

SUMMARY
Cytokinesis partitions the cell contents to complete mitosis. During cytokinesis, polo-like kinase 1 (PLK1) activates the small GTPase RhoA to assemble a contractile actomyosin ring. PLK1 is proposed to pattern RhoA activation by creating a docking site on the central spindle that concentrates the RhoA Guanine nucleotide Exchange Factor ECT2. However, ECT2 targeting to the central spindle is dispensable for cytokinesis, indicating that how PLK1 controls RhoA activation remains unresolved. To address this question, we employed an unbiased approach targeting ~100 predicted PLK1 sites in two RhoA regulators: ECT2 and the centralspindlin complex, comprised of CYK4 and kinesin-6. This comprehensive approach suggested that the only functionally critical PLK1 target sites are in a single cluster in the CYK4 N-terminus. Phosphorylation of this cluster promoted direct interaction of CYK4 with the BRCT repeat module of ECT2. However, mutational analysis in vitro and in vivo led to the surprising finding that the interaction was independent of the conserved “canonical” residues in ECT2’s BRCT repeat module that, based on structurally characterized BRCT-phosphopeptide interactions, were presumed critical for binding. Instead, we show that the ECT2 BRCT module binds phosphorylated CYK4 via a distinct conserved basic surface. Basic surface mutations mimic the effects on cytokinesis of loss of CYK4 cluster phosphorylation or inhibition of PLK1 activity. Together with evidence for ECT2 autoinhibition limiting interaction with CYK4 in the cytoplasm, these results suggest that a spatial gradient of phosphorylated CYK4 around the central spindle patterns RhoA activation by interacting with ECT2 on the adjacent plasma membrane.
Keywords: cytokinesis, cell division, RhoA, small GTPase, ECT2, MgcRacGAP, RACGAP1, centralspindlin
eTOC
Gomez-Cavazos et al. show that the key function of PLK1 kinase in RhoA activation during cytokinesis is phosphorylation of the CYK4 N-terminus. Phospho-CYK4 binds to a conserved basic surface in the guanine nucleotide exchange factor ECT2. This site is essential for cytokinesis and is distinct from the canonical BRCT phospho-peptide binding site.
Graphical Abstract
INTRODUCTION
During cytokinesis, constriction of a cortical actomyosin contractile ring partitions the cell contents [1–3]. To ensure coordination with chromosome segregation, the equatorial zone of active RhoA that directs contractile ring assembly is dynamically positioned by the anaphase spindle [4–7]. Positioning is achieved in part by signals controlling RhoA activation that emanate from antiparallel microtubule bundles, collectively called the central spindle, that form between the segregating chromosomes. Central spindle assembly requires the microtubule bundling protein PRC1 (SPD-1 in C. elegans), the chromosomal passenger complex (CPC), and the centralspindlin complex, which consists of the kinesin-6 motor MKLP1 (ZEN-4 in C. elegans) and CYK4/RACGAP1/MgcRacGAP (CYK-4 in C. elegans) [1, 7–9].
Polo-like kinase 1 (PLK1) localizes to the central spindle [10] and is essential for RhoA activation and contractile ring assembly [11–13]. The current model is that PLK1 phosphorylates CYK4 to create a central spindle docking site that binds and activates the RhoA Guanine nucleotide Exchange Factor (GEF) ECT2 [14–16]. However, two pieces of evidence argue against this ECT2-docking model. First, ECT2 does not localize to the central spindle in all metazoan species with RhoA-dependent cytokinesis; for example, ECT2 does not localize to the central spindle in C. elegans [8]; see Figure 2A). Second, mutations in human ECT2 that prevent it from localizing to the central spindle do not perturb cytokinesis [17, 18]. In human cells, phosphorylation of the CYK4 N-terminus by PLK1 promotes its interaction with the ECT2 BRCT repeat module [14, 16]. Mutation of residues in the second ECT2 BRCT domain (T153A/K195M) analogous to residues that form the phosphate binding site in the tandem BRCT domains of BRCA1 and MDC1 [19], abolish the ability of ECT2 to immunoprecipitate phosphorylated CYK4 and to localize to the central spindle [16, 18]. However, the ECT2 T153A/K195M mutant remains fully capable of supporting cytokinesis [18]. Thus, even in human cells, concentration of ECT2 at the central spindle is not required for cytokinesis, suggesting that how PLK1 activates RhoA during cytokinesis remains unresolved.
Figure 2. An unbiased screen identifies a single PLK-1 target site cluster important for cytokinesis in the CYK-4 N-terminus.

(A) Stills from a timelapse sequence of an embryo expressing transgene-encoded NeonGreen::ECT-2 and ZEN-4::mScarlet, endogenous ECT-2 and ZEN-4 were depleted. Panels below are 3X-higher magnification views of the central spindle. Scale bars are 10 μm and 2 μm. (B) Plot of mean fluorescence intensity for both markers, done as in Figure 1A. (C) Summary of candidate PLK-1 site screening in ZEN-4, CYK-4 and ECT-2. (D–F) (top) Schematics of ZEN-4, CYK-4 and ECT-2. Roman numerals mark clusters of candidate PLK-1 sites mutated in each transgene. (bottom) Plot of embryonic lethality (mean ± SD) after depletion of endogenous ZEN-4, CYK-4 or ECT-2 in strains expressing indicated mutants. N is number of worms and n the number of embryos scored. (G) Sequence alignment of the N-terminal region of C. elegans CYK-4 (162–188aa) containing the 4 functionally important PLK-1 sites. Green arrows mark PLK1 sites previously identified in human CYK4 [16]. (H) Plot of embryonic lethality (mean ± SD) after depletion of endogenous CYK-4 in strains expressing indicated mutants. N is number of worms and n the number of embryos scored. See also Figures S2 and S3, Video S1 and Table S1.
Here, we investigate how PLK1 controls RhoA activation in cytokinesis by taking an unbiased approach in the C. elegans embryo. Our results identify a non-canonical interaction mode between PLK1-phosphorylated CYK4 and a conserved basic surface in the ECT2 BRCT repeat module that is essential for cytokinesis in C. elegans and human cells. In C. elegans, autoinhibition prevents ECT-2 from interacting with phosphorylated CYK-4 at the central spindle. Our results suggest that a spatial gradient of phosphorylated CYK4 around the central spindle activates ECT2 on the equatorial plasma membrane to promote RhoA activation and cytokinesis.
RESULTS
PLK-1 localizes to the central spindle and is essential for RhoA activation during cytokinesis in C. elegans
Like its human homolog [10], in situ-tagged C. elegans PLK-1 [20] localizes to the central spindle (Figure 1A; Video S1). To enable acute PLK-1 perturbation in cytokinesis, we introduced a pair of mutations into the endogenous plk-1 locus that render it sensitive to the ATP analog 1NM-PP1 (PLK1AS, Figure S1A; [21]). After addition of 1NM-PP1 to permeabilized embryos (perm-1(RNAi); [22]) immediately prior to anaphase onset, embryos expressing PLK-1WT completed cytokinesis with normal kinetics. In contrast, embryos expressing PLK-1AS exhibited severe cytokinesis impairment (Figure 1B,C; Figure S1B–D; Video S2) with furrows frequently failing to bisect the central spindle (Figure S1D). In human cells, PLK1 activates RhoA for contractile ring assembly [11–13]. If C. elegans PLK-1 is similarly required for RhoA activation, its function could be partially masked by the presence of the nematode-specific RhoA activator NOP-1. NOP-1 is required for cortical contractility that reinforces polarity establishment prior to mitosis [23, 24]. NOP-1 is not essential for cytokinesis, but its presence augments contractility by providing a central spindle-independent pool of active RhoA [24]. Depleting NOP-1 had no effect on furrow ingression in 1NM-PP1-treated PLK-1WT embryos but completely blocked furrowing in PLK-1AS embryos (Figure 1B,C). Thus, furrow ingression in PLK-1-inhibited embryos is due to RhoA activation by NOP-1.
Figure 1. PLK-1 localizes to the central spindle and is essential for RhoA activation during cytokinesis in C. elegans.

(A) (left) Stills from a timelapse sequence of an embryo expressing in situ-tagged PLK-1::GFP and the central spindle marker ZEN-4::Scarlet. Panels below are 3X-higher magnification views of the central spindle. Scale bars: 10 μm and 2 μm. (right) Plot of mean fluorescence intensity along a 25 pixel-wide line scan of the central spindle 160 seconds after anaphase onset. Error bars are the SE. (B) Images from timelapse sequences of permeabilized one-cell embryos expressing untagged wild-type (WT) or analog-sensitive (AS) PLK-1 and the indicated fluorescent markers. 1-NM-PP1, which inhibits analog-sensitive PLK-1, was added at metaphase (red arrowhead). Embryos were imaged without (top rows) or with (bottom rows) depletion of NOP-1. Scale bar, 10 μm. (C) Individual traces of furrow width in 1-NM-PP1-treated embryos expressing PLK-1WT (grey) or PLK-1AS (red), without (left) or with (right) NOP-1 co-depletion. Asterisks mark furrow diameter before regression. See also Figure S1 and Videos S1 and S2.
An unbiased screen identifies a single essential PLK-1 target site cluster in the CYK-4 N-terminus
The primary function proposed for PLK1 is phosphorylating CYK4 to create a central spindle docking site that binds and activates ECT2 [14, 16]. However, this model has been challenged by recent work in human cells [18] and the suggestion that ECT-2 does not localize to the central spindle in C. elegans [8]. Consistent with this prior report, a functional mNeonGreen::ECT-2 fusion (Figure S2A) localized to the plasma membrane but not to the central spindle marked by ZEN-4::mScarlet (Figure 2A,B; Video S1). The association of human ECT2 with the plasma membrane, mediated by its C-terminal pleckstrin homology (PH) domain and polybasic cluster, is essential for cytokinesis [15]. Similarly, deleting the PH domain of C. elegans ECT-2 prevented its plasma membrane targeting and abrogated furrowing (Figure S2B–D). Thus, C. elegans ECT-2 localizes to the plasma membrane but not the central spindle, indicating that PLK-1 does not promote RhoA activation by recruiting ECT-2 to the central spindle.
