

m6A controls the inflammatory response through cross-talk with H3K27me3, indicating the importance of m6A in immune homeostasis.
Abstract
While N6-methyladenosine (m6A) is the most prevalent modification of eukaryotic messenger RNA (mRNA) involved in various cellular responses, its role in modulating bacteria-induced inflammatory response remains elusive. Here, we showed that loss of the m6A reader YTH-domain family 2 (YTHDF2) promoted demethylation of histone H3 lysine-27 trimethylation (H3K27me3), which led to enhanced production of proinflammatory cytokines and facilitated the deposition of m6A cotranscriptionally. Mechanistically, the mRNA of lysine demethylase 6B (KDM6B) was m6A-modified and its decay mediated by YTHDF2. YTHDF2 deficiency stabilized KDM6B to promote H3K27me3 demethylation of multiple proinflammatory cytokines and subsequently enhanced their transcription. Furthermore, we identified H3K27me3 as a barrier for m6A modification during transcription. KDM6B recruits the m6A methyltransferase complex to facilitate the methylation of m6A in transcribing mRNA by removing adjacent H3K27me3 barriers. These results revealed cross-talk between m6A and H3K27me3 during bacterial infection, which has broader implications for deciphering epitranscriptomics in immune homeostasis.
INTRODUCTION
Inflammation is a biological response of an organism to pathogens, including bacteria (1, 2). Immediately after the microbial invasion, the host immune system can recognize various microbial macromolecules, such as lipopolysaccharide (LPS) or DNA containing methylated CpG motifs through pattern recognition receptors including Toll-like receptors (TLRs) and various DNA sensors (3, 4). The activation of TLRs induces antibacterial responses through downstream signal transduction pathways, especially the activation of nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK), which results in the increased production of proinflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor–α (TNF-α) (5).
Host cells have multilevel mechanisms to negatively regulate inflammation and subsequently avoid harmful pathology associated with uncontrolled inflammatory responses (6). Previous studies including our research, have indicated that posttranslational modifications, such as ubiquitination and phosphorylation, play an important role in ensuring optimal immune responses (7–11). Recent studies suggest that epigenetic regulators, including KDMs (lysine-specific demethylases), are potent modulators of innate immunity (12). Aberrant histone modifications, such as trimethylation of histone H3 at lysine-27 (H3K27me3), are closely associated with inflammation (13). Generally, H3K27me3 modification at gene loci represses gene expression (14, 15). Demethylation of H3K27me3 is catalyzed by KDM6B (also called JMJD3) or KDM6A (also called UTX). KDM6B serves as a response gene to microbial products and inflammatory cytokines and regulates macrophage polarization (16–18). Previously, we demonstrated KDM6B-mediated ubiquitination in cellular reprogramming (19). The biological role of KDM6B in bacterial infection–induced inflammation and the molecular basis for interplay with other epigenetic modifications require further investigations.
As the most prevalent RNA epigenetic (epitranscriptomic) modification, N6-methyladenosine (m6A) modification is reversible and occurs cotranscriptionally (20–25). The m6A methylation is catalyzed by the methyltransferase complex including METTL3, METTL14, and WTAP, whereas its demethylation is mediated by FTO and ALKBH5 (26–28). The m6A modification is functionally interpreted by the m6A “reader” proteins, such as YTH-domain family 1-3 (YTHDF1-3) (24, 29). The m6A is important for the regulation of cell fate and many biological processes. A recent study has demonstrated that METTL3-mediated m6A methylation promotes dendritic cell activation (30). Viral infection can increase the levels of host m6A modification, which negatively regulates the type I interferon response (31, 32). Whether m6A also controls bacteria-induced inflammation response in host cells remains poorly understood.
In this study, we report that m6A exhibited a marked increase during bacterial infection, and was predominantly enriched in transcripts related to histone modification. Knockout (KO) of YTHDF2, an m6A reader, markedly enhanced demethylation of H3K27me3 on the promoters of proinflammatory cytokines (e.g., IL-6 and IL-12B), resulting in increased production of these cytokines during bacterial infection. In addition, we demonstrate that the mRNA of the H3K27me3 demethylase, KDM6B, showed m6A modification and could be degraded by YTHDF2. We defined H3K27me3 as an epigenetic barrier for m6A modification during inflammation. KDM6B facilitates the deposit of m6A via removal of the H3K27me3 barrier and recruitment of the m6A methyltransferase complex. Thus, our results not only reveal cross-talk between m6A and H3K27me3 during antibacterial response but also provide mechanistic insights into the potential use of m6A for preventing excessive inflammation.
RESULTS
YTHDF2 deficiency enhances proinflammatory cytokine production during bacterial infection
The role of m6A modification in bacterial infection was evaluated using an enzyme-linked immunosorbent assay (ELISA)–based m6A quantification assay. We quantified the m6A level in a heat-killed Salmonella typhimurium (HKST) infection model. The global m6A level of mRNA in THP-1 cells notably increased after HKST infection (fig. S1A). To define how bacteria enhance m6A modification of mRNA, we detected the expression level of m6A-modulated genes and found that the expression of WTAP increased in THP-1 cells during bacterial infection (fig. S1, B to D). In addition, the m6A modification of mRNAs in WTAP KO THP-1 cells did not change upon HKST treatment compared with untreated KO cells (fig. S1, E and F), suggesting that WTAP is responsible for the increased m6A modification upon bacterial infection. These findings drive us to explore the consequences of increased m6A modification caused by bacterial infection. The m6A-binding protein YTHDF2 has a critical role in the regulation of the decay of mRNAs containing m6A modification. Hence, we knocked out YTHDF2 in the THP-1 cells (a human monocyte cell line) via the CRISPR-Cas9 technology (fig. S2A) and analyzed the gene expression profiles of wild-type (WT) or YTHDF2 KO THP-1 cells in response to HKST treatment (Fig. 1A). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed that the up-regulated genes in the YTHDF2 KO THP-1 cells were enriched in the cytokine production and the inflammatory signaling pathways (Fig. 1B, fig. S2, B and C, and data file S1). Specifically, YTHDF2 KO cells exhibited up-regulated expression levels of various proinflammatory cytokine genes, such as IL6, IL12B, CCL22, and ICAM1 (Fig. 1C and fig. S2D). These data suggested that YTHDF2 KO promoted bacterial infection–induced inflammatory response.
Fig. 1. YTHDF2 deficiency enhances bacteria-induced inflammatory responses.
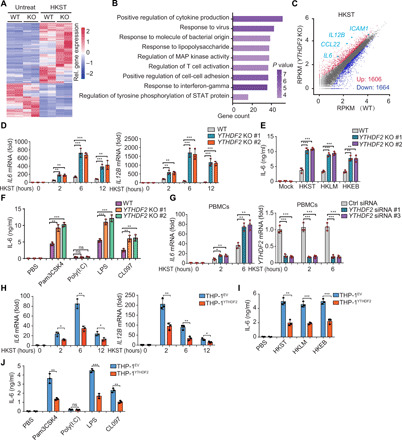
(A) Heatmap of RNA-seq analysis showing differently expressed genes in WT versus YTHDF2 KO THP-1 cells in response to HKST. (B) GO enrichment analysis of genes with substantially up-regulated transcription in YTHDF2 KO THP-1 cells than that in WT THP-1 cells after treatment with HKST. (C) Scatterplot shows that IL6, IL12B, CCL22, and ICAM1are among the most notably up-regulated genes in YTHDF2 KO THP-1 cells versus WT THP-1 cells. (D) WT or YTHDF2 KO THP-1 cells were infected with HKST for indicated periods, and then IL6 and IL12B mRNA expressions were analyzed. (E) WT or YTHDF2 KO THP-1 cells were exposed to HKST, HKLM, or HKEB, and then the production of IL-6 was measured by ELISAs. (F) WT or YTHDF2 KO THP-1 cells were stimulated with Pam3CSK4 (100 ng/ml), poly(I:C) (10 μg/ml), LPS (200 ng/ml), or CL097 (1 μg/ml) for 24 hours, and then the production of IL-6 was measured by ELISAs. (G) PBMCs that were transfected with YTHDF2 siRNA or control siRNA for 48 hours were stimulated with HKST for indicated periods, and then IL6 and YTHDF2 mRNA expressions were analyzed. (H) THP-1EV and THP-1YTHDF2 cells were stimulated with HKST for indicated periods, and then IL6 and IL12B mRNA expressions were analyzed. (I) THP-1EV and THP-1YTHDF2 cells were infected with HKST, HKLM, or HKEB for 24 hours, and the production of IL-6 was analyzed by ELISAs. (J) THP-1EV and THP-1YTHDF2 cells were treated with LPS, poly(I:C), Pam3CSK4, or CL097 for 24 hours before the supernatants were collected. The production of IL-6 was analyzed by ELISAs. Data in (D) to (J) are presented as means ± SEM combined from three independent experiments with triplicate; ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control cells. STAT, signal transducers and activators of transcription.
