

Computational redesign of chondroitinase ABC introduces 37 point mutations to improve enzyme stability and activity.
Abstract
Maintaining biocatalyst stability and activity is a critical challenge. Chondroitinase ABC (ChABC) has shown promise in central nervous system (CNS) regeneration, yet its therapeutic utility is severely limited by instability. We computationally reengineered ChABC by introducing 37, 55, and 92 amino acid changes using consensus design and forcefield-based optimization. All mutants were more stable than wild-type ChABC with increased aggregation temperatures between 4° and 8°C. Only ChABC with 37 mutations (ChABC-37) was more active and had a 6.5 times greater half-life than wild-type ChABC, increasing to 106 hours (4.4 days) from only 16.8 hours. ChABC-37, expressed as a fusion protein with Src homology 3 (ChABC-37-SH3), was active for 7 days when released from a hydrogel modified with SH3-binding peptides. This study demonstrates the broad opportunity to improve biocatalysts through computational engineering and sets the stage for future testing of this substantially improved protein in the treatment of debilitating CNS injuries.
INTRODUCTION
A proteoglycan-rich glial scar forms after traumatic injury to the central nervous system (CNS), resulting in a growth-inhibitory microenvironment (1). Despite the importance of this glial scar in tissue protection immediately following injury, chondroitin sulfate (CS) and dermatan sulfate (DS) proteoglycans inhibit long-term axonal regrowth. The enzyme chondroitinase ABC (ChABC) degrades both CS and DS proteoglycans through a unique dual endo-lyase versus exo-lyase catalytic mechanism (2), which has inspired numerous researchers to investigate ChABC as a therapeutic.
By degrading the proteoglycans in the glial scar following CNS injury, ChABC attenuates growth-inhibitory biochemical cues and enhances recovery by promoting plasticity, axonal sprouting, and neuroprotection in animal models of both spinal cord injury and stroke (3–5). There is evidence that delivery of ChABC—either on its own or in combination with other therapeutic agents—can stimulate the recovery of sensorimotor function (6–9), with recent studies progressing to primates (10).
Notwithstanding progress in preclinical models of CNS injury, widespread application of ChABC has been hindered by its instability at physiological temperature and pH, resulting in rapid unfolding, aggregation, and inactivation (11). To overcome this limitation, some have used repeated bolus injections of the enzyme (5, 8), while others have used osmotic pumps for constant infusion (12). These strategies are invasive and introduce risks of infection, especially as the protective scar tissue is degraded. Several groups (including our own) have had limited success reengineering ChABC, with only marginal impact on stability, activity, and functional half-life. The most common strategies include introducing point mutations, truncations, formulations with different solvents, tethering with poly(ethylene glycol) (PEG) chains, and/or attachment to nanoparticles (table S1). Stabilizing proteins through mutagenesis is challenging because most mutations are destabilizing, and even those that are stabilizing typically have only a minor impact on overall protein stability. Furthermore, for large proteins like ChABC, with 1021 amino acids and an approximate molecular weight of 115 kDa, deep mutational scanning can only explore a fraction of the sequence space (13).
To reengineer ChABC, we leveraged the consensus effect hypothesis in combination with computational protein design (14, 15) and produced an evolutionary-predicted, more stable, and more active ChABC. The consensus hypothesis proposes that the amino acids most frequently observed in nature increase stability (16), because amino acids that disrupt structure and function are evolutionarily disfavored, and ancestral proteins may be more thermally stable due to the cooling of Earth (17). We used a hybrid consensus hypothesis and forcefield-based optimization approach to predict three combinations of 37, 55, and 92 mutations to ChABC, which we hypothesized would improve the overall stability and activity of the enzyme. To test this hypothesis, we expressed ChABC mutants and assessed both their stability by measuring protein aggregation and enzymatic activity for CS and DS over time. Since we were interested in controlled protein release, we characterized fusion proteins of ChABC with the Src homology 3 (SH3) domain (i.e., ChABC-SH3) and their affinity-controlled release from methylcellulose (MC) hydrogels modified with SH3-binding peptides.
RESULTS
Predictions to stabilize ChABC
We started with the 1.9-Å x-ray crystal structure for the ChABC enzyme from Proteus vulgaris (1HN0) (18) and used the Protein Repair One-Stop Shop (PROSS) method to predict stabilizing mutations (14). PROSS predicts the stability of all mutations at each position using Rosetta local conformation sampling and energetic scoring with a sequence conservation bias from a multiple sequence alignment of 71 extant ChABC enzymes (fig. S1). Using three separate energy thresholds, PROSS combines all independently stabilizing mutations in a global optimization of structure and sequence (see the “Prediction of stabilizing ChABC-SH3 mutations” section in Materials and Methods), yielding mutant sets containing 37, 55, and 92 cumulative mutations (Fig. 1).
Fig. 1. Computational modeling of designed ChABC mutants.
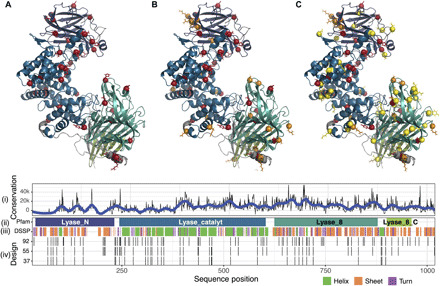
Top: Designed ChABC from P. vulgaris using PROSS, with mutated residues for (A) ChABC-37-SH3, (B) ChABC-55-SH3, and (C) ChABC-92-SH3 in red, orange, and yellow balls, respectively, highlighting additional mutations between designs. Bottom: Tracks include (i) ChABC sequence conservation where lower values indicate more conserved, defined by the sum pairwise BLOSUM64 substitution scores across all pairs of amino acids and 71 bacterial sequences in a ClustalW alignment, (ii) Pfam domains, (iii) secondary structure based on DSSP (define secondary structure of proteins) algorithm, and (iv) positions of mutations for ChABC-37-SH3, ChABC-55-SH3, and ChABC-92-SH3 (green is α-helix; orange is β-sheet; and purple is turn).
All mutants maintained close agreement in terms of root mean square deviation with the 1HN0 crystal structure and demonstrated lower predicted folding free energies than the wild-type enzyme when globally relaxed in Rosetta, indicative of increased stability (fig. S2).
