

Abstract
Background & Aims:
Autophagy maintains cellular homeostasis and plays a critical role in the development of non-alcoholic fatty liver and steatohepatitis. The pseudokinase mixed lineage kinase domain-like (MLKL) is a key downstream effector of receptor interacting protein kinase 3 (RIP3) in the necroptotic pathway of programmed cell death. However, recent data reveal that MLKL also regulates autophagy. Herein, we tested the hypothesis that MLKL contributes to the progression of Western diet-induced liver injury in mice by regulating autophagy.
Methods:
Rip3+/+, Rip3−/−, Mlkl+/+ and Mlkl−/− mice were fed a Western diet (FFC diet, high in fat, fructose and cholesterol) or chow for 12 weeks. AML12 and primary mouse hepatocytes were exposed to palmitic acid (PA).
Results:
The FFC diet increased expression, phosphorylation and oligomerization of MLKL in the liver. Mlkl, but not Rip3, deficiency protected mice from FFC diet-induced liver injury. The FFC diet also induced accumulation of p62 and LC3-II, as well as markers of endoplasmic reticulum stress, in Mlkl+/+ but not Mlkl−/− mice. Mlkl deficiency in mice also prevented the inhibition of autophagy by a protease inhibitor, leupeptin. Using an mRFP-GFP-LC3 reporter in cultured hepatocytes revealed that PA blocked the fusion of autophagosomes with lysosomes. PA triggered MLKL expression and translocation, first to autophagosomes and then to the plasma membrane, independently of Rip3. Mlkl, but not Rip3, deficiency prevented inhibition of autophagy in PA-treated hepatocytes. Overexpression of Mlkl blocked autophagy independently of PA. Additionally, pharmacologic inhibition of autophagy induced MLKL expression and translocation to the plasma membrane in hepatocytes.
Conclusions:
Taken together, these data indicate that MLKL-dependent, but RIP3-independent, signaling contributes to FFC diet-induced liver injury by inhibiting autophagy.
Keywords: NAFLD, NASH, MLKL, RIPK3, Necroptosis, Autophagic flux
Graphical abstract
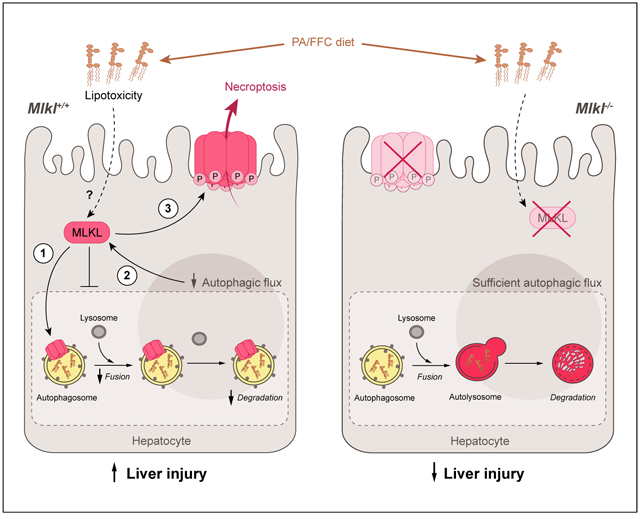
Lay summary
Autophagy is a regulated process that maintains cellular homeostasis. Impaired autophagy contributes to cell injury and death, thus playing a critical role in the pathogenesis of a number of diseases, including non-alcohol-associated fatty liver and steatohepatitis. Herein, we show that Mlkl-dependent, but Rip3-independent, signaling contributed to diet-induced liver injury and inflammatory responses by inhibiting autophagy. These data identify a novel co-regulatory mechanism between necroptotic and autophagic signaling pathways in non-alcoholic fatty liver disease.
Introduction
Regulated cell death is a crucial and active process, serving to maintain tissue homeostasis in multicellular organisms. There are 4 major forms of regulated cell death: apoptosis, necroptosis, ferroptosis and pyroptosis.1 Each pathway is defined by their specific regulatory mechanisms and molecular components. Death receptor-mediated cell death is particularly important in liver disease and is triggered by ligands binding to tumor necrosis factor (TNF) family death domain receptors, viral sensors or pattern recognition receptors. Hepatocellular fate in response to death receptor activation depends on the cellular environment.2 For example, when caspase-8 activity is high, cells will undergo apoptosis. In contrast, if caspase-8 is low, necroptosis prevails.3,4
Multiple forms of cell death are associated with non-alcoholic fatty liver and steatohepatitis (NAFL/NASH). The contribution of apoptosis has been most well studied in animal models of NAFL/NASH. For example, deletion of caspase-8 in hepatocytes reduced methionine- and choline-deficient (MCD) diet-induced hepatic injury, inflammatory response and oxidative stress.5 However, treatment with VX166, a pan-caspase inhibitor, only partially protected against high-fat diet (HFD)-induced liver steatosis and injury,6 suggesting that additional programmed cell death pathways, such as necroptosis, are involved in the pathogenesis of NAFL/NASH.
More recent work has investigated the role of the necroptotic pathway in murine models of NAFL/NASH. Necroptosis classically depends on receptor interacting protein kinase 3 (RIP3 or RIPK3), which binds to RIP1 through their RHIM (RIP homotypic interaction motif) domains to form a protein complex (necrosome). RIP3 then phosphorylates mixed lineage kinase domain like pseudokinase (MLKL), leading to its translocation to the plasma membrane, where it oligomerizes and forms pores that mediate necroptotic cell death.7,8 Interestingly, studies utilizing Rip3−/− mice identified differential contributions of RIP3 to the progression of liver injury in multiple murine models of liver diseases.9,10 Rip3−/− mice were protected from acetaminophen-induced hepatotoxicity,11,12 MCD diet-induced NAFL/NASH,13 alcohol-induced liver injury,14,15 as well as concanavalin-induced autoimmune hepatitis.16 However, work by our lab17 and Gautheron, et al.18 found that Rip3−/− mice were not protected from HFD-induced liver injury. Taken together, these data suggest very specific roles for RIP3 in mediating liver injury in murine models of NAFL/NASH.
Since the molecular machinery in the necroptotic pathway is complex, with multiple non-canonical mechanisms identified for RIP3 and MLKL,10,19,20 it is also critical to understand the contributions of MLKL to liver injury in murine models of NAFL/NASH. One report indicated that Mlkl−/− mice were protected from HFD-induced hepatic insulin resistance21; however, the role of MLKL in hepatic inflammation and injury has not been well investigated. Therefore, we investigated the role of MLKL in Western diet-induced liver injury (FFC diet, high in fat, fructose and cholesterol) in Mlkl−/− mice and their littermate controls. FFC diet increased MLKL expression, phosphorylation and oligomerization in the liver independently of Rip3. Furthermore, Mlkl, but not Rip3, deficiency protected mice from FFC diet-induced liver injury and inflammatory response. These data suggest that MLKL contributes to FFC diet-induced liver injury via a Rip3-independent, non-canonical mechanism of activation.
Multiple non-classical roles of MLKL have been identified in other model systems, including functions in regulation of inflammasomes, exosome formation, and regulation of autophagy.20,22–24 Since impaired autophagy was associated with both the initial development and progression of hepatic steatosis in NAFL/NASH,25,26 we further investigated the interactions between FFC diet, MLKL and autophagy. FFC diet induced accumulation of p62 and microtubule-associated protein 1 light chain 3 II (LC3-II) in WT, but not Mlkl−/−, mice. Exposure of cultured hepatocytes to palmitic acid (PA) increased MLKL expression and translocation to autophagosomes prior to its transit to the cell surface. Further, using an mRFP-GFP-LC3 reporter, we found that PA inhibited autophagic flux in an Mlkl-dependent, but Rip3-independent, mechanism. Taken together, these data identify an unexpected non-canonical role for MLKL in the pathogenesis of FFC diet-induced liver injury through inhibition of autophagy.