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis revealed that the mRNA expression levels of IL6 and IL12B in the YTHDF2 KO THP-1 cells were markedly higher than those in the WT THP-1 cells upon stimulation with HKST (Fig. 1D). Furthermore, we obtained similar results in the YTHDF2 KO THP-1 cells after treatment with heat-killed Listeria monocytogenes (HKLM) or heat-killed Escherichia coli O111:B4 (HKEB) (fig. S2E). ELISA analysis revealed that the YTHDF2 KO THP-1 cells exhibited significant up-regulation of IL-6 production in response to HKST, HKLM, or HKEB treatment (Fig. 1E). Furthermore, YTHDF2 KO markedly enhanced the mRNA and protein expression levels of IL-6 in the THP-1 cells stimulated with LPS (a TLR4 ligand), Pam3CSK4 (a TLR2 ligand), or CL097 (a TLR7 ligand), but had little effect on polyinosinic:polycytidylic acid [poly(I:C)] (a TLR3 ligand) treatment (Fig. 1F and fig. S2F).
Next, we investigated the effect of YTHDF2 depletion on the inflammatory response in primary cells. We infected peripheral blood mononuclear cells (PBMCs) with HKST, LPS, or Pam3CSK4 and measured the expression levels of IL6. YTHDF2 small interfering RNA (siRNA)–transfected PBMCs exhibited enhanced IL6 mRNA expression levels under these stimuli when compared with the control cells (Fig. 1G and fig. S2, G to I).
Furthermore, we generated a YTHDF2-overexpressing THP-1 cell line (THPYTHDF2) (fig. S2J) and observed that the ectopic expression of YTHDF2 substantially reduced the IL-6 mRNA and protein expression levels under inflammatory stimulation (Fig. 1, H to J, and fig. S2, J to L). These results indicate that YTHDF2 inhibits a subset of proinflammatory cytokines (e.g., IL-6) during bacterial infection in human cells.
Bacterial infection increases m6A level of the histone modification–related transcripts
To investigate the molecular mechanisms of YTHDF2 in regulating the bacterial infection–induced inflammatory response, we analyzed the activation of NF-κB and MAPK signaling pathways. As shown in fig. S3A, the phosphorylation and expression levels of IκB kinase (IKK), p65, p38, c-Jun N-terminal kinase (JNK), and extracellular signal–regulated kinase (ERK) were similar in the WT and YTHDF2 KO cells in response to HKST. These findings indicated that YTHDF2 negatively regulates inflammation through a mechanism independent of NF-κB and MAPK signaling pathways.
Next, we examined whether YTHDF2 recognition of m6A RNA methylation directly contributes to the proinflammatory cytokine mRNA decay. We expressed the YTHDF2 truncation mutants (YTHDF2-N and YTHDF2-C) and m6A recognition site mutant (YTHDF2-WA) in the THP-1 cells (Fig. 2A and fig. S3B). Overexpression of full-length YTHDF2 (YTHDF2-WT), but not truncated or mutated YTHDF2, reduced the mRNA and protein expression level of IL-6 (Fig. 2B and fig. S3C). This result indicated that YTHDF2 negatively regulates the proinflammatory cytokine production via m6A recognition.
Fig. 2. m6A level of histone modification–related genes is increased during bacterial infection.
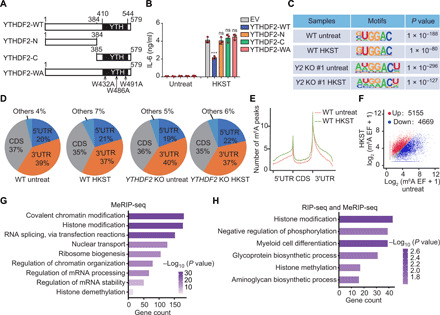
(A) A structural diagram of WT YTHDF2 as well as schematic representation of truncation mutants and m6A recognition site mutations of YTHDF2. The numbers indicate amino acid positions. (B) The constructed YTHDF2 (WT) as well as YTHDF2 truncations (N and C termini) or site mutation (WA) THP-1 cell lines were exposed to HKST for 24 hours, and then the production of IL-6 was measured by ELISAs. (C) Sequence motif was identified within m6A peaks by HOMER analysis of WT and YTHDF2 KO THP-1 cells with or without HKST treatment. (D) Pie charts depicting the proportion of m6A peak distribution in the 5′UTR, CDS, and 3′UTR regions across mRNA transcriptome. (E) Metagene profiles of m6A peak distribution across the 5′UTR, CDS, and 3′UTR in THP-1 cells with or without HKST treatment. (F) Scatterplot showing the m6A peaks in mRNA of HKST-treated and untreated THP-1 cells. The abundance of m6A peaks was calculated as enrichment fold (EF; immunoprecipitate/input) from the m6A-seq data. The m6A-containing transcripts with notably increased and decreased peaks are highlighted in red and blue, respectively. (G) GO analysis of the genes that contained hypermethylated m6A peaks in HKST-infected THP-1 cells versus the uninfected cells. MeRIP-seq, m6Aspecific methylated RNA immunoprecipitation combined with high-throughput sequencing. (H) GO analysis of a group of overlapped genes that bound to YTHDF2 based on the YTHDF2 RIP-seq data and contained hypermethylated m6A peaks after exposure to HKST in THP-1 cells.
Using m6A sequencing (m6A-seq) analysis, we mapped the transcriptome-wide m6A methylation of WT and YTHDF2-deficient THP-1 cells with or without HKST stimulation. The consensus m6A core motifs were enriched within the m6A peaks in all samples (Fig. 2C). There was a similarity in the total m6A distribution between the WT and YTHDF2 KO cells with or without HKST treatment, which coincided with the typical m6A peak distribution features (Fig. 2D). The m6A peaks were especially abundant around the start and stop codons (Fig. 2E). HKST stimulation resulted in an elevated m6A peak across the entire gene bodies (Fig. 2E). Meanwhile, the m6A-seq analysis revealed that the m6A peaks of 5155 transcripts increased in the THP-1 cells after treatment with HKST (Fig. 2F). No m6A of IL6 mRNA was found with or without HKST treatment. GO enrichment analysis indicated that these genes with increased m6A peaks were associated with the covalent chromatin and histone modifications (Fig. 2G).
RNA immunoprecipitation sequencing (RIP-seq) was performed to explore the correlation among the YTHDF2-bound mRNAs (fig. S3D), particularly the mRNAs with elevated m6A modification after bacterial infection. Two biological replicates of RIP-seq exhibited a strong correlation (fig. S3E). We observed that 61.9% of the YTHDF2-bound mRNAs contain m6A peaks (fig. S3F). YTHDF2-bound mRNAs were specifically enriched in histone modification GO term (fig. S3G and data file S2). Intriguingly, YTHDF2 did not directly bind proinflammatory cytokine IL6 mRNA (fig. S3H).
