

Inherited blindness can be treated with a safe gene therapy approach using activation of functionally equivalent genes.
Abstract
Catalytically inactive dCas9 fused to transcriptional activators (dCas9-VPR) enables activation of silent genes. Many disease genes have counterparts, which serve similar functions but are expressed in distinct cell types. One attractive option to compensate for the missing function of a defective gene could be to transcriptionally activate its functionally equivalent counterpart via dCas9-VPR. Key challenges of this approach include the delivery of dCas9-VPR, activation efficiency, long-term expression of the target gene, and adverse effects in vivo. Using dual adeno-associated viral vectors expressing split dCas9-VPR, we show efficient transcriptional activation and long-term expression of cone photoreceptor-specific M-opsin (Opn1mw) in a rhodopsin-deficient mouse model for retinitis pigmentosa. One year after treatment, this approach yields improved retinal function and attenuated retinal degeneration with no apparent adverse effects. Our study demonstrates that dCas9-VPR–mediated transcriptional activation of functionally equivalent genes has great potential for the treatment of genetic disorders.
INTRODUCTION
Various inherited disorders are caused by mutations in genes for which counterparts with similar function but distinct expression pattern exist. CRISPR-Cas–mediated transcriptional activation (transactivation) of such functionally equivalent genes is one attractive therapeutic strategy to compensate for the function of their mutant counterparts. Different transcriptional activators have been fused to catalytically inactive Cas9 (dCas9) proteins and evaluated regarding their transactivation efficiency. Among the tested candidates, the catalytically inactive dCas9 fused to transcriptional activators (dCas9-VPR) transactivating module shows high efficiency across different species and cell types (1). However, because of its size (5.8 kb), dCas9-VPR exceeds the genome packaging capacity of recombinant adeno-associated viral (rAAV) vectors, which are currently the gold standard for gene delivery to native tissues and for gene therapy. A previous study provided a proof of principle for reconstitution of split dCas9-VPR using dual rAAVs in vitro and in vivo in wild-type (WT) mice (2). Nonetheless, the therapeutic potential of this tool has not been evaluated in disease models so far. In particular, the long-term effects, such as efficiency and expression of the transactivated gene as well as the potential adverse effects, remained largely unexplored.
Inherited retinal dystrophies (IRDs) affect several million people worldwide. Retinitis pigmentosa (RP) is the most common IRD subtype and primarily affects rod photoreceptors (3). By contrast, achromatopsia (ACHM) is among the most frequent IRDs affecting the cones (4). Many genes associated with RP or ACHM encode members of the phototransduction cascade in rods or cones. The key phototransduction molecules in these cells are encoded by distinct yet functionally equivalent genes, and mutations in many of these genes, such as the visual pigments (opsins) or cyclic nucleotide-gated (CNG) ion channels, are associated with different types of blinding disorders. Mutations in the rhodopsin gene (RHO) are the leading cause for RP, whereas mutations in the cone CNG channel genes (CNGA3 and CNGB3) are the most frequent cause for ACHM. While rods only express rhodopsin, most mammals including mice express two opsin types in cones, the short wavelength–sensitive S-opsin (Opn1sw) and the medium wavelength–sensitive M-opsin (Opn1mw). CNG channels are heterotetrameric complexes composed of the channel function defining CNG A and the modulatory CNG B subunit. The native rod CNG channels contain CNGA1 and CNGB1 and their cone counterparts CNGA3 and CNGB3 subunits, respectively. A previous study has shown that rod and cone CNG A subunits can also form functional units with the CNG B subunits from the other photoreceptor type, i.e., CNGA1 with CNGB3 and CNGA3 with CNGB1 (5). Given the functional similarity between native and chimeric CNG channels, activation of the respective functionally equivalent gene in rods or cones appears an attractive treatment option. Recent work on mouse models has shown that rhodopsin and cone opsins are also functionally equivalent (6–9). This suggests that activation of genes encoding for cone opsins in rods could compensate for the defective rhodopsin in the respective animal models.
Here, using dCas9-VPR, we were able to efficiently transactivate the Rho homolog Opn1mw and the rod-specific Cnga3 homolog Cnga1 in vitro. Using a dual rAAV vector system for split dCas9-VPR–mediated Opn1mw activation in a rhodopsin-deficient mouse model for RP, we also demonstrate that this treatment results in safe and efficient long-term expression, gain in retinal function, and delay of retinal degeneration.
RESULTS
Cnga1 and Opn1mw transactivation in transiently transfected 661W and MEF cells
To test for the feasibility and efficiency of ectopic activation of nonexpressed or poorly expressed genes, we transfected different mouse cell lines with dCas9-VPR in combination with single guide RNAs (sgRNAs) binding to the promoter region of either murine Cnga1 or Opn1mw (Fig. 1A). As the transactivation efficiency can be increased with a growing number of sgRNAs (1), we used a combination of three sgRNAs for each of the genes. Transactivation of Cnga1 was addressed in 661W cells, immortalized derivatives of murine cones lacking Cnga1 expression (10). As 661W cells express Opn1mw endogenously, we used mouse embryonic fibroblast (MEF) cells for transactivation of this gene.
Fig. 1. Transactivation of Cnga1 and Opn1mw via dCas9-VPR.
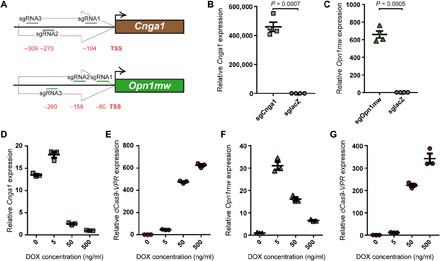
(A) Binding position of the three sgRNAs used for targeting dCas9-VPR to the promoter of the Cnga1 or Opn1mw gene, respectively. The relative distance of each sgRNA to the transcription start site (TSS) of the target gene is given in base pairs. (B) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) from 661W cells cotransfected with dCas9-VPR and either Cnga1 or lacZ sgRNAs. Cnga1 expression was normalized to the lacZ control. (C) qRT-PCR from MEF cells cotransfected with dCas9-VPR and either Opn1mw or lacZ sgRNAs. Opn1mw expression was normalized to the lacZ control. A two-tailed unpaired t test with Welch’s correction was used for statistical analysis in (B) and (C). (D to G) qRT-PCR from 661W-pb (D and E) or MEF-pb cells (F and G) cultured at different doxycycline (DOX) concentrations as indicated. Cnga1 expression was normalized to DOX (500 ng/ml), and Opn1mw and dCas9-VPR expression was normalized to DOX (0 ng/ml).
In 661W cells transiently cotransfected with the dCas9-VPR cassette and Cnga1-specific sgRNAs, we observed efficient transactivation of Cnga1 on the transcript level, which was absent in control cells expressing the lacZ sgRNA (Fig. 1B and fig. S1A). Nevertheless, no Cnga1 protein signal was detectable in cells labeled with a specific antibody under these conditions.