Methods and materials
Animals and FFC diet feeding
All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Mlkl−/− mice were purchased from Taconic Biosciences (#TF2780, Germantown, NY). A strain of Rip3−/− mice, in which the neomycin element used to generate the original Rip3 knock-out was deleted, was a generous gift from Vishva Dixit (Genentech, San Francisco, CA).27 Five to six week-old male Rip3+/+, Rip3−/−, Mlkl+/+ and Mlkl−/− littermates were allowed free access to standard chow or Western (FFC) diet.28
Subcellular localization of MLKL in primary and AML12 hepatocytes
Primary hepatocytes, isolated from male/female mice, and AML12 hepatocytes were cultured as described in the Supplementary Materials. After 20 h in culture, primary and AML12 hepatocytes were exposed to 500 lM PA complexed to BSA or BSA alone for up to 16 h. Subcellular localization of MLKL was assessed by confocal microscopy using specific markers.
Autophagic flux assay in hepatocytes
Autophagic flux was assessed in primary and AML12 hepatocytes using the Premo™ Autophagy Tandem Sensor mRFP-GFP-LC3 Kit (P36239, Thermo Fisher), following the manufacturer’s instructions. Appearance of LC3-positive autophagosomes and autolysosomes was assessed by confocal microscopy. Detailed methods can be found in the Supplementary Information.
Biochemical assays, immunohistochemistry and western blot
Detailed methods can be found in the Supplementary Information.
Statistical analysis
Values shown in all figures represent the means ± SEM. Analysis of variance was performed using the general linear models procedure (SAS, Carey, IN). Data were log-transformed as necessary to obtain a normal distribution. Two group comparisons were made by unpaired t test. Follow-up comparisons in 3 groups were made by least square means testing. P <0.05 were considered significant.
Results
Mlkl, but not Rip3, deficiency protected mice from FFC diet-induced liver injury, hepatocyte apoptosis and inflammatory response
Previous studies reported that Rip3−/− mice were not protected from HFD-induced liver injury.29 Here we tested the hypothesis that Mlkl−/− mice would also not be protected from FFC diet-induced liver injury. All genotypes gained more body weight on FFC diet compared to chow diet and there was no genotype effect on body weight change on either chow or FFC diet (Fig. 1A,B and Fig. S1). Unexpectedly, FFC diet feeding for 12 weeks increased aminotransferase concentrations in the plasma, as well as concentrations of hepatic triglycerides in Rip3+/+ and Mlkl+/+ littermates (WT) and Rip3−/− mice, but not in Mlkl−/− mice (Fig. 1A,B). Furthermore, histologic staining showed that liver tissues of WT and Rip3−/− mice on FFC diet displayed macro-vesicular and microvesicular steatosis, which was ameliorated in Mlkl−/− mice (Fig. 1C,D).
Fig. 1. Differential role of Rip3 and Mlkl deficiency on FFC diet-induced liver injury, steatosis, inflammatory response and hepatocyte apoptosis.
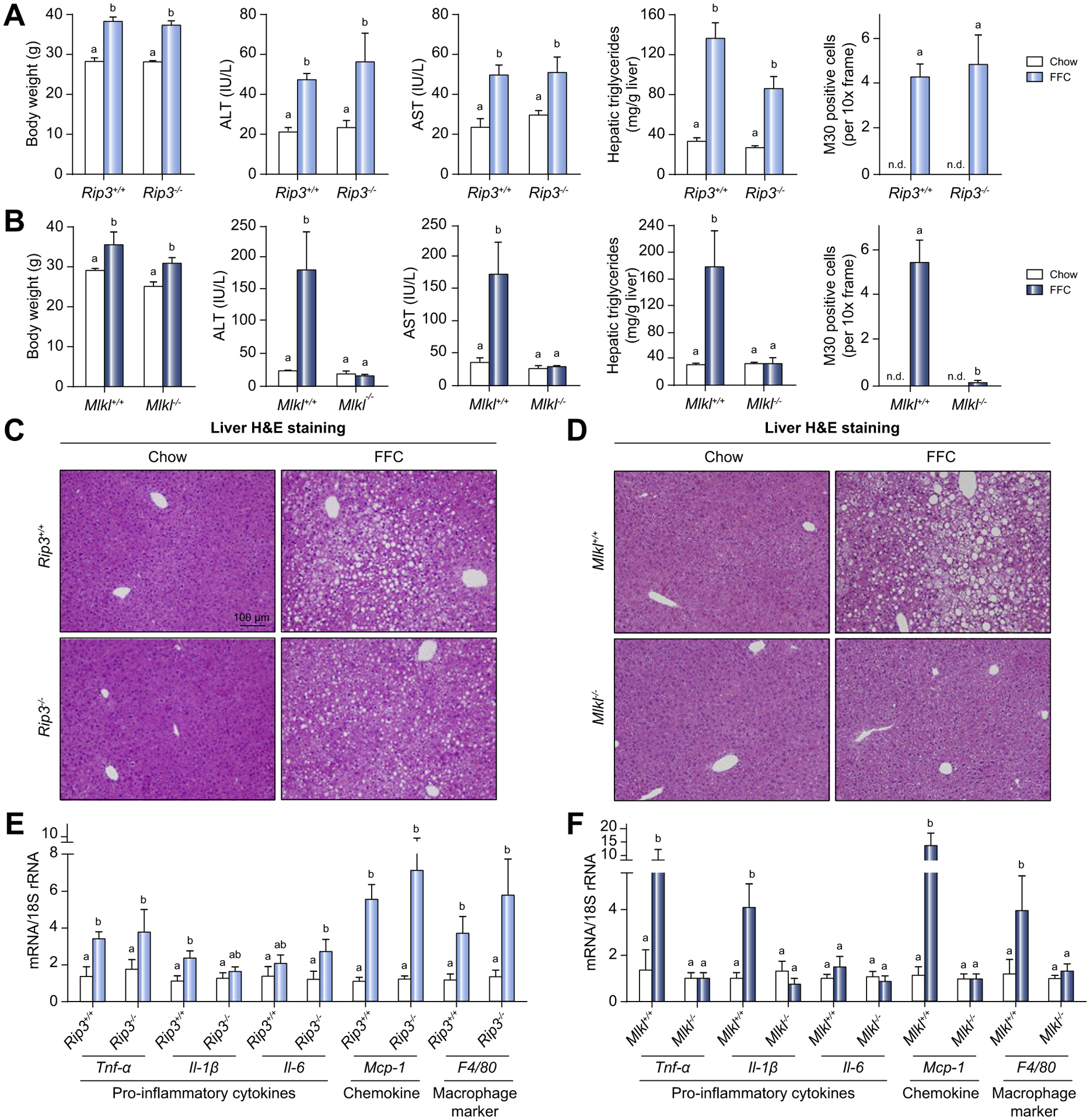
Rip3+/+, Rip3−/−, Mlkl+/+ and Mlkl−/− mice were allowed free access to FFC or chow diet for 12 weeks. Body weight, ALT and AST concentration in plasma, hepatic triglyceride content in whole liver homogenate and M30 positive cells (total number of cells per 10× frame) in formalin-fixed paraffin-embedded sections of liver from (A) Rip3+/+ and Rip3−/− and (B) Mlkl+/+ and Mlkl−/− littermates. Images of M30 are shown in Fig. S2. N.D.: M30-positive cells were not detectable in livers from chow-fed mice. H&E staining of livers from (C) Rip3+/+ and Rip3−/− and (D) Mlkl+/+ and Mlkl−/− mice on FFC or chow diet. Images were acquired at 10× magnification. Expression of mRNA for pro-inflammatory cytokines, chemokine and macrophage markers was detected in livers from (E) Rip3+/+ and Rip3−/− and (F) Mlkl+/+ and Mlkl−/− littermates using qRT-PCR and normalized to 18S rRNA. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3–6 per group. p <0.05, assessed by ANOVA. ALT, alanine aminotransferase; AST, aspartate aminotransferase; FFC, high-fat, high-fructose, high-cholesterol; qRT-PCR, quantitative reverse transcription PCR.