To understand how PLK-1 activates RhoA, we performed an unbiased screen mutating candidate PLK-1 sites in three proteins implicated in RhoA activation during cytokinesis: ECT-2 and the centralspindlin components CYK-4 and ZEN-4 (Figure 2C–F). Strains with untagged single-copy RNAi-resistant transgenes encoding WT ECT-2, CYK-4, and ZEN-4 were generated, along with 23 additional strains mutating 98 candidate PLK-1 sites in the three proteins to non-phosphorylatable alanines in regional clusters (Figure 2C–F; Figure S3A–H; Table S1). The functionality of mutant transgenes was assessed by measuring embryonic lethality after endogenous protein depletion. Our screen identified two putative PLK-1 site clusters that compromised embryonic viability when mutated, one in the N-terminal half of ZEN-4 (ZEN-4 cluster III; Figure 2D) and one in the N-terminal half of CYK-4 (CYK-4 cluster II; Figure 2E). The ZEN-4 cluster III transgene, which mutates eight S/T residues in the ZEN-4 motor domain, had only a minor effect on furrow closure (Figure S3J) despite reducing ZEN-4 transfer from chromosomes to the central spindle (Figure S2I), suggesting compromised motor function rather than a defect in PLK-1-dependent RhoA activation, so we did not analyze it further. The second candidate, CYK-4 cluster II, was positioned in an N-terminal location similar to the PLK1 sites identified as important for cytokinesis in human CYK-4 (Figure 2G; [14, 16]).
To assess the functional importance of the four putative PLK-1 phosphorylation sites in CYK-4 cluster II, we mutated each residue to alanine individually and made all possible double and triple mutants (Figure 2H). Analysis of the triple mutants, in which only one of the four putative target sites can be phosphorylated, revealed that either T177 or S180 was largely sufficient for function, whereas neither T163 nor S170 were sufficient on their own (Figure 2H). Consistent with this, simultaneous mutation of T177 and S180 was the only double mutant that exhibited significant lethality. While phosphorylation of two sites in human CYK4 is proposed to be required to promote interaction with ECT-2 [25], one phosphorylated site (T177 or S180) may suffice in the context of C. elegans CYK-4, where the region spanning the putative PLK-1 sites is significantly more acidic.
In summary, the CYK-4 N-terminus is likely the only critical PLK-1 target in the three main proteins implicated in RhoA activation during cytokinesis; CYK-4 is the functionally critical target despite the fact that C. elegans ECT-2 is not recruited to the central spindle.
The putative PLK-1 sites in the CYK-4 N-terminus are essential for cytokinesis
Next, we generated RNAi-resistant transgenes encoding untagged (for functional analysis) and mNeonGreen-tagged (for localization) WT or mutant CYK-4. We tested two mutants, one in which the four cluster II residues were mutated to alanine (4A) and a second deleting the 18-amino acid region containing the four sites (Δ163–180). Like WT CYK-4, both mutants localized to the central spindle (Figure 3B; Figure S4A) and rescued the spindle snapping phenotype observed following endogenous CYK-4 depletion (data not shown), indicating that they support normal central spindle assembly. As the C. elegans-specific RhoA activator NOP-1 affects cytokinesis phenotypes following CYK-4 inhibition [24], phenotypes were analyzed in the absence (Figure 3C,D) and presence (Figure S4B–D) of NOP-1. Immunoblotting confirmed robust depletion of endogenous CYK-4 and expression of transgene-encoded proteins at levels comparable to endogenous CYK-4 (Figure 3E). WT CYK-4 supported normal furrow closure kinetics following NOP-1 depletion; in contrast, little to no furrowing was observed in embryos expressing 4A or Δ163–180 mutant CYK-4 (Figure 3C,D; Video S3). The CYK-4 mutants also compromised furrow ingression in the presence of NOP-1 (Figure S4C,D). Although furrows usually ingressed to completion in the presence of NOP-1 and the mutant CYK-4 variants, ingression was slower. The 4A and Δ163–180 mutant phenotypes were similar, although furrows ingressed slightly more slowly and were more prone to regression in Δ163–180 compared to 4A mutant embryos (Figure S4D). We conclude that the PLK-1 target site cluster in the CYK-4 N-terminus is essential for RhoA activation during cytokinesis.
Figure 3. PLK-1 phosphorylation the CYK-4 N-terminus promotes its binding to the ECT-2 BRCT repeat domain and is essential for cytokinesis.

(A) Schematics of the CYK-4 4A and Δ163–180 mutants. (B) Stills from timelapse sequences of embryos expressing NeonGreen fusions with WT, 4A, or Δ163–180 CYK-4 following endogenous CYK-4 depletion. Images shown are 200 seconds after anaphase onset. Insets are magnified 3X. Scale bar, 10μm. (C) Images of the furrow region from timelapse sequences of embryos expressing an mCherry::plasma membrane marker for the indicated conditions. Scale bar, 10μm. (D) Plots of the kinetics of contractile ring closure in individual embryos for the conditions shown in (C). The WT CYK-4 control traces are shown in gray on all three graphs. (E) Immunoblot of extracts prepared from the indicated strains in the absence (−) or presence (+) of endogenous CYK-4 depletion. α-tubulin as a loading control. (F) Schematic of the protocol for analysis of PLK-1 phosphorylation promoted binding of CYK-4 to ECT-2. (G) Schematics of CYK-4 146–190 (WT or 4A mutant) and ECT-2 1–320 proteins used in the pulldown assay (top) and analyzed by SDS-PAGE and Coomassie staining (bottom). Numbers below lanes indicate amount of ECT-2 1–320 pulled down, relative to the amount pulled down by unphosphorylated WT CYK-4 fragment. See also Figure S4 and Video S3.
PLK-1 phosphorylation promotes binding of CYK4 to the ECT2 BRCT module, but not via its canonical phospho-recognition residues
PLK1 phosphorylation of human CYK4 promotes an interaction with the ECT2 BRCT module that recruits ECT2 to the central spindle. Mutation of residues in the ECT2 BRCT module (T153A/K195M) analogous to residues that form the phosphate binding site in the tandem BRCT domains of BRCA1 and MDC1 [19] prevents ECT2 recruitment to the central spindle and interaction with CYK4 from mitotic extracts in pull-down experiments [16, 18]. Nevertheless, the ECT2 T153A/K195M mutant is capable of supporting cytokinesis. Thus, it has been suggested that while phosphorylation of the CYK4 N-terminus is critical for cytokinesis, it is for a reason other than because it promotes interaction with ECT2 [18].
To determine if phosphorylation of C. elegans CYK-4 promotes an interaction with the ECT-2 BRCT module, we took a biochemical approach (Figure 3F,G; Figure S4E). A GST fusion with a fragment of CYK-4 containing the four putative PLK-1 phosphorylation sites (aa 146–190; Figure 3G) was immobilized on beads and incubated with constitutively active PLK-1, which led to a reduction in the migration of the WT but not the 4A mutant fragment (Figure S4F). The ECT-2 BRCT module bound to the CYK-4 146–190 beads with and without PLK-1 phosphorylation. In the absence of PLK-1 phosphorylation, an equivalent amount of ECT-2 BRCT module bound to the WT and 4A CYK-4 beads, indicating that there is basal phosphorylation-independent binding. Similar basal binding has been observed in pull-down experiments with the human proteins [25, 26]. PLK-1 phosphorylation led to an ~4-fold increase in ECT-2 BRCT module binding to the WT but not the 4A mutant CYK-4 fragment (Figure 3G). We conclude that PLK-1 phosphorylation promotes CYK-4 binding to the BRCT module of C. elegans ECT-2, analogous to what has been reported for the human proteins [25].
Structural analysis of the human ECT2 BRCT module [27] suggested that T153 and K195 are analogous to residues that mediate phosphopeptide binding by the tandem BRCT domains in BRCA1 [19]. K166 in C. elegans ECT-2 is equivalent to human K195; however, T153 is not conserved and is an alanine (Figure 4A). Mutating K166 in the ECT-2 BRCT module to methionine (eliminates basic charge while minimizing loss of hydrophobic interactions) had no effect on phospho-dependent binding to CYK-4 (Figure 4B). Embryos expressing K166M mutant ECT-2 exhibited furrow closure kinetics identical to embryos expressing WT ECT-2 and were viable (Figure 4C–E). Thus, the canonical phosphor-recognition residues of C. elegans ECT-2 are not important for binding to phosphorylated CYK-4 or for the function of ECT-2 during cytokinesis.
Figure 4. PLK-1 phosphorylated CYK-4 binds to the ECT-2 BRCT repeat domain, but not via its canonical phospho-recognition residues.

(A) Sequence alignment of a section of the human ECT2 BRCT module, with the corresponding region from other vertebrate and nematode sequences. (B) (left) Schematics of proteins used in the pulldown assay, conducted as in Figure 3F. (right) Pulldown results analyzed by SDS-PAGE and Coomassie staining. Numbers below lanes indicate amount of ECT-2 1–320 pulled down, relative to the amount pulled down by unphosphorylated WT CYK-4 fragment. (C) Analysis of furrow ingression for the indicated conditions, done as in Figure 3C. Scale bar, 10 μm. (D) Plots of the kinetics of contractile ring closure in individual embryos for the conditions shown in (C). (E) Plot of embryonic lethality (mean ± SD) for the indicated conditions. N is number of worms and n the number of embryos scored. See also Figure S5.
While PLK1 phosphorylation has been shown to promote interaction of the human CYK4 N-terminus with the human ECT2 BRCT module in vitro [25], the effect of the ECT2 T153A/K195M mutations has not been assessed. Analogous to the C. elegans proteins, WT and T153A/K195M mutant human ECT2 BRCT modules exhibited comparable phospho-dependent binding to the human CYK4 N-terminus (Figure S5A,B). This result suggests that the inability of T153A/K195M mutant ECT2 to interact with phosphorylated CYK4 in mitotic extracts [16, 18] is not due to disruption of a direct binding interface.
We conclude that PLK1 phosphorylation of CYK4 promotes direct binding to the ECT2 BRCT module independent of the proposed canonical phospho-recognition residues in ECT2.