When addressing the Opn1mw transactivation in MEF cells, we detected Opn1mw transcript in both naïve and in lacZ sgRNA–expressing cells, indicating endogenous expression of Opn1mw in this cell line (fig. S1B). The efficiency of dCas9-mediated transactivation is known to correlate negatively with the basal expression level of a given gene (1). Nevertheless, despite the basal Opn1mw expression, a robust transactivation of Opn1mw was detectable in MEF cells coexpressing dCas9-VPR and Opn1mw-specific sgRNAs (Fig. 1C and fig. S1B). A combination of another set of three Opn1mw sgRNAs could not further improve the transactivation efficiency (fig. S1, C and D). These results show that, in transiently transfected cells, both Cnga1 and Opn1mw can be efficiently transactivated using dCas9-VPR.
Transactivation in cell lines stably expressing inducible dCas9-VPR sgRNA cassettes
Next, we analyzed whether Cnga1 protein can be detected in a transfection-independent system. For this, we created 661W and MEF cell lines with stable integration of expression cassettes for doxycycline (DOX)–inducible dCas9-VPR in combination with Cnga1, Opn1mw, or lacZ-specific sgRNAs [661W–piggyBac (pb) and MEF-pb, respectively]. When analyzing the Cnga1 transcript in the 661W cells, we detected a DOX concentration–dependent transactivation of this gene. However, robust Cnga1 transactivation was also present in the absence of DOX, suggesting a leaky activity of the DOX-dependent promoter driving dCas9-VPR expression. Moreover, an increase in Cnga1 expression was only obtained for the lowest DOX concentration (5 ng/ml), whereas Cnga1 levels were decreasing with further increase in drug concentration (Fig. 1D). Thus, when exceeding the optimal DOX concentration, there was an inverse correlation between dCas9-VPR transcript levels and the efficiency of Cnga1 transactivation (Fig. 1E). Very similar results were obtained in MEF cells stably expressing the dCas9-VPR cassette and Opn1mw sgRNAs, indicating that this effect was gene independent (Fig. 1, F and G). In contrast to Cnga1, there was no apparent Opn1mw transactivation in MEFs stably expressing dCas9-VPR and Opn1mw-specific sgRNAs in the absence of DOX, which is most likely due to the endogenous basal expression of this gene in this cell line.
In summary, these results suggest that an optimal window for transactivation exists in which sufficient levels of dCas9-VPR protein support maximal levels of gene expression. Upon exceeding the optimal dCas9-VPR levels, there is a gradual decrease in transactivation efficiency.
Analysis of protein expression and function in transactivated cells
Next, we assessed the Cnga1 and M-opsin protein expression in the corresponding stable cell lines treated with the optimal DOX concentration. Under these conditions, no evident increase in M-opsin protein expression was detectable when compared to the control cells (fig. S1E). For Cnga1, however, we could detect a robust signal that was absent in the lacZ sgRNA–expressing 661W cell line (Fig. 2A). To address whether functional channels can be formed from the proteins expressed from the transactivated Cnga1 locus, we conducted electrophysiological recordings in 661W cells cultured under optimal DOX concentration. In contrast to the lacZ control cell line lacking any CNG channel-like responses, several Cnga1 channel–specific characteristics including guanosine 3′,5′-cyclic monophosphate (cGMP) sensitivity, calcium, and magnesium blockage and outward rectification could be measured upon transactivation of this gene (Fig. 2, B to D). This indicates that successful transactivation by dCas9-VPR can lead to fully functional Cnga1 channels.
Fig. 2. dCas9-VPR–mediated transactivation results in functional Cnga1 channels.
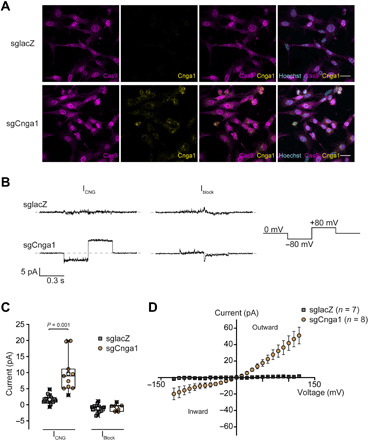
(A) Immunostainings of 661W-pb cells stably expressing dCas9-VPR and lacZ- (sglacZ, top row) or Cnga1-specific sgRNAs (sgCnga1, bottom row) in the presence of DOX (5 ng/ml) using Cas9- and Cnga1-specific antibodies. Scale bar, 30 μm. (B) Representative current traces recorded from inside out patches of DOX-induced 661W-pb cells in the presence of 300 μM cGMP (left, ICNG) or cGMP and Ca2+/Mg2+ (right, Iblock). (C) Quantification of the cGMP-induced currents in the absence (ICNG) or presence of Ca2+/Mg2+ (Iblock) (unpaired t test with Welch’s correction, two-tailed). (D) Current-voltage plot of cGMP-induced currents from sgCnga1 or sglacZ membrane patches.
Cnga1 and Opn1mw transactivation in cells expressing split intein dCas9-VPR
Owing to the limited genome packaging capacity of rAAVs, the entire dCas9-VPR cassette cannot be packaged into a single rAAV vector for in vivo delivery. To circumvent this limitation, two recent studies took advantage of the split intein technology to reconstitute (d)Cas9-VPR or its derivatives at the protein level upon codelivery of two separate dual rAAVs, each of which expressing one half of the split SpCas9 cassette (2, 11). The split intein-mediated reconstitution efficiency is known to depend on the position of the intein integration within the corresponding protein (12). The aforementioned studies addressed the transactivation of (d)Cas9-VP(R) split either after the amino acid position E573 (11) or V713 (fig. S1F) (2). Nevertheless, to our best knowledge, no quantitative or comparative data with respect to the reconstitution efficiency resulting from these two approaches are available so far. As such data would be very helpful to achieve optimal results in vivo, we first set out to compare the reconstitution efficiency for both approaches side-by-side at the protein level. To this end, we transiently cotransfected human embryonic kidney (HEK) 293 cells with plasmids encoding the SpCas9 halves intersected after the E573 or V713 position (fig. S1G) and quantified the resulting reconstitution efficiency. We found that the reconstitution efficiency of the SpCas9 variant split after V713 (56.9 ± 2.1%) was considerably higher than its counterpart split after the E573 position (33.3 ± 2.1%) (fig. S1H).
Next, we examined whether the coexpression of split dCas9-V713-VPR fragments also leads to efficient transactivation of Cnga1 and Opn1mw in the respective cells. While the transactivation efficiency for both target genes originating from the split dCas9-VPR was lower than from full-length dCas9-VPR, it still appeared robust and high enough in relation to the respective controls (fig. S1, I and J). Together, we show that dCas9-VPR split after the V713 position can transactivate both Cnga1 and Opn1mw in cell culture experiments and was therefore used for the subsequent in vivo experiments.