Consistently, combined analysis of YTHDF2-bound genes and m6A up-regulated genes revealed that these genes were enriched in histone modification (Fig. 2H), suggesting that m6A has an important role in modulating bacterial infection–induced inflammation via regulating histone modification genes. These results suggest that the m6A modifications of histone modification–related gene mRNAs were markedly increased and are recognized by YTHDF2 in response to bacterial infection.
YTHDF2 inhibits H3K27me3 demethylation of IL6 promoter during bacterial infection
In general, transcription start sites of active genes are marked with trimethylated H3K4 (H3K4me3) or acetylated H3K27 (H3K27ac), and inactive genes are marked with H3K27me3. Next, we determined whether YTHDF2 depletion affects histone modification marks (H3K27me3, H3K4me3, and H3K27ac) in the THP-1 cells. The global levels of H3K27me3 in the YTHDF2 KO THP-1 cells were markedly lower than those in the WT control, whereas those of H3K4me3 and H3K27ac were similar between the YTHDF2 KO and WT cells in response to HKST (Fig. 3A). Furthermore, chromatin immunoprecipitation sequencing (ChIP-seq) and ChIP qRT-PCR analyses revealed that the H3K27me3 modification of proinflammatory cytokine gene promoters (IL6, IL12B, ICAM1, and CCL22) in the YTHDF2 KO THP-1 cells was notably lower than that in the WT THP-1 cells (Fig. 3, B and C). Contrastingly, there were no marked differences between YTHDF2 KO and WT THP-1 cells in the H3K4me3 modification levels in the proinflammatory cytokine promoters in response to HKST stimulation (Fig. 3D). These data suggested that loss of YTHDF2 specifically enhances the expression of a subset of proinflammatory cytokines, such as IL-6 and IL-12B, during bacterial infection by facilitating H3K27me3 demethylation at their promoters.
Fig. 3. YTHDF2 inhibits H3K27me3 demethylation of IL6 promoter.
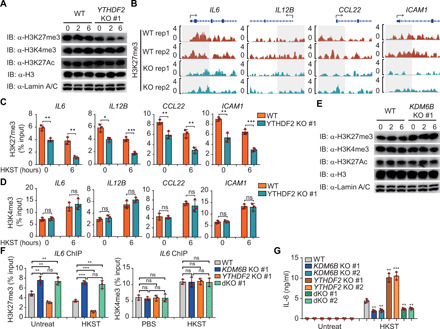
(A) WT and YTHDF2 KO THP-1 cells were infected with HKST for indicated periods, and then the nuclear extracts were subjected to immunoblot analysis with the indicated antibodies. IB, immunoblot. (B) IGV browser tracks show the distribution of H3K27me3 ChIP-seq signals at indicated loci in WT and YTHDF2 KO THP-1 cells stimulated with HKST. (C and D) ChIP-qPCR assay for H3K27me3 and H3K4me3 at the IL6, IL12B, CCL22, or ICAM1 promoter region in WT and YTHDF2 KO THP-1 cells infected with HKST or not. (E) WT and KDM6B KO THP-1 cells were exposed to HKST for indicated periods, and then the nuclear extracts were analyzed by immunoblotting with the indicated antibodies. (F) ChIP-qPCR assay for H3K27me3 and H3K4me3 at the IL-6 promoter region in WT, KDM6B KO, YTHDF2 KO, or YTHDF2 and KDM6B double knockout (dKO) THP-1 cells in response to HKST. (G) WT, YTHDF2 KO, KDM6B KO, or YTHDF2 and KDM6B dKO THP-1 cells were infected with HKST for 24 hours, and supernatants were collected to analyze the production of IL-6 by ELISAs. Data in (A) and (E) are representative of three individual experiments with similar results. Data in (C), (D), (F), and (G) are presented as means ± SEM combined from three independent experiments with triplicate; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control cells (Student’s t test). ns, not significant.
KDM6A and KDM6B were identified as H3K27 demethylases, which catalyze the demethylation of H3K27me3. As KDM6A expression could barely be detected in THP-1 cells (fig. S4A), we generated the KDM6B KO THP-1 cells to evaluate the role of KDM6B in regulating inflammatory response (fig. S4, B and C). KDM6B deficiency had no effect on the activation of NF-κB and MAPK signaling pathways (fig. S4D). As expected, immunoblotting and ChIP-qPCR analyses revealed that KDM6B depletion enhanced the global H3K27me3 modification (Fig. 3E) and H3K27me3 modification of IL6 promoter (fig. S4E) with or without HKST stimulation. Consistently, the deficiency of KDM6B substantially decreased the mRNA and protein expression levels of IL-6 in response to HKST (fig. S4, F and G).
To further confirm the role of KDM6B in YTHDF2-mediated inflammatory response, we generated YTHDF2 and KDM6B double-KO (dKO) THP-1 cells (fig. S4H). Intriguingly, KDM6B deficiency rescued YTHDF2 deficiency–attenuated H3K27me3 modification of IL6 promoter both under normal and HKST-stimulated conditions (Fig. 3F). However, H3K4me3 modification of IL6 promoter was not affected in KDM6B and YTHDF2 dKO cells (Fig. 3F). Accordingly, dKO of KDM6B and YTHDF2 failed to promote the transcription and production of IL-6 when compared with KO of YTHDF2 (Fig. 3G and fig. S4I). These results suggest that KO of KDM6B can rescue the up-regulation of proinflammatory cytokine release in the YTHDF2-deficient THP-1 cells during bacterial infection.
YTHDF2 binds and decays the KDM6B transcripts via an m6A-dependent mechanism
RIP-seq analysis revealed that YTHDF2 recognized m6A peaks in the KDM6B transcripts but not in the KDM6A transcripts after HKST stimulation (Fig. 4A and fig. S5A). The RIP-qPCR analysis revealed that YTHDF2 binds to the human KDM6B mRNA under normal condition and that this association is markedly enhanced in response to HKST stimulation (Fig. 4B). qRT-PCR and immunoblot analyses further confirmed that YTHDF2 deficiency substantially increased the mRNA and protein expression levels of KDM6B, respectively (Fig. 4, C and D, and fig. S5B). Consistently, knockdown of YTHDF2 in PBMCs (fig. S5C) increased the transcription of KDM6B both before and after HKST treatment (Fig. 4E). These findings suggested that YTHDF2 recognition of m6A-modified KDM6B mRNA inhibits KDM6B expression.
Fig. 4. YTHDF2 degrades KDM6B mRNA via an m6A-dependent manner.
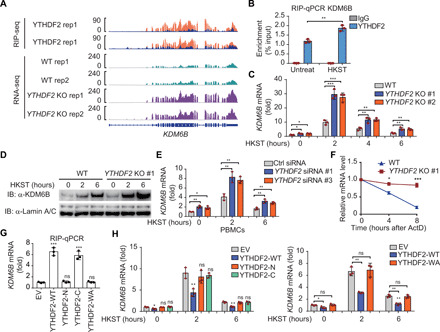
(A) IGV tracks displaying YTHDF2 RIP-seq (top) and RNA-seq (bottom) read distribution in KDM6B mRNA of WT and YTHDF2 KO THP-1 cells with HKST infection. (B) RIP-qPCR detecting the binding of YTHDF2 to the transcripts of KDM6B in THP-1 cells stimulated with and without HKST. (C) The expression of KDM6B analyzed by RT-PCR increased in YTHDF2 KO versus WT THP-1 cells after infection with HKST for indicated periods. (D) Immunoblot analysis of KDM6B in WT and YTHDF2 KO THP-1 cells exposed to HKST for indicated periods. (E) PBMCs were transfected with YTHDF2 or control siRNA for 48 hours and then treated with HKST, and KDM6B transcription was analyzed by real-time PCR. (F) Representative mRNA profile of KDM6B at indicated time points after actinomycin D (5 μg/ml) treatment in WT and YTHDF2 KO THP-1 infected with HKST. (G) RIP-qPCR detecting the binding region of YTHDF2 to the mRNA of KDM6B in THP-1 cell lines that overexpressed Flag-tagged YTHDF2 (WT) as well as YTHDF2 truncations (N and C termini) or site mutation (WA) exposed to HKST. (H) THP-1EV, THP-1YTHDF2-WT, THP-1YTHDF2-N, THP-1YTHDF2-C (left), and THP-1YTHDF2-WA (right) cells were stimulated with HKST for indicated periods, and then KDM6B expression was analyzed by real-time PCR. Data in (D) are representative of three individual experiments with similar results. Data in (B), (C), and (E) to (H) are presented as means ± SEM combined from three independent experiments with triplicate, *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control cells.