A conserved basic surface on the ECT2 triple BRCT domain module binds to PLK1-phosphorylated CYK4
The results above suggested that phosphorylated CYK4 binds to the ECT2 BRCT module, but not via the previously proposed canonical tandem BRCT phospho-recognition site. Testing the functional significance of this interaction therefore required defining the binding interface on ECT2. Using a homology model of the C. elegans ECT-2 BRCT module, we identified a conserved basic surface located ~10 Å from K166 near the interface between the first (BRCT-0) and second (BRCT-1) BRCT domains (Figure 5A). This surface feature is largely composed of residues from BRCT-1; T123, S146 and V147 (human: V149, V174, I175) lie at its center; R148, K149 and R154 (human: R176, K177, K182) line one side; and R112 (in the linker between BRCT-0 and BRCT-1) and H141 (human: R138 and H169) line the opposite side (Figure 5B,C). To determine if this basic surface is involved in binding phosphorylated CYK4, we purified mutant forms of the C. elegans ECT-2 BRCT module in which three of the conserved basic residues (R148, K149 and R154) were mutated to alanine (3A) or charge-reversed to glutamic acid (3E). Both the 3A and 3E basic surface mutations prevented the ECT-2 BRCT module from binding phosphorylated CYK-4 (Figure 5D). Notably, basal phosphorylation-independent binding was also lost. Mutation of the equivalent residues in human ECT2 also abrogated basal and phosphorylation-enhanced interaction with human CYK4 (Figure S6A).
Figure 5. A conserved basic surface in the ECT-2 BRCT module mediates interaction with PLK-1 phosphorylated CYK-4 and is essential for cytokinesis.

(A) Surface representations of the triple BRCT repeat module of human ECT2 (PDB:4n40) colored by electrostatic potential (left: blue: positive; white: neutral; red: negative) or conservation across 150 homologs (right). (B) Sequence alignment of the indicated region of the ECT2 BRCT-1 repeat. (C) Structure of the human ECT2 BRCT repeat module highlighting the “canonical phospho-recognition” residues T153 and K195 as orange/red/blue spheres and the residues that comprise the basic surface as magenta/blue spheres. The residues in the center of the surface (T123, S146 and V147 in C. elegans and V149, V174, I175 in human) are shown in light gray spheres. The side chains of K177 and K182, which are disordered in the crystal structure, were modeled into PDB:4n40. (D) (top) Schematics of proteins used in the pulldown assay, conducted as in Figure 3F. (bottom) Pulldown results analyzed by SDS-PAGE and Coomassie staining. Numbers below lanes indicate amount of ECT-2 1–320 pulled down, relative to the amount pulled down by unphosphorylated WT CYK-4 fragment. (E) Immunoblot of the indicated worm extracts. α-tubulin serves as a loading control. (F) Analysis of furrow ingression for the indicated conditions, done as in Figure 3C. Scale bar, 10 μm. Scale bar, 10μm. (G) Plots of the kinetics of contractile ring closure in individual embryos for the conditions shown in (F). The control WT ECT-2 traces are shown in gray on all three graphs. See also Figure S6 and Video S3.
Strikingly, in in vivo experiments no furrow ingression was observed in NOP-1 depleted embryos expressing either 3A or 3E basic surface mutant ECT-2 (Figure 5E–G; Video S3). Since NOP-1 activates ECT-2 in a parallel pathway to CYK-4 [24], we also analyzed embryos containing NOP-1. We found that the ECT-2 basic surface mutants phenocopied the CYK-4 PLK-1 target site mutants, with embryos exhibiting slower, but largely successful, furrow ingression (Figure S6B–D). Thus, NOP-1 activates ECT-2 via a distinct mechanism that does not involve the basic surface in the BRCT module. Combining the CYK-4 4A mutant with ECT-2 basic surface 3A mutant in the presence of NOP-1 led to a phenotype that similar to the ECT-2 basic surface 3A mutant alone (Figure S6E), suggesting that the ECT-2 basic surface mutant is nearly fully compromised in its interaction with phosphorylated CYK-4; consequently, additionally mutating the CYK-4 phosphorylation sites does not exacerbate the phenotype.
Collectively, the biochemical and in vivo data reveal that a non-canonical mode of binding underlies the ECT-2 BRCT interaction with phosphorylated CYK-4 that is critical for RhoA activation and cytokinesis.
The conserved basic surface on the ECT2 BRCT module is essential for cytokinesis in human cells
Biochemical and in vivo analysis in C. elegans embryos indicated that cytokinesis relies on a conserved basic surface in the ECT-2 BRCT module binding to phosphorylated CYK-4. To determine if the basic surface in human ECT2 is also important for cytokinesis, we generated clonal HeLa Kyoto cell lines with RNAi-resistant transgenes that expressed GFP alone or GFP fusions with wild-type (WT) or T153A/K195M (TK), Basic Surface 3A (2 clones; R176A/K177A/K182A), or Basic Surface 3E (2 clones; R176E/K177E/K182E) mutant ECT2 at comparable levels (Figure 6A,B,E; Figure S7A–C,F). After endogenous ECT2 depletion using an siRNA, we characterized the percent of multi-nucleated cells in fixed cells (Figure 6C,D; Figure S7B–E) and monitored protein localization and furrow ingression in living cells (Figure 6F–J; Figure S7F–H). WT GFP-ECT2 localized to the central spindle and the overlying equatorial plasma membrane (Figure 6G,I; Video S4), promoted furrow ingression (Figure 6H,J; Video S5), and prevented multinucleation following endogenous ECT2 depletion (Figure 6C,D). As shown previously [18], TK mutant GFP-ECT2 was compromised in targeting to the central spindle, but otherwise behaved similarly to WT GFP-ECT2 (Figure 6C–J; Videos S4 & S5). The ECT2 3A and 3E Basic Surface mutants also failed to localize to the central spindle, with inhibition of central spindle targeting being more penetrant than for the TK mutant (Figure 6G,I (clones 3A-1 and 3E-1); Figure S7G (clones 3A-2 and 3E-2)). While the Basic Surface mutants were recruited to the plasma membrane following anaphase onset, they did not concentrate on the equatorial plasma membrane (Figure 6G,I; Figure S7G; Video S4) and did not support successful cytokinesis. In cells expressing either Basic Surface mutant, furrows typically ingressed part way and then regressed, resulting in multinucleated cells (Figure 6C,D,H,J; Figure S7D,E,H; Video S5). Thus, the conserved basic surface on the ECT2 BRCT region required for binding to phosphorylated CYK4 is functionally critical for cytokinesis in both C. elegans and in human cells.
Figure 6. The conserved basic surface on the ECT2 BRCT module is essential for cytokinesis in human cells.

(A) Schematics of GFP-tagged ECT2 proteins expressed in human cells. (B) Outline of the procedure used to generate stable clonal HeLa Kyoto cell lines expressing GFP alone or GFP fusions with WT or mutant ECT2. (C) (top) Schematic outlining the experimental protocol used to analyze fixed HeLa Kyoto cell lines expressing GFP or GFP-ECT2 fusions after endogenous ECT2 depletion. (middle) Representative images of fields of cells expressing GFP after transfection with non-targeting (NT, left)) or ECT2 siRNA (right). (bottom) Representative images of fields of cells expressing the indicated GFP-ECT2 fusions after depletion of endogenous ECT2. Scale bar, 10 μm. (D) Plot of the percentage of multinucleated cells from the experiment in C (mean ± SE). Bars represent mean values of three independent experiments. n represents the total number of cells scored for each condition. On average, 150 cells were scored in each experiment. (E) Immunoblot analysis of the indicated lysates prepared 48 hours after transfection with non-targeting or ECT2 siRNA. Actin serves as a loading control. (F) Schematic of the protocol used to conduct live imaging. (G) Stills from timelapse sequences of GFP-ECT2 fusions in the indicated cell lines after depletion of endogenous ECT2. Cells were imaged 24 hours after ECT2 siRNA transfection. Time 0 is anaphase onset. (H) Images showing cytokinetic phenotypes for the indicated conditions after depletion of endogenous ECT2. Cells were labeled with SiR-DNA and overlays of brightfield and DNA signals are shown. The metaphase-to-anaphase transition is t=0 min. Dashed white lines mark cell boundaries. Scale bar, 10 μm. (I) Quantification of the experiment in G. The percentage of cells in which the indicated GFP-ECT2 fusion accumulated at the central spindle (left graph) or was enriched on the equatorial membrane (right graph) is shown. Bars represent mean values of two independent experiments; error bars are the SE. n is the total number of cells scored for each condition. (J) Quantification of cytokinetic phenotypes from the experiment in H. Phenotypes were scored between 24 and 48 hours after siRNA transfection. See also Figure S7 and Videos S4 & S5.
Autoinhibition prevents ECT-2 in the cytoplasm from binding to phosphorylated CYK-4 on the central spindle
The above data establish that phosphorylation of CYK-4 by PLK-1 promotes binding to a basic surface in the ECT-2 BRCT module. So why is ECT-2 not recruited to CYK-4 on the central spindle in C. elegans, as it is in human cells? ECT2 is catalytically autoinhibited by an interaction between its N-terminal BRCT module and its C-terminus [28]. Thus, one possibility is that autoinhibition is stronger for C. elegans ECT-2 than for the human protein, which prevents it from binding to phosphorylated CYK-4 at the central spindle. An alternative possibility is that CYK-4 at the central spindle is not phosphorylated by PLK-1. To discriminate between these possibilities, we expressed the ECT-2 BRCT module alone fused to mNeonGreen; as the C-terminus was not present, there should be no autoinhibition. Unlike full-length NG::ECT-2, the NG::ECT-2 BRCT module (aa 1–320) was robustly recruited to the central spindle (Figure 7A,B; Video S6). Recruitment was disrupted by mutation of the ECT-2 Basic Surface and by the 4A CYK-4 mutant that prevents PLK-1 phosphorylation, but not by the ECT-2 K166M mutation (Figure 7B,C; Video S6). We conclude that the ECT-2 BRCT module is competent to interact with phosphorylated CYK-4 on the central spindle but is prevented from doing so by autoinhibition by the ECT-2 C-terminus.
Figure 7. Autoinhibition prevents ECT-2 from interacting with phosphorylated CYK-4 at the central spindle.

(A) Schematics of transgenic NeonGreen-tagged full length ECT-2 and ECT-2 1–320. (B,C) Representative time-lapse images acquired 120 seconds after anaphase onset of embryos expressing NeonGreen fusions with full length ECT-2 or ECT-2 1–320 (WT or mutant as indicated) and ZEN-4::mScarlet as a central spindle marker. Panels below are higher magnification views of the central spindle. Scale bars: 10 and 2 μm. Embryos in (C) additionally express transgenic WT or 4A mutant CYK-4 after endogenous CYK-4 depletion. (D) Schematic model of the ECT-2 BRCT module, and (E) Model for cytokinesis signaling. See also Video S6.
