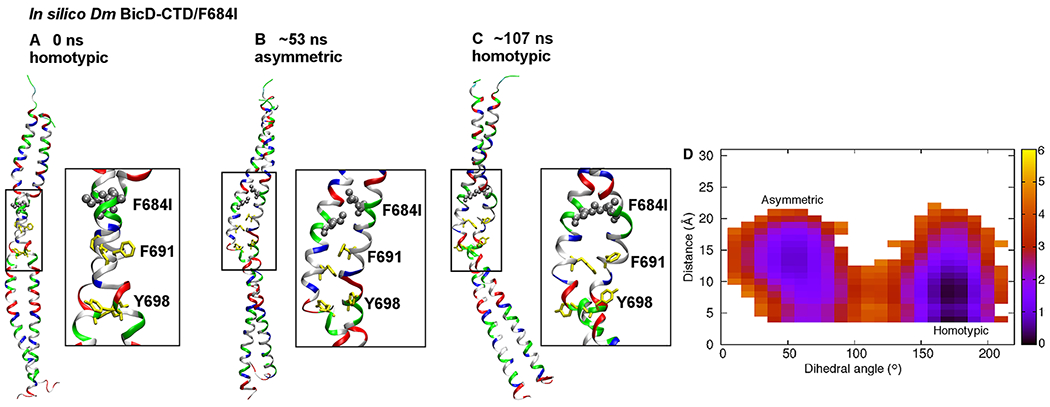
(A) Cartoon representation of the equilibrated structure of Dm BicD-CTD/F684I,30 with homotypic registry, colored by residue type (blue: positively charged, red: negatively charged, green: polar, white: non-polar). F684 was mutated to isoleucine (silver spheres). F691 and Y698 are shown in yellow stick representation. A close-up of the boxed area is shown on the right. The equilibration comprised of a 10 ns simulation with restraints on the backbone heavy atoms, and a 50 ns unrestrained simulation. (B) Structure of the F684I mutant of Dm BicD-CTD after ~53 ns of an MD simulation. Note that the N-terminal region of the coiled-coil switches to a heterotypic registry; therefore, the overall coiled-coil registry is asymmetric. (C) Structure of the F684I mutant of Dm BicD-CTD after 107 ns of the same MD simulation. Note that the structure switches back to a homotypic coiled-coil registry. However, the solvent-exposed F691 sidechains are oriented towards the same side, as opposed to opposite sides in A. This leads to a slight distortion of the coiled coil around the F691 residues. (D) Free energy in kcal/mol as a function of the C-Cα-Cβ-Cγ dihedral angle of F691 of chain A (plotted along the horizontal axis), and the distance between the sidechain N atom of K678 of chain A and the Cδ atom of E673 of chain B (plotted along the vertical axis). The distance between the sidechain N atom and Cδ was chosen, since both oxygen atoms of the carboxyl group can engage in salt bridge formation. The free energy is depicted using a color map that ranges from 0 to 6 kcal/mol. The free energy difference between the minima is ~1 kcal/mol, with a free energy barrier of ~4-5 kcal/mol. See Figure S5.
In order to gain insights into the kinetics and thermodynamics of the observed coiled-coil registry shift, we also calculated the relative free energies of the conformations with homotypic and asymmetric coiled-coiled registries, as well as the free energy barrier that separates them. Detailed analysis of the MD trajectory for Dm BicD-CTD/F684I revealed the C-C-C-C dihedral angle of F691 of chain A to be directly correlated with the registry shift. It assumed values around 175° and around 55° in conformations with homotypic and asymmetric registries, respectively. Interestingly, the corresponding dihedral angle of F691 of chain B and of I684 and Y698 of either chain were not found to be correlated with the registry shift. The different behaviors of F691 of the two chains is consistent with the disorder in the N-terminal region of only one of the chains in the crystal structure. In addition to the above mentioned dihedral angle, the salt-bridge interaction between K678 of chain A and E673 of chain B that is formed in the conformation with the homotypic registry was also found to be correlated with the registry shift, with the interaction completely broken in the conformation with asymmetric registry. Interestingly, of all the salt-bridges in the N-terminal region, only this one was found to be related to the registry shift. Our data therefore provide insights into the molecular mechanism of the coiled-coil registry shift, and reveal key roles of residue F691 from chain A as well as of the salt bridge between K678 of chain A and E673 of chain B in the mechanism.
The identification of these key coordinates related to registry shift allowed the calculation of the potential of mean force (PMF) or free energy as a function of the two coordinates (Eqn. 1, see Materials and Methods), revealing the free energy difference between the homotypic and asymmetric registries to be less than 1 kcal/mol, and the free energy barrier for transition between the registries to be ~4-5 kcal/mol (Figure 5D). Hence, the two registries have similar stability and can interconvert on a timescale of tens of ns, as observed in the MD simulations. In the crystal structure of Dm BicD-CTD WT, disorder was not observed. It is conceivable that F684 is a key residue that serves to lock the WT BicD coiled-coil in conformations with either homotypic or asymmetric registry. Replacement of this residue by isoleucine may lead to promiscuity, allowing other conformations to form. This is in line with the crystal structure of the F684I mutant and the MD simulations. It should be noted that the structure of the mutant has an elevated overall B-factor (Table 1), indicating flexibility, and a region of one chain that undergoes coiled-coil registry shifts is also disordered, further suggesting conformational variability.
In comparison, in our recent MD simulations of the human homolog of the Dm BicD-CTD/F684I mutant, Hs BicD2-CTD/F743I with asymmetric coiled-coil registry, the homologous F743I mutation induced a coiled-coil registry shift from an asymmetric to a fully heterotypic registry,30 whereas simulations starting from the homotypic registry of Hs BicD2-CTD/F743I maintained a homotypic registry (Figure S5). The results from these two simulations support the idea that the human homolog of the Dm BicD/F684I mutant can also sample conformations with distinct registries, likely resulting in conformational flexibility.
To conclude, our MD simulations of the Dm BicD-CTD/F684I mutant show that it samples conformations with homotypic and asymmetric coiled-coil registries which are of similar stability on a time scale of tens of ns (Figure 5), which would explain why one chain in the N-terminal region of the coiled-coil is disordered in the crystal structure.
The homologous F743I mutation modulates cargo selection in Hs BicD2.