To explore whether YTHDF2 regulates KDM6B expression through mRNA decay, we measured the KDM6B mRNA level in THP-1 cells after treatment with the transcription inhibitor actinomycin D. Compared with the WT THP-1 cells, the remaining KDM6B mRNA levels were markedly higher in the YTHDF2-deficient cells (Fig. 4F). The RIP-qPCR analysis revealed that the C-terminal RNA binding domain (YTHDF2-C) is responsible for the binding of YTHDF2 to KDM6B mRNA as well as full-length YTHDF2 (YTHDF2-WT), whereas the P/Q/N-rich N terminus (YTHDF2-N) did not bind to the KDM6B mRNA (Fig. 4G). Consistently, mutation of m6A recognition sites of YTHDF2 (YTHDF2-WA: W432A, W486A, and W491A) resulted in complete loss of m6A binding (Fig. 4G). Furthermore, overexpression of YTHDF2 truncation mutants or m6A recognition site mutants alone could not degrade KDM6B mRNA (Fig. 4H). These results indicated that the enhanced KDM6B mRNA expression was due to reduced mRNA decay caused by insufficient recognition of m6A modification in the YTHDF2-deficient cells.
Next, we tested whether the regulation of Il6 production by Ythdf2 is conserved in murine macrophages. Unlike what we have found in human cells, transfection of murine macrophages with murine Ythdf2 siRNA has no effect on Il6 expression (fig. S6A). Furthermore, we analyzed Ythdf2 cross-linking and immunoprecipitation sequencing (CLIP-seq) data obtained in murine cells and found that the binding mRNA of murine Ythdf2 is quite different from that in human cells (fig. S6B). Ythdf2 did not recognize m6A peaks in murine Kdm6a/6b mRNAs (fig. S6, C and D), supporting YTHDF2 specifically mediates the mRNA processing of KDM6B in human cells.
KDM6B mRNA is m6A modified and is more stable in YTHDF2 KO cells
Genome-wide mapping of m6A methylation revealed that KDM6B mRNA exhibited significant m6A peaks (at 2439, 2461, 2837, and 3065) in the coding sequence (CDS) region (Fig. 5, A to C). These m6A methylations were increased when treated with HKST. Next, we cloned the 690–nucleotide (nt)–long WT or mutant CDS truncation sequence (from 2407 to 3096) into a Renilla luciferase (Rluc) reporter (Fig. 5D). Consistent with the function of m6A modification in the regulation of KDM6B mRNA stability, the Rluc activity of mutant reporter was higher than that of the WT reporter under normal condition, even though both reporters were cloned under the same promoter (Fig. 5E). The ectopic expression of YTHDF2 substantially inhibited the Rluc activity of the WT reporter, and this inhibition was impaired by mutations in the m6A consensus sites (Fig. 5E). Accordingly, the relative Rluc mRNA level of KDM6B-WT-luc (but not KDM6B-MUT) was largely decreased by YTHDF2 overexpression (Fig. 5E). Consistently, the binding of YTHDF2 on Rluc was abrogated due to the mutations at the m6A sites (Fig. 5F). KO of YTHDF2 increased the Rluc activity and delayed mRNA degradation of WT reporter but not that of the mutant reporter (Fig. 5G).
Fig. 5. Identification of specific m6A modification sites in KDM6B mRNA.
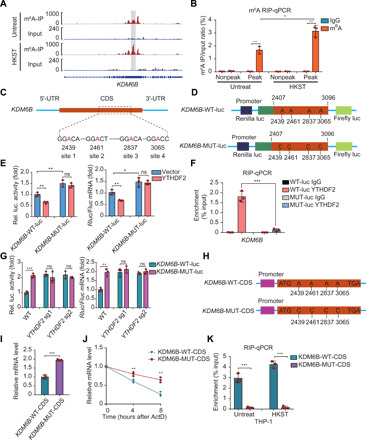
(A) IGV browser tracks showing that m6A peaks were enriched in the CDS of the KDM6B transcript from the m6A-seq data. IP, immunoprecipitation. (B) m6A-qPCR analysis of KDM6B mRNA in WT THP-1 cells with or without HKST infection. (C) Schematic representation of the position of m6A motifs within the KDM6B transcript. (D) Schematic diagram of KDM6B WT (KDM6B-WT-luc) and KDM6B mutant (KDM6B-MUT-luc) reporters. The 690-nt DNA sequence of the WT KDM6B segment sequence was inserted to the dual-luciferase reporter plasmid to give rise to the KDM6B-WT-luc reporter. For the KDM6B-MUT-luc, A–C substitutions were made within the m6A consensus. (E) Relative dual-luciferase reporter activity and mRNA level of WT (KDM6B-WT-luc) or mutated (KDM6B-MUT-luc) reporters in HeLa cells with ectopically expressed YTHDF2. (F) RIP-qPCR detecting the binding of YTHDF2 to the transcripts of KDM6B-WT-luc or KDM6B-MUT-luc reporter in HeLa cells. (G) Relative dual-luciferase reporter activity and mRNA level of WT (KDM6B-WT) or mutated (KDM6B-MUT) reporters in WT or YTHDF2 KO HeLa cells. (H) Schematic depiction of mutation of the m6A site in the KDM6B CDS region. (I and J) Full-length WT or mutant (MUT) KDM6B CDS regions were ectopically expressed in HeLa cells. After 24 hours, KDM6B mRNA levels were measured by RT-PCR (I), or cells were treated with actinomycin D (5 μg/ml) and total mRNA at indicated time points were collected to analyze the remaining KDM6B mRNA (J). (K) THP-1 cells stably expressing WT or mutant (MUT) KDM6B CDS fragment were infected with HKST for 6 hours, and then the binding of YTHDF2 was detected by RIP-qPCR. Data in (B), (E) to (G), and (I) to (K) are presented as means ± SEM combined from three individual experiments with triplicate. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control cells.
Furthermore, we assayed the stability of KDM6B transcripts by cloning the WT and mutant CDS sequence into a murine stem cell virus (MSCV)–based retroviral vector, respectively (Fig. 5H). The mutant KDM6B mRNA was more stable than the WT KDM6B mRNA, which suggested that the methylation of the four adenosines is crucial for regulating the KDM6B mRNA stability (Fig. 5, I and J). Last, the binding of YTHDF2 on KDM6B mRNA was abrogated by the mutations of the m6A sites in THP-1 cells (Fig. 5K). In summary, these data indicate that m6A modifications (at 2439, 2461, 2837, and 3065) in the KDM6B CDS are required for the binding of YTHDF2 to KDM6B mRNA and for the YTHDF2-mediated KDM6B mRNA decay.