Split dCas9-VPR efficiently transactivates cone opsins in WT mice
We next focused on activation of Opn1mw in rod photoreceptors to provide an in vivo proof of concept for transactivation of functionally equivalent genes using split dCas9-VPR dual AAVs. For this purpose, we coinjected titer-matched dual rAAVs expressing split dCas9-VPR under control of a human rhodopsin promoter and Opn1mw-specific sgRNAs (Fig. 3A). Given the high percentage of rods, an activation of Opn1mw in this cell type is expected to superimpose the endogenous Opn1mw mRNA and protein signal originating from the more sparse cones, which make up only 3% of the total photoreceptor population in mice. For initial experiments, we used WT (+/+) animals subretinally injected on postnatal day 28 (P28) with split dCas9-VPR dual rAAVs. Four weeks after injection, a robust increase in Opn1mw transcript levels was observed in eyes coinjected with dual rAAVs compared to saline-injected eyes (Fig. 3B). This up-regulation of Opn1mw transcript was also confirmed by RNA sequencing (RNA-seq) (fig. S2A). Moreover, we could detect a strong M-opsin signal using an M-opsin–specific antibody in rod photoreceptor outer segments (Fig. 3C). This rod photoreceptor–specific expression of M-opsin was absent in untreated retinas, which only show M-opsin signal in cone photoreceptors (Fig. 3D). Encouraged by this finding, we addressed whether S-opsin (Opn1sw), another Rho homolog and potential candidate for transactivation in rhodopsin-deficient mice, can also be transactivated in rods using the same approach. Akin to Opn1mw, in retinas expressing split dCas9-VPR dual rAAVs and Opn1sw-specific sgRNAs, we could also detect S-Opsin protein in rod outer segments (Fig. 3, E and F). Notably, there was an evident variability in protein expression of transactivated M-Opsin and S-Opsin from experiment to experiment (fig. S2, B to E).
Fig. 3. Split dCas9-VPR–mediated transactivation of Opn1mw and Opn1sw in WT mice.
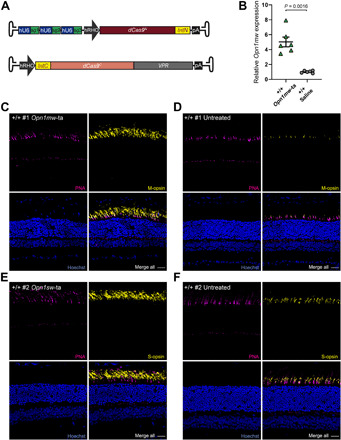
(A) Scheme of split dCas9-VPR and sgRNA encoding dual rAAV vectors used for transactivation of Opn1mw (B to D) and Opn1sw (E and F) in C57Bl/6J WT (+/+) mice. (B) qRT-PCR for Opn1mw expression upon transactivation 4 weeks after injection (Opn1mw-ta). The expression was normalized to control eyes injected with NaCl solution (saline, n = 6 eyes, paired t test, two-tailed). (C to F) Immunolabeling of two +/+ mice injected with Opn1mw- (C) or Opn1sw-transactivating dual AAVs (E). The contralateral eyes (D and F) of both mice served as control (untreated) (D and F). Peanut agglutinin (PNA; magenta) was used as marker for cones. Scale bars, 30 μm.
Opn1mw transactivation delays retinal degeneration and improves retinal function in Rho+/− mice
Next, we also tested whether Opn1mw transactivation is sufficient to ameliorate the RP phenotype in the heterozygous rhodopsin-deficient (Rho+/−) RP mouse model (13). In contrast to Rho−/− mice, which completely lack rod outer segments from birth, heterozygous mice are capable of developing shortened but functional outer segments (13, 14), which is expected to be an important prerequisite for treatments requiring injections at later time points. Rho+/− mice were subretinally injected on P14 with titer-matched dual rAAV vectors expressing the split dCas9-VPR and Opn1mw sgRNAs. The contralateral control eye was injected with a NaCl (saline) solution. As Rho+/− mice show a slow course of retinal degeneration (13), we assessed the effects of our treatment 1 year after injection. Retinal degeneration is accompanied by a reduction of photoreceptors, a condition that can be addressed noninvasively by optical coherence tomography (OCT) measuring the thickness of the outer nuclear layer (ONL). OCT recordings from eyes expressing split dCas9-VPR and Opn1mw sgRNAs revealed an increase in the ONL thickness compared to the contralateral saline-injected eye, suggesting that our treatment is capable of delaying the degeneration (Fig. 4A and fig. S2F). Similar results were obtained when comparing the ONL thickness between treated and saline-injected eyes using an independent method based on histological analysis of retinal cryosections (fig. S2G).
Fig. 4. Opn1mw activation improves retinal phenotype in Rho+/− mice.
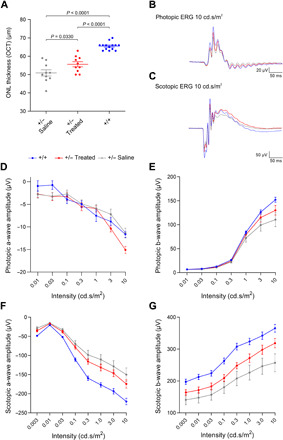
(A) OCT measurements from Rho+/− mice injected with a NaCl solution (+/− saline, n = 10 eyes) or dual rAAVs expressing split dCas9-VPR and Opn1mw-specific sgRNAs (+/− treated, n = 10 eyes). Age-matched C57Bl/6J WT mice (+/+, n = 14 eyes) were used as controls. Statistical analysis was performed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test. (B and C) Averaged photopic and scotopic traces of the same Rho+/− and WT mice at 10 cd.s/m2. (D and E) Quantification of light-adapted photopic a- and b-wave amplitudes of the same groups. (F and G) Dark-adapted scotopic a- and b-wave amplitudes of the same groups plotted against different light intensities. All P values for each comparison (+/− treated versus +/− saline, +/− treated versus +/+, and +/− saline versus +/+) are shown in fig. S5E.
To assess beneficial effects of our approach on rod-mediated (scotopic) and cone-mediated (photopic) retinal function, electroretinography (ERG) measurements were performed in dark- and light-adapted Rho+/− mice, respectively (Fig. 4, B to G, and figs. S3 and S4). The associated statistical analysis has been conducted in two different ways: (i) We performed a multiple comparison test to compare the ERG amplitudes at the different light intensities of all three groups (WT and treated or saline-injected Rho+/− mice; Fig. 4), and (ii) we made a paired comparison of ERG amplitudes between treated and saline-injected eyes only (fig. S5).