Because a registry shift is expected to re-model the surface of the coiled-coil, which harbors binding sites for cargo adaptors, we hypothesized that a coiled-coil registry shift in BicD2 plays a role in cargo selection. The binding sites for the cargo adaptors Dm Egalitarian and Dm Rab6GTP on Dm BicD have been previously mapped to residues 702-743 (Figure 6A),9,10 a region of Dm BicD that does not undergo coiled-coil registry shifts. A similar region has been mapped as minimal Rab6GTP binding site for a close homolog of human BicD2 (residues 755-802, Figure 6B, see Table 2 for residue numbering in the homologs). Indeed, the F684I mutation of the Drosophila BicD-CTD, which is located N-terminal of the mapped binding site, does not affect the interaction with Egalitarian and Rab6GTP.9
Figure 6. Role of coiled-coil registry shifts in cargo selection of human BicD2.
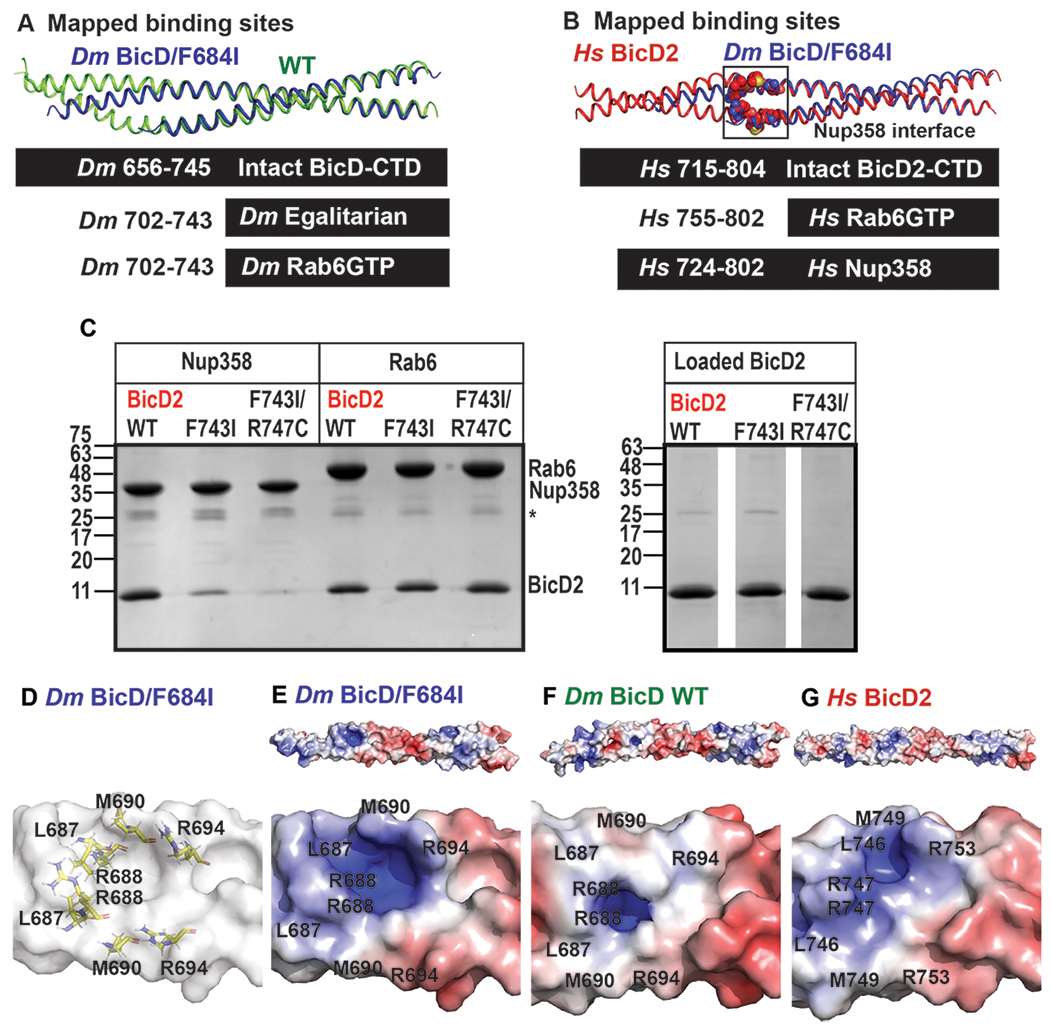
(A) Least-squares superimposition of the structures of Dm BicD-CTD/F684I (dark blue) and wild-type (green) in cartoon representation is shown. A schematic representation of the intact Dm BicD-CTD (black bar) and of the mapped cargo adaptor binding sites including residue numbers is shown below. (B) Least squares superimposition of the structures of Dm BicD-CTD/F684I (dark blue) and Hs BicD2-CTD (red) in cartoon representation, with a schematic representation of the intact protein and the mapped cargo adaptor binding sites below. Known Nup358/BicD2 interface residues (see Table 2) are shown in spheres representation.10 (C) Pull-down-assays of BicD2-CTD (wild type, F743I or F743I/R747C mutant) with the GST-tagged cargo adaptors Rab6GTP-GST and Nup358-min-GST. An asterisk indicates the location of the GST-band. An SDS-PAGE of the elution fractions is shown in the left panel. Right panel: SDS-PAGE analysis of the BicD2-CTD load fractions. Pull-down assays were repeated three times with similar results. (D) Surface representation of the structure of Dm BicD-CTD/F684I. Several important Nup358/BicD2 interface residues are known10 (see Table 2); homologous Dm residues are shown in stick representation and labeled. (E-G) The surface electrostatic potential of distinct structures is shown. Positive (blue: 5 kT/e) and negative (red: −5 kT/e) potentials are mapped on the solvent excluded molecular surface (top panel). The bottom panel shows a close-up of the known BicD2/Nup358 interface residues. The same view as in (D) is shown. (E) Dm BicD-CTD/F684I. (F) Dm BicD-CTD wild type.9 (G) Hs BicD2-CTD.30
Notably, the mammalian cargo adaptor Nup358 binds to a larger region that includes residues 724-802 of Hs BicD2 (mapped for a homolog)10 and a portion of this region undergoes a coiled-coil registry shift (Figure 6B). Several important Nup358 interface residues that are N-terminal of F743 were identified for a close homolog of human BicD2 (Table 2; residues L746, R747, M749 and R753).10 It should be noted that it is unknown whether Dm Nup358 interacts with Dm BicD43,44 and as outlined above, Dm Egalitarian and Dm Rab6GTP bind to a smaller binding site that is not expected to undergo coiled-coil registry shifts. Therefore, we investigated the effects of the Hs BicD2/CTD F743I mutation on the interaction with Hs Nup358, rather than the Drosophila homologs. We hypothesized that the homologous F743I mutation in Hs BicD2 would modulate the interaction between BicD2 and Nup358, since a portion of the interface is located in the region that is thought to undergo coiled-coil registry shifts. The BicD2/Rab6GTP interaction however is expected to be unaffected by the F743I mutation, since the binding site is located in the region that remains homotypic. This is confirmed by pull-down assays of Rab6GTP with the BicD2-CTD/F743I mutant, and the F743I/R747C mutant, which both bind with comparable strength as observed for the wild-type (Figure 6C). Interestingly, binding of Nup358 is modulated by the mutation. In a pull-down-assay, the minimal interacting domain Nup358-min30 pulls down wild-type BicD2-CTD much more strongly compared to the F743I mutant (Figure 6C). Binding is even more strongly weakened for the double mutant F743I/R747C, which combines two mutations that induced coiled-coil registry shifts in simulations (Figure 6C).30
To gain further insights into the impact of the mutation on the cargo-adaptor-binding interface, we compared the electrostatic surface potential of the structures of Dm BicD-CTD/F684I, the wild type (asymmetric registry) and Hs BicD2-CTD (homotypic registry) (Figure 6D–G). There are differences in the electrostatic surface potential in the area where Nup358 binds (L746, R747, M749 and R753)10 (Figure 6D–G). Notably, both the Dm BicD-CTD/F684I mutant and Hs BicD2-CTD (homotypic registry) have a highly positively charged surface electrostatic potential in the area of these interface residues, creating a basic pocket (blue, Figure 6E,G). In comparison, the same interface area in the Dm BicD-CTD has a much less charged electrostatic surface potential (Figure 6F). Such changes could be caused by a coiled-coil registry shift and could be responsible for the observed difference for the interaction between Hs BicD2-CTD wild type and the F743I mutant with Nup358-min. It should be noted that the crystal structures of Hs BicD2-CTD and Ms BicD1-CTD revealed homotypic coiled-coil registries. However, in both cases, crystals were obtained from crystallization of BicD/cargo adaptor complexes, and therefore it remains to be established whether the coiled-coil registry of mammalian apo-BicD is asymmetric or homotypic.
