

Abstract
Copper accumulation and deficiency are reciprocally connected to lipid metabolism. In Wilson disease (WD), which is caused by a genetic loss of function of the copper-transporting P-type ATPase beta, copper accumulates mainly in the liver and lipid metabolism is dysregulated. The underlying mechanisms linking copper and lipid metabolism in WD are not clear. Copper may impair metabolic machinery by direct binding to protein and lipid structures or by generating reactive oxygen species with consequent damage to cellular organelles vital to energy metabolism. In the liver, copper overload results in mitochondrial impairment, down-regulation of lipid metabolism, and the development of steatosis with an etiology not fully elucidated. Little is known regarding the effect of copper overload on extrahepatic energy homeostasis. This review aims to discuss alterations in hepatic energy metabolism associated with WD, highlights potential mechanisms involved in the development of hepatic and systemic dysregulation of lipid metabolism, and reviews current knowledge on the effects of copper overload on extrahepatic energy metabolism.
Keywords: Copper, Copper-transporting P-type ATPase B, (ATP7B), Metabolism, Steatosis, Lipid, Carbohydrate
1. Introduction
Current evidence indicates a reciprocal connection between copper and lipid metabolism. In both animals and humans, the dietary and genetic modulation of copper levels affect cellular and systemic lipid homeostasis.1–7 Furthermore, many chronic diseases featuring dysregulated lipid metabolism are associated with alterations in copper levels.8–10 In Wilson disease (WD), a genetic copper overload condition, lipid metabolism is dysregulated and hepatic steatosis is a common early presentation. Despite the evidence, the underlying mechanism connecting copper and lipid metabolism in WD is not clear and the etiology of hepatic steatosis is not understood. In addition, the effects of copper overload on extrahepatic energy metabolism are understudied. Recently, dietary lipid composition was shown to play a role in WD liver injury,11 further highlighting a potential interaction between copper and lipids in the progression of WD.
This review aims to discuss copper homeostasis and alterations in hepatic energy metabolism in WD. It examines the interplay between copper metabolism and hepatic steatosis. It also highlights potential mechanisms underlying the observed dysregulation in hepatic and systemic lipid metabolism. Last, it reviews the current state of knowledge regarding the effects of copper overload on extrahepatic lipid metabolism.
2. Copper metabolic dysregulation, a common feature of metabolic disorders
Copper homeostasis is altered in many chronic conditions featuring dysregulation in lipid metabolism, including metabolic syndrome, obesity, and non-alcoholic fatty liver disease (NAFLD).8–10 In NAFLD, both adults and children present with relative hepatic copper deficiency. Aigner et al.12 showed patients with NAFLD have 50% less hepatic copper content compared to healthy subjects or patients with other liver diseases. In the same study, there was an inverse correlation between hepatic copper content and severity of steatosis, fasting glucose levels, insulin resistance, and the diagnosis of diabetes and metabolic syndrome.12 Similarly, pediatric patients with NAFLD presented with lower serum ceruloplasmin and copper levels in association with severe fatty liver activity scores.13 In liver biopsies from children with non-alcoholic steatohepatitis (NASH), there was evidence of lower copper levels in association with severe steatosis.14 Interestingly, obese patients with no or minimal hepatic steatosis presented with higher hepatic copper content compared to lean subjects and a positive correlation was reported between serum copper and body mass index, leptin, and insulin.15
NAFLD patients with lower serum and hepatic copper concentrations also presented higher serum ferritin levels and increased hepatic iron concentrations.16 Transcript levels of ferroportin, a transporter responsible for hepatic iron export, were reduced in NAFLD patients and associated with reduced hepatic copper levels.16
Extensive studies in animal models from the McClain group17 indicated features of dietary-induced NASH are exacerbated by concurrent dietary copper deficiency. When receiving high amounts of fructose, a major Western diet component involved in the development of NAFLD and NASH, diet-induced copper-deficient rats presented marked hepatocyte apoptosis, hepatic steatosis, and elevated plasma ferritin and hepatic iron when compared to rats receiving the same diet but with adequate copper status.17 In addition, alterations in dietary copper and fructose content were associated with changes in gut microbiome and fecal metabolome,18,19 which could act as possible mediators of liver injury.
Epidemiological data indicate the Western diet is relatively copper deficient.20,21 Though it is difficult to make a direct association between population studies, copper intake, and NAFLD incidence, NAFLD is coincidentally becoming an epidemic, a major cause of morbidity, and a frequent indicator for liver transplant.22,23
The mechanisms connecting lower copper levels with severe NAFLD histological features are largely unknown and multiple factors are potentially involved. Hepatic lipid metabolism is regulated by multi-level interactions between nutrients, hormones, nuclear receptors, transcription factors, and post-translational modifications.24,25 Hepatocellular lipid accumulation is a reflection of imbalanced hepatic uptake of circulating free fatty acids (FFA), de novo lipogenesis (DNL), fatty acid β-oxidation, and triglyceride (TAG) export as very low-density lipoproteins (VLDL).24
3. Copper metabolism and copper homeostasis
Copper is an essential micronutrient and the third most abundant transition metal in humans.26 It is estimated close to 1% of the total eukaryotic proteome contains recognized copper-binding proteins.27 The capacity of copper to shift between oxidation states, copper (I) and (II), enables it to function as a cofactor for enzymes involved in numerous vital biological functions, including cytochrome c oxidase (COX), or complex IV of the electron transport chain, which is central in mitochondrial energy generation28; copper-zinc superoxide dismutase 1 (SOD1), which is involved in antioxidant defense29; tyrosinase, which is involved in pigmentation30; and peptidylglycyl α-amidating monooxygenase, which is part of neuropeptide processing machinery.31 In addition, copper is involved in iron transport and is a mediator in various cellular signaling processes.32–36
Under a normal physiological state, copper rarely exists in a free and unbound form,37 and copper homeostasis is tightly controlled by regulating gastrointestinal uptake, transport, targeted tissue and intracellular shuttling, and hepatic biliary excretion.38 Free copper is detrimental to many cellular sub-structures by favoring the production of reactive oxygen species (ROS) which, ultimately, damage cellular proteins, lipids, and nucleic acids.26
Following dietary acquisition and intestinal absorption, copper is loosely bound to albumin. It is then delivered to the liver where about 75% of circulating copper is cleared and the remaining is distributed to other organs.39 In the hepatocyte, copper is incorporated into ceruloplasmin and exported to systemic circulation, whereas surplus copper is eliminated with bile secretion.
At the cellular level, hepatic copper uptake is mediated by copper transporter 1 (CTR1), which is responsible for copper trafficking across the plasma membrane.40 CTR1 is also thought to be involved in intestinal copper absorption as it is highly expressed on the apical membrane located on the luminal surface of enterocytes.41 With normal copper status, CTR1 functions by creating a channel which allows copper uptake into the cell, whereas with elevated copper, it is internalized and distributed in intracellular vesicles.42
In the cytosol, copper can be either stored in a complex with metallothioneins or distributed to various intracellular targets via a system of copper “chaperone” proteins.43 In particular, the copper chaperone for superoxide dismutase (CCS) mediates the delivery of copper to SOD1, located in the cytosol and mitochondrial intermembrane space; chaperones COX11, COX17, and COX19 deliver copper to COX in the mitochondria.43–45 Therefore, the mitochondria play an important role in intracellular copper metabolism as these organelles have a high content of COX and SOD1, both with high copper-binding affinity. COX is the terminal electron-accepting complex of OXPHOS and it requires multiple copper-binding assembly factors to have copper delivered, with mitochondrial phosphate carrier SLC25A3 playing a recently discovered central role in these mechanisms.46
The copper chaperone antioxidant protein 1 delivers copper for biliary excretion via activity of the copper-transporting P-type ATPases (ATP7A and ATP7B) in the trans-Golgi network.43–45,47,48 The homeostatic mechanism of biliary copper secretion is activated in response to excess copper and includes the delivery of copper to vesicles which, by merging with the hepatocyte’s canalicular membrane, allow copper excretion into the biliary tract. Beside copper excretion, ATP7B is also responsible for the supply of copper to ceruloplasmin and the maturation of apo-ceruloplasmin into holo-ceruloplasmin.49 The ATP7A copper transporter is universally expressed, whereas ATP7B is predominantly expressed in the liver. However, ATP7B is also expressed in the intestinal epithelial cells and regulates copper vesicular storage, with potential implications for intestinal lipid metabolism.50
4. Wilson disease and associated metabolic alterations
4.1. Definition, clinical manifestations, and treatment
WD is an autosomal recessive genetic condition due to a mutation in the ATP7B gene.51 The disease-causing mutations affecting ATP7B are responsible for varying degrees of impairment of copper transporter functions with consequent copper accumulation mainly in the liver, brain, and cornea. Traditionally, patients with WD are classified according to their prevalent phenotype as hepatic and/or neurologic (or psychiatric). However, WD is a systemic disease and multiple organs are thought to be involved simultaneously; clinical manifestations are often diversely combined and can present with variable severity. In addition, organ involvement is often not correlated with the amount of accumulated copper, e.g., severe neurologic involvement is not always associated with advanced liver disease, making the prediction of disease progression and outcome very challenging.52,53
The most common neurologic presentations are tremors, parkinsonism, and dystonia. Patients can also present with gait abnormalities, ataxia, choreoathetosis, dysarthria, and dysphagia. Virtually all patients with neurologic symptoms have corneal copper depositions, or Kayser-Fleischer rings.54 Psychiatric symptoms are very common and often undiagnosed. Cognitive impairment, depression, anxiety, psychosis, and sleep disturbances are frequently observed.55 Cardiac involvement is an emerging clinical issue and consists mainly of arrhythmias and cardiac myopathy. Recent data indicate patients with WD are frequently hospitalized for signs and symptoms related to cardiac complications.56,57 Other organs affected by copper accumulation include the kidney and patients can present with renal tubular acidosis, proteinuria, aminoaciduria, and hematuria.