In light-adapted animals, an increase in the a-wave amplitude of dual rAAV–injected eyes at the highest light flash intensity was achieved compared to saline-injected eyes and untreated WT animals (Fig. 4D). Moreover, when performing a pairwise comparison of the treated eyes to the corresponding saline-injected counterparts only, an increase in photopic a-wave amplitudes could be observed for the two highest flash intensities (fig. S5A). This suggests that under these conditions, M-opsin expressing rods might respond to cone-activating intensities. A slight tendential improvement was also detectable in the photopic b-wave amplitudes in treated eyes at the highest flash intensity (Fig. 4E and fig. S5B). When addressing the rod-mediated function in dark-adapted animals, we could measure a slight tendency for improvement of the scotopic a-wave toward WT-like responses (Fig. 4F and fig. S5C). In comparison, a robust trend toward an improvement of the scotopic b-wave was observed when comparing the treated eyes to their saline-injected counterparts (Fig. 4G and fig. S5D). This trend was more pronounced with increasing light intensities, further supporting the assumption that treated rods expressing M-opsin are capable of responding to cone-activating stimuli. The individual P values for all ERG measurements shown in Fig. 4 are summarized in fig. S5E. Conclusively, these data suggest that Opn1mw transactivation can ameliorate retinal degeneration and results in partially improved retinal function in the Rho+/− RP mouse model.
Opn1mw transactivation reduces apoptosis without inducing gliosis or invasion of immune responsive cells in Rho+/− mice
In another set of experiments, we also analyzed the expression of M-opsin protein and markers for potential gliosis, apoptosis, or immune response in the retinas of the mice used for OCT and ERG measurements. Analogous to the results obtained in WT mice (cf. Fig. 3C and fig. S2), we found a considerable expression of transactivated M-opsin in the rod outer segments of injected animals, which, however, varied between animals (Fig. 5, A and B, and figs. S6 and S7). Furthermore, to assess the translational potential of this approach, we examined whether our treatment induced persistent gliosis or immune responses, which would be accompanied by proliferation of glial fibrillary acidic protein (GFAP)–positive Müller glia or ionized calcium binding adaptor molecule 1 (Iba-1)–positive microglial or mononuclear cells in the retina. Immune labeling of the retinas with these markers revealed no obvious increase in the number of glial, microglial, or mononuclear cells between the different groups in contrast to retinas of rd1 (retinal degeneration 1) mice exhibiting a fast retinal degeneration peaking on P13 (Fig. 5, C to H, and fig. S8) (15). To investigate whether photoreceptor degeneration is caused by apoptosis in the Rho+/− mouse model, we conducted a terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay on retinal sections from the treated Rho+/− mice (Fig. 6, A and B). In this assay, we could detect a low but considerable number of TUNEL-positive cells, indicating that apoptosis is the underlying mechanism for the photoreceptor loss in this mouse model. Moreover, by comparing the number of TUNEL-positive cells per area in the transduced versus untransduced part of the treated retinas, we show that Opn1mw transactivation reduces apoptosis (Fig. 6C). These data further emphasize the beneficial effects of our treatment on photoreceptor survival.
Fig. 5. Transactivation of Opn1mw in Rho+/− mice does not evoke any microglial activation or reactive gliosis.
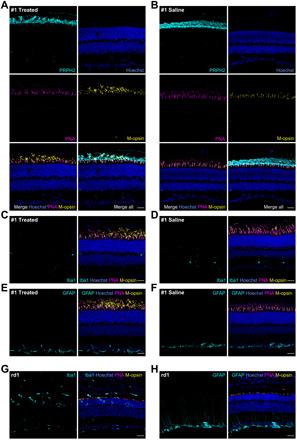
(A and B) Representative immunostainings of retinas from Rho+/− mouse #1 injected with either split dCas9-VPR and Opn1mw-specific sgRNAs (A) (treated) or NaCl (B) (saline, contralateral eye). Peripherin-2 antibody (PRPH2, cyan) was used as rod and cone outer segment marker and PNA (magenta) as marker for cones. (C to F) Immunolabeling of the same retinas with Iba1 or GFAP (cyan) to visualize microglial cells or reactive gliosis in the treated (C and E) and saline-injected contralateral eye (D and F). (G and H) Immunolabeling of retinas from Pde6b-deficient (rd1) mice on P13 with Iba1 (G, cyan) or GFAP (H, cyan) served as a positive control. Scale bars, 30 μm.
Fig. 6. Transactivation of Opn1mw in Rho+/− mice reduces apoptosis.
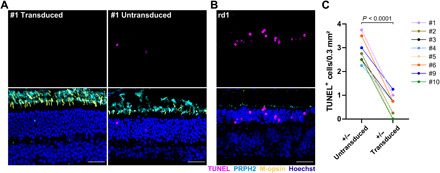
(A) Representative sections of the immunolabeled retina from Rho+/− mouse #1 injected with split dCas9-VPR and Opn1mw-specific sgRNAs showing a transduced (left) or untransduced (right) area of the same retina 1 year after injection. (B) Immunolabeling of the rd1 mouse retina on P13 served as a positive control. TUNEL staining (magenta, top) was used to visualize apoptosis, PRPH2 (cyan) was used as rod and cone outer segment marker (bottom). Scale bar, 30 μm. (C) Quantification of TUNEL+ cells in transduced versus untransduced areas of retinas from eight Rho+/− mice injected with split dCas9-VPR and Opn1mw-specific sgRNAs. A paired t test (two-tailed) was used for statistical analysis.
Characterization of untreated Rho+/− mice
The interpretation of the results obtained so far was largely based on the comparison of the treated Rho+/− mice to the saline-injected contralateral eye. To address whether the injection of saline might induce some effects on its own, we characterized the retinal degeneration and function, reactive gliosis, recruitment of immune reactive cells, and apoptosis in age-matched (1-year-old) untreated Rho+/− mice and compared these parameters side by side to those obtained from saline-injected control eyes. Except for a very slight decrease in the photopic b-wave amplitude at one single light flash intensity (0.1 cd.s/m2) upon saline injection, there were no noticeable differences in any of these parameters between the two groups (fig. S9). The injection of the saline solution thus most likely does not affect the interpretation of our results and the treatment success.
DISCUSSION
In this study, we provide the first evidence that ectopic transcriptional activation of functionally equivalent genes using split dCas9-VPR can ameliorate a disease phenotype, further expanding the spectrum of possibilities for treatment of genetic disorders. Given that many IRD-linked genes expressed in rods or cones are encoded by functionally equivalent genes, the transactivation approach provides an attractive option for therapy of these diseases for several reasons: (i) Transactivation of functionally equivalent genes can be used for the treatment of large and frequent IRD genes, such as ABCA4 or MYO7A, which cannot be efficiently reconstituted using classical dual AAV vectors. ABCA4 and MYO7A belong each to a large protein family with many functionally equivalent partners expressed across different cell types. Some of these partners, e.g., ABCA1 or MYO7B, show a very high structural conservation to ABCA4 and MYO7A, respectively, with at least 50% amino acid identity and 65% amino acid similarity and therefore represent highly attractive candidates for transactivation. (ii) A transactivation approach can also be used for therapy of genes with autosomal dominant inheritance, for which gene delivery has to be combined with simultaneous knockout of the diseased allele. Recent studies demonstrated that sgRNAs with shortened spacer lengths (<16 bases) repress the catalytic activity of Cas9-VPR (16, 17). A strategy combining catalytically active Cas9-VPR with sgRNAs of shortened spacer lengths for transactivation and with standard sgRNAs (usually 20 bases spacer length) for simultaneous down-regulation of the diseased gene can, e.g., be used for the treatment of the most common rhodopsin P23H gain-of-function mutation but should also be applicable to other mutations in RHO or to other genes with autosomal-dominant inheritance. (iii) Owing to the possibility of multiplexing in CRISPR-Cas approaches, transactivation could potentially also be used for the treatment of more complex (retinal) diseases, such as those caused by mutations in two or multiple genes (18–20). (iv) The (d)Cas9-VPR–mediated transactivation approach is mutation independent and thus allows for the treatment of a large number of patients.