To conclude, while the Dm BicD/F684I mutant shows comparable selectivity as the wild-type protein towards the cargo adaptors Egalitarian and Rab6GTP, 9 in human BicD2 the homologous F743I mutant that likely induces a coiled-coil registry shift, affects cargo selection. While binding of BicD2 to Rab6GTP is not affected by the mutation, binding to Nup358 is strongly reduced, likely because it binds to a larger binding site that contains a portion of the protein that may undergo a coiled-coil registry shift.
Discussion
In the absence of cargo, BicD forms an auto-inhibited state that is unable to recruit dynein.3–12 Here we show by single molecule processivity assays that the classical Dm BicDF684I mutant can recruit dynein/dynactin in the absence of cargo, resulting in cargo-independent activation of dynein-dynactin motility. Our results are consistent with cellular studies that show increased dynein recruitment.9,16 Our observation is significant and unexpected, since previous models based on studies in flies suggested that the BicDF684I mutation enhances dynein-mediated trafficking of BicD/cargo complexes into oocytes,9 and the authors hypothesized that the enhanced dynein recruitment still required cargo, based on a double mutant in flies (one that reduced cargo binding, L731A, in addition to F684I) that largely failed to induce anterior-posterior defects.9 It should be noted that in these in vivo studies, cargoes were invariably present, and thus it was prior to our in vitro study not known that the Dm BicDF684I mutant activates dynein motility even in the absence of cargo. It should be noted that when cargo binding was disrupted by a L731A mutation, BicDF684I did not induce more dynein/BicD dependent trafficking in fly oocytes.9 Future experiments will test whether this reflects the involvement of additional processes or protein factors (such as additional cargoes) that are present in vivo or if this is due to structural changes caused by the additional L731A mutation. To conclude, our results suggest that the Dm BicDF684I mutant is capable of activating dynein in a cargo-independent manner.
To probe the mechanism of BicD activation for dynein recruitment in the F684I mutant, we determined the X-ray structure of the C-terminal cargo-binding domain of the bicaudal mutant (Dm BicD-CTD/F684I). The bicaudal mutant assumes a conformation with homotypic registry as its predominant structural state, as the helices are aligned at equal height up to residue I684, unlike in the wild-type, where F684 residues are displaced vertically by one helical turn against each other. However, a ~20 residue region upstream of residue I684 is not resolved for one chain in the structure. While I684 is resolved, the shape of the electron density map and the high B-factors also suggest flexibility of this residue (Fig. 3). This structural flexibility of the N-terminal region is also confirmed by MD simulations and free energy calculations, in which the mutant samples conformations with homotypic and asymmetric coiled-coil registries of similar stability on a time scale of tens of ns, which would explain the observed disorder in the crystal structure. Our data suggest that the F684I mutation shifts the equilibrium of registry-shifted conformers, resulting in formation of a larger percentage of BicD with homotypic registry.
It was previously proposed that BicD undergoes coiled-coil registry shifts, which activate it for dynein recruitment upon cargo binding.9,10,30 This idea is based on the structures of distinct BicD homologs with distinct coiled-coil registries as well as our recent MD simulations, which suggest that human BicD2 can assume stable conformations with either homotypic or asymmetric coiled-coil registries.9,10,30
Here, we show that in MD simulations, the structure of the Dm BicD/F684I mutant transitions back and forth between homotypic and asymmetric coiled-coil registries on a time scale of tens of ns. These simulations also reveal key roles for the salt bridge between K678 of chain A and E673 of chain B, as well as of residue F691 from chain A in the structural transition. Notably, the previous finding that a F691I mutant causes a phenotype of double-abdomen fly embryos that is similar to the BicDF684I phenotype supports the role of residue F691 in promoting auto-inhibition.9 Our results suggest that the F684I mutation induces structural dynamicity and leads to formation of multiple conformations, which is also supported by the crystal structure. The structure has a comparatively high B-factor, suggesting flexibility and a ~20 amino acid region at the N-terminus of one chain is not resolved in the structure, whereas the second chain is resolved, and therefore the coiled-coil registry in this region cannot be determined. It is unlikely that the different crystallization conditions contribute to the distinct conformations of the coiled-coil, since the mutant and wild-type protein have the same α-helical content in different crystallization buffers. Our CD data strongly suggest that the disordered region is present in the crystal and α-helical. The disordered region likely also still interacts with the second ordered chain, since the melting temperatures in solution studies indicate similar thermodynamic stability for the mutant and the wild-type.