The hepatic manifestations in WD include an elevation of transaminases, steatosis, cirrhosis with portal hypertension, and acute liver failure. WD is not commonly associated with the development of hepatocellular carcinoma or other types of liver cancer, even in the presence of cirrhosis.58 Interestingly, hepatic steatosis is a common histological feature in WD. In a large study of patients with WD and biopsy-confirmed liver pathology, the patatin-like phospholipase domain-containing protein 3 (PNPLA3) G allele, known to be strongly associated with increased hepatic fat content, was found to be associated with moderate to severe steatosis.59 Intriguingly, WD and NASH histological features partially overlap. Similar to NASH, WD can present with both micro- and macro-vesicular steatosis and glycogenated hepatocyte nuclei, ballooning of hepatocytes, and Mallory bodies.60
Evidence from animal models of WD indicates copper does not accumulate in hepatocytes continuously during WD progression and is redirected to other cells when hepatocytes are saturated with copper to facilitate hepatic regeneration. In Atp7b−/− mice, in situ imaging indicates that in early stages, copper is sequestered by metallothioneins in the cytosol and, with further accumulation, it enters the nucleus and triggers transcriptome remodeling to up-regulate cell-cycle machinery and down-regulate lipid metabolism.61 After an excess threshold of 250- to 800-fold above the normal level, copper uptake is inhibited, possibly by a down-regulation in CTR1. The authors also suggested the presence and activation of alternative copper-export mechanisms, yet to be characterized, to prevent copper accumulation and favor the uptake of copper by inflammatory cells, extracellular deposition, or urinary excretion.
If left undiagnosed or untreated, WD can be fatal. Once diagnosis is established, adherence to treatment is required for life.62 Treatment options include copper-chelating agents (D-penicillamine, trientine, or tetrathiomolybdate) and zinc salts (zinc sulphate, gluconate, and acetate).51 These agents aim to favor copper urinary excretion and/or to block its intestinal absorption, promoting copper elimination with feces.51,63,64 Patients with liver failure or with end-stage liver disease are considered for liver transplantation, which ultimately corrects hepatic metabolic defects in WD.51 Dietary interventions include restricting foods with high copper content, such as beef liver, nuts, chocolate, shellfish, mushrooms, and soy.65
4.2. Hepatic metabolic dysregulation in Wilson disease
4.2.1. Mitochondria structural and functional defects
The mitochondrion is the cellular hub for energy metabolism and production. It houses β-oxidation, the tricarboxylic acid cycle (TCA), and adenosine triphosphate (ATP) synthesis through oxidative phosphorylation (OXPHOS) complexes, related to the electron transport chain.66 The process of ATP production is dependent on a steady copper flux to the OXPHOS enzyme complexes.37 Although copper is essential for mitochondrial function, excess copper causes mitochondria morphological and functional changes. Signs of mitochondrial impairment appear to be independent from liver function as they are evident at an early age, before signs of hepatic dysfunction manifest in humans and animal models of WD.67–72 Furthermore, a gradual mitochondrial copper overload was shown to cause a steady decline in mitochondrial function, as defined by decreased ATP production.73 Copper-chelation therapy, particularly with methanobactin, was shown to restore these alterations.71,73
Described morphological changes of hepatic mitochondria include elongations and enlarged cristae and intermembrane space,70,71,73,74 as well as cardiolipin fragmentation,75 and are linearly correlated to the amount of accumulated copper.
Functional mitochondrial alterations are also reported in both WD patients and animal models. These include abnormalities in the OXPHOS system, with decreased activities of complex IV and complexes I, II, and III.70,76 Mitochondrial fatty acid β-oxidation is also impaired in WD. Work from our group has previously shown the transcript and protein levels of peroxisome proliferator-activated receptor α (PPARα) and carnitine palmitoyl transferase 1A were lower in an untreated Jackson Laboratory toxic milk mouse model of WD (tx-j) compared to control mice.77 A similar finding of decreased hepatic Ppara transcript levels was reported from Atp7b−/−knockout mice.5 Hepatic PPARa expression was also decreased in WD patients and the magnitude of this change was related to the progression of liver damage.78 In many genetic defects with mitochondrial β-oxidation impairment, alternative rescue pathways are induced, including ω-oxidation of fatty acids that may take place in mitochondria and microsomes.79 Nevertheless, the effects of mitochondrial β-oxidation impairment and compensating mechanisms to maintain energy homeostasis in WD are not understood. Metabolomic analysis of serum and hepatic metabolites also suggest an impaired TCA cycle. In Atp7b−/−mice, there was an accumulation of TCA intermediates (oxaloacetate, citrate, fumarate, and malate) and a decrease in acetyl-CoA and α-ketoglutarate.80 In patients with WD, we have previously reported alterations in TCA cycle intermediates.81
The mechanism by which copper overload results in mitochondrial defects can be direct and indirect. At initial stages of copper overload, alterations in mitochondrial labile proteins originate via copper-mediated thiol modifications.37 At later stages, multiple lines of evidence indicate a major role of oxidative stress in the pathogenesis of WD and in inducing mitochondrial dysfunction. These include reduced aconitase activity,76,82 and an increase in transcript levels of the antioxidants thioredoxin-2 and peroxiredoxin-3 in tx-j mice.70 Moreover, patients with WD presented single or multiple hepatic mitochondrial DNA deletions, a common response to oxidative stress,83 and patients and animal models of WD presented increased mitochondrial lipid peroxidation.84,85 Among mitochondrial phospholipids, cardiolipin is a copper-sensitive target which fragments upon copper accumulation, likely via free-radicals.75 Therefore, the generation of ROS is thought to contribute to copper-mediated mitochondrial damage.84,86 Of note, compensatory mechanisms for copper-induced mitochondrial abnormalities are not fully investigated.
4.2.2. Hepatic bioenergetic defects
Adenosine monophosphate-activated protein kinase (AMPK) is a key responder to both metabolic and oxidative stimuli and its downstream signaling cascade is a chief regulator of cellular energy metabolism in hepatic and extrahepatic tissue.87 During metabolic stress and energy deprivation states, the intracellular AMP to ATP ratio is elevated and the binding of AMP to the AMPK regulatory subunit results in AMPK phosphorylation and activation.87,88 The activated AMPK initiates signal cascades aiming to restore intracellular energy by which multiple ATP-consuming anabolic pathways, e.g., isoprenoid, cholesterol, lipogenesis, TAG, phospholipid, glycogen, and protein syntheses, are down-regulated, whereas multiple ATP-producing pathways, e.g., glucose uptake and glycolysis, fatty acid uptake and oxidation, and enhanced mitochondrial biogenesis, are up-regulated.88
Beside ATP deprivation, AMPK activity is affected by pro-oxidant conditions as it is induced by ROS to initiate apoptosis and auto-phagy.89 The direct effect of copper excess on AMPK activation has been demonstrated in vitro in human neuroblastoma cells. Compared to untreated cells, treatment with the synthetic isatin-Schiff base copper (II) complexes Cu(isapn) and Cu(isaepy)2 at a concentration of 50 μM resulted in increased ROS production, apoptosis induction, and subsequent varying degrees of oxidative damage to proteins and lipids.90 Further work from the same group has shown impairment of complex I of the OXPHOS chain and enhanced apoptosis via an AMPK-dependent mechanism.91
In animals, dietary copper modulation can alter tissue AMPK activity1; the effect of progressive copper accumulation on hepatic AMPK activity and signaling was recently demonstrated in Atp7b−/− mice. While on a chow diet, Atp7b−/−mice exhibited increased hepatic phosphorylated-AMPKα with reduced gluconeogenic and lipogenic gene expression, including phosphoenolpyruvate carboxykinase, pyruvate carboxylase, fructose bisphosphatase 1, fatty acid synthase (Fasn), acetyl-CoA carboxylase, and sterol regulatory element-binding protein 1c (Srebp1c).80