Using alternative activation strategies, two recent studies demonstrated that CRISPR-Cas9–based transactivation of structurally and functionally related genes can ameliorate the phenotypes of two different mouse models for muscular dystrophy (21, 22). Additional work is necessary to compare the transactivation efficiency, long-term effects, and safety between these approaches and dCas9-VPR side by side to determine the best treatment strategy for the individual disorders. It also remains to be investigated whether other split position within dCas9-VPR (23) or other technologies for reconstitution of large coding sequences might further improve the transactivation efficiency and/or the therapeutic outcome of dCas9-VPR–based approaches. Unlike its catalytically active counterpart, dCas9 and its fusion variants do not cause single or double strand breaks at the genomic level. Studies addressing the off-targets of dCas9-VPR at the transcript level revealed that such off-target rates are very low or nondetectable (1, 2), further emphasizing the therapeutic potential of this approach. We demonstrate herein that the split dCas9-VPR technology can induce long-term expression and morphological as well as functional phenotypic changes in an animal model with no apparent adverse effects, such as gliosis or invasion of immune cells. These promising results might therefore help pushing this approach toward first clinical trials. Notably, as in previous retinal gene therapy studies in mice, we could not fully rescue the WT-like retinal function or morphology. One possible reason is that a single subretinal injection with conventional rAAV capsids usually covers only up to one third of the retina, and hence, amplitudes of up to 30% of the WT response could be expected. We suggest that functional or structural improvements can be further increased by developing more potent rAAV vectors, which show pan-retinal expression upon subretinal injection (e.g., due to lateral spreading) or which allow for other more convenient administration routes (e.g., intravitreal injection). In addition, we use a dual rAAV vector approach that requires both vectors to be expressed and reconstituted in the same cell. Accordingly, one would benefit from more efficient dual rAAV vector strategies or techniques to increase the reconstitution efficiency of the split dCas9-VPR.
Last, we also provide the first in vitro evidence that transactivation efficiency is inversely correlated with dCas9-VPR expression in a transfection-independent system. It remains to be clarified whether this unexpected dose dependency of dCas9-VPR also occurs in vivo and whether it reflects an inherent property of other catalytically active and inactive Cas9 or other Cas variants. A precise understanding of this correlation could help to further improve and fine-tune the efficiency of these enzymes and might thus also increase the therapeutic outcome of CRISPR-Cas–based approaches.
METHODS
Animals
All animal procedures were performed with the permission of local authorities (District Government of Upper Bavaria) and in accordance with the German laws on animal welfare (Tierschutzgesetz). Animals were anesthetized via an intraperitoneal injection of ketamine (40 mg/kg body weight) and xylazine (20 mg/kg body weight). Euthanasia was performed by cervical dislocation. For all experiments, C57Bl/6J or Rho+/− mice (13) backcrossed to a C57Bl/6J background for at least eight generations were used.
sgRNA design
The Eukaryotic Promoter Database (https://epd.epfl.ch//index.php) was consulted for the identification of promoter regions (24). sgRNA sequences in the genomic target region were chosen using the CRISPOR website with 20bp-NGG PAM settings for SpCas9 (25) and elimination of sgRNAs with a specificity score (26) lower than 50. All sgRNA sequences used in this study are shown in table S1.
Construction and cloning of expression plasmids
The Cas9m4-VP64, SP-dCas9-VPR, pSMVP-Cas9N, pSMVP-Cas9C, and pAAV-CMV-Cas9C-VPR plasmids were obtained from Addgene (#47319, #63798, #80934, #80939, and #80933, respectively). sgRNAs expressed via a U6 promoter were added using standard cloning techniques. Cas9N and Cas9C were rendered catalytically inactive by introducing the D10A and the H840A point mutations via a standard site-directed mutagenesis protocol using the KAPA HiFi HotStart ReadyMix PCR kit (Kapa Biosystems). For the generation of stable cell lines, dCas9-VPR was subcloned into a pb expression vector containing the Tet-On system for DOX-inducible expression of dCas9-VPR. For expression in mouse photoreceptors, the split dCas9-VPR driven by a human rhodopsin promoter and corresponding sgRNAs each driven by a human U6 promoter were subcloned into the pAAV2.1 vector (27). All transgenes were sequenced before use (Eurofins Genomics).
Cell culture and transfection
The murine 661W cell line derived from retinal tumors was provided by M. Al-Ubaidi, University of Houston (28). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) GlutaMAX medium (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Biochrom) and 1% Anti-Anti (ThermoFisher Scientific) at 37°C and 10% CO2. Immortalized MEFs were generated as previously described (29, 30). MEF cells were cultured in DMEM GlutaMAX medium supplemented with 10% FBS (Biochrom) and 1% penicillin/streptomycin (Biochrom) at 37°C and 5% CO2. Transient transfections of 661W and MEF cells were performed using the Xfect Transfection Reagent (Takara Bio) according to the manufacturer’s instructions. HEK293 cells were transfected using the standard calcium phosphate technique.
Generation of stable cell lines using pb technology
The stable cell lines were generated using the pb transposon system. Briefly, 661W or MEF cells were cotransfected with the respective dCas9-VPR and sgRNA containing pb vector and a pb transposase expression vector using the Xfect Transfection Reagent (Takara Bio) according to the manufacturer’s instructions. Fourty-eight to 72 hours after transfection, cells were selected for successful integration by addition of puromycin dihydrochloride (Gibco, Thermo Fisher Scientific) for approximately 1 week at 4.5 and 1 μg/ml concentration for 661W and MEF cells, respectively. To induce dCas9-VPR expression, DOX hyclate (Sigma-Aldrich) was added directly to the medium.
RNA isolation and (quantitative) reverse transcription polymerase chain reaction
Forty-eight hours after transfection, the cells were harvested and lysed using the mixer mill MM400 (Retsch). RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s instructions, and RNA concentration and purity were determined via the NanoDrop2000 (ThermoFisher Scientific). Complementary DNA (cDNA) was synthesized using the RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific) according to the manufacturer’s instructions for up to 1 μg of total RNA. For reverse transcription polymerase chain reaction (RT-PCR), the Herculase II polymerase (Agilent Technologies) was used following the manufacturer’s instructions. The quantitative RT-PCR (qRT-PCR) was performed on the StepOnePlus Real-Time PCR System (Applied Biosystems, ThermoFisher Scientific) using the SYBR Select Master Mix (Applied Biosystems, ThermoFisher Scientific) according to the manufacturer’s instructions. The relative expression levels of Cnga1 and Opn1mw were normalized to the housekeeping gene Alas and calculated using the 2-ΔΔC(T) method. All primers used in this study can be found in table S1.