In the wild-type, F684 likely serves as a switch to lock Dm BicD in conformations with distinct registries.9,30 In the conformation with the asymmetric registry, F684 rotates to the core of the coiled-coil and forms an edge-to-face aromatic interaction with residue F684 from the second chain, which leads to the observed vertical displacement of the helices.9,30 In the mutant, the phenylalanine side chain is replaced by a much smaller isoleucine side chain, which likely cannot lock the asymmetric registry in place, and due to its smaller size is unable to prevent conformational changes. The result is likely a dynamic mixture of several states.
The single molecule assays, which use full-length mutant BicD, are consistent with the predictions derived from the minimal cargo-binding domain structure. In the auto-inhibited conformation, full-length BicD2 forms a looped structure in which the CTD binds to the N-terminal dynein/dynactin binding site, likely causing steric interference.2–15 Notably, the very similar sedimentation coefficients of wild-type and BicDF684I (Figure 1, Table S2) imply that both wild-type BicD and the F684I mutant form auto-inhibited, looped conformation in the absence of dynein and cargo, ruling out the simple mechanism that the mutation acts via abolishing the looped structure that is characteristic for the auto-inhibited state in wild-type BicD.11 It should be noted that the latter conclusion is supported by prior yeast two hybrid studies, which suggest that the F684I and the wild-type BicD-CTD both interact with the dynein binding domain.9 Based on our data, we hypothesize that a coiled-coil registry shift in BicD, either induced by the F684I mutation or by binding of cargo to wild-type BicD would prime auto-inhibited BicD for dynein/dynactin recruitment. Binding of dynein/dynactin to registry-shifted BicD would then convert BicD to its fully activated state. The activated state of BicD would in turn activate dynein motility and could potentially be an open or extended structural state. High-resolution structural studies will be needed to further confirm this mechanism. Cargo binding or the F684I mutation could induce a local coiled-coil registry shift in the BicD-CTD, which might be sufficient to weaken binding to the N-terminal dynein/dynactin binding site and thus activate BicD for dynein recruitment. Alternatively, the registry shift could propagate through the entire coiled-coil to the N-terminal dynein-dynactin binding site. Finally, the induced flexibility and formation of multiple conformations as observed in the mutant may potentially also be an inherent structural feature of cargo-bound wild type BicD. Such structural and mechanistic details remain to be established, and studies with full-length proteins in physiological context remain to be conducted to fully understand the molecular mechanism of BicD auto-inhibition and activation.
In addition to BicD2, several other dynein adaptors have coiled-coil structures (e.g. NudE/NudEL,45–47 the Hook proteins,48,49 RILP,50 Rab11-FIP3 and Spindly51,52 and some of them, e.g. Spindly assume an auto-inhibited state in the absence of cargo.7,52 The molecular mechanism for auto-inhibition in these dynein adaptors remains to be established, and it is possible that some of them undergo coiled-coil registry shifts as well.
Cargo selection for BicD2-dependent transport events are tightly regulated, but currently known regulatory mechanisms, which include competition of cargo adaptors29 and the G2 specific kinase Cdk1,24 are insufficient to fully explain how BicD2 switches between these cargoes in a cell-cycle specific manner.29
In Drosophila, cargo adaptors Egalitarian and Rab6GTP bind to a small domain of BicD that remains homotypic and does not undergo coiled-coil registry shifts, consistent with the F684I mutation not affecting cargo selection.9,22 The F684I mutation does not affect the affinity of BicD for Egalitarian or Rab6GTP 9 but it promotes cargo-independent dynein recruitment (Figure 1) thereby resulting in increased transport frequencies. An increase in dynein-mediated transport of oskar mRNA/Egalitarian in the F684I mutant causes the double-abdomen fly phenotype.9,16–20 This likely means that dynein recruitment is more limiting to transport than the affinity of BicD towards cargo adaptors.9,53
Notably, we propose that coiled-coil registry shifts in human BicD2 modulate cargo selection. The cargo adaptor Hs Nup358 binds to a larger interface on BicD2 that includes a region which undergoes coiled-coil registry shift.10 The homologous F743I mutation which is expected to induce a coiled-coil registry shift30 diminishes binding of Nup358 to Hs BicD2, whereas the interaction of BicD2 with Rab6GTP is unaffected. These studies were performed with the human homologs, since it remains to be established whether Dm Nup358 interacts with Dm BicD.43,44 Binding of cargo-adaptors such as Nup358 is expected to induce a coiled-coil registry shift in BicD2, and in our in vitro binding assays, Nup358 remains associated with BicD2. It remains to be established by future studies, if this is due to a slow dissociation constant of the complex or whether the interaction with BicD2 induces structural changes in Nup358 that stabilize it in the complex once bound (“induced fit”). Notably, a registry shift could potentially have a regulatory role in preventing the cargo adaptor Nup358 from binding, however, it remains to be established if such mechanism indeed facilitates cargo adaptor selection in vivo. Future studies will establish, whether a coiled-coil registry shift in BicD2 may be triggered by a regulatory signal, which could modulate cargo selection by reducing the binding of selected cargo adaptors including Nup358.