Given the interplay between necroptosis and apoptosis in many diseases,30,31 we investigated whether Mlkl deficiency influenced hepatocyte apoptosis. Accumulation of M30, a caspase cleavage product of CK18, is a specific marker of hepatocyte apoptosis. FFC feeding increased M30 accumulation in Rip3−/− and WT mice; this response was attenuated in Mlkl−/− mice (Fig. 1A,B and images shown in Fig. S2A–D). Mlkl−/− mice were also protected from additional markers of FFC-induced apoptosis, including the number of TUNEL-positive cells and cleavage of caspase-3 when compared to WT mice (Fig. S2E–H).
An increased inflammatory milieu in the liver is a hallmark of NAFL/NASH. FFC feeding to WT mice markedly elevated expression of mRNA for Tnf-α, Il-1β, Mcp-1, and F4/80 in the liver. Rip3 deficiency did not suppress FFC diet-induced inflammatory responses (Fig. 1E), while Mlkl deficiency prevented these strong inflammatory responses (Fig. 1F). Inflammatory responses in adipose tissue are also critical for the progression of NAFL/NASH. The presence of crown structures was apparent in both WT and Rip3−/− mice (Fig. S3A). Mlkl−/− mice had fewer crown structures than WT mice (Fig. S3B). Furthermore, Rip3 deficiency did not protect from FFC diet-induced expression of mRNA for Tnf-α, Mcp-1 and F4/80, while Mlkl deficiency prevented increased expression of mRNA for Tnf-α and F4/80 in response to FFC diet (Fig. S3C,D).
Taken together, these data indicate that Mlkl, but not Rip3, is an important contributor to FFC diet-induced liver injury, hepatocyte apoptosis and inflammatory response. Therefore, the contribution of MLKL-mediated signaling to FFC diet-induced liver injury is independent of Rip3.
MLKL expression, phosphorylation and oligomerization in livers of FFC diet-fed mice
If MLKL contributes to FFC diet-induced liver injury, we would expect FFC diet to induce MLKL expression in the liver. MLKL mRNA and protein expression was low in chow-fed mice, but was increased by FFC diet in WT and Rip3−/− mice (Fig. 2A–D). RIP3 protein expression was also low in chow-fed mice and was induced by FFC in WT, but not Mlkl−/−, mice (Fig. 2D). Liver lysates from Mlkl−/− and Rip3−/− mice were included as negative controls for western blots (Fig. 2D). In addition, phospho-MLKL immunoreactivity was increased after FFC feeding in WT and Rip3−/− livers (Fig. 2E).
Fig. 2. MLKL expression, phosphorylation and oligomerization in livers of FFC diet fed mice.

Expression of Mlkl mRNA in livers from (A) Rip3+/+ and Rip3−/− and (B) Mlkl+/+ and Mlkl−/− mice was assessed by qRT-PCR and normalized to 18S rRNA. MLKL and RIP3 protein in liver lysates from (C) Rip3+/+ and Rip3−/− and (D) Mlkl+/+ and Mlkl−/− mice was assessed by western blot and normalized to b-actin. Liver lysates were isolated from Rip3−/− mice on chow or FFC diet as a negative control for the RIP3 antibody. (E) Paraffin-embedded livers were de-paraffinized followed by phospho-MLKL staining. Images were acquired using a 10× objective. Representative images are shown (C–E). Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3–6 per group, p <0.05, assessed by ANOVA. (F) Plasma membranes and (G) subcellular fractions (cytosol, P10: 10,000 g pellet) were isolated from liver tissues, resolved by non-reducing PAGE and probed with antibody to MLKL. A longer exposure of the PM fraction is shown in panel G. Images are representative on n = 3 isolations. FFC, high-fat, high-fructose, high-cholesterol; PM, plasma membrane; qRT-PCR, quantitative reverse transcription PCR.
Necroptosis is mediated by the formation of phospho-MLKL into oligomeric, channel-like structures, which disrupt the integrity of the plasma membrane.32 To confirm the oligomerization of MLKL in response to FFC diet, plasma membrane fractions were isolated and proteins resolved on non-reducing PAGE. MLKL oligomers were detected in FFC-fed WT, as well as Rip3−/−, mice (Fig. 2F). Interestingly, only a relatively small proportion of total MLKL protein was recruited to the plasma membrane, with the majority of MLKL remaining in intracellular compartments (Fig. 2G).
PA triggered MLKL translocation to the plasma membrane and caspase-independent cell death in hepatocytes
In order to understand the mechanistic contributions of MLKL to FFC diet-induced liver injury, we modeled lipotoxicity by exposing cultured hepatocytes to PA.33 MLKL expression was low at baseline in both AML12 and primary hepatocytes. However, after 16 h exposure to PA, MLKL expression was increased and partially localized at the cell surface in AML12 (Fig. 3A) and primary hepatocytes from both WT and Rip3−/− (Fig. 3B).
Fig. 3. PA-mediated MLKL translocation to the cell surface and caspase-independent cell death.
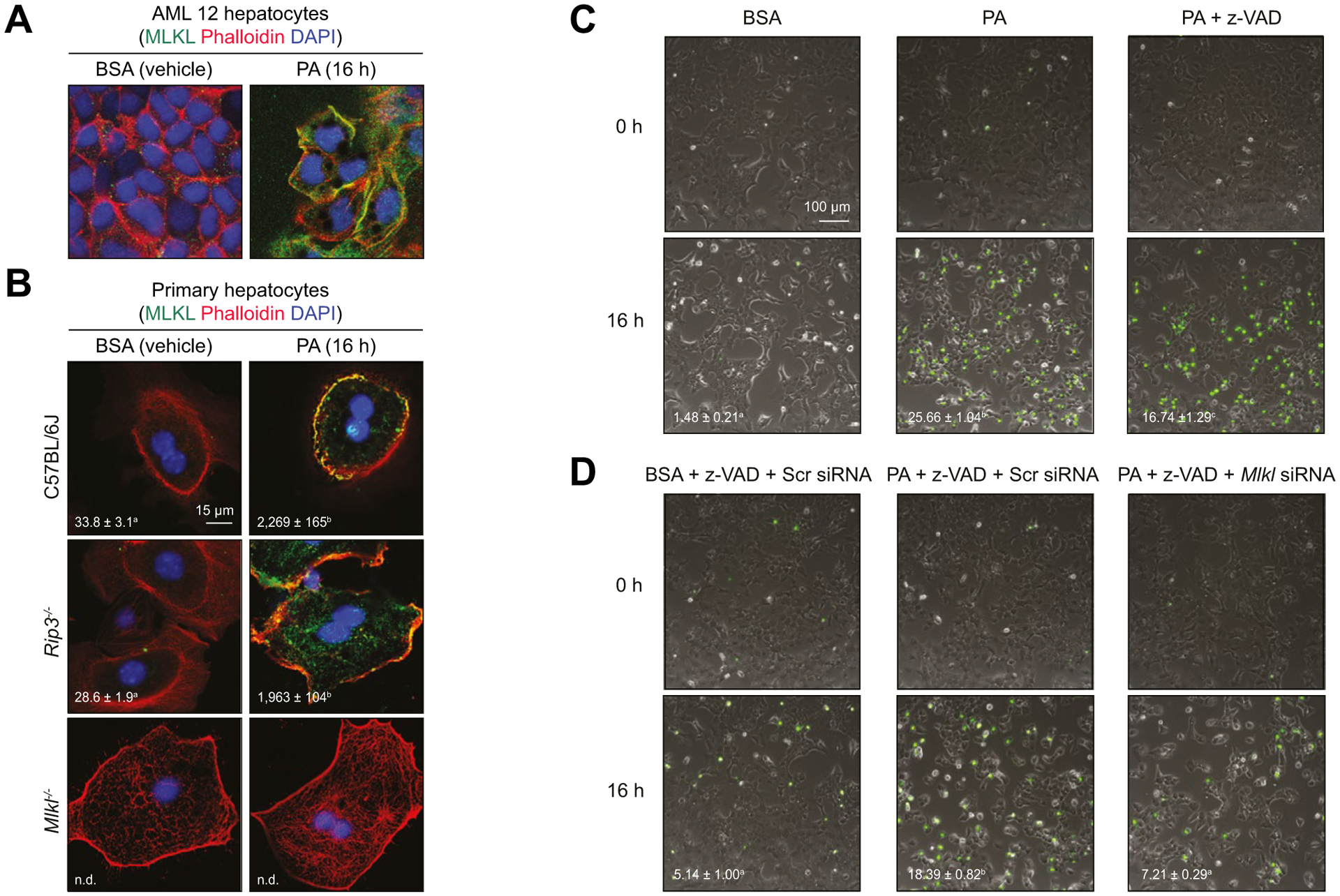
(A) AML12 and (B) primary hepatocytes isolated from C57BL/6J, Rip3−/−, and Mlkl−/− mice were exposed to 500 lM PA for 16 h. Colocalization of MLKL and Alexa Fluor-labeled phalloidin, which stains plasma membrane-associated F-actin, was examined by confocal microscopy. (C,D) Sytox Green nucleic acid staining was used to determine cell death by Incucyte live imaging analysis and quantification. (C) AML12 hepatocytes were pre-treated or not with Z-VAD and then challenged with PA or BSA (Vehicle) for 16 h. (D) AML12 hepatocytes were transfected with scrambled siRNA or siRNA targeted to knock-down Mlkl. 24 h after transfection, cells were pre-treated with z-VAD and challenged with or without PA for 16 h. All images were obtained using a 10× objective. Representative images are shown. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 4, p <0.05, assessed by t test (group = 2) or ANOVA (group ≥3). PA, palmitic acid; siRNA, small-interfering RNA.