PROSS maintained amino acids at or near conserved active site residues (H501, Y508, R560, and Q653) (19) and at metal-binding residues (D442, D444, and Y392) (20), highlighting the value of consensus design. Furthermore, individual mutations predicted by PROSS used established stabilization strategies, such as introducing charge-charge interactions (21) and rigidifying loops and helices through side-chain H-bonds and backbone prolines (Fig. 2) (22, 23). Simultaneously mutating dozens of residues not only increased the risk of introducing a highly destabilizing mutation but also opened the opportunity for multiple weak stabilizing mutations to lead to an overall stabilizing effect (24). All the amino acid mutations were previously unidentified, except for 4, which overlapped with 46 previously mutated residues (table S1) that had been introduced in various locations throughout the protein structure (fig. S3A).
Fig. 2. Example mutations from ChABC-37-SH3.
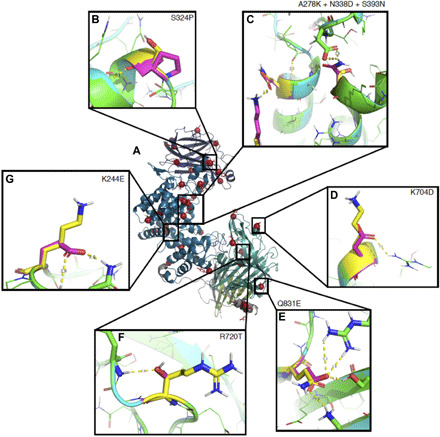
(A) 1HN0 colored by domain as Fig. 1, with residues mutated in ChABC-37-SH3 in magenta. In boxes, native residues are shown in yellow, and mutant residues are shown in magenta. (B) Introducing proline reduces conformational entropy. (C) Three mutations coordinately stabilize the helix termini while maintaining the wild-type helix capping motif. (D and E) Introducing charge-charge interactions increases resistance to aggregation. (F and G) Introducing polar H-bonds stabilizes loops.
Expression and stability of ChABC-SH3 mutants
The DNA sequences for ChABC-SH3 mutants were codon optimized for expression in Escherichia coli. We expressed N-terminal SH3 fusions of each design (i.e., ChABC-SH3; fig. S3B) in NiCo21 (DE3) E. coli cells to enable controlled release from a hydrogel containing SH3-binding peptides. Expressed proteins were purified using a nickel affinity column for the hexahistidine tag on the protein followed by size exclusion chromatography. Gel electrophoresis and staining demonstrated that each ChABC-SH3 design was expressed at the correct molecular weight (125 kDa), with minimal contaminating proteins (Fig. 3A). A slight increase in additional, lower–molecular weight protein bands was observed with each ChABC-SH3 design, but these bands were much less abundant than the intact protein band and did not appear to affect other properties, such as protein structure. Large-scale (2 liters) cultures of designs resulted in 3.5-fold more protein than wild type (an average of 5.06 ± 0.67 mg/liter for mutant designs compared to 1.44 ± 0.46 mg/liter for wild-type ChABC; Fig. 3B) under the same protein expression and purification protocols, reflecting a better functional protein production overall.
Fig. 3. ChABC-SH3 designs are highly expressed and more stable than wild type.
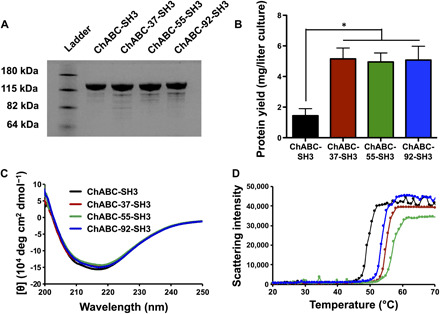
(A) Gel electrophoresis of 5 μg of ChABC-SH3 and mutated designs followed by Coomassie Brilliant Blue staining of protein bands. (B) ChABC-SH3 and mutant yield from large volume (2 liters) E. coli cultures (n = 3, means ± SD, *P < 0.05). (C) Circular dichroism spectra of ChABC-SH3 and designs from 200 to 250 nm at 25°C. (D) Protein aggregation curves between 20° and 70°C (1°C/min) measured by scattering intensity of the solution.
Circular dichroism revealed that mutants did not significantly disrupt protein structure (Fig. 3C), with similar α-helix and β-sheet composition between wild type (α-helix, 32% and β-sheet, 16%) and mutants (ChABC-37-SH3: α-helix, 32% and β-sheet, 16%; ChABC-55-SH3: α-helix, 32% and β-sheet, 17%; ChABC-92-SH3: α-helix, 31% and β-sheet, 17%). We evaluated mutant stability by measuring protein aggregation under a temperature increase (1°C/min) (Fig. 3D). The mutant aggregation temperatures increased between 4° and 8°C relative to the wild-type ChABC-SH3 aggregation temperature of 49°C: ChABC-37-SH3, 55°C; ChABC-55-SH3, 57°C; and ChABC-92-SH3, 53°C. These aggregation temperature increases reflect greater thermal stability and either matched or exceeded the shifts of other ChABC mutants that maintained full enzymatic activity (table S1).
Enzymatic activity of ChABC-SH3 mutants
Since ChABC degrades both CS and DS substrates (2, 20), it may synergistically stimulate tissue regeneration by both decreasing glial scar formation through DS degradation and increasing axonal regrowth into the injury site through CS degradation (25). Only ChABC-37-SH3 exhibited higher initial enzymatic activity against both CS and DS compared to wild type, whereas ChABC-55-SH3 and ChABC-92-SH3 exhibited significantly lower activity for CS (Fig. 4A) and DS (fig. S4). Although the activity of all ChABC proteins for CS decreased over time, this decrease was slower for mutant proteins than the wild type, with all mutant proteins maintaining between 18 and 32% of their original activity at day 7 and the wild type maintaining less than 4% of its original activity (fig. S5). While the wild type and all the mutants maintained some activity (<3 U/mg) after 7 days, only ChABC-37-SH3 displayed substantial activity (>16 U/mg) after 7 days at 37°C (Fig. 4A). All the designed ChABC mutants demonstrated higher functional half-lives (50% of initial activity, 3.2 to 6.3 days) compared to ChABC-SH3 (0.7 days) (Fig. 4B); however, only ChABC-37-SH3 significantly increased total CS degradation (Fig. 4C). Estimated kinetic parameters (kcat and Vmax) of ChABC-37-SH3 (Table 1) using the Michaelis-Menten model (fig. S6) were significantly higher than those of ChABC-55-SH3 and ChABC-92-SH3, further demonstrating the increased efficacy of ChABC-37-SH3 compared to the other designed mutants.