4.2.3. Alterations in lipid metabolism
4.2.3.1. Hepatic steatosis.
Beside hepatocyte mitochondrial insufficiency and energy deprivation, dysregulation in hepatic lipid metabolism is another hallmark alteration of WD, with hepatic steatosis being a frequent feature at diagnosis.59,67,92–94 The molecular mechanisms underlying steatosis in WD remains poorly understood. However, mitochondrial dysfunction and the marked inhibition of hepatic β-oxidation are likely contributing factors.95 Interestingly, evidence from animal models indicates hepatic DNL is impaired in WD. At the transcriptional level, lipogenesis is regulated by multiple signaling proteins including liver X receptor (LXR), SREBP1c, and carbohydrate-responsive element-binding protein.24 Work from our group has shown SREBP1c transcript and protein levels were reduced in untreated tx-j mice compared to control mice with normal copper metabolism.77 Similar findings were observed in Atp7b−/–knockout mice, showing an alteration in SREBP-2 function rather than maturation and re-localization.5 There was also a down-regulation in many genes involved in lipid metabolism, including ATP citrate lyase, Fasn, elongation of very long chain fatty acids protein 6, and lipin-1. Remarkably, when fed an obesogenic Western diet, Atp7b−/−mice exhibited lower body weight and adiposity and lower hepatic steatosis compared to wild-type control mice. When comparing chow-fed and Western diet-fed mice, no difference was observed in body weight, adiposity, and hepatic steatosis, suggesting the lack of a diet-specific effect. This reduced susceptibility to hepatic steatosis was attributed to the blunted weight gain while on the Western diet.80
Hepatic steatosis is also associated with impaired synthesis and export of VLDL, both understudied pathways in WD. The association of the PNPLA3 G allele with hepatic steatosis in WD suggests impaired lipid remodeling and involvement of VLDL lipidation and export in the etiology of WD hepatic steatosis.59,96,97 The process of VLDL synthesis and export is mediated by the microsomal triglycerides transfer protein that combines TAG and apolipoprotein B to create neutral lipid masses, which are further packaged with phospholipid monolayers enriched with phosphatidylcholine (PC) before export to circulation.98 Relevant to VLDL metabolism, we and others have described a dysregulated methionine cycle and altered choline levels in humans and animal models of WD.77,99–101 These dysregulations in methionine and choline metabolism may impact the assembly or export of VLDL in WD. Methionine is a precursor to S-adenosylmethionine (SAM), a universal methyl-group donor, which is required for transmethylation reactions.102,103 An elevated methylation potential, expressed as high SAM levels in relation to its product S-adenosylhomocysteine, is required for activation of the hepatic phosphatidylethanolamine methyltransferase (PEMT) pathway and the synthesis of PC.104–106 Choline is also a substrate for the synthesis of PC via the choline dependent cytidine-diphosphate-choline (CDP-choline) pathway, the major contributor to the PC pool.107 We recently reported a dysregulated serum phospholipid profile in WD patients with profound alterations in PC levels compared to healthy controls as well as a down-regulation of PC synthesis genes in tx-j mice, including Pemt, [phosphate cytidylyltransferase 1 choline-alpha], and choline phosphotransferase 1.101 In general, alterations in phospholipids are implicated in hepatic steatosis,108–111 and evidence indicates both pathways, hepatic PC synthesis via CDP-choline or PEMT, are critical for VLDL production.104,112
4.2.3.2. Cholesterol synthesis.
Along with copper accumulation, defects of lipid utilization in isoprenoid and cholesterol syntheses are shown in various animal models and in humans with WD. The transcript and activity levels of 3-hydroxy-3-methyl-glutaryl-CoA reductase (Hmgcr) were down-regulated in Long-Evans Cinnamon rats.113 Similar findings were described in Atp7b−/−mice at an early stage and prior to the appearance of histopathological changes.5,114 Concomitantly, there was a significant reduction in hepatic VLDL-cholesterol and dysregulation of enzymes involved in bile acid biosynthesis, including a down-regulation in cholesterol 7 α-hydroxylase and up-regulation in microsomal oxysterol 7 α-hydroxylase. In vitro, it was shown copper does not continuously accumulate in Atp7b−/−hepatocytes throughout the course of WD. However, there was a down-regulation in cholesterol metabolism and a reduction in the VLDL-cholesterol fraction that remained significantly lower throughout the course of the disease.61
Copper was shown to impair the transcriptional function of several nuclear receptors by assimilating into the DNA-binding domain and inducing conformational changes that prevent DNA binding.115 An in silico gene expression analysis indicated LXR, retinoid X receptor (RXR), and the LXR/RXR-mediated regulation of lipid metabolism are impaired in patients with WD and Atp7b−/− mice.116 In the same study, the hepatic mRNA levels of Rxr, Fasn, and Hmgcr were significantly down-regulated in Atp7b−/−livers. Treatment with an LXR agonist significantly increased Fasn expression as well as plasma total cholesterol, low-density lipoproteins (LDL), and high-density lipoproteins compared to untreated Atp7b−/−mice, suggesting alterations in nuclear receptor-mediated lipid metabolism are major contributors to dysregulated lipid metabolism and liver damage pathogenesis in WD.116 Similar findings were reported in patients with WD, with a significant reduction in hepatic transcript levels of HMGCR and LDL receptor compared to control subjects.5 Another study investigated serum lipid profiles in WD patients treated with a copper chelator compared to healthy subjects, describing significantly lower levels of circulating total cholesterol and LDL cholesterol in WD patients, with no difference in TAG levels between groups.117 Comparing preand post-treatment WD patients, copper-chelation treatment was associated with lower plasma cholesterol and no TAG change.117,118 In a different study, a significant reduction in cholesterol was reported only in WD with hepatic manifestations compared to asymptomatic, neurologic, and combined phenotype with both neurologic and hepatic manifestations.114 This reduction was restored to healthy control levels after two years of copper-chelation therapy.114 Together, these defects may reflect impaired lipid utilization as a possible contributor to the development of steatosis. A proposed mechanism for the development of hepatic steatosis in WD is depicted in Fig. 1.
Fig. 1. Hypothetical framework for the development of hepatic steatosis in Wilson disease.