Since cell surface localization of MLKL is associated with caspase-independent/necroptotic cell death,34 we next used Sytox green nucleic acid staining to investigate the role of MLKL in PA-mediated hepatocyte cytotoxicity. Challenging AML12 cells with PA resulted in 25% cytotoxicity over 16 h (Fig. 3C). Pre-treatment with the pan-caspase inhibitor z-VAD partially prevented PA-induced cytotoxicity (Fig. 3C), demonstrating that PA-induced cytotoxicity in hepatocytes occurred by both caspase-dependent and -independent signaling. Importantly, siRNA knock-down of Mlkl protected cells from caspase-independent cytotoxicity (Fig. 3D). These data indicate that PA triggers MLKL translocation to the cell surface and Mlkl-dependent, caspase-independent cell death.
MLKL translocated to autophagosomes and then to the plasma membrane in response to PA
We next sought to better understand the dynamics of MLKL induction and translocation in response to PA. Increased expression of MLKL was detected as early as 4 h after PA exposure in AML12 cells (Fig. 4A,B); PA-induced expression of MLKL in primary hepatocytes was independent of Rip3 (Fig. 4C). In our confocal analysis of MLKL expression, we observed that MLKL was transiently localized to intracellular compartments at 4 h and 8 h, prior to localization at the cell surface (Fig. 4A).
Fig. 4. Subcellular localization of MLKL in primary hepatocytes and AML12 hepatocytes in response to PA.

(A) AML12 hepatocytes were exposed to PA for different time intervals. Colocalization of MLKL and phalloidin was examined by confocal microscopy. Expression of MLKL protein in (B) AML12 or (C) primary hepatocytes isolated from C57BL/6J, Rip3−/−, and Mlkl−/− mice was assessed by western blot and normalized to HSC70. (D) Primary hepatocytes isolated from C57BL/6J mice were treated with PA for 8 h. Subcellular colocalization of MLKL with mitochondria, lysosomes, early endosomes, late endosomes and Golgi was examined by confocal microscopy. (E) Primary hepatocytes from C57BL/6J, Rip3−/− and Mlkl−/− mice and (F) AML12 hepatocytes were treated with PA for 8 h or 16 h. Colocalization of MLKL and mature autophagosomes (LC3) was examined by confocal microscopy. All images were obtained using a 40× objective (Zoom 4). Representative images are shown. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3–5, p <0.05, assessed by ANOVA. PA, palmitic acid.
Using markers for mitochondria, lysosomes, early endosomes, late endosomes, Golgi and autophagosomes, we found that MLKL only co-localized with LC3, a marker of autophagosomes, in primary hepatocytes treated with PA for 8 h (Fig. 4D,E). BSA-treated (vehicle) cells were shown in Fig. S4A. A similar subcellular distribution of MLKL was observed in PA-treated AML12 cells (Fig. S4B). After 16 h exposure to PA, MLKL still partially localized with LC3 in hepatocytes (Fig. 4E), consistent with the distribution of MLKL in mouse livers from FFC-fed mice (Fig. 2F,G). Colocalization of MLKL with LC3 was independent of Rip3 in primary hepatocytes (Fig. 4E). A similar subcellular distribution of MLKL was observed in PA-treated AML12 cells (Fig. 4F). These observations indicate that MLKL translocates to autophagosomes prior to the plasma membrane in response to PA.
Mlkl deficiency protected mice from FFC diet-induced accumulation of p62 and LC3-II and ER stress
Insufficient autophagic flux is associated with pathogenesis of NAFL/NASH.26 Since a recent study reported that, in addition to its canonical role in mediating necroptosis, MLKL also functions as an inhibitor of autophagic flux in dermal fibroblasts,23 we hypothesized that MLKL contributes to FFC diet-induced liver injury by regulating autophagy prior to necroptosis. FFC feeding increased the abundance of p62 and LC3-II in liver lysates from WT mice; this accumulation was reduced in Mlkl−/− mice (Fig. 5A,B).
Fig. 5. Mlkl deficiency protected mice from FFC diet- and leupeptin-induced accumulation of p62 and LC3II and ER stress.
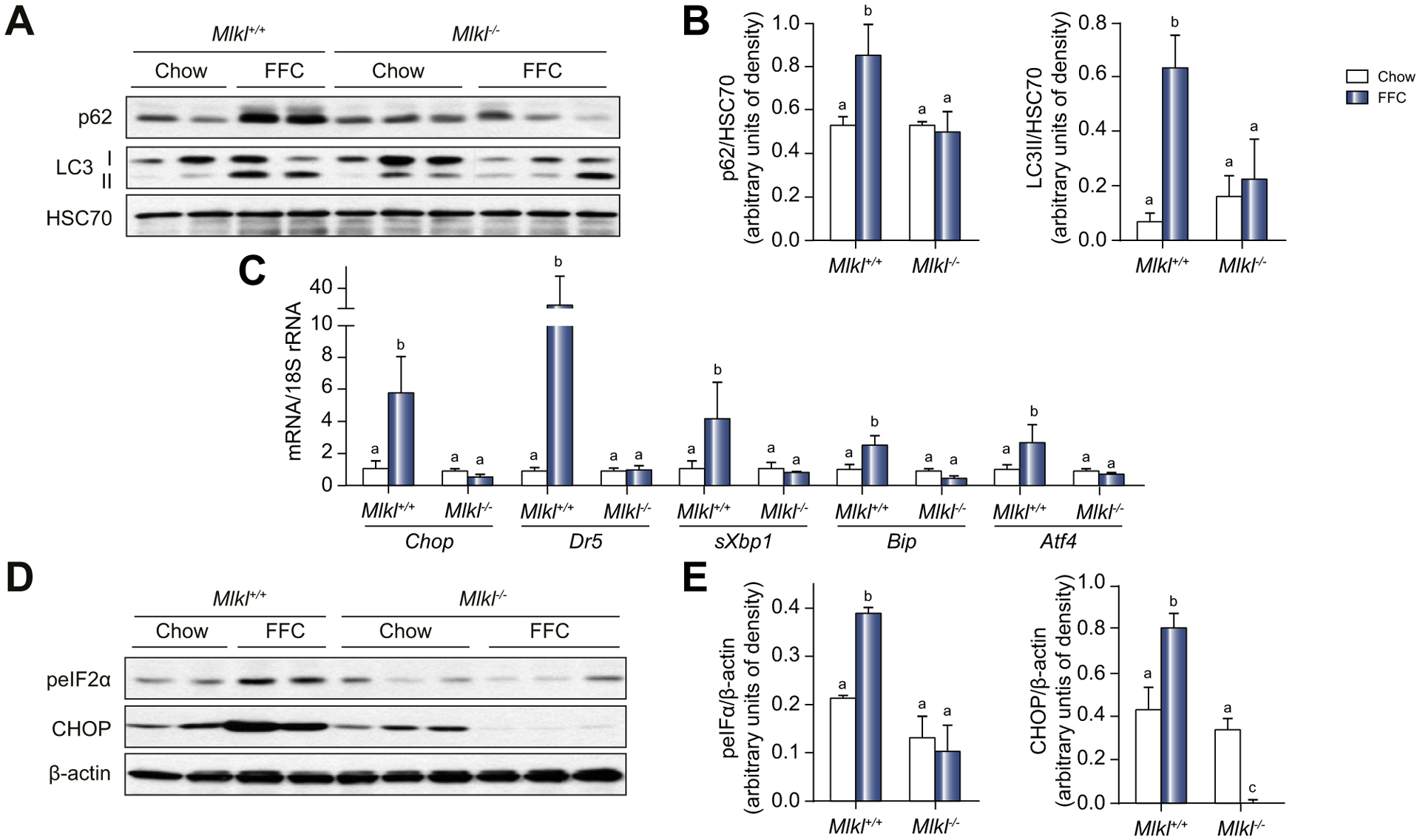
(A) Autophagy markers including p62 and LC3-II protein in liver lysates were assessed by western blot and (B) normalized to HSC70. (C) Expression of mRNA for ER stress markers including Chop, Dr5, sXbp1, Bip and Atf4 genes in the liver was assessed by qRT-PCR and normalized to 18S rRNA. (D) Phospho-eIF2a and CHOP protein in liver lysates was assessed by western blot and (E) normalized to b-actin. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3–5, p <0.05, assessed by ANOVA. ER, endoplasmic reticulum; FFC, high-fat, high-fructose, high-cholesterol; qRT-PCR, quantitative reverse transcription PCR.