In conclusion, our data provide mechanistic insights into auto-inhibition and cargo selection of the dynein adaptor BicD2. Our results suggest that the full-length Dm BicDF684I mutant is capable of activating dynein for processive transport in a cargo-independent manner. The X-ray structure of Dm BicD-CTD/F684I reveals that the mutation induces a coiled-coil registry shift, which we propose as the underlying mechanism for cargo-independent activation. A ~20 residue N-terminal region of one monomer is disordered in the structure, in line with MD simulations of the mutant which samples conformations with homotypic and asymmetric registries on a time scale of tens of ns. Free energy calculations indicate that conformations with homotypic and asymmetric registries have similar stability and are separated by a free energy barrier of ~4-5 kcal/mol. The observed structural dynamicity could either be a structural feature of activated BicD2 or it could be caused by the mutation. Notably, the human homolog of the F684I mutant (i.e. F743I) shows reduced affinity to the cargo adaptor Nup358, which recruits BicD2 to the nuclear envelope. We thus propose that a coiled-coil registry shift modulates cargo selection for BicD2-dependent transport pathways, which are important for cell cycle control and brain development.15,22–25,54
Materials and Methods
Protein expression and purification
Codon optimized human dynein for expression in Sf9 cells (DYNC1H1 (DHC), DYNC1I2 (DIC), DYNC1LI2 (DLIC), DYNLT1 (Tctex), DYNLRB1 (Robl) and DYNLL1(LC8)) was a generous gift from Simon Bullock.5 The heavy chain was modified to contain an N-terminal FLAG tag followed by a biotin tag to enable heavy chain labeling. Dynein expression in Sf9 cells and purification was as described in 11. Purified dynein was stored at −20°C in 10 mM imidazole, pH 7.4, 0.2 M NaCl, 1 mM EGTA, 2 mM DTT, 10 μM MgATP, 5 µg/ml leupeptin, 50% glycerol. Dynactin was purified from ~300 g bovine brain tissue as described,55 and stored at −20°C in the same buffer as dynein. Full-length WT and mutant Drosophila BicD (BicDWT and BicDF684I) were expressed in Sf9 cells and Drosophila BicD-coiled-coil domain 1 (BicDCC1) in bacteria as described.11
To create an expression construct of Drosophila BicD-CTD/F684I (residues 656-745), the corresponding codon optimized DNA sequence was commercially synthesized by Genscript and cloned into the pET28a vector with the NdeI and XhoI restriction sites. Drosophila Bicaudal D-CTD F684I (Dm BicD-CTD F684I, residues 656-745) was expressed in E. coli BL20(DE3)-RIL strain. His6-tagged Dm BicD-CTD/F684I was purified by Ni-NTA affinity chromatography and the tag was cleaved by thrombin, followed by second Ni-NTA affinity chromatography as described in 29. The protein was further purified by gel filtration chromatography on a HiLoad™ 16/600 Superdex 200 pg column (GE Healthcare) with the following buffer: 20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM TCEP as described.29 Wild-type Dm BicD-CTD was expressed and purified as described.30,56,57 Purified protein was analyzed by SDS-PAGE, 16% acrylamide gels and stained by Coomassie Blue.
GST-pull down assays of Rab6GTP-GST, Nup358-min-GST and Hs BicD2-CTD wild type as well as the mutants F743I and F743I/R747C were performed as described.30 For the assays, His6-tagged Hs BicD2-CTD fragments (wild type and mutants) were purified as described by a single affinity chromatography step from 1L of cell culture, whereas Rab6GTP-GST and Nup358-min-GST where purified from 0.5L of cell culture.30 For GST-pull-down with Nup358-min, human Nup358 (residues 2148-2240) was purified by glutathione sepharose as described30 but not eluted. We then proceeded with the same protocol as described for the Rab6GTP GST-pull-down,30 however, GTP was omitted.
Crystallization
Purified Dm BicD-CTD/F684I was set up for crystallization at 20°C in hanging drops. For the drop, 1 μl of the protein sample at a concentration of 8 mg/ml was mixed with 1 μl of reservoir buffer (4% PEG 3350, 0.4 M NaSCN, 5% glycerol). Crystals in space group P3121 were obtained after 2-3 days in the dimensions 0.2 mm * 0.2 mm * 0.2 mm. Crystals were soaked in a cryo-buffer consisting of the reservoir solution with addition of 30% glycerol and 10 mM HEPES pH 7.5 and flash frozen in the liquid nitrogen.
Structure determination
Data was collected from a single crystal at NE-CAT beam line 24ID-C at the Advanced Photon Source (APS), Argonne National Lab (ANL), which was equipped with a Pilatus 6M detector. X-ray intensities were processed and scaled using the RAPD software developed by F. Murphy, D. Neau, K. Perry and S. Banerjee, APS (https://rapd.nec.aps.anl.gov/login/login.html). The structure was determined by molecular replacement in the PHENIX suite,40 with the structure of the wild-type as the search model, which was truncated N-terminal of residue 692.9 An initial model was obtained from automatic model building in the PHENIX suite and completed by manual model building in the program COOT.40,58 The structure was refined through iterative cycles of manual model building and refinement40,58 to 2.35Å resolution in the PHENIX suite,40 with an Rfree of 25.99% and an Rwork of 25.06% (Table 1). The stereochemical quality of the model was assessed with MolProbity.59 The crystallographic statistics are summarized in Table 1.
Structural analysis
Structures were compared by least-squares superimposition of the coordinates in COOT.58 Dimer interfaces and knobs-into-holes interactions were analyzed by the web servers PISA and SOCKET, respectively.60,61 For identification of knobs-into-hole interactions, a cutoff of 7.5 Å was used (helix extension 1 residue). Secondary structure assignment was performed with DSSP.62 Figures were created in the PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC) and VMD.63 The program APBS was used to analyze surface electrostatic potentials of proteins. 64 Default parameters were used; neutral charges were assigned to the N- and C-termini, waters were removed and selenomethionine residues were converted to methionine.
Single molecule processivity assays with full-length Dm BicD
Dynein, dynactin and Dm BicD constructs were each diluted into 30 mM HEPES pH 7.4, 300 mM KOAc, 2 mM MgOAc, 1 mM EGTA, 20 mM DTT, clarified for 20 min at 400,000 x g to remove aggregates, and the concentration determined with the Bradford reagent (Bio-Rad). To form the dynein-dynactin-BicD (DDB) complex, dynein, dynactin and BicD (WT, CC1, or F684I) were incubated at a molar ratio of 1:1:2 (200 nM dynein, 200 nM dynactin and 400 nM BicD) on ice for 30 min in motility buffer (30 mM HEPES pH 7.4, 150 mM KOAc, 2 mM MgOAc, 1 mM EGTA, 2 mM MgATP, 20 mM DTT, 8 mg/ml BSA, 0.5 mg/ml kappa-casein, 0.5% pluronic F68, 10 mM paclitaxel and an oxygen scavenger system (5.8 mg/ml glucose, 0.045 mg/ml catalase, and 0.067 mg/ml glucose oxidase). The molar ratio of dynein:dynactin:BicD was 1:1:2 to favor recruitment of only 1 dynein to the ternary complex. Previously, we observed substantial recruitment of two dyneins at a higher molar ratio of dynein to BicD (dynein:dynactin:BicD=2:1:1).11 To label the biotin-tag at the N-terminal region of the dynein heavy chain, 400 nM streptavidin-conjugated 655 quantum dots (Invitrogen) were added to the DDB complex and incubated on ice for 15 min. The DDB complex was diluted in motility buffer containing 50 mM KOAc to a final concentration of 0.5 nM dynein for observing motion on microtubules.