Fig. 4. ChABC-37-SH3 retains activity longer than wild type.

(A) Specific activity of wild type and mutants after incubation at 37°C in 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 7 days (*P < 0.05 for ChABC-37-SH3 versus ChABC-SH3 at all time points). (B) Half-life of ChABC-SH3 and mutants based on specific activity (*P < 0.05, **P < 0.01, and ***P < 0.001). (C) Total CS degradation, as measured by AUC of ChABC-SH3 and mutants over 7 days (***P < 0.001 compared to all other groups) (n = 3, means ± SD).
Table 1. Michaelis-Menten kinetic parameters of ChABC-SH3 designs (n = 3, mean ± SD).
| Chondroitin sulfate A | Dermatan sulfate | |||||||||
| Protein | KM (μM) | Vmax (μM min−1) | kcat (min−1) | kcat/KM (μM−1 min−1) | R2 | KM (μM) | Vmax (μM min−1) | kcat (min−1) | kcat/KM (μM−1 min−1) | R2 |
| ChABC-SH3 | 2132 ± 467 | 392 ± 18 | 4894 ± 230 | 2.30 ± 0.51 | 0.94 | 2175 ± 291 | 365 ± 11 | 4560 ± 140 | 2.10 ± 0.29 | 0.98 |
| ChABC-37-SH3 | 2821 ± 657 | 387 ± 23 | 4840 ± 293 | 1.72 ± 0.41 | 0.93 | 3655 ± 565 | 461 ± 21 | 5759 ± 261 | 1.58 ± 0.25 | 0.98 |
| ChABC-55-SH3 | 16,180 ± 12,149 | 64 ± 27 | 802 ± 334 | 0.05 ± 0.04 | 0.87 | 4778 ± 530 | 222 ± 8 | 2776 ± 102 | 0.58 ± 0.07 | 0.99 |
| ChABC-92-SH3 | 2120 ± 445 | 140 ± 7 | 1753 ± 83 | 0.83 ± 0.18 | 0.94 | 3297 ± 1091 | 298 ± 28 | 3722 ± 344 | 1.14 ± 0.39 | 0.90 |
Resistance of ChABC-SH3 mutants to proteolytic degradation
All mutant proteins displayed higher resistance to proteolytic degradation than the wild type (fig. S7). After incubation with trypsin (2 μg/ml) for 45 min, mutant ChABC-(37, 55, and 92)-SH3 displayed a higher proportion of intact protein (125-kDa band) compared to wild-type ChABC-SH3 (fig. S7A). Furthermore, impressively, all mutant proteins retained 100% of their original activity following trypsin treatment, whereas wild-type ChABC-SH3 only retained 31% of its original activity (fig. S7B). This difference in stability may be due to either changes in the number of basic residues (lysines and arginines), which are typically cleaved by trypsin, or the conformational changes in the protein structure, which increased protein rigidity and decreased accessibility to basic residues (26).
Affinity-controlled release of ChABC-SH3
To achieve sustained release of the ChABC-SH3 designs using an injectable, affinity-controlled hydrogel delivery system (3, 27, 28), we modified 5% (w/v) thiolated MC hydrogels cross-linked with PEG bis-maleimide with (or without) SH3-binding peptides (100:1 molar ratio of SH3-binding peptide to fusion protein) and separately incorporated ChABC-37-SH3 and ChABC-SH3 therein (Fig. 5A). Protein release into artificial cerebrospinal fluid (aCSF) was measured over 7 days using a custom-designed enzyme-linked immunosorbent assay (ELISA) to detect the hexahistidine and FLAG tags expressed on either ends of the ChABC-SH3 enzymes. Hydrogels containing SH3-binding peptides reduced protein release at 2, 4, and 7 days compared to hydrogels without binding peptides (Fig. 5B), with approximately 40% of the ChABC-37-SH3 loaded into MC-peptide gels released within the first 2 days and an additional 20% released between days 2 and 7 (P < 0.05). These data confirm that the sustained protein release via the SH3-binding domain was achieved with both wild-type and mutant proteins. Released ChABC-37-SH3 demonstrated better long-term proteolytic activity than the wild-type enzyme over 7 days (Fig. 5C), as demonstrated by significant differences in enzymatic activity in the supernatant at 1 and 2 days (fig. S8) and an increased area under the activity curve (AUC) (Fig. 5D). ChABC activity was detectable in the supernatant for the entire 7-day period (fig. S8).
Fig. 5. Release of bioactive ChABC-SH3 and ChABC-37-SH3 is sustained over 7 days from cross-linked MC hydrogels covalently modified with SH3-binding peptides.
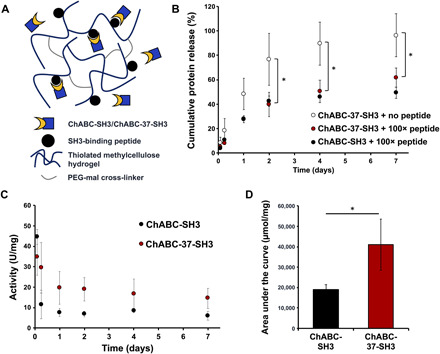
(A) Cross-linked MC-peptide hydrogels for affinity-controlled release. (B) Twenty micrograms of ChABC-SH3 or ChABC-37-SH3 was mixed into 100 μl of MC hydrogel alone or modified with SH3-binding peptides. The hydrogels released protein into artificial cerebrospinal fluid (aCSF) over 7 days. Release was plotted as percentage of total protein loaded (*P < 0.05). (C) Specific activity of ChABC-SH3 and ChABC-37-SH3 released from MC hydrogels over 7 days. (D) AUC of ChABC-SH3 and ChABC-37-SH3 mutant over 7-day release from hydrogels (*P < 0.05) (n = 3, means ± SD).