KDM6B recruits m6A methyltransferase complex and facilitates m6A modifications cotranscriptionally
Previously, we have reported that H3K36me3 guides m6A modification cotranscriptionally (25, 33). To identify the role of H3K27me3 demethylation in the dynamic regulation of m6A modification, we compared the genome-wide distribution of H3K36me3 and H3K27me3 modifications to that of the m6A in the transcriptome of the THP-1 cells. We found that the position of H3K27me3 peaks was exclusive with that of m6A and H3K36me3 peaks in HKST-treated THP-1 cells (Fig. 6A and fig. S7A). Distributions of m6A were mostly in the region of H3K27me3-negative chromatin (Fig. 6B). KO of H3K27me3 demethylase KDM6B resulted in a decrease in total m6A modification of mRNAs after HKST treatment (fig. S7B). Further genome-wide m6A-seq analyses showed that m6A signal around the stop codon of KDM6B KO THP-1 cells was significantly down-regulated compared with WT control during bacterial infection (Fig. 6C and fig. S7C). Transcripts (9703) whose m6A modifications were significantly decreased in KDM6B KO cells (Fig. 6D) were mainly enriched in the regulation of chromatin modification (fig. S7D). Moreover, inhibition of H3K27me3 demethylases by GSK-J4 also led to a global reduction in m6A modification on mRNAs (Fig. 6E). In addition, the mRNA expression of H3K27me3 demethylases KDM6B is positively related to the m6A methyltransferase complex genes (METTL3, METTL14, and WTAP) in normal tissues (fig. S7E). These results imply H3K27me3 as a barrier for m6A deposition and suggest that the loss of H3K27me3 facilitates m6A methylation.
Fig. 6. KDM6B facilitates m6A deposition by recruiting m6A methyltransferase complex.
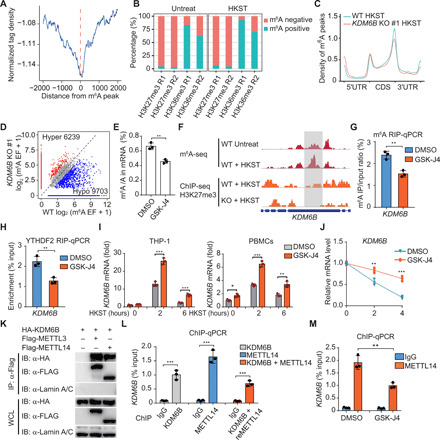
(A) Distance of H3K27me3 to the nearest m6A peaks in HKST-infected THP-1 cells. (B) Percentages of m6A peaks in different regions in THP-1 cells with or without HKST infection. The m6A peaks were normalized with exomePeak. (C) Metagene profiles of m6A distribution in WT or KDM6B KO THP-1 cells treated with HKST. (D) Scatterplot of m6A in KDM6B KO or WT THP-1 cells infected with HKST. (E) m6A levels on poly(A) RNA of HKST-stimulated THP-1 cells treated with or without GSK-J4. (F) (Top) Distribution of m6A peaks across the KDM6B mRNA transcript in THP-1 cells treated with or without HKST and (bottom) distribution of H3K27me3 peaks across the KDM6B mRNA transcript in WT and YTHDF2 KO THP-1 cells treated with HKST. (G and H) m6A RIP-qPCR (G) and YTHDF2 RIP-qPCR (H) analyses of KDM6B mRNA in DMSO- or GSK-J4–treated THP-1 cells in response to HKST. (I) KDM6B mRNA expression in HKST-stimulated THP-1 cells or PBMCs with or without GSK-J4 treatment. (J) HKST-stimulated THP-1 cells were treated with DMSO or GSK-J4 for 24 hours, and then total mRNA were collected at 0, 2, or 4 hours after actinomycin D treatment to analyze the remaining KDM6B mRNA. (K) Coimmunoprecipitation of KDM6B and METTL3/14. WCL, whole cell lysate. (L) ChIP and ChIP-re-ChIP analysis of the association of KDM6B and METTL14 with the KDM6B genome DNA in HeLa cells transfected with Flag-tagged KDM6B or/and hemagglutinin (HA)–tagged METTL14. (M) Reduction in METTL14 at KDM6B gene body in HA-tagged METTL14-transfected HeLa cells with or without GSK-J4 treatment. Data in (K) are representative of three individual experiments with similar results. Data in (E), (G) to (J), (L), and (M) are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control.
This interplay of H3K27me3 demethylation and m6A deposition was found in the individual representative genes, such as KDM6B, in HKST-stimulated THP-1 cells (Fig. 6F). We further used GSK-J4 to inhibit H3K27me3 demethylation and investigated the casual regulation of H3K27me3 demethylation on m6A deposition at the KDM6B locus. GSK-J4 treatment inhibited m6A deposition (Fig. 6G) and subsequently impaired YTHDF2 binding on KDM6B mRNA (Fig. 6H). The mRNA expression levels are determined by the interplay of RNA production and degradation. Although GSK-J4 suppressed the transcription of KDM6B (fig. S7F), GSK-J4 treatment still increased the mRNA level of KDM6B in THP-1 cells and PBMCs (Fig. 6I), owing to the inhibition of m6A and YTHDF2-mediated mRNA decay (Fig. 6J). Consistently, we found that the H3K27me3 demethylation is required for the transcriptional activation of IL6 during HSKT-induced inflammation in THP-1 cells and PBMCs through GSK-J4 treatment experiments (fig. S7G).
Mechanistically, we found that KDM6B interacted with the m6A methyltransferase complex components METTL3 and METTL14 (Fig. 6K and fig. S7H). The binding seemed to be specific during transcription, as the interaction between KDM6B and METTL14 was also observed at the KDM6B gene locus using ChIP and re-ChIP assays (Fig. 6L). Furthermore, inhibition of H3K27me3 demethylation by GSK-J4 showed that depletion of KDM6B demethylase activity impaired the binding of the METTL4 on the KDM6B gene locus (Fig. 6M), suggesting that H3K27me3 demethylation is critical for the recruitment of the m6A methyltransferase complex cotranscriptionally.
DISCUSSION
The m6A is the most prevalent mRNA modification in eukaryotic cells, but the interplay of m6A and histone modifications remains elusive. In this study, we revealed that m6A negatively regulated the inflammatory response through modulating H3K27me3, indicating the importance of m6A in maintaining immune homeostasis. Depleting m6A reader YTHDF2 expression limits the mRNA decay of KDM6B and promotes H3K27me3 demethylation, which is required for the activation of proinflammatory cytokine gene (e.g., IL6) transcription (fig. S7I, left). Meanwhile, H3K27me3 demethylation facilitates m6A deposition on histone modification genes (e.g., KDM6B), which enhances the mRNA decay to restrain inflammation (fig. S7I, right). The findings on the interplay of m6A RNA modification and histone modification also provide mechanistic insight for the regulation of proinflammatory cytokine gene expression.
Both m6A methylation and histone modification are reported to be essential for cell state transition. In most cases, bacteria elicit histone modifications and chromatin remodeling to inhibit the host innate immune response (34–36). Therefore, studying the regulatory role of m6A in histone modification during the bacterial infection would provide new insights into the mechanisms of pathogen altering host epigenome. Here, we found that various nascent transcripts gained m6A modification in the process of inflammation. Our YTHDF2 RIP-seq and m6A-seq data revealed that the gained m6A peaks induced by bacterial infection and bound by YTHDF2 are predominantly enriched in the histone modification transcripts (e.g., KDM6B). We noted that the KO of YTHDF2 markedly reduced the H3K27me3 modification through decay of KDM6B mRNA in the THP-1 cells infected with bacteria, leading to markedly increased production of proinflammatory cytokines. Notably, although YTHDF2 mediated the degradation of the KDM6B transcript, the overall expression of KDM6B was increased due to its markedly increased transcription during the bacterial infection. Thus, our findings uncover a mechanism that reveals the role of m6A in modulating H3K27me3 modification in the host cells and provide the basis for explaining the up-regulated IL-6 production in the YTHDF2-deficient cells. The contribution of the m6A on other histone modification transcripts in the regulation of inflammation needs to be further determined.