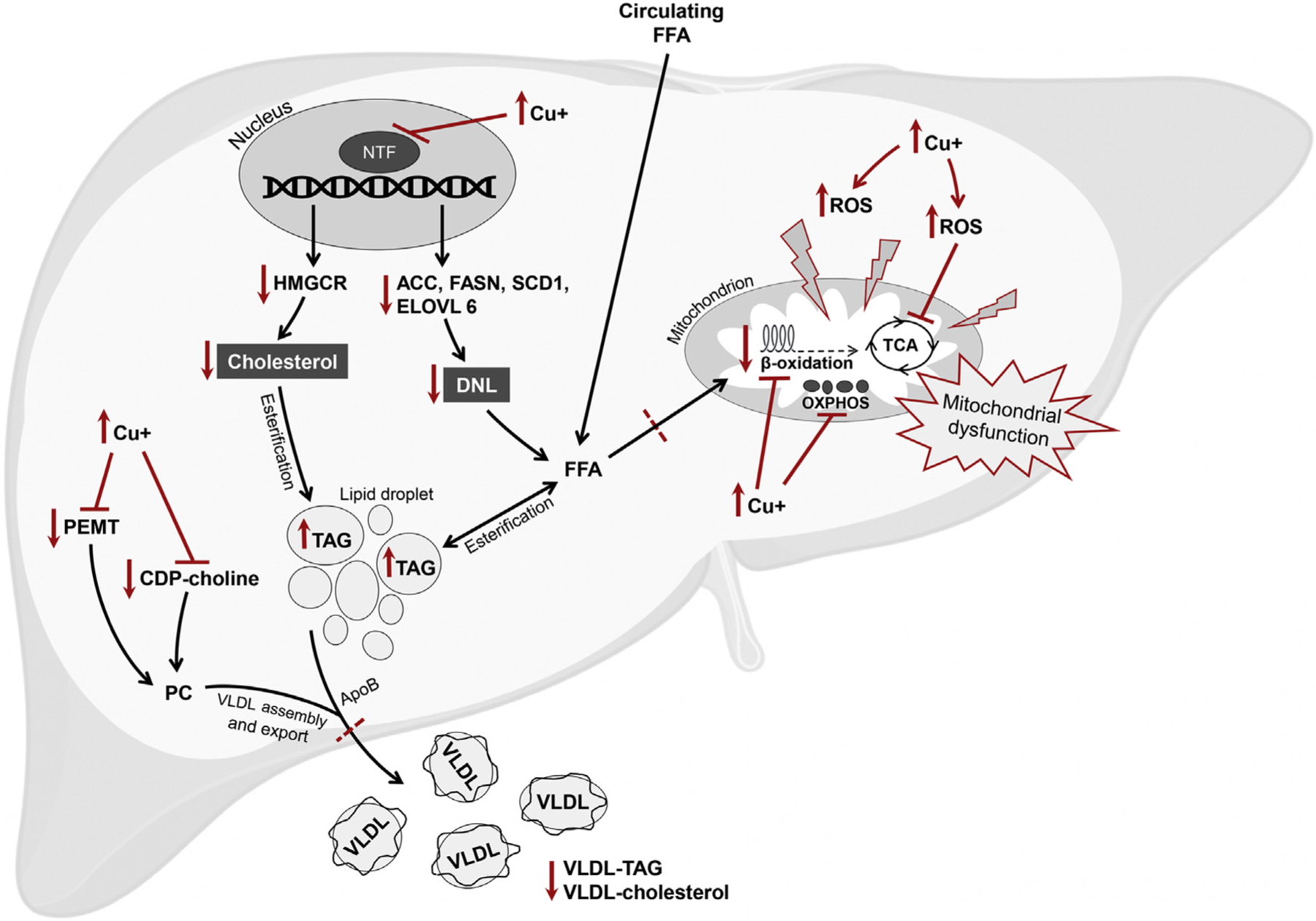
Copper overload induces the production of ROS which interacts with subcellular protein structures, including the mitochondria, to cause structural and functional mitochondrial alteration and bioenergetic defects. Copper may also directly impair enzymes involved in the TCA cycle, fatty acid β-oxidation, and OXPHOS chain. Copper accumulation impairs the function of multiple nuclear transcription factors including retinoid X receptor, liver X receptor, carbohydrate-responsive element-binding protein, and sterol regulatory element-binding protein 1c. The transcript and protein levels of genes involved in cholesterol synthesis and lipogenesis are down-regulated, including HMGCR, ACC, FASN, SCD1, and ELOVL6. The impairment of PC synthesis pathways results in impaired assembly and/or export of VLDL. Together, the uptake of FFA and subsequent esterification into TAG, combined with impaired utilization of FFA and impaired assembly and/or export of VLDL, result in the net effect of hepatic lipid accumulation, or steatosis. ACC, acetyl-CoA carboxylase; ApoB, apolipoprotein B; CDP-choline, cytidine-diphosphate-choline; Cu+, free copper; DNL, de novo lipogenesis; ELOVL6, elongation of very long chain fatty acids protein 6; FASN, fatty acid synthase; FFA, free fatty acids; HMGCR, 3-hydroxy-3-methyl-glutaryl-CoA reductase; NTF, nuclear transcription factor; OXPHOS, oxidative phosphorylation; PC, phosphatidylcholine; PEMT, phosphatidylethanolamine methyltransferase; ROS, reactive oxygen species; SCD1: stearoyl-CoA desaturase; TAG, triglycerides; TCA, tricarboxylic acid; VLDL, very low-density lipoproteins.
4.2.4. Dysregulation in carbohydrate metabolism
The effect of copper overload on hepatic glucose metabolism is understudied. However, dysregulation in hepatic glucose metabolism is reported in subjects with WD. Hypoglycemia, a typical complication of advanced liver disease, is also present as an early clinical sign in WD. In a case report, two siblings with WD and abnormal liver histology in the absence of cirrhosis, exhibited fasting hypoglycemia, mild glucose intolerance, and hyper-insulinemia that were normalized with penicillamine treatment.119
In another case report of a young female with normal liver morphology as confirmed by imaging, hypoglycemia was reported and resolved with copper-chelation treatment.120 The described hypoglycemia could be due to impaired glycolysis or gluconeogenesis. In light of mitochondrial defects, particularly impairment in mitochondrial β-oxidation, copper overload may be associated with impairment in the hepatic metabolic capacity to transition between fat and glucose metabolism. During the fasting state, the liver maintains glucose homeostasis by glycogenolysis from glycogen and by gluconeogenesis using lactate, glycerol, and amino acids as substrates for glucose production.121 In a rat model of WD, the circulating level of glucose is decreased and lactate is increased,122 possibly indicating the glycolysis by-product, pyruvate, accumulates and is further converted to lactate and exported to circulation.123 However, elevated lactate may be due to defects in gluconeogenesis. In fasted Atp7b−/−mice, an elevation in several hepatic glycolysis intermediates and decreased hepatic glucose, pyruvate, and lactate levels are reported. Furthermore, insulin administration resulted in hypoglycemia, indicating an impaired glucose counterregulatory response, a finding supported with marked down-regulation of hepatic genes involved in gluconeogenesis.80 Interestingly, Atp7b−/−mice were more glucose-tolerant and insulin-sensitive compared to their wild-type counterpart.80
In Atp7b−/− mice and WD patients, there is evidence of decreased nuclear receptor binding to promoter response elements, including farnesoid X receptor (FXR), RXR, hepatic nuclear factor 4 alpha, and liver receptor homolog-1, and decreased transcript levels of nuclear receptor target genes, indicating impaired nuclear receptor signaling.115 FXR signaling is a modulator of glucose homeostasis and insulin sensitivity.124–126 Hepatic Fxr expression is regulated by insulin and glucose in rodent hepatocytes in vitro.127 Moreover, Fxr−/−mice display an accelerated hepatic response to high carbohydrate re-feeding, mainly by induction of glycolytic and lipogenic genes and a repression of gluconeogenic genes.128 Therefore, impaired nuclear receptor signaling in copper overload can have implications in carbohydrate metabolism and homeostasis.
In a metabolomic analysis of WD patients, we reported elevated serum sorbitol levels,81 further corroborating the dysregulation in WD carbohydrate metabolism. Sorbitol is an intermediate of the polyol pathway by which glucose is converted to sorbitol by aldose reductase, followed by the conversion of sorbitol to fructose by sorbitol dehydrogenase.129 The hepatic polyol pathway is activated under hyperglycemic conditions. However, further work is needed to understand the relevance of elevated sorbitol with regard to carbohydrate metabolism in WD. Of note, in type 2 diabetes, a state in which alternating use of carbohydrates and lipids as metabolic fuel is impaired, fasting serum sorbitol is reported to be significantly elevated compared to healthy controls, with a significant elevation in sorbitol persisting in the post-feeding state.130
4.3. Metabolic dysregulation in extrahepatic tissues
The substantial role of ATP7B-mediated copper homeostasis is well-recognized in the liver. However, it is poorly studied in extrahepatic tissues, including adipose tissue, muscle, and intestine.
4.3.1. Adipose tissue
White adipose tissue (WAT) is an active endocrine organ regulating body fat mass and energy homeostasis. Lipogenesis and lipolysis are central to these functions and are tightly regulated.131 Lipogenesis takes place mainly in adipose tissue, liver, and muscle, where esterification of FFA results in the formation of TAG. In contrast, lipolysis mobilizes metabolic fuel to peripheral tissues, mainly from adipose tissue, in response to energy needs.