DISCUSSION
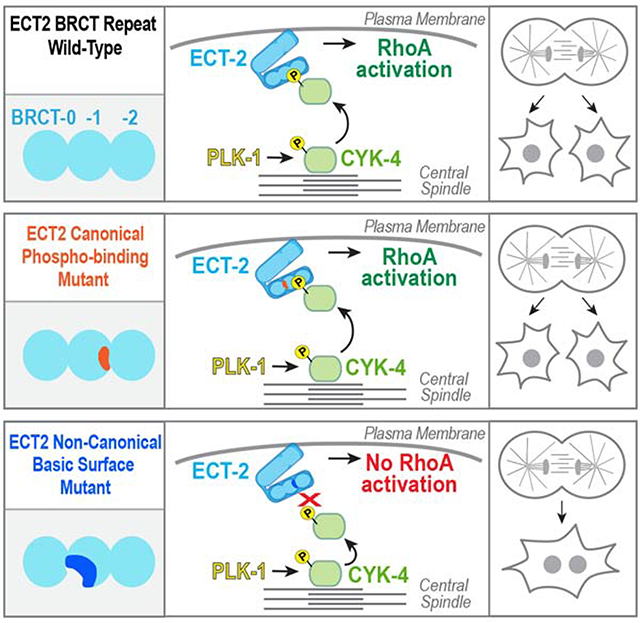
The prevalent model for cytokinesis signaling in metazoans posits that PLK1 phosphorylation of CYK4 generates a central spindle binding site for ECT2, resulting in a gradient of active ECT2 that leads to equatorially-biased RhoA activation. However, a problem with this model is that ECT2 does not universally localize to the central spindle in metazoans and, even in systems where it does, mutations that disrupt central spindle targeting do not inhibit cytokinesis [17, 18]. Thus, how PLK1 promotes RhoA activation has been unclear. Here, we show that the essential function of PLK1 in cytokinesis is to promote an interaction between the CYK4 N-terminus and a conserved basic surface in the ECT2 BRCT module that is distinct from the previously proposed canonical phospho-recognition site (Figure 7D). Our observations suggest that autoinhibition prevents C. elegans ECT-2 from localizing to the central spindle, leading us to propose a model in which ECT-2 localizes to the membrane following anaphase onset [15] and the central spindle generates a gradient of phosphorylated CYK-4 that locally activates plasma membrane-bound ECT-2 (Figure 7E). The fact that human ECT2 can support cytokinesis without localizing to the central spindle [18] suggests that the proposed signaling mechanism is likely to be widely conserved.
A non-canonical ECT2 BRCT domain – phosphorylated CYK4 interaction underlies RhoA activation in cytokinesis
Prior experiments analyzing the interaction between CYK4 and ECT2 were based on the assumption that the ECT2 BRCT module interacted with phosphorylated CYK4 via threonine and lysine (TK) residues analogous to those that form the phosphate-binding site in the tandem BRCT domains of BRCA1 and MDC1 [19]. Although mutation of the ECT2 TK residues in human cells prevented phosphorylated CYK4 from co-immunoprecipitating with ECT2 and blocked ECT2 central spindle targeting [16, 18], it had no discernible effect on cytokinesis. Thus, it was suggested that the importance of CYK4 phosphorylation for cytokinesis [14, 16] might be for reasons other than promoting interaction with ECT2 [18]. Our results show that this conclusion is incorrect because the TK residues are not required for the ECT2 BRCT module to interact with CYK4.
We define an evolutionarily conserved basic surface on the ECT2 BRCT module that constitutes the critical interface for interaction with phosphorylated CYK4. In contrast to mutation of the TK residues, mutations disrupting this basic surface led to penetrant cytokinesis failure. The basic surface is primarily composed of residues from the second domain (BRCT-1) of the triple BRCT module (BRCT-0,1,2), suggesting that this single domain mediates interaction with phosphorylated CYK4. This binding mode is distinct from that observed in tandem BRCT modules where the phosphopeptide binds across the interface between two BRCT domains. Intriguingly, the DNA damage response scaffolding protein TOPBP1 also contains a triple BRCT module [27, 29, 30]. Biochemical and structural studies of TOPBP1 family proteins indicate that phosphopeptide ligands can bind to either BRCT-1 or BRCT-2. While the TK residues in each repeat contribute to binding, the phosphopeptide interacts primarily with a single BRCT domain rather than across an interface generated by a tandem pair [29, 30], making the interactions similar to the one that we describe. To our knowledge, ECT2 represents the first case where a BRCT module does not depend on the canonical TK phospho-recognition site to bind its phosphorylated partner. Given the presence of BRCT modules in many signaling proteins, our results raise the possibility that non-canonical interaction modes may mediate BRCT module functions in diverse contexts.
A conserved mode of cytokinesis signaling
The aim of cytokinesis signaling is to activate ECT2 on the equatorial cortex. Prior biochemical and structural studies suggest that ECT2 activation requires relief of two modes of autoinhibition. ECT2 was shown to be held in an autoinhibited conformation by an interaction between the N-terminal BRCT module and the C-terminal catalytic and membrane association domains [28]. Our finding that removal of the ECT-2 C-terminus results in robust central spindle localization of the N-terminal BRCT region provides strong support for this autoinhibitory mechanism. Recent structural work suggests that ECT2 is also autoinhibited via a second mechanism in which the PH domain blocks the RhoA-binding site involved in catalysis by the GEF domain [31]. This second mode of autoinhibition was suggested to be released by association of the PH domain with an allosteric GTP-bound RhoA molecule at the plasma membrane [31]. Thus, we propose that ECT2 activation occurs in two steps and requires: (1) interaction with the plasma membrane where its PH domain associates with an allosteric GTP-bound RhoA, and (2) interaction of its BRCT region with phosphorylated CYK4. In C. elegans, we propose that interaction of ECT-2’s C-terminal polybasic region and PH domain with the plasma membrane and GTP-bound RhoA partially relieves autoinhibition, thereby generating a state permissive for interaction with phosphorylated CYK4 (Figure 7E). In this model, a gradient of PLK1-phosphorylated CYK4, which has its own essential plasma membrane targeting C1 domain [32, 33], is generated by the central spindle and stimulates localized RhoA activation by interacting with and completing the activation of plasma membrane-bound ECT2 (Figure 7E).
The above model is consistent with experiments in Drosophila cells in which mislocalization of CYK4 to the entire plasma membrane using an artificial membrane-tethering motif resulted in dramatic ectopic furrowing [34]. Recent optogenetic experiments showing that RhoA-dependent furrowing can be induced at any location on the cell cortex by targeting the catalytic domain of the RhoA GEF LARG [35]. suggest that there are no spindle-based mechanisms that act downstream of RhoA activation to prevent furrowing of non-equatorial cortex. Notably, optogenetic experiments targeting ECT2 to the plasma membrane found that ECT2 could only induce furrowing of the equatorial cortex [18]. Thus, the equatorial cortex is uniquely competent to activate ECT2 and we propose this is due to relief of autoinhibition by a spatially confined gradient of PLK1-phosphorylated CYK4.
We note that, in the C. elegans embryo, ECT-2 is detected at the plasma membrane but CYK-4 labeled with the same fluorophores (GFP or NeonGreen) is difficult to detect at the membrane when expressed at endogenous levels. CYK-4 in situ-tagged with a bright fluorophore (mScarlet, [36]) can be detected at the equatorial membrane when the central spindle is disrupted (Figure S6F). The relative abundance of plasma membrane-associated ECT-2, compared to the limited amount of CYK-4, has also been noted in human cells [15], raising the possibility that phosphorylated CYK4 may act catalytically to activate ECT2. Alternatively, it is possible that only a small fraction of ECT2 at the plasma membrane needs to be active to promote furrow formation.
Functional implications of evolutionary variation in ECT2 association with phosphorylated CYK4 at the central spindle
Similar to C. elegans ECT2, Drosophila ECT2 (Pebble) does not localize to the central spindle [37]. By contrast, sea urchin ECT2 and vertebrate ECT2s localize to the central spindle [38]. This pattern suggests that the ability of ECT2 homologs to target to the central spindle may have emerged in deuterostomes, the group of bilaterally symmetric animals that includes echinoderms and vertebrates. Analysis of ECT2 localization in more species will be needed to test if this is indeed the case. We speculate that the mechanism we describe for C. elegans, in which ECT2 associates with the plasma membrane first and is then activated by phosphorylated CYK4, represents the ancestral mechanism present in all animal cells. In species in where ECT2 interacts with CYK4 at the central spindle, the order of relief of ECT2 autoinhibition modes may be reversed: ECT2 BRCT interacts first with phosphorylated CYK4 at the central spindle and then the ECT2-CYK4 complex diffuses to the plasma membrane where the second autoinhibition mode is released by RhoA-GTP. Notably, this pathway is not essential, as removal of ECT2 from the central spindle does not significantly perturb cytokinesis, at least in HeLa cells. Identification of physiological contexts where central spindle association of ECT2 makes a more substantial functional contribution is necessary to address its significance. An additional interesting question is why mutating the TK residues, which are not part of the essential binding interface between ECT2 and phosphorylated CYK4, prevents ECT2 from targeting to the central spindle in human cells. Although our data do not provide an answer to this question, one speculation might be that mutating the TK residues could enhance autoinhibition, making the behavior of human ECT2 more similar to its C. elegans homolog.
In conclusion, our data define a non-canonical mode of BRCT interaction, involving a basic surface in the BRCT module of ECT2 and PLK1 phosphorylated CYK4, that is critical for cytokinesis signaling. This mode of interaction is conserved between C. elegans and human cells, and explains the function of PLK1 in RhoA activation during cytokinesis. Precisely how the interaction of phosphorylated CYK4 with ECT2’s BRCT module activates ECT2 is the key mechanistic question that emerges from this effort and will need to be addressed in future work.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karen Oegema (koegema@health.ucsd.edu).
Materials Availability
All strains, cell lines, plasmids and other materials are available upon request.