Accumulation of p62 and LC3-II in vivo cannot distinguish between increased or impaired autophagic flux.26 However, insufficient autophagic flux promotes endoplasmic reticulum (ER) stress in the liver.35 Therefore, we investigated the role of Mlkl in FFC diet-induced ER stress as a surrogate indicator of impaired autophagy. FFC diet induced expression of Chop, Dr5, sXbp1, Bip and Atf4 mRNA (Fig. 5C), as well as increased phosphorylation of eIF2a and expression of CHOP protein in WT mice, but not in Mlkl−/− mice (Fig. 5D,E).
Inhibition of autophagic flux in PA-treated hepatocytes
Making use of cultured AML12 hepatocytes, we investigated the impact of PA on autophagy. PA markedly elevated p62 and LC3-II levels within 8 h (Fig. 6A). Bafilomycin A1 (Baf A) suppresses lysosomal enzyme activity and blocks the fusion of autophagosomes with lysosomes. As expected, Baf A treatment increased p62 and LC3-II; however, Baf A did not further increase p62 or LC3-II in PA-treated cells (Fig. 6A). To monitor autophagic flux per se, a tandem labeled mRFP-GFP-LC3 reporter was used (Fig. 6B). In AML12 hepatocytes challenged with chloroquine (CQ), an inhibitor of flux, the number of autophagosomes (yellow-fluorescent puncta) increased, while with rapamycin (Rapa), an enhancer of flux, autolysosomes (red-fluorescent puncta) were predominant (Fig. 6C,D). Treatment of hepatocytes with PA increased yellow-fluorescent puncta and decreased red-fluorescent puncta after 4 h exposure (Fig. 6C,D). These data indicate that accumulation of p62 and LC3-II in PA-challenged hepatocytes is primarily due to impaired autophagic degradation, rather than increased autophagosome formation.
Fig. 6. Inhibition of autophagy PA-treated hepatocytes.
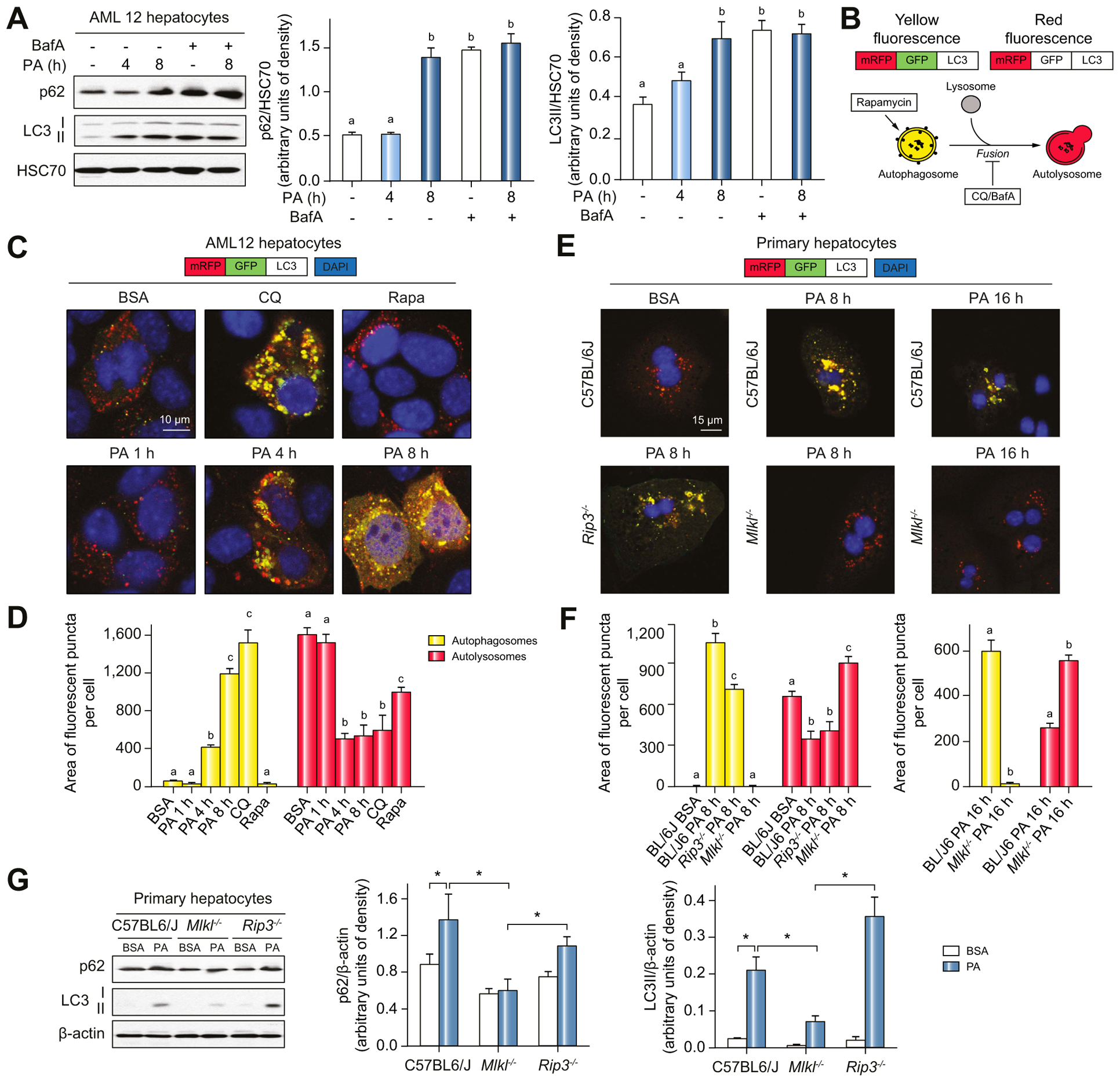
(A) AML12 hepatocytes were exposed to PA for 4–8 h in the presence or absence of BafA. p62 and LC3-II protein in liver lysates was assessed by western blot and normalized to HSC70. (B) Schematic of the different possible outcomes for the mRFP-GFP-LC3 autophagic flux reporter. (C) Confocal analysis and (D) quantification of AML12 cells infected with mRFP-GFP-LC3. Cells were then treated with PA, CQ or Rapa. (E) Primary hepatocytes isolated from C57BL/6J, Rip3−/− and Mlkl−/− mice were transduced with mRFP-GFP-LC3 and then exposed to PA for 8 h or 16 h. (E) Confocal analysis and (F) quantification of LC3 reporter. All images were obtained using a 40× objective (Zoom 4). (G) p62 and LC3-II protein in lysates of primary hepatocytes was assessed by western blot and normalized to b-actin. Values represent means ± SEM. Values with different superscripts are significantly different from each other within the same color bars, n = 3–5, p <0.05, assessed by t test (group = 2) or ANOVA (group≥3). BafA, Bafilomycin A1; CQ, chloroquine; PA, palmitic acid; Rapa, rapamycin.