The role of copper-mediated lipid metabolism in adipocytes and its contribution to whole-body energy metabolism are not well-understood. We observed tx-j mice to be lean and protected from obesity, despite dysregulated lipid metabolism. While on the same chow diet, tx-j mice gained less weight and fat mass compared to their wild-type littermates.77,100 Similar observations were reported in Atp7b−/−mice. Compared to wild-type littermates, chow-fed Atp7b−/−mice had lean body weight and less total body weight gain, fat mass, and epididymal WAT weight, with markedly reduced weight gain when fed the Western diet. Although this variation was attributed to decreased food intake and, to an extent, increased activity, a significant increase in respiratory exchange ratio was only observed in the active (dark) cycle. In addition, the total energy expenditure was comparable between the two genotypes in both active (dark) and inactive (light) cycles, suggesting the involvement of a metabolic derangement component.80
On the other hand, the hepatocyte-specific deletion of Atp7b in mice resulted in steatosis before evident signs of liver damage and, interestingly, increased adiposity with an up-regulation in Hmgcr attributed to nonparenchymal liver cell signaling.132 It is worth mentioning Atp7b−/−rats exhibited increased visceral fat and hepatic fat content when fed the Western diet, contradicting the obesity-protected phenotype.11
Evidence demonstrates copper has a signaling role in the regulation of lipolysis, by which TAG are broken down to FFA and glycerol via the 3′,5′-cyclic AMP (cAMP) pathway. In rabbits, dietary copper supplementation inhibited lipid accumulation in adipose tissue via increased lipolysis.1 In Atp7b−/−mice compared to wild-type, hepatic copper overload was associated with a significant decrease in WAT copper levels and lipolytic activity.33 When lipolysis was induced by a β-adrenergic receptor agonist, cAMP levels were found to be lower and less glycerol was released from WAT, indicating impaired lipolysis. In the same study, using 3T3-L1 white adipocytes, cellular copper content increased lipolysis in a dose-dependent manner, and treatment with a copper chelator decreased FFA and glycerol release upon inducing lipolysis. Furthermore, the cAMP-degrading phosphodiesterase 3B (PDE3B) was identified as a target of copper accumulation; PDE3B activity is inhibited when bound to copper resulting in decreased cAMP degradation and increased lipolysis. These findings were confirmed in mice with an adipocyte-specific deletion of Atp7a that showed increased adipose copper levels compared to control mice and progressive loss of WAT due to increased lipolysis.48 This lipolytic state was associated with decreased levels of serum leptin and adiponectin and increased levels of TAG and insulin, suggesting adipose dysfunction, in addition to pronounced glucose intolerance, insulin resistance, and hepatic steatosis, indicating systemic metabolic impairment. Collectively, these data provide in vivo evidence that alteration in adipose tissue copper levels influence WAT lipolytic activity and lipid homeostasis. Yet, the mechanism by which tx-j and Atp7b−/−mice are protected from obesity remains unclear and multiple factors need to be considered, including lipogenesis, energy expenditure, and physical activity.
It is possible that the impaired lipolytic activity in Atp7b−/−mice could reflect adipose dysfunction. Recently, the role of copper in adipocyte morphology, metabolism, and fat storage was identified through the activity of the copper-dependent enzyme, semicarbazide-sensitive amine oxidase (SSAO).133 SSAO synchronizes changes in metabolic fuel availability via proteome-remodeling to regulate the uptake of glucose and long-chain fattyacids. Inparticular, copper-activated SSAO increases glucose uptake and glycolysis; in states of copper deficiency, SSAO is inactivated and fatty acid uptake is up-regulated, leading to adipocyte hypertrophy and lipid deposition. Therefore, this evidence, together with decreased WAT copper levels and lipolytic activity,33 highlights potential impairment in adipose function and metabolic adaptability in Atp7b−/−mice.
4.3.2. uscle
Data examining the effect of ATP7B mutations on extrahepatic copper levels and lipid metabolism are limited, and discrepancies in muscle copper levels have been reported in WD. In one small study, patients with WD had a significant increase in muscle copper level compared to healthy subjects.134 On the other hand, using functional imaging aided with administration of 64CuCl2 as a tracer, there was no significant difference in 64Cu levels between Atp7b−/−and control mice in multiple extrahepatic tissues, including the muscle.135 Interestingly, in rabbits, dietary copper supplementation resulted in muscle growth and inhibited lipid accumulation in skeletal muscle via elevated β-oxidation.1 More studies are needed to investigate the effect of hepatic copper overload on adipose tissue energy metabolism.
4.3.3. Intestine
Recent evidence suggests copper modulates processing of chylomicrons in the intestine. In enterocytes, copper-transporter ATP7A facilitates dietary copper export into portal circulation.47,136–138 The intestine also expresses ATP7B, which appears to modulate enterocyte vesicular copper storage and regulate chylomicron formation and dietary fat absorption.139 The Atp7b−/−mouse model presents duodenal copper deficiency and dysregulated fat-processing in enterocytes.50 Intriguingly, Atp7b−/−mice also presented duodenal villi with increased microvilli length and enterocytes with mitochondrial ballooning and lipid accumulation, indicating impaired dietary fat absorption and aberrant chylomicron maturation which most likely require both adequate copper levels and ATP7B function. These alterations were not observed in mice with the hepatocyte-specific deletion of Atp7b, which showed normal intestinal morphology with no enterocyte lipid droplet accumulation.
Alterations in the intestinal microbiome are reported in many diseases, including obesity and type 2 diabetes.140,141 A great body of evidence supports a role of gut microbiota in modulating host metabolism. This was demonstrated in germ-free mice, which are more glucose-tolerant, insulin-sensitive, and have reduced adiposity compared to conventional-colonized mice.142–144 When fed the Western-diet, germ-free mice are protected from obesity.145 Diet, among other environmental factors, can modulate the gut microbiota.146–148 In WD, copper intestinal content is significantly altered as the genetic mutation in ATP7B leads to impaired biliary copper excretion into the intestine and subsequent fecal elimination. In addition, WD therapy with zinc salts may modulate dietary copper absorption. Studies of the intestinal microbiome composition in WD patients are limited. One study reported a significant difference in the diversity and composition of WD patients’ intestinal flora. Compared to healthy subjects, Bacteroidetes was significantly more abundant in WD patients, while Firmicutes, Proteobacteria, and Fusobacteria were less abundant.149 However, the relation between the observed microbiome alterations with energy metabolism in WD is not clear and worth further investigation. It is thought the gut microbiome can affect host lipid metabolism by modulating the production of secondary bile acids which, after being absorbed into systemic circulation, target multiple nuclear receptors to regulate hepatic and/or systemic lipid and glucose metabolism.150,151 Another potential mechanism involves the production of short-chain fatty acids, such as acetate, propionate, and butyrate, derived from the fermentation of non-digestible carbohydrates. Short-chain fatty acids affect energy expenditure and insulin sensitivity via mechanisms involving G protein-coupled receptors.152,153 Also, the hepatic production of trimethylamine N-oxide, derived from the oxidation of the gut-produced trimethylamine, can exert direct effects on reverse cholesterol sterol and bile acids metabolism.152,153
5. Conclusion
The mechanisms linking copper to dysregulated lipid and energy metabolism in WD are not clear and may include direct copper binding and modifications to labile proteins that impair enzymes and nuclear receptor function. Copper, via the generation of ROS, can also damage subcellular protein and lipid structures of vital organelles, including the mitochondria.
Hepatic copper accumulation is accompanied by mitochondrial bioenergetic defects, with evident mitochondria structural and functional abnormalities that appear to be independent from liver function. However, the hepatic response to this energetic defect is not clear.
Dysregulation of hepatic lipid metabolism and the development of steatosis is an early feature of copper accumulation in WD. The molecular mechanisms underlying hepatic steatosis in WD remains poorly understood. However, hepatic DNL is down-regulated and steatosis appears to be a net result of impaired mitochondrial β-oxidation, reduced VLDL assembly or export, decreased utilization of lipids, and a marked down-regulation in cholesterol synthesis. Altered carbohydrate metabolism is also suggested, mainly via defects in the alternating use of carbohydrates and fat as metabolic fuel. This was evident with hypoglycemia and impaired gluconeogenesis in animal models of WD. These metabolic alterations may be explained by the impaired function of nuclear transcription factors, a feature reported in both animal models and human subjects with WD.
There is a gap in current knowledge regarding the effects of ATP7B mutations and copper accumulation on metabolic dysregulation and energy homeostasis in extrahepatic tissue. It is plausible the metabolic effects of copper overload in WD are tissue-dependent, as copper accumulation is uneven across different tissues. In the liver, where copper selectively accumulates, copper overload is profound and is reflected by down-regulated lipid metabolism and lipid droplet accumulation. In enterocytes, copper is excessively stored in vesicles and lipid droplets accumulate in the cytosol, suggesting impaired chylomicron formation and dietary lipid absorption. In adipose tissue, copper is reduced, lipolysis is impaired, and lipogenesis and hypertrophy are down-regulated.
Ultimately, the systemic dysregulation of energy metabolism in WD may reflect a process of metabolic adaptation to synchronize metabolic fuel availability and tissue energy demands.
Acknowledgements
This work was supported by the National Institutes of Health -National Institute of Diabetes and Digestive and Kidney Diseases, USA, through grant number R01DK104770 to V. Medici. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
☆ Edited by Peiling Zhu and Genshu Wang.