Data and Code Availability
All primary data associated with the paper is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans Strains
C. elegans strains (listed in the Key Resources Table) were maintained at 20°C. Single-copy transgenes were generated by using the transposon-based MosSCI method [39]. Transgenes were cloned into pCFJ151 and injected into strains with specific Mos transposon insertions to recombine them into one of the following chromosomal sites: the ttTi5605 site on Chr II; the Uni I oxTi185 site on Chr I or the Uni V oxTi365 site on Chr V. Transgenes were generated by injecting a mixture of the pCFJ151-derived repairing plasmid containing the Cb-unc-119 selection marker and appropriate homology arms (50 ng/μL), transposase plasmid (pCFJ601 encoding the Mos1 transposase under the Peft-3 promoter, 50 ng/μL) and four plasmids encoding markers for negative selection against chromosomal arrays (pMA122 [Phsp-16.41::peel-1, 10 ng/μL], pCFJ90 [Pmyo-2::mCherry, 2.5 ng/μL], pCFJ104 [Pmyo-3::mCherry, 5 ng/μL] and pGH8 [Prab-3::mCherry, 10 ng/μL]) into strains EG6429 (outcrossed from EG4322; ttTi5605, Chr II), EG8078 (oxTi185, Chr I), or EG8082 (oxTi365, Chr V). After one week, the progeny of injected worms were heat-shocked at 34°C for 3 hours to induce the expression of PEEL-1 to kill worms containing extra chromosomal arrays. Moving worms without fluorescent markers were identified as candidates, and PCR across the junctions on both sides of the integration site was used to confirm transgene integration in their progeny.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ECT2 | This study | OD324 |
| Mouse monoclonal anti-Actin | Milipore | MAB1501 |
| Goat anti GFP | Hyman Lab | OD194 |
| Mouse monoclonal anti alpha-tubulin (clone DM1A) | Sigma-Aldrich | Cat #T9026; RRID AB_477593 |
| Goat anti rabbit IgG, HRP-conjugated | Jackson Immunoresearch | Cat #111-035-003; RRID AB_2313567 |
| Donkey anti mouse IgG, HRP-conjugated | Jackson Immunoresearch | Cat #715-035-150; RRID AB_2340770 |
| Bacterial and Virus Strains | ||
| Retrovirus: CMV promoter-AcGFP-FLAG | This study | N/A |
| Retrovirus: CMV promoter-AcGFP-FLAG-ECT2-WT | This study | N/A |
| Retrovirus: CMV promoter-AcGFP-FLAG-ECT2-TK | This study | N/A |
| Retrovirus: CMV promoter-AcGFP-FLAG-ECT2-3A | This study | N/A |
| Retrovirus: CMV promoter-AcGFP-FLAG-ECT2-3E | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GST-(PreScission site)-CYK-4 aa 146–190 | This study | pOD3454 (Plasmid) |
| GST-(PreScission site)-CYK-4 aa 146–190 4A (T163A/S170A/T177A/S180A) | This study | pOD3460 (Plasmid) |
| GST-6His-(PreScission site)-ECT-2 aa 1–320 | This study | pOD3411 (Plasmid) |
| GST-6His-(PreScission site)-ECT-2 aa 1–320 3A (R148A/K149A/R154A) | This study | pOD3455 (Plasmid) |
| GST-6His-(PreScission site)-ECT-2 aa 1–320 3E (R148E/K149E/R154E) | This study | pOD3456 (Plasmid) |
| GST-6His-(PreScission site)-ECT-2 aa 1–320 K166M | This study | pOD3422 (Plasmid) |
| GST-(PreScission site)-hCYK4 aa 1–288 | This study | pOD3483 (Plasmid) |
| GST-6His-(PreScission site)-hECT2 aa 22–326 | This study | pOD3468 (Plasmid) |
| GST-6His-(PreScission site)-hECT2 aa 22–326 3A (R176A/K177A/K182A) | This study | pOD3471 (Plasmid) |
| GST-6His-(PreScission site)-hECT2 aa 22–326 3E (R176E/K177E/K182E) | This study | pOD3472 (Plasmid) |
| GST-6His-(PreScission site)-hECT2 aa 22–326 TK (T153A/K195M) | This study | pOD3475 (Plasmid) |
| Experimental Models: Cell Lines | ||
| Parental Cell Line: RCL028: HeLa Kyoto | Gerlich Lab | RCL028 |
| Engineered Clonal Cell Line: ODCL0123: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG | This study | ODCL0123 |
| Engineered Clonal Cell Line: ODCL0124: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 WT | This study | ODCL0124 |
| Engineered Clonal Cell Line: ODCL0125: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 TK mutant (T153A & K195M) | This study | ODCL0125 |
| Engineered Clonal Cell Line: ODCL0126: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 3A-1 (R176A, K177A, K182A) | This study | ODCL0126 |
| Engineered Clonal Cell Line: ODCL0127: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 3A-2 (R176A, K177A, K182A) | This study | ODCL0127 |
| Engineered Clonal Cell Line: ODCL0128: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 3E-1 (R176E, K177E, K182E) | This study | ODCL0128 |
| Engineered Clonal Cell Line: ODCL0129: Modification to Parent Line HeLa Kyoto: CMVpro - AcGFP FLAG hECT2 3E-2 (R176E, K177E, K182E) | This study | ODCL0129 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain wild type N2 (ancestral) | Caenorhabditis Genetics Center | N2 |
| C. elegans: Strain OD1970: ltSi835[pKL62; Pcyk-4::CYK-4reencoded; cb-unc-119(+)]II; unc-119(ed3)III | [42] | OD1970 |
| C. elegans: Strain OD1984: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; unc-119(ed3)III | This study | OD1984 |
| C. elegans: Strain OD2873: ltSi1013[pSG015; Pect-2::ect-2 RE-encoded-exon8::ect-2 3’-UTR; cb unc-119(+)]II;unc-119(ed3)III | This study | OD2873 |
| C. elegans: Strain OD2899: ltSi1014[pSG016; Pzen-4::zen-4 RE-encoded-exon6::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD2899 |
| C. elegans: Strain OD3009: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1013[pSG015; Pect-2::ect-2 RE-encoded-exon8::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3009 |
| C. elegans: Strain OD3010: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1014[pSG016; Pzen-4::zen-4 RE-encoded-exon6::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3010 |
| C. elegans: Strain OD3064: ltSi1239[pSG033; Pect-2::ect-2 RE-encoded-exon8 S4A S64A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3064 |
| C. elegans: Strain OD3159: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1014[pSG016; Pzen-4::zen-4 RE-encoded-exon6::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3159 |
| C. elegans: Strain OD3222: ltSi1246[pSG042; Pect-2::ect-2 RE-encoded-exon8 S380A, S478A, S791A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3222 |
| C. elegans: Strain OD3223: ltSi1247[pSG043; Pect-2::ect-2 RE-encoded-exon8 S171A, S236A, S315A, S362A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3223 |
| C. elegans: Strain OD3224: ltSi1248[pSG044; Pect-2::ect-2 RE-encoded-exon8 S405A S491A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3224 |
| C. elegans: Strain OD3225: ltSi1249[pSG045; Pect-2::ect-2 RE-encoded-exon8 S787A S836A S866A S869A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3225 |
| C. elegans: Strain OD3226: ltSi1250[pSG046; Pect-2::ect-2 RE-encoded-exon8 S917A S918A S922A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3226 |
| C. elegans: Strain OD3328: ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3328 |
| C. elegans: Strain OD3433: ltSi1251[pSG047; Pzen-4::zen-4 RE-encoded-exon6 S2A, S21A, T25A, T92A, S93A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3433 |
| C. elegans: Strain OD3434: ltSi1252[pSG048; Pzen-4::zen-4 RE-encoded-exon6 S51A, S93A, S106A, T157A, T189A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3434 |
| C. elegans: Strain OD3435: ltSi1256[pSG051; Pzen-4::zen-4 RE-encoded-exon6 T189A, S257A, S259A, T278A, S284A, S285A, S289A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3435 |
| C. elegans: Strain OD3436: ltSi1257[pSG052; Pzen-4::zen-4 RE-encoded-exon6 T310A, S314A, S347A, S351A, S368A, S369A, S370A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3436 |
| C. elegans: Strain OD3437: ltSi1255[pSG053; Pzen-4::zen-4 RE-encoded-exon6 S415A, S423A, S425A, S455A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3437 |
| C. elegans: Strain OD3438: ltSi1253[pSG049; Pzen-4::zen-4 RE-encoded-exon6 S494A, S495A, S499A, S500A, T507A, S532A, S556A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3438 |
| C. elegans: Strain OD3439: ltSi1254[pSG050; Pzen-4::zen-4 RE-encoded-exon6 S499A, S500A, S607A, S609A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3439 |
| C. elegans: Strain OD3440: ltSi1258[pSG054; Pzen-4::zen-4 RE-encoded-exon6 T652A, S656A, S661A, S723A, S740A, S770A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3440 |
| C. elegans: Strain OD3441: ltSi1259[pSG055; Pcyk-4::CYK-4reencoded S3A, S4A, S6A, S15A, S57A, S89A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3441 |
| C. elegans: Strain OD3443: ltSi1261[pSG057; Pcyk-4::CYK-4reencoded S209A, S211A, S224A, S226A, T232A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3443 |
| C. elegans: Strain OD3444: ltSi1262[pSG058; Pcyk-4::CYK-4reencoded T259A, T268A, S269A, S273A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3444 |
| C. elegans: Strain OD3445: ltSi1263[pSG059; Pcyk-4::CYK-4reencoded S293A, S298A, S325A, T326A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3445 |
| C. elegans: Strain OD3446: ltSi1264[pSG060; Pcyk-4::CYK-4reencoded T587A, S589A, S622A, S646A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3446 |
| C. elegans: Strain OD3447: ltSi1265[pSG061; Pect-2::ect-2 RE-encoded-exon8 S310A, S315A, S318A, S321A, S323A, S325A, S326A, S331A, S362A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3447 |
| C. elegans: Strain OD3505: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi835[pKL62; Pcyk-4::CYK-4reencoded::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3505 |
| C. elegans: Strain OD3507: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1256[pSG051; Pzen-4::zen-4 RE-encoded-exon6 T189A, S257A, S259A, T278A, S284A, S285A, S289A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3507 |
| C. elegans: Strain OD3619: ltSi1124 [pSG092; Pcyk-4::CYK-4reencoded::mNeongreen::cyk-4::cyk-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3619 |
| C. elegans: Strain OD3626: ltSi1269[pSG085; Pcyk-4::CYK-4reencoded T163A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3626 |
| C. elegans: Strain OD3627: ltSi1270[pSG086; Pcyk-4::CYK-4reencoded T177A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3627 |
| C. elegans: Strain OD3628: ltSi1271[pSG087; Pcyk-4::CYK-4reencoded S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3628 |
| C. elegans: Strain OD3665: ltSi1272[pSG084; Pcyk-4::CYK-4reencoded T163A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3665 |
| C. elegans: Strain OD3677: ltSi1284[pSG041; Pect-2::ect-2 RE-encoded-exon8 S123A, S331A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3677 |