The mechanism that underlies the dynamic deposition of m6A modification during transcription is largely unknown. Our previous work showed that H3K36me3 guides m6A methylation during the process of gene transcription, which suggested the importance of histone modification for the establishment of mRNA m6A methylation (33). Note that H3K36me3 and H3K27me3 occupy exclusive domains of the chromatin, and H3K27me3 rarely coexisted with H3K36me3 on the same histone H3 tail (37–39). We hypothesize H3K27me3 as a barrier for m6A modification. We found that m6A modifications are reduced globally when H3K27me3 demethylase is depleted or inhibited. Mechanistically, we first reveal the important role of the interaction between KDM6B and METTL14 in the control of m6A deposition. It is known that manipulation of the H3K36me3 methyltransferase SET domain containing 2 (SETD2) changes H3K36me3 and corresponding m6A modification in human and mouse transcriptomes. Moreover, the METTL14 could interact with H3K36me3 and the transcribing RNA polymerase II (Pol II) (33). A previously recognized model showed that Pol II recruits components of the histone modification complexes, such as KDM6B, which can directly interact with SETD2, to mediate methylation of H3K36me3 and H3K27me3 demethylation (40). Although it is still unknown whether KDM6B interacted with METTL14 or SETD2 when encountering RNA Pol II, our study suggested that METTL14 recognizes H3K36me3 through KDM6B-mediated recruitment and removal of H3K27me3 barrier.
There are diverse roles of YTHDF2 under different circumstances. YTHDF2 is responsible for transporting mRNAs from translation pools to P bodies (41). YTHDF2 mediates two degradation pathways of mRNAs with m6A modifications: (i) deadenylation via the YTHDF2-CCR4/NOT complex (42) and (ii) endoribonucleolytic cleavage by the YTHDF2–HRSP12–ribonuclease P/mitochondrial RNA–processing complex (43, 44). Another report showed that YTHDF2 promotes cap-independent translation initiation by increasing 5′ untranslated region (5′UTR) methylation in m6A modification (45). We believe that the RNA degradation machinery enables YTHDF2 to negatively regulate inflammatory response by degrading the m6A-modified transcripts selectively during bacterial infection. Although it has been reported that the C-terminal domain of YTHDF2 binds to mRNA with m6A methylation, the mechanism underlying the target specificity of YTHDF2 on the m6A mRNAs is still unclear. In murine cells, Ythdf2 target transcripts revealed enrichment of genes related to regulation of oxidative phosphorylation as well as regulation of histone acetylation (46). Notably, in human cells, we found that YTHDF2 mainly bound to histone modulator mRNAs, such as KDM6B, on sites largely overlapping with m6A peaks. We demonstrated that binding of YTHDF2 to KDM6B was only observed in human cells but not in murine cells. These results reveal that the YTHDF2-targeted mRNAs are species specific. Future studies are needed to clarify the details of this species-specific YTHDF2 function in human and mouse inflammation.
Bacterial infection induces the expression of hundreds of genes in the host cells, including proinflammatory cytokines, chemokines, antimicrobial proteins, and coagulation factors. Both IL6 and TNF act as proinflammatory cytokine genes, which can be activated by NF-κB and MAPK signaling pathways. In our study, we observed that bacterial infection–induced proinflammatory cytokine genes in the YTHDF2-deficient cells fall into two categories: IL-6, whose production was enhanced, and TNF-α, whose production was unaffected upon depletion of YTHDF2 when compared to those in the WT control. We reasoned that different categories of the proinflammatory cytokine gene loci should have different histone modification patterns to meet their regulatory requirements. KDM6B-mediated H3K27me3 demethylation is crucial for IL6 gene transcriptional activation (47). In this study, we showed that many proinflammatory cytokines, such as IL12B, are also increased in YTHDF2 KO THP-1 cells upon bacterial infection. Because IL-6 plays a central role in the integrated immune defense network against infection, we chose IL-6 as a proof of concept. Moreover, we observed that, in contrast to the IL6 locus, which was enriched with H3K27me3 in the HKST-untreated THP-1 cells, H3K27me3 modifications at the TNF locus were undetectable. This finding suggested that the activation of TNF transcription does not require removal of the H3K27me3 barrier after bacterial infection, which can elicit a rapid antibacterial response. As many proinflammatory cytokine promoters are enriched with H3K27me3, the inhibition of H3K27me3 demethylation mediated by YTHDF2 will decrease the efficiency of innate host defenses. Although our data highlight the importance of m6A and YTHDF2 in the negative regulation of antibacterial responses, further studies are needed on the safe manipulation of the innate immune response and inflammation by targeting m6A and YTHDF2 for the clinical treatment of infectious diseases.
MATERIALS AND METHODS
Antibodies, reagents, and plasmids
Antibodies against YTHDF2 (catalog number 24744-1-AP), β-tubulin (catalog number 10094-1-AP), lamin A/C (catalog number 10298-1-AP), YTHDF1 (catalog number 17479-1-AP), METTL3 (catalog number 15073-1-AP), and β-actin (catalog number 60008-1-Ig) were purchased from Proteintech. Antibody against YTHDF3 (catalog number A8395) was purchased from ABclonal. Antibody against Flag (catalog number A8592) was purchased from Sigma-Aldrich. Antibodies against WTAP (catalog number sc-374280) were purchased from Santa Cruz. Antibodies against the following proteins were purchased from Cell Signaling Technology: KDM6B (catalog number 3457), p-IKKα/β (catalog number 2697), p-IκBα (catalog number 9246), IκBα (catalog number 4814), p-p38 (catalog number 9211), p38 (catalog number 9212), p-JNK (catalog number 9251), JNK (catalog number 9252), p-ERK (catalog number 9101), ERK (catalog number 9102), FTO (catalog number 14386), H3K27me3 (catalog number 9733), and H3K4me3 (catalog number 9751). The anti–N6-methyladenosine (m6A) antibody (catalog number 202303) was purchased from Synaptic Systems. Antibodies against METTL14 (catalog number ab98166), ALKBH5 (catalog number ab234528), and H3K27ac (catalog number ab4729) were purchased from Abcam. LPSs (100 ng/ml; catalog number L4391-1MG) were purchased from Sigma-Aldrich. Pam3CSK4 (100 ng/ml), poly(I:C) (10 μg/ml), CL097 (1 μg/ml), HKST (106 cells/ml), HKEB (107 cells/ml), and HKLM (5 × 107 cells/ml) were purchased from InvivoGen. Plasmid pCMV-HA-KDM6B (catalog number 24167) was purchased from Addgene. All plasmids were verified by Sanger sequencing before further analysis.
Cell culture and transfection
The THP-1 cells [American Type Culture Collection (ATCC), catalog number TIB-202] were cultured in RPMI 1640 (Corning) supplemented with 10% (v/v) fetal bovine serum (FBS), 4 nM l-glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml) (RPMI 1640 complete medium). The human embryonic kidney (HEK) 293T cells (ATCC, catalog number CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium (Corning) supplemented with 10% (v/v) FBS, 4 nM l-glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml). The PBMCs were separated from the peripheral blood of healthy donors. The isolated PBMCs were washed with Dulbecco’s phosphate-buffered saline (DPBS; Corning) and cultured in RPMI 1640 complete medium. All cells were cultured at 5% (v/v) CO2 at 37°C in a CO2 incubator. The transfection of HEK293T cells was performed using Lipofectamine 2000 (Invitrogen). The transfection of THP-1 cells was performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions.