RNA isolation and mRNA-seq library preparation and sequencing
Total RNA was isolated from retinas using the RNeasy Plus Micro Kit (QIAGEN) according to the manufacturer’s protocol. For mRNA library production, we used a scaled-up version of the SMARTseq2 protocol. Briefly, from ca. 7 to 90 ng of total RNA, mRNA was captured with a mix of 0.5 μl of 20 μM oligo dT primer and 0.5 μl of 20 mM dNTPs, followed by heating to 72°C for 3 min and immediately putting into ice-water bath. Then, in a 10-μl reaction, double-stranded cDNA was generated by adding 2 μl of 5× Superscript II first-strand buffer (ThermoFisher Scientific), 2 μl of 5 M Betaine, 0.6 μl of 100 mM MgCl2, 0.5 μl of 100 mM dithiothreitol (DTT), 0.4 μl of RNAsin (Promega), 0.5 μl of 20 μM template-switch oligo (20 μM), and 0.5 μl of SuperScript II reverse transcriptase (200 U/μl; ThermoFisher Scientific) and incubation for 90 min at 42°C, followed by 14 cycles (50°C for 2 min, 42°C for 2 min) and heat inactivation (70°C for 15 min). Pre-amplification was performed by addition of 12.5 μl of 2× KAPA HiFi HotStart Ready mix, 0.25 μl of 10 μM IS PCR primers, and 2.25 μl of nuclease-free water in a thermos protocol of 98°C for 3 min, 10 pre-amplification cycles (98°C for 20 s, 67°C for 15 s, 72°C for 6 min), followed by 5 min at 72°C and hold at 4°C. Purification was performed with AMPure XP beads (Beckman Coulter), and cDNA was quantified with Qubit (ThermoFisher Scientific) and checked for fragment length distribution on an Agilent Bioanalyzer chip. Next, 7 ng of cDNA was fragmented in a 20-μl reaction by incubation with 1 μl of Tn5 enzyme from Illumina Nextera library preparation kit and 10 μl of 2× tagmentation DNA buffer for 10 min at 55°C. Tagmented cDNA was purified with MinElute columns (QIAGEN) and PCR-amplified with NEBNext High-Fidelity 2× PCR Mastermix, 1 μl of each 10 μM Nextera index 1 and Nextera index 2 primer (Illumina) with a thermos protocol of 72°C for 5 min, 98°C for 30 s, 7 cycles (98°C for 10 s, 63°C for 30 s, and 72°C for 1 min), 72°C for 5 min, and hold at 4°C. The final library was purified with AMPure beads, quantified by Qubit, and sequenced for 100 base pairs using a V3 single read flow cell on a HiSeq 2500 (Illumina). The generated data were trimmed for quality and adapter reads with TrimGalore! and then mapped with STAR aligner. Duplicates were marked with the MarkDuplicates function from Picard tools. Reads were summarized with RSEM (RNA-seq by expectation maximization) software, and FPM (fragments per million mapped fragments) count matrix was generated with DESeq2.
Immunocytochemistry
For immunocytochemistry, 661W-pb or MEF-pb cells were seeded onto sterile 12-mm-diameter cover slips coated with poly-l-lysine hydrobromide (Sigma-Aldrich). After 48 hours of DOX application, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) and permeabilized for 30 min in 0.3% Triton X-100. Next, the coverslips were incubated with blocking solution (5% ChemiBLOCKER, Merck Millipore). To stain for Cnga1 and SpCas9 in 661W-pb cells, an anti-Cnga1 mouse monoclonal antibody (1:30; gift from R. Molday) (31) and the anti-SpCas9 rabbit polyclonal antibody (1:1000; C15310258, Diagenode) were used, respectively. To stain for M-opsin and SpCas9 in MEF-pb cells, the anti-opsin red/green (M-opsin) rabbit polyclonal antibody (1:300; AB5405, Merck) and an anti-SpCas9 mouse monoclonal antibody (1:500; SAB4200751, Sigma-Aldrich) were used, respectively. Hoechst 33342 solution (5 μg/ml; Invitrogen, ThermoFisher Scientific) was used as a nuclear staining. Images were obtained via the Leica TCS SP8 spectral confocal laser scanning microscope (Leica), acquired with the LASX software (Leica), and further processed with the ImageJ software (National Institutes of Health).
Patch-clamp measurements
Inside-out patches were excised from 661W stable cells that were maintained at a DOX concentration of 5 ng/ml. Currents were recorded using an EPC-10 double patch-clamp amplifier (HEKA Elektronik, Harvard Bioscience) and PatchMaster acquisition software (HEKA Elektronik, Harvard Bioscience). Data were digitized at 20 kHz and filtered at 2.9 kHz. All recordings were obtained at room temperature. The extracellular solution was composed of 140 mM NaCl, 5 mM KCl, 10 mM Hepes, and 1 mM EGTA (pH 7.4). The intracellular solution contained 140 mM KCl, 5 mM NaCl, 10 mM Hepes, and 1 mM EGTA (pH 7.4). The effect of cGMP was examined by perfusing the patch with extracellular solution supplemented with 300 μM cGMP. To investigate channel blocking, perfusion with a symmetric Ca2+/Mg2+ solution composed of 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM Hepes was performed, followed by perfusion with a cGMP-supplemented Ca2+/Mg2+ solution as a control. Currents were evoked from a holding potential of 0 mV by applying a 300-ms pulse of −80 mV followed by a 300-ms pulse of 80 mV every 3 s.
TX lysates and Western blot analysis
Transiently transfected HEK293 cells were harvested 48 hours after transfection and lysed in TX lysis buffer (0.5% Triton X-100, v/v) using the mixer mill MM400 (Retsch). The lysis buffer was supplemented with cOmplete ULTRA Protease Inhibitor Cocktail tablets (Roche). For Western blotting, 30 μg of the whole-cell protein was incubated in 1× Laemmli sample buffer supplemented with DTT at 72°C for 10 min. The proteins were separated on a 6 to 12% SDS-polyacrylamide gel via gel electrophoresis. For immunoblotting, the anti-SpCas9 antibody (1:1000; C15310258, Diagenode) was used. The relative band intensities were quantified using the ImageLab software (Bio-Rad).
rAAV production and subretinal injections
rAAV vectors containing the N- or C-terminal part of dCas9-VPR and the sgRNA expression cassette were produced using the 2/8YF capsid (32) as described previously (33, 34). C57Bl/6J mice were injected subretinally at P28 with a single injection (1 μl) of titer-matched rAAVs (total 1011 vg/μl), and the retinas were harvested for immunohistochemistry 4 weeks after injection. For 10 Rho+/− mice, one eye was injected via a single injection (1 μl) of titer-matched rAAVs (total 1011 vg/μl) on P14. The contralateral eye was control injected with 1 μl of NaCl (saline). Twelve months after injection, ERG and OCT were performed on both eyes of all Rho+/− mice, and all retinas were harvested for immunohistochemistry.