Declaration of competing interest
The authors declare they have no conflict of interest.
References
- 1.Lei L, Xiaoyi S, Fuchang L. Effect of dietary copper addition on lipid metabolism in rabbits. Food Nutr Res. 2017;61:1348866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carr TP, Lei KY. High-density lipoprotein cholesteryl ester and protein catabolism in hypercholesterolemic rats induced by copper deficiency. Metabolism. 1990;39:518–524. [DOI] [PubMed] [Google Scholar]
- 3.Engle TE. Copper and lipid metabolism in beef cattle: a review. J Anim Sci. 2011;89:591–596. [DOI] [PubMed] [Google Scholar]
- 4.Lei KY. Alterations in plasma lipid, lipoprotein and apolipoprotein concentrations in copper-deficient rats. J Nutr. 1983;113:2178–2183. [DOI] [PubMed] [Google Scholar]
- 5.Huster D, Purnat TD, Burkhead JL, et al. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem. 2017;282:8343–8355. [DOI] [PubMed] [Google Scholar]
- 6.Huster D, Lutsenko S. Wilson disease: not just a copper disorder. Analysis of a Wilson disease model demonstrates the link between copper and lipid metabolism. Mol Biosyst. 2007;3:816–824. [DOI] [PubMed] [Google Scholar]
- 7.Burkhead JL, Svetlana L. The role of copper as a modifier of lipid metabolism In: Baez RV, ed. Lipid Metabolism. London, UK: IntechOpen Publishers; 2013: 39–60. Available at www.intechopen.com/books/lipid-metabolism/the-role-of-copper-as-a-modifier-of-lipid-metabolism. Accessed January 14 , 2020. [Google Scholar]
- 8.Lima SC, Arrais RF, Sales CH, et al. Assessment of copper and lipid profile in obese children and adolescents. Biol Trace Elem Res. 2006;114:19–29. [DOI] [PubMed] [Google Scholar]
- 9.Saari JT. Copper deficiency and cardiovascular disease: role of peroxidation, glycation, and nitration. Can J Physiol Pharmacol. 2000;78:848e855. [DOI] [PubMed] [Google Scholar]
- 10.Tang Z, Gasperkova D, Xu J, Baillie R, Lee JH, Clarke SD. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J Nutr. 2000;130:2915–2921. [DOI] [PubMed] [Google Scholar]
- 11.Einer C, Leitzinger C, Lichtmannegger J, et al. A high-calorie diet aggravates mitochondrial dysfunction and triggers severe liver damage in Wilson disease rats. Cell Mol Gastroenterol Hepatol. 2019;7:571–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aigner E, Strasser M, Haufe H, et al. A role for low hepatic copper concentrations in nonalcoholic fatty liver disease. Am J Gastroenterol. 2010;105: 1978–1985. [DOI] [PubMed] [Google Scholar]
- 13.Nobili V, Siotto M, Bedogni G, et al. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2013;56:370–375. [DOI] [PubMed] [Google Scholar]
- 14.Mendoza M, Caltharp S, Song M, et al. Low Hepatic tissue copper in pediatric non-alcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2017;65:89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H, Liu CN, Wolf RM, et al. Obesity is associated with copper elevation in serum and tissues. Metallomics. 2019;11:1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aigner E, Theurl I, Haufe H, et al. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology. 2008;135:680–688 (e681). [DOI] [PubMed] [Google Scholar]
- 17.Song M, Schuschke DA, Zhou Z, et al. Modest fructose beverage intake causes liver injury and fat accumulation in marginal copper deficient rats. Obesity. 2013;21:1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song M, Li X, Zhang X, et al. Dietary copper-fructose interactions alter gut microbial activity in male rats. Am J Physiol Gastrointest Liver Physiol. 2018;314:G119–G130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei X, Song M, Yin X, et al. Effects of dietary different doses of copper and high fructose feeding on rat fecal metabolome. J Proteome Res. 2015;14: 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas D A study on the mineral depletion of the foods available to us as a nation over the period 1940 to 1991. Nutr Health. 2003;17:85–115. [DOI] [PubMed] [Google Scholar]
- 21.White PJ, Broadley M. Historical variation in the mineral composition of edible horticultural products. J Hortic Sci Biotech. 2005;80:660–667. [Google Scholar]
- 22.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—dmeta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 23.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16:678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–964. [DOI] [PubMed] [Google Scholar]
- 26.Bertinato J1, L’Abbé MR. Maintaining copper homeostasis: regulation of copper-trafficking proteins in response to copper deficiency or overload. J Nutr Biochem. 2004;15:316–322. [DOI] [PubMed] [Google Scholar]
- 27.Andreini C, Banci L, Bertini I, Rosato A. Occurrence of copper proteins through the three domains of life: a bioinformatic approach. J Proteome Res. 2007;7: 209–216. [DOI] [PubMed] [Google Scholar]
- 28.Nair PM, Mason HS. Reconstitution of cytochrome c oxidase from a copper-depleted enzyme and cui. J Biol Chem. 1967;242:1406–1415. [PubMed] [Google Scholar]
- 29.Lewandowski Ł, Kepinska M, Milnerowicz H. The copper-zinc superoxide dismutase activity in selected diseases. Eur J Clin Invest. 2019;49, e13036. [DOI] [PubMed] [Google Scholar]
- 30.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84: 1155–1228. [DOI] [PubMed] [Google Scholar]
- 31.Prigge ST, Mains RE, Eipper BA, Amzel LM. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell Mol Life Sci. 2000;57:1236–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Askwith C, Kaplan J. Iron and copper transport in yeast and its relevance to human disease. Trends Biochem Sci. 1998;23:135–138. [DOI] [PubMed] [Google Scholar]
- 33.Krishnamoorthy L, Cotruvo JA Jr, Chan J, et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat Chem Biol. 2016;12:586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brady DC, Crowe MS, Turski ML, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turski ML, Brady DC, Kim HJ, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32:1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grubman A, White AR. Copper as a key regulator of cell signalling pathways. Expert Rev Mol Med. 2014;16:e11. [DOI] [PubMed] [Google Scholar]
- 37.Zischka H, Sabine B. Mitochondrial Copper Toxicity with a Focus on Wilson Disease Clinical And Translational Perspectives on Wilson Disease. New York: Academic Press; 2019:65–75. [Google Scholar]
- 38.Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107–115. [DOI] [PubMed] [Google Scholar]
- 39.Harvey LJ, Dainty JR, Hollands WJ, et al. Use of mathematical modeling to study copper metabolism in humans. Am J Clin Nutr. 2005;81:807–813. [DOI] [PubMed] [Google Scholar]
- 40.Zhou B, Gitschier. hCTR1: a human gene for copper uptake identified by complementation in yeast. Proc Natl Acad Sci U S A. 1997;94:7481–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimnicka AM, Ivy K, Kaplan JH. Acquisition of dietary copper: a role for anion transporters in intestinal apical copper uptake. Am J Physiol Cell Physiol. 2011;300:C588–C599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohrvik H, Thiele DJ. How copper traverses cellular membranes through the mammalian copper transporter 1, Ctr1. Ann N Y Acad Sci. 2014;1314:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lalioti V, Muruais G, Tsuchiya Y, Pulido D, Sandoval IV. Molecular mechanisms of copper homeostasis. Front Biosci (Landmark Ed). 2009;14:4878–4903. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ. Advances in the understanding of mammalian copper transporters. Adv Nutr. 2011;2:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker ZN, Cobine PA, Leary SC. The mitochondrion: a central architect of copper homeostasis. Metallomics. 2017;9:1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boulet A, Vest KE, Maynard MK, et al. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J Biol Chem. 2018;293:1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monty JF, Llanos RM, Mercer JF, Kramer DR. Copper exposure induces trafficking of the menkes protein in intestinal epithelium of ATP7A transgenic mice. J Nutr. 2005;135:2762–2766. [DOI] [PubMed] [Google Scholar]
- 48.Tao C, Wang Y, Zhao Y, et al. Adipocyte-specific disruption of ATPase copper transporting α in mice accelerates lipoatrophy. Diabetologia. 2019;62: 2340–2353. [DOI] [PubMed] [Google Scholar]
- 49.Pierson H, Lutsenko S. Chapter 3-ATP7B function In: Weiss KH, Schilsky M, eds. Wilson Disease. Netherlands: Elsevier,Inc; 2019:23–32. [Google Scholar]