| C. elegans: Strain OD3679: ltSi1286[pSG063; Pect-2::ect-2 RE-encoded-exon8 K166M::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3679 |
| C. elegans: Strain OD3685: ltSi1256[pSG051; Pzen-4::zen-4 RE-encoded-exon6 T189A, S257A, S259A, T278A, S284A, S285A, S289A::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3) III; ltIs37 [pAA64; pie-1/mCHERRY::his-58; unc-119 (+)] IV | This study | OD3685 |
| C. elegans: Strain OD3734: ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)III; plk-1((lt106[plk-1 C52V] lt108[plk-1 L115G])III | This study | OD3734 |
| C. elegans: Strain OD3745: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I;ltSi1292[pSG096; Pcyk-4::CYK-4reencoded T163A S170A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3745 |
| C. elegans: Strain OD3749: ltSi1296[pSG090; Pcyk-4::CYK-4reencoded Δ210-244aa::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3749 |
| C. elegans: Strain OD3850: ltSi1298[pSG0104; Pcyk-4::CYK-4reencoded Δ163-180aa::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3850 |
| C. elegans: Strain OD3861: ltSi1472 [pSG104; Pcyk-4::CYK-4reencoded::mNeongreen Δ163-180aa::cyk-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3861 |
| C. elegans: Strain OD3862: ltSi1473 [pSG097; Pcyk-4::CYK-4reencoded T163A T177A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3862 |
| C. elegans: Strain OD3863: ltSi1474 [pSG098; Pcyk-4::CYK-4reencoded T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3863 |
| C. elegans: Strain OD3864: ltSi1475 [pSG099; Pcyk-4::CYK-4reencoded T163A S170A T177A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3864 |
| C. elegans: Strain OD3865: ltSi1476 [pSG100; Pcyk-4::CYK-4reencoded T163A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3865 |
| C. elegans: Strain OD3866: ltSi1477 [pSG101; Pcyk-4::CYK-4reencoded S170A T177A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3866 |
| C. elegans: Strain OD3867: ltSi1478 [pSG102; Pcyk-4::CYK-4reencoded T163A S170A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3867 |
| C. elegans: Strain OD3868: ltSi1479 [pSG103; Pcyk-4::CYK-4reencoded S170A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3868 |
| C. elegans: Strain OD3870: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1290[pSG068; Pect-2::ect-2 RE-encoded-exon8 Δ559-729aa (PH domain deIetion)::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3870 |
| C. elegans: Strain OD3872: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1288[pSG064; Pect-2::ect-2 RE-encoded-exon8 Δ116-190aa (BRCT-1 domain deletion)::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD3872 |
| C. elegans: Strain OD4077: ltSi1480 [pSG113; Pect-2::mNeonGreen::ect-2 RE-encoded-exon8::ect-2 3’-UTR; cb unc-119(+)]II;unc-119(ed3)III | This study | OD4077 |
| C. elegans: Strain OD4080: ltSi1483 [pSG116; Pect-2::mNeonGreen::ect-2 RE-encoded-exon8:: Δ559-726aa (PH domain deIetion)::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4080 |
| C. elegans: Strain OD4129: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I;ltSi1298[pSG0104; Pcyk-4::CYK-4reencoded Δ163-180aa::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4129 |
| C. elegans: Strain OD4131: ltSi1292[pSG096; Pcyk-4::CYK-4reencoded T163A S170A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4131 |
| C. elegans: Strain OD4132: ltSi1485[pSG118; Pect-2::ect-2 RE-encoded-exon8 R148A K149A R154A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4132 |
| C. elegans: Strain OD4133: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1485[pSG0118; Pect-2::ect-2 RE-encoded-exon8 R148A K149A R154A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4133 |
| C. elegans: Strain OD4134: ltSi1486[pSG119; Pect-2::ect-2 RE-encoded-exon8 R148E K149E R154E::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4134 |
| C. elegans: Strain OD4135: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1486[pSG0119; Pect-2::ect-2 RE-encoded-exon8 R148E K149E R154E::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4135 |
| C. elegans: Strain OD4338: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1480 [pSG113; Pect-2::mNeonGreen::ect-2 RE-encoded-exon8::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4338 |
| C. elegans: Strain OD4382: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; plk-1(lt18[plk-1::sGFP]::loxp) | This study | OD4382 |
| C. elegans: Strain OD4555: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1286[pSG063; Pect-2::ect-2 RE-encoded-exon8 K166M::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4555 |
| C. elegans: Strain OD4559: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1490 [pSG120; Pect-2::mNeonGreen::ect-2 1-320aa::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III; unc-119(ed3)III; ltSi1489[pKL62; Pcyk-4::CYK-4reencoded::cyk-4 3’UTR; cb-unc-119(+)]V | This study | OD4559 |
| C. elegans: Strain OD4560: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1490 [pSG120; Pect-2::mNeonGreen::ect-2 1-320aa::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III; unc-119(ed3)III; ltSi1487[pSG096; Pcyk-4::CYK-4reencoded T163A S170A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]V | This study | OD4560 |
| C. elegans: Strain OD4561: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1490 [pSG120; Pect-2::mNeonGreen::ect-2 1-320aa::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4561 |
| C. elegans: Strain OD4563: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1492 [pSG122; Pect-2::mNeonGreen::ect-2 1-320aa R148A K149A R154A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4563 |
| C. elegans: Strain OD4645: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi835[pKL62; Pcyk-4::CYK-4reencoded::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4645 |
| C. elegans: Strain OD4650: ltSi1491[pSG121; Pzen-4::zen-4 RE-encoded-exon6::Scarlet::zen-4 3’-UTR; cb unc-119(+)]I; ltSi1494 [pSG127; Pect-2::mNeonGreen::ect-2 1-320aa K166M::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4650 |
| C. elegans: Strain OD4653: ltSi1495[pSG124; Pcyk-4::CYK-4 reencoded S170A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4653 |
| C. elegans: Strain OD4654: ltSi1496[pSG125; Pcyk-4::CYK-4 reencoded T163A S170A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4654 |
| C. elegans: Strain OD4655: ltSi1497[pSG126; Pcyk-4::CYK-4 reencoded S170A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD4655 |
| C. elegans: Strain OD4656: ltSi1014[pSG016; Pzen-4::zen-4 RE-encoded-exon6::zen-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3) III; ltIs37 [pAA64; pie-1/mCHERRY::his-58; unc-119 (+)] IV | This study | OD4656 |
| C. elegans: Strain OD4658: ltSi1124 [pSG092; Pcyk-4::CYK-4reencoded T163A S170A T177A S180A::mNeongreen::cyk-4::cyk-4 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III | This study | OD4658 |
| C. elegans: Strain OD4835: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1485[pSG118; Pect-2::ect-2 RE-encoded-exon8 R148A K149A R154A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III; unc-119(ed3)III; ltSi1489[pKL62; Pcyk-4::CYK-4reencoded::cyk-4 3’UTR; cb-unc-119(+)]V | This study | OD4835 |
| C. elegans: Strain OD4836: ltSi849[pKL120; Pmex-5::mCh-PH::tbb-2 3’UTR; cb-unc-119(+)]I; ltSi1485[pSG118; Pect-2::ect-2 RE-encoded-exon8 R148A K149A R154A::ect-2 3’-UTR; cb unc-119(+)]II; unc-119(ed3)III; unc-119(ed3)III; ltSi1487[pSG096; Pcyk-4::CYK-4reencoded T163A S170A T177A S180A::cyk-4 3’UTR; cb-unc-119(+)]V | This study | OD4836 |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting siRNA #1 | Dharmacon | D-001810-01-05 |
| siGENOME Human ECT2 siRNA | Thermo Scientific | D-006450-02 |
| Primer pair for synthesis of dsRNA targeting cyk-4 (K08E3.6): (Oligo 1: AATTAACCCTCACTAAAGGGATGT, Oligo 2: TAATACGACTCACTATAGGCTTCGAATTGGCAGCAGC); Template: N2 genomic DNA | This paper | N/A |
| Primer pair for synthesis of dsRNA targeting ect-2 (T19E10.1): (Oligo 1: AATTAACCCTCACTAAAGGCAAAGAAGCTCTGGAATGTGAG, Oligo 2: TAATACGACTCACTATAGGCAAAACTTCGTCAATCGCTTTTG); Template: N2 genomic DNA | This paper | N/A |
| Primer pair for synthesis of dsRNA targeting zen-4 (M03D4.1): (Oligo 1: AATTAACCCTCACTAAAGGTCAACTCTTCTTACTATGATTCGCC, Oligo 2: TAATACGACTCACTATAGGTGTACGAGACTGAAGAACCG); Template: N2 genomic DNA | This paper | N/A |
| Primer pair for synthesis of dsRNA targeting spd-1 (Y34D9A.4): (Oligo 1: TAATACGACTCACTATAGGTCGTTGACGCGTACTCAACT, Oligo 2: AATTAACCCTCACTAAAGGGAATTCGAAATCCGACTCCA); Template: N2 cDNA | This paper | N/A |
| Primer pair for synthesis of dsRNA targeting nop-1 (F25B5.2): (Oligo 1: TAATACGACTCACTATAGGCAAACGAAAAAGGAGAAACATTG, Oligo 2: AATTAACCCTCACTAAAGGCTAACATTCCGAAGGTGATCAAG); Template: N2 cDNA | This paper | N/A |
| Primer pair for synthesis of dsRNA targeting perm-1 (T01H3.4): (Oligo 1: TAATACGACTCACTATAGGAATTTTCTAGGTCGTCAATCTTCA, Oligo 2: AATTAACCCTCACTAAAGGCGAAAACGCGATCATTTTTA); Template: N2 cDNA | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pOD3889: CMV promoter-AcGFP-FLAG - pQCXIB | This study | pOD3889 |
| Plasmid: pOD3890: CMV promoter-AcGFP-FLAG-ECT2-WT (RNAi-resistant) - pQCXIB | This study | pOD3890 |
| Plasmid: pOD3891: CMV promoter-AcGFP-FLAG-ECT2-TK (RNAi-resistant) - pQCXIB | This study | pOD3891 |
| Plasmid: pOD3892: CMV promoter-AcGFP-FLAG-ECT2-3A (RNAi-resistant) - pQCXIB | This study | pOD3892 |
| Plasmid: pOD3893: CMV promoter-AcGFP-FLAG-ECT2-3E (RNAi-resistant) - pQCXIB | This study | pOD3893 |
| Software and Algorithms | ||
| Fiji | [46] | RRID: SCR_002285 |
| Prism | Graphpad | RRID: SCR_002798 |
To generate the plk-1 analogue sensitive allele (C52V, L115G; [40]), CRISPR-Cas9 was used as previously described [41]. The C52V mutation was introduced at the endogenous plk-1 locus by injecting adult worms with a mixture containing 27 μM of ribonucleoprotein particle (RNP) containing a crRNA targeting plk-1 (GGACGATTTTTGGGCAAGGG), and an oligonucleotide to repair the cut and generate the C52V mutation (CCACTTTTCCAGCGACAACCTCGCGTGTTGCTCGATTCGTAAGCTCATAAACGTGAGCGAATCCTCCtTTGCCCAAAAATCGTCCTTTCTCATAATAGGTCCCACGATCCTTGTCGGC). The progeny of injected adults were screened by PCR followed by sequencing to identify edited organisms. After backcrossing once, the same procedure was used on single mutant worms to introduce the second L115G mutation, using a different crRNA (TCTCAACGTGTATATCACTT) and repairing oligonucleotide (TTCGGTGACCGCCTTTCTTCTTTTGTGCAACTCCATCAGCGACCGTCTTGCACATAACTCACCAGT GATATACACGTTGAGATTGTCCTCGAAGAAGTGAATAACTTCACGATATTGATGTG).