DISCUSSION
Although all mutants were more stable and displayed increased proteolytic resistance compared to wild-type ChABC-SH3, only one design (ChABC-37-SH3) demonstrated the desired increase in both stability and enzymatic activity toward CS and DS. This indicates that, while PROSS successfully predicted increased protein structural stability, one limitation of this approach is that it cannot reliably predict enzymatic activity. We hypothesize that mutations that reduced enzymatic activity may interfere with other functionally relevant states that facilitate substrate binding to the active site, thereby reducing the catalytic efficiency. This is supported by the high Km (or low substrate affinity) of ChABC-55-SH3 compared to the other enzymes. A similar effect was observed by Shirdel et al. (29), where structurally stabilized ChABC mutants demonstrated a lower enzymatic efficiency; this was attributed to decreased flexibility of the protein since substrate binding typically results in a conformational change. Notably, increasing the number of mutations did not necessarily lead to decreased function. ChABC-92-SH3, which included an additional 37 unique mutations to the 55 changes already in ChABC-55-SH3, demonstrated a higher initial activity, indicating that additional stabilizing mutations can counteract destabilizing mutations and rescue lost enzymatic activity.
The success of ChABC-37-SH3 with 37 mutations highlights the value of integrating both evolutionary data and native-state energy optimization into a single approach (14, 15), given the high likelihood of destabilizing mutations that reduce enzyme activity. Unlike other efforts to stabilize ChABC, which focused on optimizing specific aspects of the protein structure with point mutations, PROSS considers all possible amino acid changes biasing toward those that independently contribute stability and are often observed at the given position. The fact that 71 ChABC variants are naturally occurring in a variety of bacteria species provides ample evolutionary data to increase the predictive power of this approach.
The use of PROSS to generate ChABC-37-SH3, with 3.5 times higher protein yield, a 6.5 times greater functional half-life, and 6°C increase in aggregation temperature with increased enzymatic activity, demonstrates the notable advantage of this strategy compared to traditional site-directed mutagenesis. Stabilization via mutagenesis often leads to decreased catalytic efficiency (kcat/Km). For example, H700N/L701T, the most stabilizing mutation to date (29), has an aggregation temperature 16°C greater than wild-type ChABC but is 40% less catalytically efficient. In contrast, ChABC-37-SH3 has an aggregation temperature 6°C greater than wild type, is more active overall, and is only 25% less catalytically efficient for CS degradation. To our knowledge, ChABC-37-SH3 exhibits the highest long-term activity and total substrate degradation of any documented wild-type or modified form of ChABC. Moreover, the marked improvement in proteolytic resistance demonstrated by all mutants has never been reported in literature. While only resistance to trypsin degradation was investigated in this study, future studies investigating the resistance of mutant ChABC-SH3 to other proteases are warranted and may yield similar results. A study by Nazari-Robati et al. (30) demonstrated similar loss of ChABC activity following proteolysis by either trypsin or chymotrypsin, despite the fact that these proteases cleave different residues. It was hypothesized that fewer chymotrypsin cleavage sites were accessible to proteases, resulting in similar degradation patterns.
Widespread use and commercialization of ChABC have been hampered by its thermal instability, limited activity, and poor sustained, local delivery. Our reengineered ChABC mutant, ChABC-37-SH3, overcomes these challenges by significantly extending the bioactive life span of the enzyme and enabling sustained release from a hydrogel via the SH3 fusion domain. Considering the fragile nature of this protein, a hydrogel that maintains protein bioactivity will substantially improve the feasibility of its clinical use. This affinity-based delivery strategy maintains ChABC activity at levels similar to that of soluble protein over 7 days. Furthermore, we expect that degradation of proteoglycans using ChABC-37-SH3 will promote axonal outgrowth in the CNS (6–9, 31, 32).
While this affinity-based delivery strategy only resulted in release of ~62% of the loaded ChABC-37-SH3 over 7 days in vitro, we expect full protein release to be achieved in vivo due to gradual dissolution of the hydrogel. A prolonged effect of a less stable form of ChABC-SH3, delivered using this strategy, was observed for up to 2 and 4 weeks in the spinal cord and brain, respectively (3, 28). To facilitate complete protein release, we are further engineering the MC delivery vehicle to expedite degradation and resorption in vivo. We have recently synthesized a resorbable MC hydrogel with a disulfide-containing cross-linker, which enables material degradation in the presence of naturally occurring thiols in vivo (33).
The modifications to ChABC described here have improved its production, stability, and long-term bioactivity, overcoming many challenges required for clinical translation and facilitating its future use as a viable therapeutic in treating CNS injuries. More broadly, with this first demonstration of the use of PROSS to optimize a therapeutic agent, such as ChABC, we highlight the versatility of this method and open up this approach for others to use in optimizing the stability and activity of other particularly sensitive proteins.
MATERIALS AND METHODS
Prediction of stabilizing ChABC-SH3 mutations
Using the PROSS protocol (14), we predicted stabilizing mutations for an apo structure of ChABC from P. vulgaris (UniProt entry: CABC1_PROVU): chain A of Protein Data Bank 1HN0 (18), solved using x-ray diffraction to a resolution of 1.9 Å. The structure has 1021 residues, with three adjacent domains Lyase_N-lyase_catalyt-Lyase_8, as identified by Pfam 31.0 (34).