- 50.Pierson H, Muchenditsi A, Kim BE, et al. The function of ATPase copper transporter ATP7B in intestine. Gastroenterology. 2018;154:168–180 (e165). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferenci P, Ott P. Wilson’s disease: fatal when overlooked, curable when diagnosed. J Hepatol. 2019;71:222–224. [DOI] [PubMed] [Google Scholar]
- 53.Soltanzadeh A, Soltanzadeh P, Nafissi S, Ghorbani A, Sikaroodi H, Lotfi J. Wilson’s disease: a great masquerader. Eur Neurol. 2007;57:80e85. [DOI] [PubMed] [Google Scholar]
- 54.Członkowska A, Litwin T, Dziezyc K, Karliński M, Bring J, Bjartmar C. Characteristics of a newly diagnosed Polish cohort of patients with neurological manifestations of Wilson disease evaluated with the unified Wilson’s disease rating scale. BMC Neurol. 2018;18:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munjal S, Zimbrean PC. Psychiatric aspects of wilson In: Schilsky M, ed. Management of Wilson Disease: A Pocket Guide. Switzerland: Humana Press; 2018:121. [Google Scholar]
- 56.Buksińska-Lisik M, Litwin T, Pasierski T, Członkowska A. Cardiac assessment in Wilson’s disease patients based on electrocardiography and echocardiography examination. Arch Med Sci. 2019;15:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grandis DJ, Nah G, Whitman IR, et al. Wilson’s disease and cardiac myopathy. Am J Cardiol. 2017;120:2056e2060. [DOI] [PubMed] [Google Scholar]
- 58.Pfeiffenberger J, Mogler C, Gotthardt DN, et al. Hepatobiliary malignancies in Wilson disease. Liver Int. 2015;35:1615–1622. [DOI] [PubMed] [Google Scholar]
- 59.Stättermayer AF, Traussnigg S, Dienes HP, et al. Hepatic steatosis in Wilson diseaseerole of copper and PNPLA3 mutations. J Hepatol. 2015;63:156–163. [DOI] [PubMed] [Google Scholar]
- 60.Müller T, Langner C, Fuchsbichler A, et al. Immunohistochemical analysis of Mallory bodies in Wilsonian and non-Wilsonian hepatic copper toxicosis. Hepatology. 2004;39:963–969. [DOI] [PubMed] [Google Scholar]
- 61.Ralle M, Huster D, Vogt S, et al. Wilson disease at a single cell level intracellular copper trafficking activates compartment-specific responses in hepatocytes. J Biol Chem. 2010;285, 30875–20883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089–2111. [DOI] [PubMed] [Google Scholar]
- 63.European Association For the Study of the Liver(EASL). EASL clinical practice guidelines: wilson’s disease. J Hepatol. 2012;56:671–685. [DOI] [PubMed] [Google Scholar]
- 64.Farinati F, Cardin R, Mestriner C, Sturniolo GC, Naccarato R. Mechanisms of pennicillamine and zinc in the treatment of Wilson’s disease. Am J Gastroenterol. 1995;90:2264–2265. [PubMed] [Google Scholar]
- 65.Russell K, Gillanders LK, Orr DW, Plank LD. Dietary copper restriction in Wilson’s disease. Eur J Clin Nutr. 2018;72:326–331. [DOI] [PubMed] [Google Scholar]
- 66.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitochondrial Sternlieb I. and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology. 1968;55:354–367. [PubMed] [Google Scholar]
- 68.Sternlieb I Fraternal concordance of types of abnormal hepatocellular mitochondria in Wilson’s disease. Hepatology. 1992;16:728–732. [DOI] [PubMed] [Google Scholar]
- 69.Sternlieb I Electron microscopy of mitochondria and peroxisomes of human hepatocytes. Prog Liver Dis. 1979;6:81. [PubMed] [Google Scholar]
- 70.Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metabol. 2008;93:54–65. [DOI] [PubMed] [Google Scholar]
- 71.Zischka H, Lichtmannegger J, Schmitt S, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu J, Forbes JR, Chen HS, Cox DW. The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene. Nat Genet. 1994;7:541. [DOI] [PubMed] [Google Scholar]
- 73.Lichtmannegger J, Leitzinger C, Wimmer R, et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest. 2016;126: 2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huster D, Finegold MJ, Morgan CT, et al. Consequences of copper accumulation in the livers of the Atp7b−/−(Wilson disease gene) knockout mice. Am J Pathol. 2006;168:423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yurkova IL, Arnhold J, Fitzl G, Huster D. Fragmentation of mitochondrial cardiolipin by copper ions in the Atp7b−/−mouse model of Wilson’s disease. Chem Phys Lipids. 2011;164:393–400. [DOI] [PubMed] [Google Scholar]
- 76.Gu M, Cooper JM, Butler P, et al. Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. Lancet. 2000;356:469–474. [DOI] [PubMed] [Google Scholar]
- 77.Medici V, Shibata NM, Kharbanda KK, et al. Wilson’s disease: changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology. 2013;57:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagasaka H, Miida T, Inui A, et al. Fatty liver and anti-oxidant enzyme activities along with peroxisome proliferator-activated receptors γ and α expressions in the liver of Wilson’s disease. Mol Genet Metabol. 2012;107: 542–547. [DOI] [PubMed] [Google Scholar]
- 79.Wanders RJ, Komen J, Kemp S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011;278: 182–194. [DOI] [PubMed] [Google Scholar]
- 80.Wooton-Kee CR, Robertson M, Zhou Y, et al. Metabolic dysregulation in the Atp7b−/−Wilson’s disease mouse model. Proc Natl Acad Sci U S A. 2020;117: 2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarode GV, Kim K, Kieffer DA, et al. Metabolomics profiles of patients with Wilson disease reveal a distinct metabolic signature. Metabolomics. 2019;15: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sauer SW, Merle U, Opp S, et al. Severe dysfunction of respiratory chain and cholesterol metabolism in Atp7b−/−mice as a model for Wilson disease. Biochim Biophys Acta. 2011;1812:1607–1615. [DOI] [PubMed] [Google Scholar]
- 83.Mansouri A, Gaou I, Fromenty B, et al. Premature oxidative aging of hepatic mitochondrial DNA in Wilson’s disease. Gastroenterology. 1997;113:599–605. [DOI] [PubMed] [Google Scholar]
- 84.Sokol RJ, Twedt D, McKim JM Jr, et al. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology. 1994;107:1788–1798. [DOI] [PubMed] [Google Scholar]
- 85.Sokol RJ, Devereaux M, Mierau GW, Hambidge KM, Shikes RH. Oxidant injury to hepatic mitochondrial lipids in rats with dietary copper overload: modification by vitamin E deficiency. Gastroenterology. 1990;99:1061–1071. [DOI] [PubMed] [Google Scholar]
- 86.Zischka H, Lichtmannegger J. Pathological mitochondrial copper overload in livers of Wilson’s disease patients and related animal models. Ann N Y Acad Sci. 2014;1315:6–15. [DOI] [PubMed] [Google Scholar]
- 87.Viollet B, Guigas B, Leclerc J, et al. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf). 2009;196:81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hardie DG. AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cardaci S, Filomeni G, Ciriolo MR. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci. 2012;125:2115–2125. [DOI] [PubMed] [Google Scholar]
- 90.Filomeni G, Cerchiaro G, Da Costa Ferreira AM, et al. Pro-apoptotic activity of novel Isatin-Schiff base copper (II) complexes depends on oxidative stress induction and organelle-selective damage. J Biol Chem. 2007;282: 12010e12021. [DOI] [PubMed] [Google Scholar]
- 91.Filomeni G, Piccirillo S, Graziani I, et al. The isatin-Schiff base copper (II) complex Cu (isaepy) 2 acts as delocalized lipophilic cation, yields widespread mitochondrial oxidative damage and induces AMP-activated protein kinase-dependent apoptosis. Carcinogenesis. 2009;30:1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alt ER, Sternlieb I, Goldfischer S. The cytopathology of metal overload. Int Rev Exp Pathol. 1990;31:165–188. [DOI] [PubMed] [Google Scholar]
- 93.Ludwig J, Moyer TP, Rakela J. The liver biopsy diagnosis of Wilson’s disease: methods in pathology. Am J Clin Pathol. 1994;102:443–446. [DOI] [PubMed] [Google Scholar]
- 94.Langner C, Denk H. Wilson disease. Virchows Arch. 2004;445:111–118. [DOI] [PubMed] [Google Scholar]
- 95.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101e154. [DOI] [PubMed] [Google Scholar]
- 96.Ruhanen H, Perttilä J, Hölttä-Vuori M, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab. 2010;7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Delgado M, Pérez-Miguelsanz J, Garrido F, Rodríguez-Tarduchy G, Pérez-Sala D, Pajares MA. Early effects of copper accumulation on methionine metabolism. Cell Mol Life Sci. 2008;65:2080–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Le A, Shibata NM, French SW, et al. Characterization of timed changes in hepatic copper concentrations, methionine metabolism, gene expression, and global DNA methylation in the Jackson toxic milk mouse model of Wilson disease. Int J Mol Sci. 2014;15:8004–8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mazi TA, Sarode GV, Czlonkowska A, et al. Dysregulated choline, methionine, and aromatic amino acid metabolism in patients with wilson disease: exploratory metabolomic profiling and implications for hepatic and neurologic phenotypes. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mudd SH, Cantoni G. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J Biol Chem. 1958;231: 481–492. [PubMed] [Google Scholar]