Human Cell Lines
The HeLa Kyoto cell line was obtained from the Gerlich lab and a series of clonal engineered cell lines that express GFP alone or fusions of GFP with WT or mutant ECT2 (listed in the Key Resources Table) was generated from this parental line. All cell lines were maintained in DMEM Dulbecco’s Modified Eagle Medium (Gibco), supplemented with 10% Fetal Bovine Serum and 100 μg/ml streptomycin, and 100 U/ml penicillin in a humidified incubator at 37°C with 5% CO2. Plasmids containing GFP-tagged and siRNA-resistant WT and TK mutant human ECT2 were obtained from Mark Petronczki [18]. ECT2 constructs with the 3A and 3E Basic Surface mutations were generated by synthesizing a DNA fragment containing the desired mutations in residues R176, K177 and K182. DNA fragments containing the basic surface mutations were cloned into ECT2 in pIRES-Puro 3.1 using Gibson assembly. To generate ECT2 retroviral constructs, ECT2 WT, ECT2 TK and ECT2 Basic Surface mutants were first amplified from pIRES-Puro 3.1 by PCR, cloned into pDONR227 (pENTRY) and recombined into the retroviral destination vector CMV-pQCXIB using the Gateway Cloning System (Invitrogen). For retroviral production, 293T cells were transfected in 10 cm plates with 4 μg of retroviral vector and 4 μg of Ampho retroviral packaging vector using 4 μl of Fugene. In a 6-well plate, Hela Kyoto cells were infected with retrovirus supernatant for two consecutive days. Hela Kyoto cells were then split and cultured in media containing 2 μg/ml blasticidin for selection. Selected cells were sorted in order to enrich for GFP positive cells. To obtain single clones, cells were diluted and seeded in 96-well plates. Five clones from each ECT2 transgene were then expanded, characterized and frozen.
METHOD DETAILS
C. elegans single-copy transgenes
An RNAi-resistant transgene encoding CYK-4 was previously described ([42]; Figure S3B). The ECT-2 and ZEN-4 replacement systems were generated by PCR amplification of their respective genomic loci. The ect-2 transgene included 2134 bp region upstream of the start codon and 1301 bp downstream of the stop codon. The zen-4 transgene included 2953 bp region upstream of the start codon and 1500 bp downstream of the stop codon. Segments of the ect-2 and zen-4 transgenes were modified to make the transgenes RNAi-resistant without altering coding information (Figure S3A,C).
C. elegans RNA-mediated interference (RNAi)
Single-stranded RNAs (ssRNAs) were synthesized in 50 μL T3 and T7 reactions (MEGAscript, Invitrogen) using gel purified DNA templates generated by PCR from N2 genomic DNA or cDNA using oligonucleotides containing T3 or T7 promoters (Key Resources Table). Reactions were cleaned using the MEGAclear kit (Invitrogen), and the 50 μL T3 and T7 reactions were mixed with 50 μL of 3x soaking buffer (32.7 mM Na2HPO4, 16.5 mM KH2PO4, 6.3 mM NaCl, 14.1 mM NH4Cl) and annealed (68°C for 10 minutes followed by 37°C for 30 minutes). For single depletions, double-stranded RNAs (dsRNAs) were injected at a concentration of at least 1 μg/μl. For double depletions, dsRNAs were mixed at equal concentrations (~1 μg/μl for each dsRNA). L4 hermaphrodites were injected with dsRNA and incubated at 16°C or 20°C depending on the experiment. To assess embryonic viability after RNAi-mediated depletion, L4 hermaphrodites were injected with dsRNA and incubated at 20°C for 24 hours. Worms were singled and allowed to lay embryos at 20°C for 24 hours. Adult worms were removed and all embryos and hatchlings were counted after an additional 24 hours. For live imaging of early embryos after RNAi, L4 hermaphrodites were injected with dsRNAs and incubated at 16°C for 18 hours for CYK-4 and ECT-2 depletions or at 20°C for 24 hours for ZEN-4 depletions (or as indicated in specific experiments) before dissection to isolated embryos for imaging.
Live imaging of C. elegans embryos
Embryos for live imaging experiments were obtained by dissecting gravid adult hermaphrodites in M9 buffer (42 mM Na2HPO4, 22 mM KH2PO4, 86 mM NaCl, and 1mM MgSO4). One-cell embryos were transferred with a mouth pipette onto a 2% agarose pad, overlaid with an 18 X 18 `mm coverslip, and imaged using a spinning disk confocal system (Andor Revolution XD Confocal System; Andor Technology) with a confocal scanner unit (CSU-10; Yokogawa) mounted on an inverted microscope (TE2000-E; Nikon) equipped with a 60X 1.4 Plan-Apochromat objective, solid-state 100-mW lasers, and an electron multiplication back-thinned charge-coupled device camera (iXon; Andor Technology), or an inverted microscope (Axio Observer.Z1; Carl Zeiss) equipped with a spinning-disk confocal head (CSU-X1; Yokogawa) and a 63X 1.4 NA Plan Apochromat lens (Zeiss), in a temperature-controlled room at 20˚C. For imaging furrow closure, an 8 plane z-series at 2.0 μm intervals was acquired every 20s. Imaging was initiated in one-cell embryos upon nuclear envelope breakdown and was terminated when embryos reached the four-cell stage. If the first cell division failed, imaging was terminated after cleavage furrow regression.
Protein expression and purification
GST-tagged CYK-4 proteins were expressed in BL21(DE3)pLysS E. coli from DNA constructs cloned into a pGEX-6P-1 vector. GST control protein was expressed in BL21(DE3)pLysS E. coli from a blank pGEX-6P-1 vector. GST-His6-tagged ECT-2 proteins were expressed in Rosetta 2(DE3)pLysS E. coli from DNA constructs cloned into a pET42a vector that was modified to contain a PreScission protease cleavage site after the dual GST-His6 tags.
For all constructs, when the bacterial cultures reached an OD600 of 0.4–0.6, protein expression was induced for 5 hours at 25°C by addition of IPTG to 0.1 mM. Cells were washed once with cold PBS and flash frozen in liquid nitrogen. Pelleted cells were resuspended in lysis buffer (25 mM Hepes pH 7.5, 300 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, 10 mM β-mercaptoethanol), supplemented with 10 μg/ml pepstatin A, 1 mM PMSF and EDTA-free protease inhibitor cocktail (cOmplete, Roche). Cells were lysed by incubation with 1 mg/ml lysozyme on ice for 10 minutes followed by sonication. Cleared cell lysates were incubated with glutathione agarose (Sigma) for 2 hours at 4°C. The resin was then washed with lysis buffer (30x bed volume). For GST and GST-CYK-4 (both C. elegans and human), glutathione resin with purified proteins was stored as 50% slurry in storage buffer (25 mM Hepes pH 7.5, 200 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, 10 mM β-mercaptoethanol, 50% glycerol) at –20°C. For GST-His6-tagged ECT-2 (both C. elegans and human), glutathione resin with purified proteins was further washed with elution buffer (25 mM Hepes pH 7.5, 200 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, 10 mM β-mercaptoethanol, 5% glycerol) (10x bed volume). To remove the GST-His6 tag, resin was incubated with PreScission protease (Eton Bioscience) overnight at 4°C and the ECT2 proteins were eluted the next day in elution buffer, concentrated using an Amicon Ultra centrifugal filter unit (MilliporeSigma), and snap frozen in liquid nitrogen. PLK-1 T194D, purified from Sf9 cells, was a gift from Jeffrey Woodruff (UT Southwestern).
Kinase and pulldown assays
For pulldown assays in Figure 3G, 4B, 5D and S4E using C. elegans proteins, 2 μM GST or GST-tagged CYK-4 proteins were immobilized on glutathione beads, mixed with 100 nM constitutively active PLK-1 T194D (gift from Jeffrey Woodruff) in kinase buffer (25 mM Hepes pH 7.5, 50 mM NaCl, 10 mM MgCl2, 0.2 mM ATP, 0.1% Triton X-100, 1 mM β-mercaptoethanol), and incubated for 1 hour at room temperature. After washing twice with binding buffer (25 mM Hepes pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, 1 mM β-mercaptoethanol) to remove PLK-1, bead-bound GST or GST-CYK-4 proteins were mixed with soluble ECT-2 proteins in binding buffer and incubated for 1 hour at 4°C. The final concentration of each protein was 2 μM. The beads were washed three times with binding buffer and resuspended in sample buffer before analysis on SDS-PAGE.