Electroretinography
Corneal electroretinograms were recorded from 10 Rho+/− and 10 age-matched C57Bl/6J WT mice using the Celeris full-field ERG system from Diagnosys (model D430). Scotopic and photopic electroretinograms were carried out for each animal. Mice were dark adapted overnight, and scotopic ERG measurements were conducted first under dim red light conditions followed by photopic tests. Pupils were dilated using 1% atropine- and 0.5% tropicamide-containing eye drops (Mydriaticum Stulln, Pharma Stulln GmbH). As contact fluid, hydroxylpropyl methylcellulose (Methocel 2%, OmniVision GmbH) was applied on both eyes before placing the light guide electrodes. During the whole protocol, animals were kept warm by the integrated platform heater of the Celeris system. ERG responses were obtained simultaneously from both eyes. For scotopic measurements, single flash recordings were performed at light intensities of 0.003 (blue light, 455 nm), 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 cd.s/m2 (all remaining intensities 6500 K white light). Background intensity was 0 cd/m2. For photopic measurements, mice were light adapted (3 cd.s/m2) for 5 min, and single flash recordings were obtained at light intensities of 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 cd.s/m2. The background intensity for all photopic recordings was 9 cd/m2. For both, scotopic and photopic assessments, measurements were recorded from 50 ms before stimulus onset to 300 ms poststimulus. Voltage signals were sampled at 1 Hz, and recorded responses were averages of 5 (scotopic) or 10 (photopic) sweeps depending on signal-to-noise ratios. The measurements were analyzed using the provided Espion V6 software from Diagnosys with the a-wave amplitude measured from stimulus onset to the trough of the a-wave and the b-wave amplitude ranging from the trough of the a-wave to the peak of the b-wave.
Optical coherence tomography
Retinal morphology of the Rho+/− mice was assessed with OCT using an adapted Spectralis HRA + OCT system (Heidelberg Engineering) in combination with contact lenses as described previously (35). Pupils were dilated using 1% atropine- and 0.5% tropicamide-containing eye drops (Mydriaticum Stulln, Pharma Stulln GmbH), and hydroxylpropyl methylcellulose (Methocel 2%, OmniVision GmbH) was applied to keep the eyes moist. OCT scans (20 frames per retina) were performed in high-resolution mode with the scanner set to 30° field of view.
Immunohistochemistry and confocal microscopy
Retinas of 13-month-old injected Rho+/− and age-matched WT mice were dissected and processed for immunohistochemistry as described previously (36). The mouse monoclonal anti–PRPH2 2B7 antibody (1:100; gift from M. Naash) (37) served as marker for rod and cone outer segments. The guinea pig anti-iba1 antibody (1:100; 234 004, Synaptic Systems) was used to visualize microglia. As marker for potential reactive gliosis served the mouse anti–GFAP-Cy3 antibody (1:1000; C9205, Sigma-Aldrich). Fluorescein isothiocyanate–conjugated lectin from Arachis hypogaea [peanut agglutinin (PNA)] (1:100; L7381, Sigma-Aldrich) was applied to stain cone photoreceptors. To detect cone-specific and activated M-opsin in rod photoreceptors, the rabbit anti-opsin red/green (M-opsin) antibody (1:300; AB5405, Merck) was used. The cell nuclei were visualized with Hoechst 33342 solution. To detect apoptosis, the TUNEL assay was performed using the In Situ Cell Death Detection Kit, Fluorescein (11684795910; Roche) according to the manufacturer’s instruction. The TUNEL assay was conducted for 8 of 10 injected Rho+/− mice except for mouse #7 and #8 (Fig. 6) as there were no cryosections left to stain from these two mice. Retinal images were obtained via the Leica TCS SP8 spectral confocal laser scanning microscope (Leica), acquired with the LASX software (Leica), and further processed with the ImageJ software (National Institutes of Health). Postmortem analysis of the ONL thickness in stained retinal sections was performed using the ImageJ software. Areas with equal distance to the optic nerve were chosen for analysis. At least three measurements were averaged per retina.
Statistics
All values are given as means ± SEM. The number of replicates (n) and the used statistical tests are indicated in each figure legend for each experiment.
Supplementary Material
Acknowledgments
We thank B. Noack, J. Koch, and K. Skokann for excellent technical support. We also thank M. Naash for the gift of the peripherin-2 antibody and R. Molday for the gift of the CNGA1 antibody. Moreover, we want to thank M. Al-Ubaidi for the gift of the 661W cells. Funding: This work was supported by the Deutsche Forschungsgemeinschaft, SPP2127 (to E.B., M.B., and S.M.). K.J.V.N. was funded by the BMBF grant (031L0101D) for de.NBI and is currently hired by AstraZeneca. This work was also supported, in part, by German Research Foundation Grants SFB 870 B05. Author contributions: E.B. designed the study and supervised the project with input from S.M. and M.B. S.B. conducted the in vivo experiments including AAV preparation, subretinal injections, OCT, immunohistochemistry, and ERG experiments with contributions from V.S., L.M.R., J.E.W., and K.S.H. V.S. designed and generated the stable cell lines, performed immunocytochemistry, and the molecular biology experiments including sgRNA design, qRT-PCR, and statistical analyses with contributions from S.B. and L.M.R. R.D.R. performed and analyzed the patch-clamp measurements with contributions from C.W.-S. and S.F. G.G. performed the RNA-seq experiment and K.J.V.N. analyzed the RNA-seq data. E.B., V.S., S.B., and L.M.R. wrote the manuscript with contributions from S.M. and M.B. E.B., S.M., M.B., C.W.-S., and J.W. acquired funding. E.B., S.M., M.B., S.B., V.S., and L.M.R. analyzed and discussed the data with input from all authors. Competing interests: E.B., M.B., and S.M. are authors on a patent application related to this work (no. EP19198830, filed 23 September 2019). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba5614/DC1
REFERENCES AND NOTES
- 1.Chavez A., Tuttle M., Pruitt B. W., Ewen-Campen B., Chari R., Ter-Ovanesyan D., Haque S. J., Cecchi R. J., Kowal E. J. K., Buchthal J., Housden B. E., Perrimon N., Collins J. J., Church G., Comparison of Cas9 activators in multiple species. Nat. Methods 13, 563–567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chew W. L., Tabebordbar M., Cheng J. K., Mali P., Wu E. Y., Ng A. H., Zhu K., Wagers A. J., Church G. M., A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 13, 868–874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daiger S. P., Sullivan L. S., Bowne S. J., Genes and mutations causing retinitis pigmentosa. Clin. Genet. 84, 132–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michalakis S., Schön C., Becirovic E., Biel M., Gene Therapy for Achromatopsia. J. Gene Med. 19, e2944 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Finn J. T., Krautwurst D., Schroeder J. E., Chen T. Y., Reed R. R., Yau K. W., Functional co-assembly among subunits of cyclic-nucleotide-activated, nonselective cation channels, and across species from nematode to human. Biophys. J. 74, 1333–1345 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Y., Kefalov V., Luo D.-G., Xue T., Yau K.-W., Quantal noise from human red cone pigment. Nat. Neurosci. 