- 103.Mato JM, Alvarez L, Ortiz P, Pajares MA. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73: 265–280. [DOI] [PubMed] [Google Scholar]
- 104.van der Deen JN, Kennelly JP, Wan S, et al. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. BBA - Biomembranes. 2017;1859:1558–1572. [DOI] [PubMed] [Google Scholar]
- 105.Vance DE, Walkey CJ, Cui Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim Biophys Acta. 1997;1348:142–150. [DOI] [PubMed] [Google Scholar]
- 106.Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J Biol Chem. 2000;275: 29318–29323. [DOI] [PubMed] [Google Scholar]
- 107.DeLong CJ, Shen YJ, Thomas MJ, Cui Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J Biol Chem. 1999;274:29683–29688. [DOI] [PubMed] [Google Scholar]
- 108.Ma DW, Arendt BM, Hillyer LM, et al. Plasma phospholipids and fatty acid composition differ between liver biopsy-proven nonalcoholic fatty liver disease and healthy subjects. Nutr Diabetes. 2016;6, e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arendt BM, Ma DW, Simons B, et al. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl Physiol Nutr Metabol. 2012;38:334–340. [DOI] [PubMed] [Google Scholar]
- 110.Whiley L, Sen A, Heaton J, et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol Aging. 2014;35:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Demirkan A, Isaacs A, Ugocsai P, et al. Plasma phosphatidylcholine and sphingomyelin concentrations are associated with depression and anxiety symptoms in a Dutch family-based lipidomics study. J Psychiatr Res. 2013;47: 357–362. [DOI] [PubMed] [Google Scholar]
- 112.Cole LK, Vance JE, Vance DE. Phosphatidylcholine biosynthesis and lipoprotein metabolism. Biochim Biophys Acta. 2012;1821:754–761. [DOI] [PubMed] [Google Scholar]
- 113.Levy E, Brunet S, Alvarez F, et al. Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci. 2007;80:1472–1483. [DOI] [PubMed] [Google Scholar]
- 114.Seessle J, Gohdes A, Gotthardt DN, et al. Alterations of lipid metabolism in Wilson disease. Lipids Health Dis. 2011;10:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wooton-Kee CR, Jain AK, Wagner M, et al. Elevated copper impairs hepatic nuclear receptor function in Wilson’s disease. J Clin Invest. 2015;125: 3449–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hamilton JP, Koganti L, Muchenditsi A, et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B−/−(Wilson disease) mice. Hepatology. 2016;63:1828–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rodo M, Czonkowska A, Pulawska M, Swiderska M, Tarnacka B, Wehr H. The level of serum lipids, vitamin E and low density lipoprotein oxidation in Wilson’s disease patients. Eur J Neurol. 2000;7:491–494. [DOI] [PubMed] [Google Scholar]
- 118.Brewer GJ, Yuzbasiyan-Gurkan V, Johnson V. Treatment of Wilson’s disease with zinc IX: response of serum lipids. J Lab Clin Med. 1991;118:466–470. [PubMed] [Google Scholar]
- 119.Johansen K, Gregersen G. Glucose intolerance in Wilson’s disease: normalization after treatment with penicillamine. Arch Intern Med. 1972;129: 587–590. [PubMed] [Google Scholar]
- 120.Krysiak R, Okopień B. Whipple’s triad as a clinical manifestation of hepatolenticular degeneration. Pol Arch Med Wewn. 2007;117:53. [PubMed] [Google Scholar]
- 121.Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci. 2018;1411:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu J, Jiang H2, Li J, Cheng KK, Dong J, Chen Z. 1H NMR-based metabolomics investigation of copper-laden rat: a model of Wilson’s disease. PloS One. 2015;10, e0119654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maaswinkel-Mooij PD, Van den Bogert C, Scholte HR, Onkenhout W, Brederoo P, Poorthuis BJ. Depletion of mitochondrial DNA in the liver of a patient with lactic acidemia and hypoketotic hypoglycemia. J Pediatr. 1996;128:679–683. [DOI] [PubMed] [Google Scholar]
- 124.Cariou B, van Harmelen K, Duran-Sandoval D, et al. Transient impairment of the adaptive response to fasting in FXR-deficient mice. FEBS Lett. 2005;579: 4076–4080. [DOI] [PubMed] [Google Scholar]
- 125.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Prawitt J, Abdelkarim M, Stroeve JH, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60: 1861–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Duran-Sandoval D, Mautino G, Martin G, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;5:890–898. [DOI] [PubMed] [Google Scholar]
- 128.Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, et al. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–29979. [DOI] [PubMed] [Google Scholar]
- 129.Lanaspa MA, Ishimoto T, Li N, Cicerchi C, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Preston GM, Calle RA. Elevated serum sorbitol and not fructose in type 2 diabetic patients. Biomark Insights. 2010;5:33e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Saponaro C, Gaggini M, Carli F, Gastaldelli A. The subtle balance between lipolysis and lipogenesis: a critical point in metabolic homeostasis. Nutrients. 2015;7:9453–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muchenditsi A, Yang H, Hamilton JP, et al. Targeted inactivation of copper transporter Atp7b in hepatocytes causes liver steatosis and obesity in mice. Am J Physiol Gastrointest Liver Physiol. 2017;313:G39–G49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang H, Ralle M, Wolfgang MJ, et al. Copper-dependent amino oxidase 3 governs selection of metabolic fuels in adipocytes. PLoS Biol. 2018;16, e2006519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leu ML, Strickland GT, Beckner WM, Chen TS, Wang CC, Yeh SJ. Muscle copper, zinc, and manganese levels in Wilson’s disease: studies with the use of neutron-activation analysis. J Lab Clin Med. 1970;76:432–438. [PubMed] [Google Scholar]
- 135.Peng F, Lutsenko S, Sun X, Muzik O. Imaging copper metabolism imbalance in Atp7b−/−knockout mouse model of Wilson’s disease with PET-CT and orally administered 64 CuCl 2. Mol Imag Biol. 2012;14:600–607. [DOI] [PubMed] [Google Scholar]
- 136.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. [DOI] [PubMed] [Google Scholar]
- 137.Nyasae L, Bustos R, Braiterman L, Eipper B, Hubbard A. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: copper-dependent redistribution between two intracellular sites. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1181–G1194. [DOI] [PubMed] [Google Scholar]
- 138.Ravia JJ, Stephen RM, Ghishan FK, Collins JF. Menkes Copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J Biol Chem. 2005;280:36221–36227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weiss KH, Wurz J, Gotthardt D, Merle U, Stremmel W, Füllekrug J. Localization of the Wilson disease protein in murine intestine. J Anat. 2008;213:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588:4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. [DOI] [PubMed] [Google Scholar]
- 142.Velagapudi VR, Hezaveh R, Reigstad CS, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Derrien M, Van Baarlen P, Hooiveld G, et al. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucindegrader Akkermansia muciniphila. Front Microbiol. 2011;2:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Everard A, Belzer C, Geurts L, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979e984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588. [DOI] [PubMed] [Google Scholar]
- 148.Hibberd MC, Wu M, Rodionov DA, et al. The effects of micronutrient deficiencies on bacterial species from the human gut microbiota. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Geng H, Shu S, Dong J, et al. Association study of gut flora in Wilson’s disease through high-throughput sequencing. Medicine. 2018;97:e11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439: 484–489. [DOI] [PubMed] [Google Scholar]
- 151.Ghazalpour A, Cespedes I, Bennett BJ, Allayee H. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metabol. 2009;10:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ghazalpour A, Cespedes I, Bennett BJ, Allayee H. Expanding role of gut microbiota in lipid metabolism. Curr Opin Lipidol. 2016;27:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56e64. [DOI] [PMC free article] [PubMed] [Google Scholar]