PEGylated slides were coated with 0.3 mg/ml rigor kinesin for attachment of rhodamine-labeled microtubules as described in 11. Motility assays for all three DDB complexes (BicDWT, BicDCC1 and BicDF684I) were performed on three lanes of a single slide. Total Internal Reflection Fluorescence (TIRF) microscopy was used to capture images of Qdot labeled-dynein and microtubule tracks. Imaging was performed on a Nikon ECLIPSE Ti microscope equipped with through-objective type TIRF and run by the Nikon NIS Elements software. Images were captured at 200 ms temporal and 6 nm spatial resolution. Rhodamine-labeled microtubules and Qdot (655nm)-labeled dynein were excited with the 488 and 561 nm laser lines, respectively, and images simultaneously recorded at five frames/s using two Andor EMCCD cameras (Andor Technology USA, South Windsor, CT). Run-length was total travel distance, and speed was total travel distance divided by time. Binding frequency was normalized to number of events per time per μm microtubule, and statistical significance determined by one way ANOVA followed by Tukey’s post-hoc test. For speed data, an unpaired t-test with 95% confidence interval was performed. For run length data, the Kolmogorov-Smirnov test with a 95% confidence interval was performed.
Molecular dynamics simulations
In the absence of a fully resolved homotypic coiled-coil registry structure for Dm BicD-CTD/F684I, the X-ray crystallographic structure of Hs BicD2-CTD with homotypic coiled-coil registry30 was chosen as a starting point, and amino acid mutations were carried out to match the sequence of Dm BicD-CTD/F684I. MD simulations with implicit solvent were carried out using the CPU implementation of the PMEMD program in the AMBER16 package65 as described.30 The use of an implicit solvent model was justified by comprehensive comparisons of the results to those from explicit solvent simulations. An initial geometry optimization using the steepest descent method, followed by the conjugate gradient method, was carried out with the backbone heavy atoms restrained to their initial positions using a harmonic force constant of 10.0 kcal mol−1 Å−2. Subsequently, three independent simulations at 300 K with different initial velocities of the atoms were carried out for ~210 ns, ~210 ns and ~305 ns, respectively, with the first 10 ns of each simulation executed in the presence of harmonic restraints on the backbone heavy atoms using a force constant of 10 kcal mol−1 Å−2. The PMF or free energy associated with registry shift for the Dm BicD-CTD/F684I mutant was calculated using Eqn. (1) from the last 250 ns of the third trajectory in which both the homotypic and asymmetric registries were sampled.
| (1) |
Here, W is the PMF as a function of a dihedral angle ω, more specifically, the C-Cα-Cβ-Cγ dihedral angle of F691 of chain A, and a distance r, more specifically, the distance between the sidechain N atom of K678 of chain A and the C atom of E673 of chain B (see Figure 5). kB is the Boltzmann constant, T is temperature, and P is a two-dimensional probability distribution function. In the construction of the two-dimensional probability distribution and the corresponding PMF, a bin size of 10° for the dihedral angle ω and a bin size of 1 Å for the distance r were used. Any bins with zero probability were assigned a value of 100 kcal/mol for the PMF. Setting the minimum value of the overall PMF to zero changed this value to ~98.1 kcal/mol. For the bins with non-zero probability, the maximum value of PMF was found to be ~5 kcal/mol. Values of ω greater than 220° were sampled with a total probability of only ~0.03, and hence were excluded from the depiction of the PMF. At all other values of ω, all sampled values of r were included in the PMF depiction. The plot of PMF as a function of ω and r was generated using Gnuplot.66
CD spectroscopy
Purified Dm BicD-CTD/F684I (0.3 mg/ml) was dialyzed in the following buffer: 150 mM NaCl, 10 mM Tris pH 8 and 0.2 mM TCEP. CD spectra were recorded with a Jasco J-810 CD Spectrometer equipped with a thermoelectric control device, using a cuvette with a path length of 0.1cm. CD spectra shown in Figure S4 A, B were recorded with a Jasco J-1100 CD instrument. After the buffer baseline subtraction, CD signals were normalized to the protein concentration and converted to mean residue molar ellipticity [Θ]. CD spectra from 190 to 260nm were measured at 4°C or 90°C as described.30 Thermal unfolding profiles of proteins were recorded by CD spectroscopy at 222 nm as described.30 All protein denaturation profiles were sigmoidal; therefore, a two-state denaturation model (α-helix to random coil) was used to analyze the data. The apparent melting temperature (TM) was determined from the peak of smoothened differential melting curves d[Θ222](T)/dT using Origins Pro software. The fraction of folded peptide (Ffolded) was calculated from the equation Ffolded = ([Θ]-[Θ]unfolded)/([Θ]folded-[Θ]unfolded), where [Θ] is the observed molar ellipticity at any particular temperature and [Θ]unfolded and [Θ]folded are the molar ellipticities of the denatured (unfolded) and native (folded).
Supplementary Material
Synopsis:
The dynein adaptor Drosophila Bicaudal D (BicD) is auto-inhibited and activates dynein motility only after cargo is bound. In contrast, we show that the full-length BicD/F684I mutant activates dynein processivity even in the absence of cargo. Our X-ray structure of the C-terminal domain of the BicD/F684I mutant and our molecular dynamics simulations provide insights into the molecular mechanism that activates the adaptor Bicaudal D for dynein recruitment; these transport pathways are important for brain development and cell cycle control.
Acknowledgements
We thank Jonathan P Schuermann from APS and Richard Vallee from Columbia University for helpful discussions. We also thank Carol Bookwalter and Elena Krementsova for cloning, protein expression and purification, and analytical ultracentrifugation. All figures related to the computational studies were made using the Visual Molecular Dynamics (VMD) program.63 Furthermore, we thank S. Bane & B. Callahan (SUNY Binghamton), for access to equipment.