The method constructs a multiple-sequence alignment from sequences in the National Center for Biotechnology Information nonredundant database (nr) with BlastP E value of less than 1 × 10−4 to the query (data file S1) (35). The sequences were then aligned using MUSCLE (36) with the default parameters. Homologs with insertions or deletions in define secondary structure of proteins (DSSP)–labeled loops (36) were removed, yielding 70 sequences with a minimum and median percent coverage of 59.4 and 92.2% and minimum and median percent sequence identity of 23.5 and 41.7% (Fig. 2). A position-specific scoring matrix (PSSM) was then computed using Protein-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) (35), giving the log probability of each amino acid at each position. Amino acids at positions with a PSSM score ≤0 were rejected. To bias toward mutations that independently provide stability, for each candidate point mutation at each position, the wild-type structure was optimized using the Rosetta mutational scanning protocol (37), which repacks side chains within 8Å of the candidate point mutation and globally minimizes all torsion angles using the Talasis2014 weight set combined with a native-coordinate constraint with a weight of 0.5 and a Boltzmann PSSM constraint weight of 1. The mutational scanning protocol rejects candidate point mutations if the predicted change in free energy (ΔΔGcalc) was not less than −1.8, −1.25, or −0.45 Rosetta energy units to explore stringent, medium, and permissive mutation sets. Then, for each energy level, the wild-type and remaining mutations were jointly considered using a protocol that applies the following sequence of movers: soft_design, soft_min, soft_design, hard_design, hard_min, hard_design, hard_min, hard_design, RT_min, RT_min, and hard_min. The movers with the soft prefix use soft_rep weight set, which dampens repulsive van der Waals forces, and those with the hard prefix use the Talaris2014 weight sets. The “design” movers design and repack the side-chain amino acid type and torsion angles while keeping the backbone fixed, whereas the “min” movers minimize all torsion angles. The “RT_min” mover does rotamer trials, which sequentially considers each rotamer at each position without design. The design stage uses a constraint weight of 0.4 for the native sequence and 0.4 for the PSSM profile. General Rosetta flags were -ex1, -ex2, -use_input_sc, -extrachi_cutoff 5, ignore_unrecognized_res, -use_occurrence_data, -linmem_ig 10, -ignore_zero_occupancy false, and -restore_talaris_behavior. The stringent, medium, and permissive energy thresholds resulted in design sets with 37, 55, and 92 total mutations, which we named ChABC-37-SH3, ChABC-55-SH3, and ChABC-92-SH3.
Assembly of mutant ChABC-SH3 constructs
Mutant ChABC amino acid sequences (data file S2) were codon optimized for the E. coli gene expression using the Integrated DNA Technologies DNA codon optimization web tool (www.idtdna.com/CodonOpt). Gene sequences were purchased from Twist Bioscience (San Francisco, CA) as two halves containing 1499 and 1603 base pairs, and each was cloned into the Twist standard vector using Asc I, Dra I, and Xho I (New England Biolabs, Ipswich, MA). The pieces of each design were cut from the pTwist vector, and both halves were ligated and inserted into a pET28b+ vector containing a kanamycin resistance cassette after the isopropyl-β-d-1-thiogalactopyranoside (IPTG)–inducible T7 promoter between the existing hexahistidine (HHHHHH) and FLAG (DYKDDDDK) tags. The complete sequences read as follows: hexahistidine tag, SH3 domain, flexible linker, ChABC, and FLAG tag. Plasmids were cloned into NEB 5-α high-efficiency competent cells (New England Biolabs, Ipswich, MA) and validated by Sanger sequencing (ACGT Corporation, Toronto, ON).
ChABC-SH3 expression and purification
Mutant ChABC-SH3 was expressed and purified, as previously described for ChABC-SH3 (3). Plasmids were transformed into NiCo21 (DE3) E. coli cells for protein expression (New England Biolabs). For small-scale verification of protein expression from single colonies, cells were grown overnight at 37°C in 10 ml of Luria Bertani (LB) broth supplemented with kanamycin (50 μg/ml), followed by induction of protein expression with 0.8 mM IPTG during the log phase of growth [OD600nm (optical density at 600 nm) ≥ 0.8] for an additional 5 hours. Cell cultures were centrifuged, treated with BugBuster Protein Extraction Reagent (MilliporeSigma, Burlington, MA) for bacterial lysis, and centrifuged again to separate the soluble and insoluble fractions. The soluble fraction of the bacterial lysate was denatured and separated by mass using SDS–polyacrylamide gel electrophoresis (PAGE), followed by Coomassie Brilliant Blue staining for nonspecific staining of all protein bands.
For large-scale protein production, NiCo21 cells were grown overnight at 37°C in 20 ml of LB broth with kanamycin (50 μg/ml) and then transferred to 1.8 liters of terrific broth (TB) supplemented with 0.4% glycerol, kanamycin (50 μg/ml), and 500 μl of antifoaming agent (Antifoam 204). TB cultures were grown at 37°C in a LEX-10 bubbler system (Epiphyte3, Toronto, ON), with constant air sparging until OD600nm ≥ 0.8 was reached, upon which 0.8 mM IPTG was added and protein expression was allowed to proceed at 22°C for 18 hours. TB cultures were centrifuged for 15 min at 6000 rpm and 4°C (Avanti JXN-26 centrifuge, Beckman Coulter, Brea, CA). Cell pellets were resuspended in 40 ml of lysis buffer [50 mM tris (pH 7.5), 500 mM NaCl, and 5 mM imidazole] and lysed using a 500-W sonicator (Qsonica, Newtown, CT) at 30% amplitude for 5 min at 10-s intervals. The soluble fraction of the cell lysate was incubated with 1.8 ml of nickel–nitrilotriacetic acid (Ni-NTA) resin for 15 min at 4°C to promote binding between the nickel and hexahistidine tag on ChABC-SH3. The cell lysate was poured through a glass chromatography column, and the Ni-NTA resin was washed with 10 × 10 ml of wash buffer [50 mM tris (pH 7.5), 500 mM NaCl, and 30 mM imidazole]. Nickel-bound proteins were eluted using a high concentration of imidazole [40 mM tris (pH 7.5), 500 mM NaCl, and 250 mM imidazole] and subsequently incubated with 5 ml of prewashed chitin resin (New England Biolabs) in elution buffer for 1 hour. The solution was poured through another glass chromatography column and concentrated to 1 to 2 ml with a 10,000-kDa cutoff Vivaspin 20 centrifugal concentrator (Sartorius, Gottingen, Germany). Size exclusion chromatography was performed using a HiLoad 16/600 Superdex 200 column on an AKTA Purifier 10 (GE Healthcare Life Sciences, Budapest, Hungary) in 50 mM sodium acetate and 10 mM phosphate buffer (pH 8.0). Ten consecutive 1-ml fractions, corresponding to unaggregated protein based on 280-nm signal and time of elution from the Superdex 200 column, were collected and concentrated to approximately 1 to 2 ml with a centrifugal concentrator. Purified ChABC-SH3 was filter sterilized (Amicon Ultrafree-MC 0.22-μm Centrifugal Filter Units, MilliporeSigma) and stored in 50 mM sodium acetate in phosphate-buffered saline (PBS) (pH 8.0) at −80°C until use. Protein concentration was quantified by measuring sample absorbance at 280 nm using the molecular weight (125 kDa) and extinction coefficient (211,000 M−1 cm−1) of ChABC-SH3.