Mlkl, but not Rip3, deficiency prevented inhibition of autophagy by PA in hepatocytes
Using the mRFP-GFP-LC3 reporter, we next investigated the impact of MLKL on autophagic flux in PA-treated hepatocytes. In primary hepatocytes, Mlkl, but not Rip3, deficiency prevented inhibition of autophagic flux (Fig. 6E,F) and accumulation of p62 and LC3-II by PA (Fig. 6G).
Inhibition of autophagic flux by PA was also prevented in AML12 cells transfected with Mlkl siRNA compared to scrambled siRNA (Fig. 7A,C), while overexpression of Mlkl autonomously blocked autophagy in AML12 hepatocytes without PA (Fig. 7B,D). In addition, accumulation of LC3-II by PA in AML12 cells was reduced when Mlkl was knocked-down (Fig. 7E). Conversely, expression of LC3-II protein was elevated in AML12 cells transfected with Mlkl overexpression plasmid compared to empty vector (Fig. 7F). These data suggest that PA-impaired autophagy in hepatocytes is driven by Mlkl-dependent signaling.
Fig. 7. Inhibition of autophagic flux by PA in hepatocytes was dependent on MLKL.
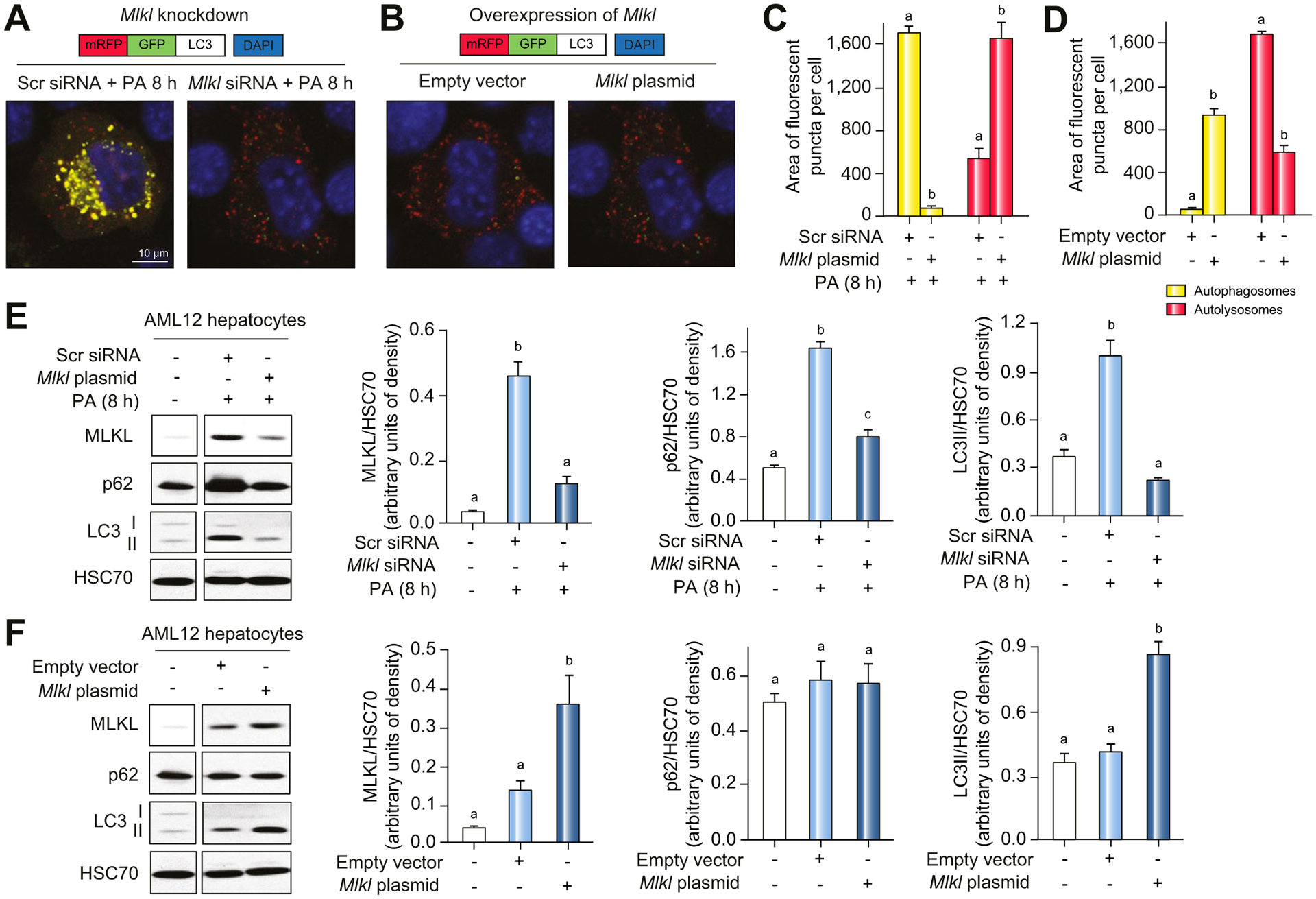
AML12 hepatocytes were transduced with mRFP-GFP-LC3 and transfected with scrambled siRNA or Mlkl siRNA empty vector or Mlkl overexpression plasmid. LC3 localization was visualized by confocal microscopy (A,B) and quantified (C,D). All images were obtained using a 40× objective (Zoom 4). (E,F) p62 and LC3-II protein in cell lysates was assessed by western blot and normalized to HSC70. Values represent means ± SEM. Values with different superscripts are significantly different from each other within the same color bars, n = 3, p <0.05, assessed by t test (group = 2) or ANOVA (group ≥3). PA, palmitic acid; siRNA, small-interfering RNA.
Interrelationship between autophagy and MLKL expression
Accumulating evidence suggests that autophagic and necroptotic pathways can influence each other.36,37 Since our in vivo data suggest that MLKL contributes to FFC diet-induced liver injury by inhibiting hepatic autophagy (Fig. 5), we investigated whether pharmacological regulators of of autophagy would also impact expression of MLKL. In AML12 hepatocytes, pharmacologic inhibition of autophagy by CQ induced MLKL expression (Fig. 8A) and movement to the cell surface (Fig. 8B), while induction of autophagy by rapamycin in hepatocytes had no effect on MLKL expression (Fig. 8A,B). Interestingly, the absence of MLKL also prevented accumulation of yellow-fluorescent puncta by CQ in primary hepatocytes (Fig. 8C), suggesting that inhibition of autophagic flux by CQ is, at least partially, dependent on Mlkl.
Fig. 8. Interrelationship between autophagy and MLKL expression.