Funding
This paper was supported by the following grants: National Institute of Health, National Institute of General Medical Sciences (NIH NIGMS) grant 1R15GM128119-01 awarded to S.R.S. and GM078097 to KMT. Additional funds came from the Chemistry Department at State University of New York at Binghamton and the Research Foundation of the State University of New York.
X-ray diffraction data was collected at Northeastern Collaborative Access Team (NE-CAT) beam line 24ID-C at the Advanced Photon Source (APS), Argonne National Lab (ANL), which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165) and by a NIH-ORIP HEI grant (S10 RR029205). APS is operated by ANL for the DOE under contract No. DE-AC02-06CH11357. This work used the Extreme Science and Engineering Discovery Environment (XSEDE)67, which is supported by NSF grant ACI-1548562. The purchase of the Jasco J-1100 CD instrument at Binghamton University was supported by NIGMS 1R01GM125853-02S1 and 3R35GM130207-01S1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicting interests
The authors declare that they have no conflicting interests.
Data sets
Coordinates and structure factors were deposited to the protein data bank (https://www.wwpdb.org, under PDB ID 6TZW).
References
- 1.Cianfrocco MA, DeSantis ME, Leschziner AE, Reck-Peterson SL. Mechanism and regulation of cytoplasmic dynein. Annu Rev Cell Dev Biol. 2015;31(1):83–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoogenraad CC, Akhmanova A, Howell SA, et al. Mammalian Golgi associated Bicaudal D2 functions in the dynein dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Splinter D, Razafsky DS, Schlager MA, et al. BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol Biol Cell. 2012;23(21):4226–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlager MA, Serra-Marques A, Grigoriev I, et al. Bicaudal D family adaptor proteins control the velocity of dynein-based movements. Cell Rep. 2014;8(5):1248–1256. [DOI] [PubMed] [Google Scholar]
- 5.Schlager MA, Hoang HT, Urnavicius L, Bullock SL, Carter AP. In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J. 2014;33:1855–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urnavicius L, Zhang K, Diamant AG, et al. The structure of the dynactin complex and its interaction with dynein. Science 2015;347(6229):1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenney RJ, Huynh W, Tanenbaum ME, Bhabha G, Vale RD. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science. 2014;345(6194):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdhury S, Ketcham SA, Schroer TA, Lander GC. Structural organization of the dynein-dynactin complex bound to microtubules. Nat Struct Mol Biol. 2015;22(4):345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Salter HK, Holding AN, et al. Bicaudal-D uses a parallel, homodimeric coiled coil with heterotypic registry to coordinate recruitment of cargos to dynein. Genes Dev. 2013;27(11):1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terawaki S, Yoshikane A, Higuchi Y, Wakamatsu K. Structural basis for cargo binding and autoinhibition of Bicaudal-D1 by a parallel coiled-coil with homotypic registry. Biochem Biophys Res Commun. 2015;460(2):451–456. [DOI] [PubMed] [Google Scholar]
- 11.Sladewski TE, Billington N, Ali MY, et al. Recruitment of two dyneins to an mRNA-dependent Bicaudal D transport complex. eLife. 2018;7:e36306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClintock MA, Dix CI, Johnson CM, et al. RNA-directed activation of cytoplasmic dynein-1 in reconstituted transport RNPs. eLife. 2018;7:e36312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoogenraad CC, Wulf P, Schiefermeier N, et al. Bicaudal D induces selective dynein mediated microtubule minus end directed transport. EMBO J. 2003;22:6004–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh W, Vale RD. Disease-associated mutations in human BICD2 hyperactivate motility of dynein–dynactin. J Cell Biol. 2017;216(10):3051–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matanis T, Akhmanova A, Wulf P, et al. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat Cell Biol. 2002;4(12):986–992. [DOI] [PubMed] [Google Scholar]
- 16.Mohler J, Wieschaus EF. Dominant maternal-effect mutations of Drosophila melanogaster causing the production of double-abdomen embryos. Genetics. 1986;112(4):803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarro C, Puthalakath H, Adams JM, Strasser A, Lehmann R. Egalitarian binds dynein light chain to establish oocyte polarity and maintain oocyte fate. Nat Cell Biol. 2004;6(5):427–435. [DOI] [PubMed] [Google Scholar]
- 18.Zimyanin VL, Belaya K, Pecreaux J, et al. In vivo imaging of oskar mRNA transport reveals the mechanism of posterior localization. Cell. 2008;134(5):843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mach JM, Lehmann R. An Egalitarian-BicaudalD complex is essential for oocyte specification and axis determination in Drosophila. Genes Dev. 1997;11(4):423–435. [DOI] [PubMed] [Google Scholar]
- 20.Bull AL. Bicaudal, a genetic factor which affects the polarity of the embryo in Drosophila melanogaster. J Exp Zool. 1966;161(2):221–241. [Google Scholar]
- 21.Wharton RP, Struhl G. Structure of the Drosophila Bicaudal D protein and its role in localizing the posterior determinant nanos. Cell. 1989;59(5):881–892. [DOI] [PubMed] [Google Scholar]
- 22.Dienstbier M, Boehl F, Li X, Bullock SL. Egalitarian is a selective RNA-binding protein linking mRNA localization signals to the dynein motor. Genes Dev. 2009;23(13):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Splinter D, Tanenbaum ME, Lindqvist A, et al. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010;8(4):e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baffet AD, Hu DJ, Vallee RB. Cdk1 activates pre-mitotic nuclear envelope dynein recruitment and apical nuclear migration in neural stem cells. Dev Cell. 2015;33(6):703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu DJ, Baffet AD, Nayak T, Akhmanova A, Doye V, Vallee RB. Dynein recruitment to nuclear pores activates apical nuclear migration and mitotic entry in brain progenitor cells. Cell. 2013;154(6):1300–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peeters K, Litvinenko I, Asselbergh B, et al. Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am J Hum Gen. 2013;92(6):955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Synofzik M, Martinez-Carrera LA, Lindig T, Schöls L, Wirth B. Dominant spinal muscular atrophy due to BICD2: a novel mutation refines the phenotype. J Neurol Neurosurg Psychiatry. 2014;85(5):590–592. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Carrera LA, Wirth B. Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein. Front Neurosci. 