11, 565–571 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kefalov V. J., Rod and cone visual pigments and phototransduction through pharmacological, genetic, and physiological approaches. J. Biol. Chem. 287, 1635–1641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakurai K., Onishi A., Imai H., Chisaka O., Ueda Y., Usukura J., Nakatani K., Shichida Y., Physiological properties of rod photoreceptor cells in green-sensitive cone pigment knock-in mice. J. Gen. Physiol. 130, 21–40 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi G., Yau K.-W., Chen J., Kefalov V. J., Signaling properties of a short-wave cone visual pigment and its role in phototransduction. J. Neurosci. 27, 10084–10093 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitzgerald J. B., Malykhina A. P., Al-Ubaidi M. R., Ding X.-Q., Functional expression of cone cyclic nucleotide-gated channel in cone photoreceptor-derived 661W cells. Adv. Exp. Med. Biol. 613, 327–334 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Moreno A. M., Fu X., Zhu J., Katrekar D., Shih Y.-R. V., Marlett J., Cabotaje J., Tat J., Naughton J., Lisowski L., Varghese S., Zhang K., Mali P., In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Mol. Ther. 26, 1818–1827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y., Split-inteins and their bioapplications. Biotechnol. Lett. 37, 2121–2137 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Humphries M. M., Rancourt D., Farrar G. J., Kenna P., Hazel M., Bush R. A., Sieving P. A., Sheils D. M., McNally N., Creighton P., Erven A., Boros A., Gulya K., Capecchi M. R., Humphries P., Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat. Genet. 15, 216–219 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Lem J., Krasnoperova N. V., Calvert P. D., Kosaras B., Cameron D. A., Nicolò M., Makino C. L., Sidman R. L., Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 736–741 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sancho-Pelluz J., Arango-Gonzalez B., Kustermann S., Romero F. J., van Veen T., Zrenner E., Ekström P., Paquet-Durand F., Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol. Neurobiol. 38, 253–269 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Dahlman J. E., Abudayyeh O. O., Joung J., Gootenberg J. S., Zhang F., Konermann S., Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 33, 1159–1161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiani S., Chavez A., Tuttle M., Hall R. N., Chari R., Ter-Ovanesyan D., Qian J., Pruitt B. W., Beal J., Vora S., Buchthal J., Kowal E. J., Ebrahimkhani M. R., Collins J. J., Weiss R., Church G., Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 12, 1051–1054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajiwara K., Berson E. L., Dryja T. P., Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 264, 1604–1608 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Burkard M., Kohl S., Krätzig T., Tanimoto N., Brennenstuhl C., Bausch A. E., Junger K., Reuter P., Sothilingam V., Beck S. C., Huber G., Ding X.-Q., Mayer A. K., Baumann B., Weisschuh N., Zobor D., Hahn G.-A., Kellner U., Venturelli S., Becirovic E., Issa P. C., Koenekoop R. K., Rudolph G., Heckenlively J., Sieving P., Weleber R. G., Hamel C., Zong X., Biel M., Lukowski R., Seeliger M. W., Michalakis S., Wissinger B., Ruth P., Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel–associated retinopathy. J. Clin. Invest. 128, 5663–5675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R. G., Marinho F. P., Iwabe S., Santana E., McDaid K. S., Aguirre G. D., Miyadera K., Variabilities in retinal function and structure in a canine model of cone-rod dystrophy associated with RPGRIP1 support multigenic etiology. Sci. Rep. 7, 12823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kemaladewi D. U., Bassi P. S., Erwood S., Al-Basha D., Gawlik K. I., Lindsay K., Hyatt E., Kember R., Place K. M., Marks R. M., Durbeej M., Prescott S. A., Ivakine E. A., Cohn R. D., A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature 572, 125–130 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Liao H.-K., Hatanaka F., Araoka T., Reddy P., Wu M.-Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D. D., Núñez-Delicado E., Guillen P., Campistol J. M., Wu C.-J., Lu L.-F., Esteban C. R., Izpisua Belmonte J. C., In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171, 1495–1507.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmelas C., Grimm D., Split Cas9, not hairs – advancing the therapeutic index of CRISPR technology. Biotechnol. J. 13, e1700432 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Dreos R., Ambrosini G., Groux R., Cavin Périer R., Bucher P., The eukaryotic promoter database in its 30th year: Focus on non-vertebrate organisms. Nucleic Acids Res. 45, D51–D55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J.-S., Concordet J.-P., Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalakis S., Mühlfriedel R., Tanimoto N., Krishnamoorthy V., Koch S., Fischer M. D., Becirovic E., Bai L., Huber G., Beck S. C., Fahl E., Büning H., Paquet-Durand F., Zong X., Gollisch T., Biel M., Seeliger M. W., Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 18, 2057–2063 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.al-Ubaidi M. R., Font R. L., Quiambao A. B., Keener M. J., Liou G. I., Overbeek P. A., Baehr W., Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J. Cell Biol. 119, 1681–1687 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jat P. S., Cepko C. L., Mulligan R. C., Sharp P. A., Recombinant retroviruses encoding simian virus 40 large T antigen and polyomavirus large and middle T antigens. Mol. Cell. Biol. 6, 1204–1217 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J., Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr. Protoc. Mol. Biol. 70, Unit 28.1 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Cheng C. L., Molday R. S., Interaction of 4.1G and cGMP-gated channels in rod photoreceptor outer segments. J. Cell Sci. 126, 5725–5734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrs-Silva H., Dinculescu A., Li Q., Min S.-H., Chiodo V., Pang J.-J., Zhong L., Zolotukhin S., Srivastava A., Lewin A. S., Hauswirth W. W., High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol. Ther. 17, 463–471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch S., Sothilingam V., Garcia Garrido M., Tanimoto N., Becirovic E., Koch F., Seide C., Beck S. C., Seeliger M. W., Biel M., Mühlfriedel R., Michalakis S., Gene therapy restores vision and delays degeneration in the CNGB1−/− mouse model of retinitis pigmentosa. Hum. Mol. Genet. 21, 4486–4496 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Becirovic E., Böhm S., Nguyen O. N., Riedmayr L. M., Hammelmann V., Schön C., Butz E. S., Wahl-Schott C., Biel M., Michalakis S., AAV Vectors for FRET-based analysis of protein-protein interactions in photoreceptor outer segments. Front. Neurosci. 10, 356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schön C., Hoffmann N. A., Ochs S. M., Burgold S., Filser S., Steinbach S., Seeliger M. W., Arzberger T., Goedert M., Kretzschmar H. A., Schmidt B., Herms J., Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLOS ONE 7, e53547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michalakis S., Geiger H., Haverkamp S., Hofmann F., Gerstner A., Biel M., Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest. Ophthalmol. Vis. Sci. 46, 1516–1524 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Conley S. M., Stricker H. M., Naash M. I., Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry 49, 905–911 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba5614/DC1