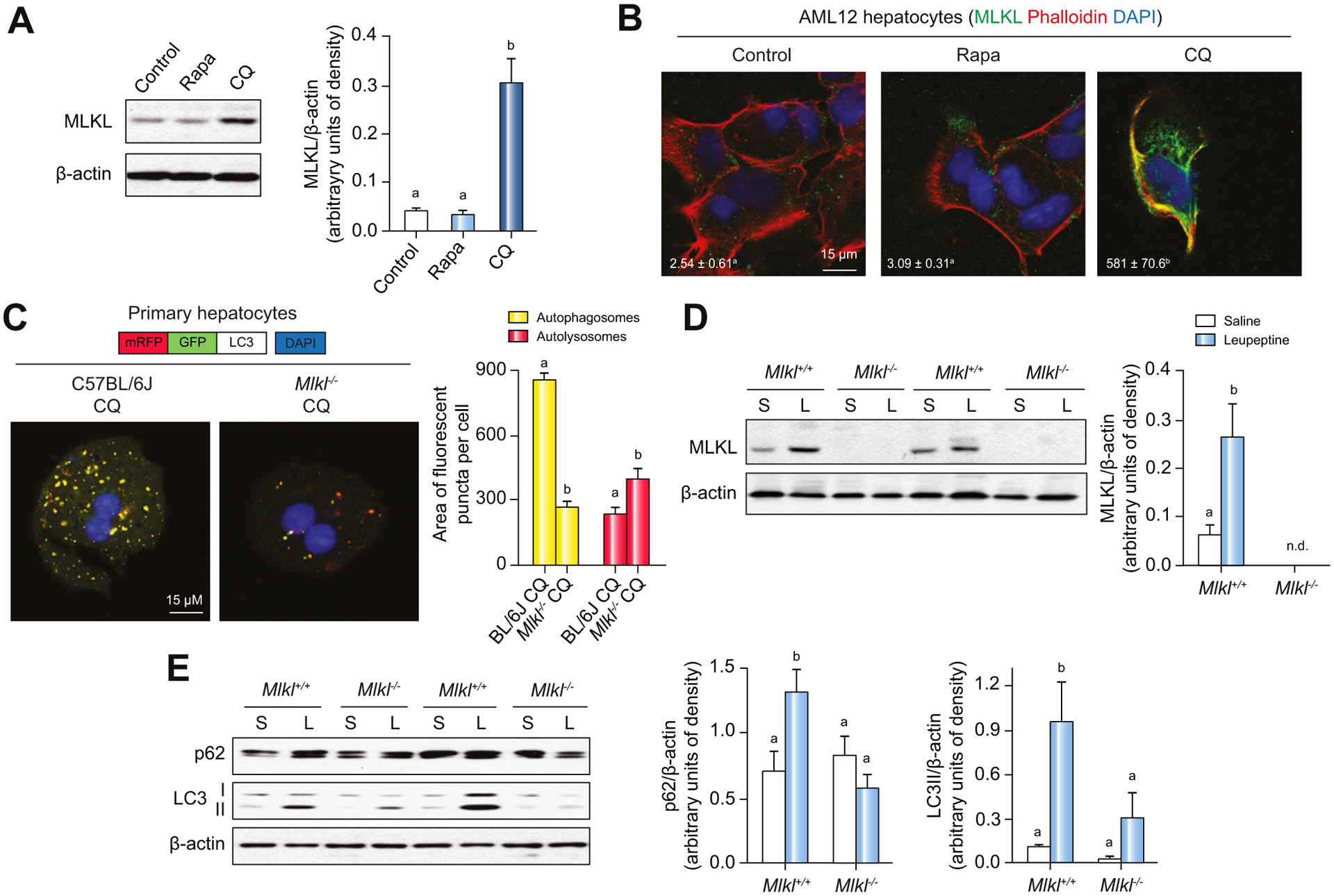
(A,B) AML12 hepatocytes were treated with CQ or Rapa for 24 h. (A) Expression of MLKL protein in cell lysates was assessed by western blot and normalized to b-actin. (B) Colocalization of MLKL and phalloidin in AML12 hepatocytes was examined by confocal microscopy. (C) Primary hepatocytes isolated from C57BL/6J and Mlkl−/− mice were transduced with mRFP-GFP-LC3 and exposed to CQ. Yellow and red fluorescent puncta were visualized by confocal microscopy and quantified. All images were obtained using a 40× objective (Zoom 4). (D,E) Mlkl−/− mice and their littermates were intraperitoneally injected with leupeptin (L) or saline (S) 4 h prior to euthanasia. MLKL protein (D) as well as accumulation of p62 and LC3-II (E) in liver lysates was assessed by western blot and normalized to b-actin. Representative images are shown. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3, p <0.05, assessed by t test (group = 2) or ANOVA (group ≥3). CQ, chloroquine; Rapa, rapamycin.
Injection of WT mice with leupeptin, a protease inhibitor that reduces autophagic flux, also induced MLKL expression in the liver (Fig. 8D). As expected, when WT mice were exposed to leupeptin, both p62 and LC3-II accumulated in the liver (Fig. 8E). In contrast, inhibition of autophagy by leupeptin was reduced in Mlkl−/− mice (Fig. 8E), similar to the involvement of Mlkl in CQ-mediated inhibition of autophagic flux in primary hepatocytes (Fig. 8C). Taken together, with impaired autophagic flux when Mlkl was overexpressed (Fig. 7B,D,F), these data indicate that increased expression of MLKL is linked with inhibition of autophagy and that, conversely, MLKL contributes to inhibition of autophagy in response to authophagy inhibitors, including CQ and leupeptin, as well as overexpression plasmid.
Discussion
Necroptosis is a form of programmed cell death; the canonical pathway requires activation of MLKL by RIP3.7,8 Here we report that an unexpected function of MLKL contributes to the development of liver injury in FFC dietary model of NAFL/NASH. Mlkl-dependent injury was independent of Rip3 and associated with an inhibition of autophagy. Utilizing a hepatocyte culture model of PA-induced lipotoxicity, we observed that PA induced MLKL expression and translocation, first to autophagosomes and then to the cell surface; some MLKL remained at the autophagosomes even as MLKL localized at the plasma membrane and cells underwent caspase-independent cell death. Lipotoxicity was associated with an inhibition of autophagic flux that was dependent on Mlkl, but not Rip3. Importantly, pharmacologic inhibition of autophagy also increased MLKL expression and translocation to the cell surface, suggesting a critical link between regulation of autophagic flux, expression of MLKL and hepatocellular injury.
In canonical RIP3-MLKL signaling, MLKL is phosphorylated by RIP3 and subsequently translocates to the plasma membrane where it oligomerizes to trigger necroptosis.38 Here we found that the FFC diet induced expression, phosphorylation and oligomerization of MLKL in the liver, even in the absence of Rip3, suggesting a non-canonical pathway of activation. MLKL also mediates a number of non-canonical functions that are largely dependent on the subcellular localization of MLKL.39 For example, MLKL can associate with mitochondria, likely stimulating the generation of mitochondrial reactive oxygen species.39 MLKL, along with RIP1 and RIP3, transiently localized to the nucleus in HT29 cells; however, the function of MLKL in the nucleus is not known.40 Importantly, MLKL inhibited autophagy in several cell models. For example, translocation of MLKL to autolysosomal membranes in response to necroptotic stimuli inhibited autophagic flux in mouse dermal fibroblasts and HT29 cells23 and MLKL suppressed autophagic flux in endothelial and smooth muscle cells in response to challenge with oxidized low density lipo-protein.41 In both primary and AML12 hepatocyte cultures, MLKL was predominantly localized to autophagosomes at 8 h, while at 16 h after PA challenge, MLKL was also localized to the cell surface. While a minor fraction of MLKL was detected in nuclei and peri-nuclear compartments, MLKL did not localize to mitochondria or other intracellular membrane compartments in response to PA. Taken together, PA-induced colocalization of MLKL with autophagosomes is consistent with a role of MLKL in regulating autophagy in response to lipotoxicity.
Importantly, we found that interventions that inhibited autophagy, including FFC diet and leupeptin in vivo, as well as PA or CQ in cultured cells, were associated with increased expression of MLKL. These data are consistent with the increase in TNF-induced necroptosis in L929 cells observed when autophagy was inhibited with 3-methyladenine or Beclin 1 siRNA.42 However, very little is known about the regulation of MLKL expression either at the transcriptional or post-transcriptional level. Long non-coding RNA (lncRNA)-FA2H-2 was found to interact with the Mlkl promoter, downregulating its expression in endothelial and smooth muscle cells in models of atherosclerosis.41 In contrast, interferon (IFN)-γ-dependent signals stimulated transcription of MLKL in a STAT1-dependent manner in hepatocytes43 and cancer cells.44 The interplay between autophagy and MLKL activation is likely complex and cell and/or stimulus specific. For example, Atg5 or Atg7 deficiency has no effect on MLKL activation and necroptotic cell death in mouse dermal fibroblasts (21), but Atg5 was key for efficient necrosome activation in TRAIL-treated Tak1-null mouse prostate epithelial cells.45
Autophagy is a critical physiological process that serves to remove potentially injurious intracellular components and maintain homeostasis. Impaired autophagy contributes to various pathophysiological processes including inflammatory responses, ER stress, cell injury and death.37,46,47 Dynamic regulation of autophagic flux is associated with the evolution of NAFL/NASH, but the impact of autophagic flux in disease progression is not well understood.26,48 However, activation of autophagy, by either hepatic overexpression of Atg749 or compounds including the mTOR inhibitor rapamycin,50 is considered as a potential therapeutic strategy against hepatic injury. Here we reported that FFC diet-induced obesity in mice led to accumulation of p62 and LC3-II in an Mlkl-dependent mechanism. Using cell culture models of lipotoxicity, we found that PA blocked the fusion of autophagosomes with lysosomes in hepatocytes. Inhibition of autophagy in cultured hepatocytes in response to PA was dependent on Mlkl. It is also likely that MLKL disrupts autophagy outside of the liver. For example, autophagy plays an important role in adipose homeostasis.51 Extrahepatic functions of MLKL could contribute to organ-organ interactions important for the progression of NAFL/NASH, such as interactions with adipose tissue and the intestine, which require further investigation.