Circular dichroism
For circular dichroism readings, proteins were diluted to 0.2 mg/ml in PBS (pH 7.4), and 150 μl was pipetted into a glass cuvette with a path length of 1 cm. The far-ultraviolet (UV) (200 to 260 nm) circular dichroism spectra of ChABC-SH3 and mutants were measured using a JASCO J-810 circular dichroism spectrophotometer (JASCO, Easton, MD) and expressed as molar ellipticity (deg cm2 dmol−1). α-helix and β-sheet content was determined via deconvolution using DichroWeb (38).
Static light scattering
Proteins were diluted to 1 mg/ml in PBS (pH 7.4), and 9 μl was pipetted into glass capillaries for static light scattering (SLS) (UNit, Unchained Labs, Pleasanton, CA). SLS readings were taken at 466 nm between 25° and 70°C using a temperature scan rate of 1°C/min to evaluate protein unfolding and subsequent aggregation. Melting temperatures (Tm) were determined using Boltzmann regression for the midpoint of the linear denaturation curve.
ChABC-SH3 activity
The enzymatic activity of ChABC-SH3 was evaluated by measuring the degradation of the substrates CS A or DS, which exhibit an absorbance change at 232 nm following cleavage by chondroitinases. Ten microliters of ChABC-SH3 (0.1 mg/ml) was mixed with 90 μl of CS A or DS (10 mg/ml) in a UV-Star microplate (Greiner Bio-One, Monroe, NC) and immediately read on a plate reader (Tecan Infinite M200 Pro) at 232 nm. Readings were taken at room temperature every 20 s for 20 min, and the slope of the resultant linear relationship between absorbance and time was used to calculate the kinetic activity of ChABC-SH3 in units of activity (millimolar substrate degraded per minute) per milligram of protein (unit per milligram). To evaluate long-term enzymatic activity, solutions of ChABC-SH3 (0.1 mg/ml) in PBS (pH 7.4) with 0.1% (w/v) bovine serum albumin (BSA) and protease inhibitor tablets (cOmplete Mini Protease Inhibitor Cocktail, Roche, Switzerland) were incubated at 37°C for up to 7 days and then frozen at −80°C until analysis. The half-life of active ChABC-SH3 was calculated by fitting activity data over time to a one-phase exponential decay curve, and the AUC over 7 days was calculated using the trapezoid rule.
Kinetic parameters for CS A and DS were determined using the initial rates of product formation when 10 μl of ChABC-SH3 (0.1 mg/ml) was mixed with 90 μl of substrate (1 to 10 mg/ml). Data were fit to the Michaelis-Menten equation, and Vmax, Km, and kcat were determined by nonlinear regression using GraphPad Prism.
Proteolytic degradation
Proteolysis was carried out as detailed by Kheirollahi et al. (22). Briefly, wild-type and mutant ChABC-SH3 (0.4 mg/ml) were incubated with trypsin (2 μg/ml) in 20 mM tris buffer containing 10 mM CaCl2 at pH 7.5 at room temperature. The reaction was inhibited with 1 mM phenylmethylsulfonyl fluoride after 45 min. Proteins were denatured and separated by SDS-PAGE, followed by Coomassie Brilliant Blue staining. Specific activity for CS A was measured, as detailed above. Specific activity was compared to ChABC-SH3 that had been incubated in buffer without trypsin for 45 min at room temperature.
Affinity release of ChABC-SH3 from MC hydrogels
Cross-linked MC hydrogels for affinity-controlled release of ChABC-SH3 were fabricated, as previously described (3). Briefly, 100 μl of 5% (w/v) MC containing 20 μg of ChABC-SH3 or ChABC-37-SH3 and 0.1 μmol of thiol were cross-linked with 3000-Da PEG-bismaleimide cross-linker at a ratio of 0.75:1 maleimide to thiol. To control release, binding peptides for the SH3 domain of the fusion protein (KPPVVKKPHYLS) with a dissociation constant of 2.7 × 10−5 M were incorporated into the hydrogel at 100 times molar excess to the protein. Hydrogels were speed mixed into solution (SpeedMixer DAC 150 FV2, FlackTek, Landrum, SC), cross-linked, and incubated at 4°C overnight before incubation in 400 μl of PBS (pH 7.4) with 0.1% (w/v) BSA and protease inhibitors for 7 days at 37°C. PBS was removed and replaced at 0, 2, and 6 hours and 1, 2, 4, and 7 days. ChABC-SH3 and ChABC-37-SH3 release was quantified using a custom ELISA, as previously described (28). Activity for CS A was quantified and normalized to protein release.
Statistical analysis
All data are reported as means ± SD. In vitro experiments were performed with a minimum of three biological replicates for each experimental group. Statistical significance was determined using one-way or two-way analysis of variance (ANOVA) as appropriate, followed by Bonferroni’s post hoc analysis (GraphPad Prism, version 7.0, La Jolla, CA). P < 0.05 was considered statistically significant. One-phase exponential decay, Michaelis-Menten curve fitting and AUC determination were also performed using GraphPad Prism.
Sequence analysis
Sequence conservation for ChABC shown in Fig. 2 is computed as the sum of pairwise scores for all wild-type positions of the alignment computed by msaConservationScore (BLOSUM64) from the R msa package (39) and smoothed using the LOESS method from the R stats package, with span = 0.05. Domain structure was defined by Pfam 31.0 (34). Secondary structure was defined from the dictionary of protein secondary structure (DSSP) codes (40). Figure 1 was generated using the R the ggplot2 package (41).
Structure analysis
Figures 1 and 2 and fig. S3 were generated using PyMOL (42).