Generation of KO cells by CRISPR-Cas9
Guide RNA (gRNA), which was designed using an online gRNA design tool (by Zhang Feng laboratory, http://crispr.mit.edu/), was subcloned into the pLentiCRISPRv2 vector for expressing gRNA and Cas9. This vector was transfected into the HEK293T cells along with the following two lentiviral packing plasmids: VSVG and Δ8.9. Next, the culture supernatants were collected at 48 hours after transfection. The culture supernatant was concentrated by ultracentrifugation before use for infection. The infection-positive cells were selected and enriched on the basis of the resistance to puromycin (InvivoGen) or Zeocin (InvivoGen). Monoclonal cells were screened by a limiting dilution assay and confirmed by sequencing of PCR fragments. Immunoblotting analysis of cell lysates was performed with the corresponding antibody. The gRNA sequences used for generating the KO cells are listed in table S1.
Immunoblotting
The proteins in the cell lysates were resolved by SDS–polyacrylamide gel electrophoresis using an 8 to 12% (w/v) gel. The resolved proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad). The membrane was blocked with 5% (w/v) skim milk prepared in TBS-T buffer (50 mM tris-HCl, 150 mM NaCl, and 0.05% Tween 20). The membrane was washed thrice with TBS-T and incubated with an appropriate primary antibody diluted in TBS-T buffer with 5% (w/v) skim milk. The membrane was subsequently visualized after incubating with the secondary antibody [a horseradish peroxidase–conjugated anti-mouse or anti-rabbit immunoglobulin G (IgG) antibody] in the ChemiDoc XRS system (Bio-Rad).
Small interfering RNAs (siRNAs) and RNA interference
The control nontargeting and YTHDF2-specific siRNAs were designed and synthesized by Bioneer Corporation. The sequences of siRNAs are listed in table S1. The cells (5 × 105 cells/ml) were transfected with siRNA (final concentration of 20 μM) using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions. The cells were used for further experiments at 48 to 72 hours after transfection.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using the TRIzol reagent (Invitrogen). The complementary DNA (cDNA) was synthesized using the StarScript II First-strand cDNA Synthesis Mix (GenStar Biosolutions, China). The qRT-PCR analysis was performed on a QuantStudio 6 Flex System (Applied Biosystems) with the SYBR Green qPCR SuperMix (GenStar Biosolutions). The relative expression levels were determined by the comparative threshold cycle (Ct) quantification method. β-Actin was used as an internal control. The primers for qRT-PCR are shown in table S1.
RNA-seq and data analysis
The whole-cell total RNA was isolated using TRIzol reagent (Invitrogen) and quantified using NanoDrop 2000. The cDNA library was constructed using Annoroad Gene Technology. The sequencing was performed on an Illumina HiSeq 2500 platform. The high-quality reads were mapped to the human reference genome (hg19) using HISAT2. DESeq, an R package, was applied for differential gene expression analysis. We filtered the differentially expressed genes based on the false discovery rate (FDR) of <0.05.
Enzyme-linked immunosorbent assay
The culture supernatants of the cells treated with the stimuli were collected at the indicated time points after stimulation. The protein expression levels of human TNF-α and IL-6 were assessed using the BD OptEIA ELISA Kits (BD Biosciences) following the manufacturer’s instructions.
ChIP-seq and ChIP-qPCR
The cells cultured with or without stimuli were cross-linked with 1% (v/v) formaldehyde for 10 min at room temperature and processed according to the ChIP protocol described in the SimpleChIP Enzymatic Chromatin IP Kit (catalog number 91820, Cell Signaling Technology). ChIP-seq libraries were sequenced on a HiSeq 2000 platform at Annoroad Gene Technology. The primers used for ChIP-qPCR are listed in table S1.
RIP-seq and RIP-qPCR
The RIP assay was performed following a previously described procedure with minor modifications. Briefly, the cells were seeded in a 150-mm dish at a density of 1 × 106 cells/ml. The cells were harvested in RIP buffer [150 mM KCl, 25 mM tris (pH 7.4), 5 mM EDTA, 0.5 mM dithiothreitol, 0.2% CA630, ribonuclease inhibitor (100 U/ml), and 1X protease inhibitors]. The cell lysates were incubated with 4 μg of anti-YTHDF2 antibody or a control IgG at 4°C overnight. Next, the cell lysates were incubated with 50 μl of protein A/G-agarose beads at 4°C for 2 hours. The beads were washed thrice and resuspended in 150 μl of RIP buffer with 0.1% SDS and 30 μg of proteinase K and incubated at 55°C for 30 min. The immunoprecipitated and input RNAs were isolated using the TRIzol reagent and subjected to RIP-qPCR or RNA-seq (RIP-seq) analysis.
RNA methylation quantification by ELISA
Total RNA was isolated using TRIzol reagent, and mRNA was purified using a PolyATtract mRNA Isolation Systems Kit (Promega, Z5310). Then, the m6A levels were quantified in a 100-ng aliquot of mRNA with an ELISA-based EpiQuik m6A RNA Methylation Quantification Kit (EpiGentek, #P-9008) according to the manufacturer’s instructions.
m6A-seq assay
Total RNA was isolated using TRIzol reagent. m6A immunoprecipitation and library preparation were performed following the published protocol. Briefly, mRNA was purified using the Dynabeads mRNA Purification Kit (Thermo Fisher). The purified mRNA was fragmented using an RNA fragmentation reagent (Invitrogen). The anti-m6A antibody (Synaptic Systems) was used for immunoprecipitation of m6A-modified mRNA. The mRNAs were washed and eluted with m6A sodium salt (Sigma-Aldrich, M2780). The input and immunoprecipitation samples were both used to construct the libraries. The libraries were sequenced on an Illumina HiSeq 2500 platform.
RNA decay assays
The cells were treated with actinomycin D (5 μg/ml; Sigma-Aldrich, catalog number A9415) or dimethyl sulfoxide (DMSO) for 2 or 6 hours, respectively. The total RNA was isolated at indicated time points for reverse transcription and qRT-PCR analysis. The mRNA decay rate was calculated by normalizing the gene expression in the actinomycin D–treated cells to that in the DMSO-treated cells.
Dual-luciferase reporter assay
The fragment of exon 11 from the human KDM6B CDS containing the predicted m6A modification sites was amplified via PCR and cloned into the psiCheck2 plasmid (Promega) to form the dual-luciferase reporter vector KDM6B-WT (WT KDM6B). To mutate the putative m6A sites of KDM6B, site-directed mutations were introduced to generate the KDM6B-mutated type (MUT KDM6B). The HEK293T cells were seeded in a 96-well plate (4 × 104 per well) and transfected with the reporter vectors. At 24 hours after transfection, the cells were lysed with a lysis buffer (Promega), and luciferase activity was measured using the Dual-Luciferase Reporter Assay Kit (Promega) on a Synergy 2 microplate reader (BioTek). The results were calculated by normalization of firefly luciferase activity to Renilla luciferase activity.
Sequencing data analysis
To process the ChIP-seq data, we used Trim galore (v.0.5.0) and fastqc (v.011.2) for trimming the adaptors and filtering the raw reads. The high-quality reads were mapped to the UCSC (University of California Santa Cruz) hg19 reference genome using bowtie2 (version 2.3.4.3). Next, MACS2 (version 2.1.1) was used for peak calling. The genome coverage files for visualization were generated using deepTools. Peak browsing and representative snapshots capturing were performed using the Integrative Genomics Viewer (IGV; IGV2.4.10, Broad Institute).