Interestingly, overexpression of Mlkl autonomously blocked autophagy in cultured hepatocytes. MLKL was also involved in the regulation of autophagy by pharmacologic inhibitors, including leupeptin in vivo and CQ in hepatocytes. These data suggest that induction of MLKL by these agents (Fig. 8) leads to the association of MLKL with autolysosomes, as observed in dermal fibroblasts23 and/or autophagosome, resulting in a vicious circle that exacerbates the inhibition of autophagy. Taken together, these data demonstrate for the first time a critical role of MLKL in the regulation of autophagy in response to FFC diet, the protease inhibitor leupeptin or hepatocyte lipotoxicity.
Consistent with our observations of a RIP3-independent role of MLKL in FFC diet-induced liver injury, accumulating evidence indicates that MLKL can be activated by alternative, RIP3-independent pathways. In a mouse model of encephalitis, while Rip3−/− mice were not protected from mortality, either Mlkl−/− mice or Mlkl−/−Casp8−/− mice were protected.52 Models of autoimmune hepatitis provided another example that MLKL triggered IFN-γ-mediated programmed hepatocellular necrosis even in the absence of Rip3.43 Interestingly, one study identified that ubiquitination of MLKL by E3 ligases was associated with MLKL activation.19 Given the important role of E3 ligases and ubiquitination in regulation of autophagy, it will be interesting to determine if changes in MLKL ubiquitination are required for its activation when autophagy is impaired.
In summary, these data indicate that Mlkl-dependent, but Rip3-independent, signaling contributes to FFC diet-induced liver injury and inflammatory response, by inhibiting autophagy and inducing necroptotic cell death. This study thus identifies a novel co-regulatory mechanism between necroptotic and autophagic pathways in the development of NAFL/NASH.
Supplementary Material
Highlights.
MLKL-mediated signaling contributes to FFC diet-induced liver injury.
FFC diet or palmitic acid treatment induces MLKL expression in hepatocytes.
Palmitic acid drives MLKL translocation to autophagosomes independently of Rip3.
Mlkl, but not Rip3, regulates autophagic flux in a murine model of NAFL/NASH.
Pharmacologic inhibition of autophagy induces MLKL expression.
Acknowledgments
We thank John Peterson from the Cleveland Clinic Lerner Research Institute Imaging Core, who provided microscopy and image analysis services.
Financial support
This work was supported in part by National Institutes of Health (NIH) grants, United States; P50 AA024333 (LEN), R21 AA020941 and P30 DK097348 (Pilot project) (SR), R21 AR071046, RO1 GM119174, RO1 DK113196 and UO1 AA021890 (SD) and K99 AA026648 (KLP). This work utilized the Leica SP5 confocal/multi-photon microscope that was purchased with partial funding from NIH SIG grant 1S10RR026820–01.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BafA
Bafilomycin A1
- CQ
chloroquine
- ER
endoplasmic reticulum
- FFC
high-fat, high-fructose, high-cholesterol
- HFD
high-fat diet
- IFN
interferon
- LC3-II
microtubule-associated protein 1 light chain 3 II
- MCD
methionine- and choline-deficient
- MLKL
mixed lineage kinase domain like pseudokinase
- NAFL
non-alcoholic fatty liver
- NASH
non-alcoholic steatohepatitis
- PA
palmitic acid
- PM
plasma membrane
- qRT-PCR
quantitative reverse transcription PCR
- Rapa
rapamycin
- RIP3
receptor interacting protein kinase 3
- siRNA
small-interfering RNA
- TNF
tumor necrosis factor
Footnotes
Conflict of interest
The authors declare no Conflicts of interest that pertain to this work.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2020.03.023.
References
- [1].Gudipaty SA, Conner CM, Rosenblatt J, Montell DJ. Unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu Rev Cell Dev Biol 2018;34:311–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583–594. [DOI] [PubMed] [Google Scholar]
- [3].Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008;133:693–703. [DOI] [PubMed] [Google Scholar]
- [4].Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015;15:362–374. [DOI] [PubMed] [Google Scholar]
- [5].Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gabetaler N, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology (Baltimore, Md) 2013;57:2189–2201. [DOI] [PubMed] [Google Scholar]
- [6].Anstee QM, Concas D, Kudo H, Levene A, Pollard J, Charlton P, et al. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol 2010;53:542–550. [DOI] [PubMed] [Google Scholar]
- [7].Linkermann A, Green DR. Necroptosis. N Engl J Med 2014;370:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 2017;18:127–136. [DOI] [PubMed] [Google Scholar]
- [9].Dara L The receptor interacting protein kinases in the liver. Semin Liver Dis 2018;38:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kondylis V, Pasparakis M. RIP kinases in liver cell death, inflammation and cancer. Trends Mol Med 2019;25:47–63. [DOI] [PubMed] [Google Scholar]
- [11].Kaplowitz N, Win S, Than TA, Liu ZX, Dara L. Targeting signal transduction pathways which regulate necrosis in acetaminophen hepatotoxicity. J Hepatol 2015;63:5–7. [DOI] [PubMed] [Google Scholar]
- [12].Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013;58:2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) 2015;129:721–739. [DOI] [PubMed] [Google Scholar]
- [14].Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013;57:1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, et al. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016;7:17681–17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 2015;6:e1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology (Baltimore, Md) 2016;64:1518–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 2017;47:51–65.e57. [DOI] [PubMed] [Google Scholar]
- [21].Xu H, Du X, Liu G, Huang S, Du W, Zou S, et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab 2019;23:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A 2017;114:E961–E969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Frank D, Vaux DL, Murphy JM, Vince JE, Lindqvist LM. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J Cell Sci 2019;132:jcs220996. [DOI] [PubMed] [Google Scholar]
- [24].Vandenabeele P, Riquet F, Cappe B. Necroptosis: (Last) message in a bubble. Immunity 2017;47:1–3. [DOI] [PubMed] [Google Scholar]
- [25].Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab 2016;27:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci 2016;61:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014;343:1357–1360. [DOI] [PubMed] [Google Scholar]
- [28].Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig Dis Sci 2016;61:1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun 2016;7:11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Green DR. The coming decade of cell death research: five riddles. Cell 2019;177:1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A 2014;111:15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007;46:823–830. [DOI] [PubMed] [Google Scholar]
- [34].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of nonapoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135–147. [DOI] [PubMed] [Google Scholar]
- [35].Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 2014;5:e1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 2015;16:329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev 2012;249:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014;54:133–146. [DOI] [PubMed] [Google Scholar]
- [40].Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ 2016;23:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guo FX, Wu Q, Li P, Zheng L, Ye S, Dai XY, et al. The role of the LncRNAFA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ 2019;26:1670–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2011;2:e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest 2016;126:4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Knuth AK, Rosler S, Schenk B, Kowald L, van Wijk SJL, Fulda S. Interferons transcriptionally up-regulate MLKL expression in cancer cells. Neoplasia 2019;21:74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell 2016;37:337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 2019;8:e44452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol 2011;5:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 2013;58:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009;119:3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 2017;169:301–313.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.