Supplementary Material
Acknowledgments
We are grateful to A. Goldenzweig (Weizmann Institute of Science) for the help with the PROSS method and B. Shoichet (UCSF) for the valuable discussion and advice, as well as members of Dr. Molly Shoichet’s lab for thoughtful review of this manuscript. Funding: This work was funded by the Natural Sciences and Engineering Council (NSERC) of Canada (Discovery Grant to M.S.S. and Postdoctoral Fellowship to M.H.H.) and Canada First Research Excellence Fund to the University of Toronto (Medicine by Design). Author contributions: M.H.H., M.J.O., T.R.O., and M.S.S. designed the research. M.H.H., M.J.O., T.R.O., A.J.P., and N.L.-K. performed the experiments. M.H.H., M.J.O., A.J.P., and N.L.-K. analyzed the data. M.H.H., M.J.O., T.R.O., and M.S.S. wrote the paper. Competing interests: Several of the authors (M.H.H., M.J.O., T.R.O., and M.S.S.) have filed a provisional patent related to this work. The other authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eabc6378/DC1
REFERENCES AND NOTES
- 1.Hamai A., Hashimoto N., Mochizuki H., Kato F., Makiguchi Y., Horie K., Suzuki S., Two distinct chondroitin sulfate ABC lyases: An endoeliminase yielding tetrasaccarides and an exoeliminase preferentially acting on oligosaccharides. J. Biol. Chem. 272, 9123–9130 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Shaya D., Hahn B.-S., Bjerkan T. M., Kim W. S., Park N. Y., Sim J.-S., Kim Y.-S., Cygler M., Composite active site of chondroitin lyase ABC accepting both epimers of uronic acid. Glycobiology 18, 270–277 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Hettiaratchi M. H., O’Meara M. J., Teal C. J., Payne S. L., Pickering A. J., Shoichet M. S., Local delivery of stabilized chondroitinase ABC degrades chondroitin sulfate proteoglycans in stroke-injured rat brains. J. Control. Release 297, 14–25 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Soleman S., Yip P. K., Duricki D. A., Moon L. D. F., Delayed treatment with chondroitinase ABC promotes sensorimotor recovery and plasticity after stroke in aged rats. Brain 135, 1210–1223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin R., Kwok J. C. F., Crespo D., Fawcett J. W., Chondroitinase ABC has a long-lasting effect on chondroitin sulphate glycosaminoglycan content in the injured rat brain. J. Neurochem. 104, 400–408 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Warren P. M., Steiger S. C., Dick T. E., MacFarlane P. M., Alilain W. J., Silver J., Rapid and robust restoration of breathing long after spinal cord injury. Nat. Commun. 9, 4843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nori S., Khazaei M., Ahuja C. S., Yokota K., Ahlfors J.-E., Liu Y., Wang J., Shibata S., Chio J., Hettiaratchi M. H., Führmann T., Shoichet M. S., Fehlings M. G., Human oligodendrogenic neural progenitor cells delivered with chondroitinase ABC facilitate functional repair of chronic spinal cord injury. Stem Cell Rep. 11, 1433–1448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradbury E. J., Moon L. D. F., Popat R. J., King V. R., Bennett G. S., Patel P. N., Fawcett J. W., McMahon S. B., Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416, 636–640 (2002). [DOI] [PubMed] [Google Scholar]
- 9.García-Alías G., Barkhuysen S., Buckle M., Fawcett J. W., Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat. Neurosci. 12, 1145–1151 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Rosenzweig E. S., Salegio E. A., Liang J. J., Weber J. L., Weinholtz C. A., Brock J. H., Moseanko R., Hawbecker S., Pender R., Cruzen C. L., Chondroitinase improves anatomical and functional outcomes after primate spinal cord injury. Nat. Neurosci. 22, 1269–1275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tester N. J., Plaas A. H., Howland D. R., Effect of body temperature on chondroitinase ABC’s ability to cleave chondroitin sulfate glycosaminoglycans. J. Neurosci. Res. 85, 1110–1118 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Harris N. G., Mironova Y. A., Hovda D. A., Sutton R. L., Chondroitinase ABC enhances pericontusion axonal sprouting but does not confer robust improvements in behavioral recovery. J. Neurotrauma 27, 1971–1982 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler D. M., Araya C. L., Fleishman S. J., Kellogg E. H., Stephany J. J., Baker D., Fields S., High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldenzweig A., Goldsmith M., Hill S. E., Gertman O., Laurino P., Ashani Y., Dym O., Unger T., Albeck S., Prilusky J., Lieberman R. L., Aharoni A., Silman I., Sussman J. L., Tawfik D. S., Fleishman S. J., Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musil M., Konegger H., Hon J., Bednar D., Damborsky J., Computational design of stable and soluble biocatalysts. ACS Catal. 9, 1033–1054 (2019). [Google Scholar]
- 16.Lehmann M., Pasamontes L., Lassen S. F., Wyss M., The consensus concept for thermostability engineering of proteins. Biochim. Biophys. Acta 1543, 408–415 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Akanuma S., Nakajima Y., Yokobori S.-i., Kimura M., Nemoto N., Mase T., Miyazono K.-i., Tanokura M., Yamagishi A., Experimental evidence for the thermophilicity of ancestral life. Proc. Natl. Acad. Sci. U.S.A. 110, 11067–11072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang W., Lunin V. V., Li Y., Suzuki S., Sugiura N., Miyazono H., Cygler M., Crystal structure of Proteus vulgaris chondroitin sulfate ABC lyase I at 1.9Å resolution. J. Mol. Biol. 328, 623–634 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Prabhakar V., Capila I., Bosques C. J., Pojasek K., Sasisekharan R., Chondroitinase ABC I from Proteus vulgaris: Cloning, recombinant expression and active site identification. Biochem. J. 386, 103–112 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prabhakar V., Capila I., Raman R., Srinivasan A., Bosques C. J., Pojasek K., Wrick M. A., Sasisekharan R., The catalytic machinery of chondroitinase ABC I utilizes a calcium coordination strategy to optimally process dermatan sulfate. Biochemistry 45, 11130–11139 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Loladze V. V., Ibarra-Molero B., Sanchez-Ruiz J. M., Makhatadze G. I., Engineering a thermostable protein via optimization of charge−charge interactions on the protein surface. Biochemistry 38, 16419–16423 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Kheirollahi A., Khajeh K., Golestani A., Rigidifying flexible sites: An approach to improve stability of chondroitinase ABC I. Int. J. Biol. Macromol. 