The RIP-seq reads were mapped to the human genome (hg19) using HISAT2 with default settings. Differential gene expression was calculated using an R package, DEGSeq. The RIP targets were defined as genes with reads per kilobase per million reads (RPKM) of ≥1 and immunoprecipitation/input log2(fold change) of ≥1.5. For m6A-seq data, filtered reads were aligned to the human (hg19) genome using HISAT2, and all nonunique mapped reads or PCR duplicates were removed. Next, exomePeak, an exome-based peak-calling R package, was used to detect a significantly enriched m6A modification site (FDR, <0.05; fold enrichment, ≥1.5) with default parameters. The changes in the expression level of m6A-modified gene in the control and treatment groups were analyzed using exomePeak. The consensus motif was determined using HOMER. The GO and KEGG analyses were performed on the gene lists obtained from the methods described above using an R/Bioconductor package clusterProfiler (version 3.10.0).
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Key Research and Development Program of China (2017YFA0103800, 2019YFA0802202), the National Natural Science Foundation of China (81901601, 81972651, 31771630, 81702784, and 31970700), the Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S029), the Natural Science Foundation of Guangdong Province (2019A1515011111 and 2017A030312009), and Guangdong Basic and Applied Basic Researh Foundation (2018A030313370). Author contributions: C.W, W.C., and J.H. performed the experiments and analyzed the results. S.J., Y.L., Y.Y., Z.G., and JY.Y. provided technical help. J.C., W.Z, JH.Y., and C.W. initiated and designed the project and directed the research. J.C., W.Z, JH.Y., and C.W. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba0647/DC1
REFERENCES AND NOTES
- 1.Medzhitov R., Origin and physiological roles of inflammation. Nature 454, 428–435 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Kotas M. E., Medzhitov R., Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O., Akira S., Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Barrat F. J., Elkon K. B., Fitzgerald K. A., Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 67, 323–336 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kawai T., Akira S., The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Sugimoto M. A., Vago J. P., Perretti M., Teixeira M. M., Mediators of the resolution of the inflammatory response. Trends Immunol. 40, 212–227 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Jiang X., Chen Z. J., The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 12, 35–48 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harhaj E. W., Dixit V. M., Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 21, 22–39 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karin M., Ben-Neriah Y., Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 18, 621–663 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Cui J., Zhu L., Xia X., Wang H. Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z. J., Wang R. F., NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141, 483–496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C., Su Z., Lin M., Ou J., Zhao W., Cui J., Wang R.-F., NLRP11 attenuates Toll-like receptor signalling by targeting TRAF6 for degradation via the ubiquitin ligase RNF19A. Nat. Commun. 8, 1977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q., Cao X., Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 19, 417–432 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Daskalaki M. G., Tsatsanis C., Kampranis S. C., Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J. Cell. Physiol. 233, 6495–6507 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Burchfield J. S., Li Q., Wang H. Y., Wang R.-F., JMJD3 as an epigenetic regulator in development and disease. Int. J. Biochem. Cell Biol. 67, 148–157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosselut R., Pleiotropic functions of H3K27Me3 demethylases in immune cell differentiation. Trends Immunol. 37, 102–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Santa F., Totaro M. G., Prosperini E., Notarbartolo S., Testa G., Natoli G., The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Satoh T., Takeuchi O., Vandenbon A., Yasuda K., Tanaka Y., Kumagai Y., Miyake T., Matsushita K., Okazaki T., Saitoh T., Honma K., Matsuyama T., Yui K., Tsujimura T., Standley D. M., Nakanishi K., Nakai K., Akira S., The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Kruidenier L., Chung C. W., Cheng Z., Liddle J., Che K., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H., Eberhard D., Hutchinson S., Jones E., Katso R., Leveridge M., Mander P. K., Mosley J., Ramirez-Molina C., Rowland P., Schofield C. J., Sheppard R. J., Smith J. E., Swales C., Tanner R., Thomas P., Tumber A., Drewes G., Oppermann U., Patel D. J., Lee K., Wilson D. M., A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao W., Li Q., Ayers S., Gu Y., Shi Z., Zhu Q., Chen Y., Wang H. Y., Wang R.-F., Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell 152, 1037–1050 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu Y., Dominissini D., Rechavi G., He C., Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Meyer K. D., Jaffrey S. R., The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15, 313–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao B. S., Roundtree I. A., He C., Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer K. D., Jaffrey S. R., Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 33, 319–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H., Wei J., He C., Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H., Weng H., Chen J., The biogenesis and precise control of RNA m6A methylation. Trends Genet. 36, 44–52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roundtree I. A., Evans M. E., Pan T., He C., Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.-G., He C., N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C.-M., Li C. J., Vagbo C. B., Shi Y., Wang W.-L., Song S.-H., Lu Z., Bosmans R. P., Dai Q., Hao Y.-J., Yang X., Zhao W.-M., Tong W.-M., Wang X.-J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H. E., Klungland A., Yang Y.-G., He C., ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patil D. P., Pickering B. F., Jaffrey S. R., Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol. 28, 113–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., Hu X., Huang M., Liu J., Gu Y., Ma L., Zhou Q., Cao X., Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 10, 1898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler R., Gillis E., Lasman L., Safra M., Geula S., Soyris C., Nachshon A., Tai-Schmiedel J., Friedman N., Le-Trilling V. T. K., Trilling M., Mandelboim M., Hanna J. H., Schwartz S., Stern-Ginossar N., m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Liu Y., You Y., Lu Z., Yang J., Li P., Liu L., Xu H., Niu Y., Cao X., N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Huang H., Weng H., Zhou K., Wu T., Zhao B. S., Sun M., Chen Z., Deng X., Xiao G., Auer F., Klemm L., Wu H., Zuo Z., Qin X., Dong Y., Zhou Y., Qin H., Tao S., Du J., Liu J., Lu Z., Yin H., Mesquita A., Yuan C. L., Hu Y. C., Sun W., Su R., Dong L., Shen C., Li C., Qing Y., Jiang X., Wu X., Sun M., Guan J. L., Qu L., Wei M., Muschen M., Huang G., He C., Yang J., Chen J., Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567, 414–419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Browning D. F., Busby S. J. W., Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 14, 638–650 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Marazzi I., Greenbaum B. D., Low D. H. P., Guccione E., Chromatin dependencies in cancer and inflammation. Nat. Rev. Mol. Cell Biol. 19, 245–261 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Hamon M. A., Cossart P., Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 4, 100–109 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K., High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B., H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang H., Howard M., Dean C., Antagonistic roles for H3K36me3 and H3K27me3 in the cold-induced epigenetic switch at Arabidopsis FLC. Curr. Biol. 24, 1793–1797 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S., Ma J., Wu F., Xiong L. J., Ma H., Xu W., Lv R., Li X., Villen J., Gygi S. P., Liu X. S., Shi Y., The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 26, 1364–1375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C., N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du H., Zhao Y., He J., Zhang Y., Xi H., Liu M., Ma J., Wu L., YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park O. H., Ha H., Lee Y., Boo S. H., Kwon D. H., Song H. K., Kim Y. K., Endoribonucleolytic cleavage of m6A-containing RNAs by RNase P/MRP complex. Mol. Cell 74, 494–507.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Lee Y., Choe J., Park O. H., Kim Y. K., Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 36, 177–188 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Zhou J., Wan J., Gao X., Zhang X., Jaffrey S. R., Qian S.-B., Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z., Qian P., Shao W., Shi H., He X. C., Gogol M., Yu Z., Wang Y., Qi M., Zhu Y., Perry J. M., Zhang K., Tao F., Zhou K., Hu D., Han Y., Zhao C., Alexander R., Xu H., Chen S., Peak A., Hall K., Peterson M., Perera A., Haug J. S., Parmely T., Li H., Shen B., Zeitlinger J., He C., Li L., Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 28, 904–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K., Na W., Lee J. Y., Na J., Cho H., Wu H., Yune T. Y., Kim W. S., Ju B.-G., Molecular mechanism of Jmjd3-mediated interleukin-6 gene regulation in endothelial cells underlying spinal cord injury. J. Neurochem. 122, 272–282 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba0647/DC1