2015;9:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noell CR, Loftus KM, Cui H, et al. A quantitative model for BicD2/cargo interactions. Biochemistry. 2018;57(46):6538–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noell CR, Loh JY, Debler EW, et al. Role of Coiled-Coil Registry Shifts in the Activation of Human Bicaudal D2 for Dynein Recruitment upon Cargo Binding. J Phys Chem Lett. 2019;10(15):4362–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macheboeuf P, Buffalo C, Fu C-y, et al. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature. 2011;472(7341):64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter AP, Garbarino JE, Wilson-Kubalek EM, et al. Structure and functional role of dynein’s microtubule-binding domain. Science. 2008;322(5908):1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbons IR, Garbarino JE, Tan CE, Reck-Peterson SL, Vale RD, Carter AP. The Affinity of the Dynein Microtubule-Binding Domain is Modulated by the Conformation of its Coiled-Coil Stalk. J Biol Chem. 2005;280(25):23960–23965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Croasdale R, Ivins FJ, Muskett F, et al. An Undecided Coiled Coil: the Leucine Zipper of Nek2 Kinase Exhibits Atypical Conformational Exchange Dynamics. J Biol Chem. 2011;286(31):27537–27547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xi Z, Gao Y, Sirinakis G, Guo H, Zhang Y. Single-molecule observation of helix staggering, sliding, and coiled coil misfolding. Proc Natl Acad of Sci U S A. 2012;109(15):5711–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snoberger A, Brettrager EJ, Smith DM. Conformational switching in the coiled-coil domains of a proteasomal ATPase regulates substrate processing. Nat Commun. 2018;9:2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi J, Park H, Seok C. How Does a Registry Change in Dynein’s Coiled-Coil Stalk Drive Binding of Dynein to Microtubules? Biochemistry. 2011;50(35):7629–7636. [DOI] [PubMed] [Google Scholar]
- 38.Kon T, Imamula K, Roberts AJ, et al. Helix sliding in the stalk coiled coil of dynein couples ATPase and microtubule binding. Nat Struct & Mol Biol. 2009;16(3):325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stathopulos PB, Schindl R, Fahrner M, et al. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat commun. 2013;4:2963–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66(2):213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crick F The packing of alpha-helices: simple coiled-coils. Acta Cryst. 1953;6:689–697. [Google Scholar]
- 42.O’Shea E, Klemm J, Kim P, Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254(5031):539–544. [DOI] [PubMed] [Google Scholar]
- 43.Hampoelz B, Schwarz A, Ronchi P, et al. Nuclear Pores Assemble from Nucleoporin Condensates During Oogenesis. Cell. 2019;179(3):671–686.e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui H, Noell CR, Behler RP, et al. Adapter Proteins for Opposing Motors Interact Simultaneously with Nuclear Pore Protein Nup358. Biochemistry. 2019;58(50):5085–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niethammer M, Smith DS, Ayala R, et al. NUDEL Is a Novel Cdk5 Substrate that Associates with LIS1 and Cytoplasmic Dynein. Neuron. 2000;28(3):697–711. [DOI] [PubMed] [Google Scholar]
- 46.Efimov VP, Morris NR. The Lis1-Related Nudf Protein of Aspergillus nidulans Interacts with the Coiled-Coil Domain of the Nude/Ro11 Protein. J Cell Biol. 2000;150(3):681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stehman SA, Chen Y, McKenney RJ, Vallee RB. NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J Cell Biol. 2007;178(4):583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olenick MA, Dominguez R, Holzbaur ELF. Dynein activator Hook1 is required for trafficking of BDNF-signaling endosomes in neurons. J Cell Biol. 2019;218(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bielska E, Schuster M, Roger Y, et al. Hook is an adapter that coordinates kinesin-3 and dynein cargo attachment on early endosomes. J Cell Biol. 2014;204(6):989–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20(4):683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffis ER, Stuurman N, Vale RD. Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol. 2007;177(6):1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosalaganti S, Keller J, Altenfeld A, et al. Structure of the RZZ complex and molecular basis of its interaction with Spindly. J Cell Biol. 2017;216(4):961–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bullock SL, Nicol A, Gross SP, Zicha D. Guidance of Bidirectional Motor Complexes by mRNA Cargoes through Control of Dynein Number and Activity. Curr Biol. 2006;16(14):1447–1452. [DOI] [PubMed] [Google Scholar]
- 54.Bianco A, Dienstbier M, Salter HK, Gatto G, Bullock SL. Bicaudal-D Regulates Fragile X Mental Retardation Protein Levels, Motility, and Function during Neuronal Morphogenesis. Curr Biol. 2010;20(16):1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bingham JB, King SJ, Schroer TA. Purification of dynactin and dynein from brain tissue. Methods Enzymol. 1998;298:171–184. [DOI] [PubMed] [Google Scholar]
- 56.Cui H, Loftus K, Noell C, Solmaz S. Identification of cyclin-dependent kinase 1 specific phosphorylation sites by an in vitro kinase assay. J Vis Exp. 2018;135 10.3791/57674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loftus KM, Coutavas E, Cui H, et al. Mechanism for G2 phase-specific nuclear export of the kinetochore protein CENP-F. Cell Cycle. 2017;16(15):1414–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen VB, Arendall WB, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walshaw J, Woolfson DN. SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol. 2001;307(5):1427–1450. [DOI] [PubMed] [Google Scholar]
- 61.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 62.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12). [DOI] [PubMed] [Google Scholar]
- 63.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. [DOI] [PubMed] [Google Scholar]
- 64.Jurrus E, Engel D, Star K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Science. 2018;27(1):112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Case DA, Betz RM, Cerutti DS, et al. AMBER 2016, University of California, San Francisco: 2016. [Google Scholar]
- 66.Williams T, Kelley C. Gnuplot 5.2: an interactive plotting program. http://gnuplot.info. 2019. [Google Scholar]
- 67.Towns J, Cockerill T, Dahan M, et al. XSEDE: Accelerating Scientific Discovery. Comput Sci Eng. 2014;16(5):62–74. [Google Scholar]
- 68.Philo JS. A Method for Directly Fitting the Time Derivative of Sedimentation Velocity Data and an Alternative Algorithm for Calculating Sedimentation Coefficient Distribution Functions. Anal Biochem. 2000;279(2):151–163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.