97, 270–278 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Matthews B. W., Nicholson H., Becktel W. J., Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl. Acad. Sci. U.S.A. 84, 6663–6667 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H., Arnold F. H., Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng. Des. Sel. 12, 47–53 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Li H.-P., Komuta Y., Kimura-Kuroda J., van Kuppevelt T. H., Kawano H., Roles of chondroitin sulfate and dermatan sulfate in the formation of a lesion scar and axonal regeneration after traumatic injury of the mouse brain. J. Neurotrauma 30, 413–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nazari-Robati M., Khajeh K., Aminian M., Mollania N., Golestani A., Enhancement of thermal stability of chondroitinase ABC I by site-directed mutagenesis: An insight from ramachandran plot. Biochim. Biophys Acta 1834, 479–486 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Vulic K., Shoichet M. S., Tunable growth factor delivery from injectable hydrogels for tissue engineering. J. Am. Chem. Soc. 134, 882–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pakulska M. M., Tator C. H., Shoichet M. S., Local delivery of chondroitinase ABC with or without stromal cell-derived factor 1α promotes functional repair in the injured rat spinal cord. Biomaterials 134, 13–21 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Shirdel S. A., Khalifeh K., Golestani A., Ranjbar B., Khajeh K., Critical role of a loop at C-terminal domain on the conformational stability and catalytic efficiency of chondroitinase ABC I. Mol. Biotechnol. 57, 727–734 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Nazari-Robati M., Golestani A., Asadikaram G., Improvement of proteolytic and oxidative stability of chondroitinase ABC I by cosolvents. Int. J. Biol. Macromol. 91, 812–817 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Zuo J., Neubauer D., Dyess K., Ferguson T. A., Muir D., Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp. Neurol. 154, 654–662 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Corvetti L., Rossi F., Degradation of chondroitin sulfate proteoglycans induces sprouting of intact purkinje axons in the cerebellum of the adult rat. J. Neurosci. 25, 7150–7158 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delplace V., Pickering A. J., Hettiaratchi M. H., Zhao S., Kivijärvi T., Shoichet M. S., Inverse electron-demand diels-alder methylcellulose hydrogels enable the co-delivery of chondroitinase ABC and neural progenitor cells. Biomacromolecules 21, 2421–2431 (2020). [DOI] [PubMed] [Google Scholar]
- 34.El-Gebali S., Mistry J., Bateman A., Eddy S. R., Luciani A., Potter S. C., Qureshi M., Richardson L. J., Salazar G. A., Smart A., Sonnhammer E. L. L., Hirsh L., Paladin L., Piovesan D., Tosatto S. C. E., Finn R. D., The pfam protein families database in 2019. Nucleic Acids Res. 47 ( D1), D427–D432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitehead T. A., Chevalier A., Song Y., Dreyfus C., Fleishman S. J., De Mattos C., Myers C. A., Kamisetty H., Blair P., Wilson I. A., Baker D., Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30, 543–548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitmore L., Wallace B. A., DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bodenhofer U., Bonatesta E., Horejš-Kainrath C., Hochreiter S., Msa: An R Package for Multiple Sequence Alignment. Bioinformatics 31, 3997–3999 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W., Sander C., Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 41.H. Wickham, Ggplot2: Elegant Graphics for Data Analysis (Springer, 2016). [Google Scholar]
- 42.L. Schrodinger, The PyMOL Molecular Graphics System. Version 1.3r1 (2010).
- 43.Nazari-Robati M., Khajeh K., Aminian M., Fathi-Roudsari M., Golestani A., Co-solvent mediated thermal stabilization of chondroitinase ABC I form proteus vulgaris. Int. J. Biol. Macromol. 50, 487–492 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Chen Z., Li Y., Feng Y., Chen L., Yuan Q., Enzyme activity enhancement of chondroitinase ABC I from Proteus Vulgaris by site-directed mutagenesis. RSC Adv. 5, 76040–76047 (2015). [Google Scholar]
- 45.Shamsi M., Shirdel S. A., Jafarian V., Jafari S. S., Khalifeh K., Golestani A., Optimization of conformational stability and catalytic efficiency in chondroitinase ABC I by protein engineering methods. Eng. Life Sci. 16, 690–696 (2016). [Google Scholar]
- 46.Moradi K., Shirdel S. A., Shamsi M., Jafarian V., Khalifeh K., Investigating the structural and functional features of representative recombinants of chondroitinase ABC I. Enzyme Microb. Technol. 107, 64–71 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Shahaboddin M. E., Khajeh K., Maleki M., Golestani A., Improvement of activity and stability of chondroitinase ABC I by introducing an aromatic cluster at the surface of protein. Enzyme Microb. Technol. 105, 38–44 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Shahaboddin M. E., Khajeh K., Golestani A., Establishment of aromatic pairs at the surface of chondroitinase ABC I: The effect on activity and stability. Appl. Biochem. Biotechnol. 186, 358–370 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Maleki M., Khajeh K., Amanlou M., Golestani A., Role of His-His interaction in Ser474-His475-Tyr476 sequence of chondroitinase ABC I in the enzyme activity and stability. Int. J. Biol. Macromol. 109, 941–949 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Kheirollahi A., Khajeh K., Golestani A., Investigating the role of loop 131–140 in activity and thermal stability of chondroitinase ABC I. Int. J. Biol. Macromol. 116, 811–816 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Mohammadyari H., Shirdel S. A., Jafarian V., Khalifeh K., Designing and construction of novel variants of chondroitinase ABC I to reduce aggregation rate. Arch. Biochem. Biophys. 668, 46–53 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Madeira F., Park Y. M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A. R. N., Potter S. C., Finn R. D., Lopez R., The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res., 47 (W1), W636–W641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.A. Rambaut, FigTree v1. 4.2, A Graphical Viewer of Phylogenetic Trees. (2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eabc6378/DC1