For the pulldown assays in Figure S5B, S6A using the human proteins, 2 μM GST or GST-tagged CYK4 proteins were immobilized on glutathione beads, mixed with 150 nM PLK1 (Invitrogen #PV3501) in kinase buffer (25 mM Hepes pH 7.5, 50 mM NaCl, 10 mM MgCl2, 0.2 mM ATP, 0.1% Triton X-100, 1 mM DTT), and incubated for 1 hour at 30°C. After washing twice with binding buffer (20 mM Hepes pH 7.2, 150 mM NaCl, 5 mM MgCl2, 0.1% Triton X-100, 1 mM DTT) to remove PLK1, bead-bound GST or GST-CYK4 proteins were mixed with soluble ECT2 proteins in binding buffer and incubated for 1.5 hours at 4°C. The final concentration of each protein was 2 μM. The beads were washed three times with binding buffer and resuspended in sample buffer before analysis on SDS-PAGE.
PLK-1 inhibition experiments
For experiments in Figure 1B,C and S1D, L4 hermaphrodites were injected with perm-1 dsRNA alone (~1 μg/μl) or with equal concentrations of perm-1 and nop-1 dsRNA (~1 μg/μl for each dsRNA). Injected worms were then incubated at 16°C for 18 hours before dissection and imaging of their embryos. Gravid adult hermaphrodites containing permeable embryos were dissected into 0.8X egg salts made fresh from an egg salt solution (1X=118 mM NaCl, 40 mM KCl, 3.4 mM MgCl2, 3.4 CaCl2, and 5 mM Hepes, pH 7.4) in microdevices ([22]; www.muwells.com/celegans-embryo-imaging.html) designed to allow simultaneous imaging during drug addition and buffer exchange. Worms were placed on the dissection chamber and dissected using tweezers and a scalpel. Upon nuclear envelope breakdown, the one-cell embryos were swept towards the wells using an eyelash tool. To inhibit PLK-1 in cytokinesis, medium in the well was exchanged for buffer containing 20 μM 1-NM-PP1 (Cayman chemical company, Cat#13330) during metaphase. An 8 X 2 μm z-series was collected every 20s using a 63X, 1.4NA objective. To confirm embryos were permeabilized, embryos were submerged in 0.8X egg salts media containing lipophilic dye FM4–64 (3.3 μM).
Screen for PLK-1 phosphorylation sites
Potential PLK-1 phosphorylation sites in ZEN-4, CYK-4, and ECT-2 were identified using the kinase-specific phosphorylation site prediction system GPS (Group-based Prediction System) Polo 1.0 (http://polo.biocuckoo.org/down.php). Protein sequences in FASTA format were entered and threshold setting was set to ALL. A list of candidate PLK-1 sites for each target protein was generated utilizing the algorithm shown in Figure S3E and the selected sites were mutated in regional clusters.
ECT2 BRCT repeat surface representations
The surface representation of the human ECT2 BRCT module colored by electrostatic potential was generated with APBS [43], PDB2PQR [44], and PyMOL- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.). The surface representation of the ECT2 BRCT module colored by conservation across 150 homologs was generated with ConSurf [45] and PyMOL.
Human cell live imaging & immunofluorescence
For Immunofluorescence, clonal HeLa Kyoto cell lines were plated at 7500 cells per well into 96-well plates. The next day, cells were transfected (Fugene) with non-targeting (Dharmacon ON-TARGETplus Non-targeting siRNA #1 D-001810–01-05) or ECT2 siRNA (Thermo Scientific siGENOME Human ECT2 siRNA D-006450–02) at a final concentration of 30 nM. 48 hrs after transfection, cells were washed twice in 1X PBS and fixed in 4% formaldehyde in PBS for 10 minutes. Fixed cells were washed two times in 1X PBS, blocked for 1 hour at RT in IF buffer (1X PBS, 10 mg/ml BSA, 0.02% SDS and 0.1% Triton X-100), and incubated with primary antibody (anti-GFP and anti-tubulin) in IF buffer for 1 hour at RT. Cells were washed in IF buffer three times followed by incubation with secondary antibodies diluted in IF buffer for an additional hour at RT. Cells were washed in IF buffer three times, then in 1X PBS, incubated with Hoechst 33342 (1 μg/ml) dye in 1X PBS for 5 minutes and then washed into 1X PBS for imaging. Images were acquired using a CQ1 spinning disk confocal system (Yokogawa Electric Corporation) with a 20X 0.75 NA U-PlanApo objective at RT. Image acquisition was performed using CellVoyager software and data analysis was conducted using Fiji software [46]. For each well, 16–20 fields were imaged, acquiring 5 sections at 2.5 μm z-intervals. For live cell imaging, the same transfection conditions as for IF were used. Cells were incubated with SiR-DNA (1:2000; Cytoskeleton) for two hours and then imaged at 37°C and 5% CO2 with a CQ1 spinning disk system from 24–48hrs post-siRNA transfection. For each well, 4 fields were imaged, acquiring 5 sections at 2.5 μm z intervals every 5 minutes. Images were analyzed using Fiji software [46].
Western blot analysis of human cell lysates
Asynchronous Hela Kyoto cells from 10 cm plates were harvested at 70–80% confluence and lysed by sonication in RIPA buffer plus protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Cell lysates were normalized based on a Bio-Rad Protein Assay (Bio-Rad Laboratories). For every sample, 15–20 μg protein/lane was run on NuPAGE Novex 4–12% Bis-Tris Protein Gels (Invitrogen) and transferred to nitrocellulose membranes. Membranes were then probed with primary antibodies (anti-ECT2 and anti-actin) and detected using horseradish (HRP)–conjugated secondary antibodies with SuperSignal West Femto Maxiumum Sensitivity Substrate (Thermo Fisher). Membranes were imaged on a ChemiDoc MP system (Bio-Rad Laboratories)
QUANTIFICATION AND STATISTICAL ANALYSIS
All images were processed, scaled, and analyzed using Fiji software (National Institutes of Health). For the measurements of cleavage furrow closure, the largest distance between opposing furrow tips was measured at each time point and normalized by dividing by the starting width at anaphase onset (t=0). Quantification of fluorescence intensity was performed using maximum intensity projections as indicated in each experiment. For Figures 1A and 2B, a 7 μm long, 25-pixel wide line was drawn across the spindle midzone on maximum intensity projections of images of embryos acquired 160 seconds after anaphase onset. Linescan values for each embryo were normalized by subtracting the minimum value from all points and dividing by the difference between maximum and minimum values. The mean normalized intensity value for each position across the set of embryos was determined and plotted. Statistical analysis was performed using Prism (Graphpad). In Figures 6D, 6I, S7E, S7G, asterisks denote statistical significance as calculated by unpaired t-tests. p-values are labeled as follows: p>0.05 (ns), p<0.05 (*), p<0.01 (**) and p<0.001(***).
Supplementary Material
Video S1. PLK-1 localizes to the central spindle but C. elegans ECT-2 does not, Related to Figure 1A and 2A. Spinning disk confocal optics were used to image one-cell stage C. elegans embryos expressing either in situ-tagged PLK-1::GFP (top) or NeonGreen::ECT-2 (bottom) along with the central spindle marker ZEN-4::mScarlet. In the lower set of panels, endogenous ECT-2 and ZEN-4 were depleted to maximize the detected signal of the probes. Time stamp indicates time elapsed after anaphase onset in seconds. For PLK-1::GFP, images are maximum intensity projections of 8 x 2 μm z-stacks acquired every 40 seconds. For NG::ECT-2 images are single central sections. Playback rate is 7 frames per second. Embryos were filmed at 20°C.
Video S2. PLK-1 is essential for RhoA activation during cytokinesis in C. elegans, Related to Figure 1B. Spinning disk confocal optics were used to image permeabilized C. elegans one-cell embryos expressing H2B::mCherry and a GFP-tagged Plasma Membrane marker along with untagged wild-type (WT; left) or analog sensitive (AS; right) PLK-1. The analog-sensitive kinase inhibitor 1-NM-PP1 was added at metaphase. Embryos were imaged without (top) or with (bottom) depletion of NOP-1. Time stamp indicates time elapsed after anaphase onset in seconds. 8 x 2 μm z-stacks were acquired every 20 seconds and a single slice was selected for each time frame. Playback rate is 7 frames per second. Embryos were filmed at 20°C.
Video S3. The PLK-1 target sites in the CYK-4 N-terminus and a conserved basic surface on the ECT-2 BRCT repeat module are essential for cytokinesis, Related to Figures 3C and 5E. A comparison of the phenotypes resulting from preventing phosphorylation of the CYK-4 N-terminus (top row) and mutating the conserved basic surface on the ECT-2 BRCT repeat module that binds phosphorylated CYK-4 (bottom row). Spinning disk confocal optics were used to image C. elegans one-cell embryos expressing an mCherry::plasma membrane marker. Embryos were depleted of endogenous NOP-1 and CYK-4 (top) or NOP-1 and ECT-2 (bottom). (top row) Representative embryos with no transgene or reconstituted with transgenes expressing untagged WT, 4A or Δ163–180 CYK-4. (bottom row) Representative embryos with no transgene or reconstituted with transgenes expressing untagged WT or Basic Surface 3A or Basic Surface 3E mutant ECT-2. Time stamp indicates time elapsed after anaphase onset in seconds. 8 x 2 μm z-stacks were acquired every 20 seconds and a single central slice was selected for each time point. Embryos were filmed at 20°C. Playback rate is 7 frames per second.
Video S4. ECT2 Basic Surface Mutations compromise targeting to the spindle midzone and enrichment at the equatorial cell membrane during cytokinesis in human cells, Related to Figure 6G. HeLa Kyoto cells stably expressing GFP fusions with WT ECT2 or TK, Basic Surface 3A-1, or Basic Surface 3E-1 mutant ECT-2 from siRNA-resistant transgenes were transfected with ECT-2 siRNA and imaged from 24 to 48 hours after transfection. Time stamp indicates time elapsed after anaphase onset in minutes. Maximum intensity projections were generated for the 5 x 2.5 μm z-stacks acquired every 5 minutes and are shown at a playback rate of 2 frames per second.
Video S5. ECT2 Basic Surface Mutations lead to cytokinesis failure in human cells, Related to Figure 6H. HeLa Kyoto cells stably expressing GFP alone or GFP fusions with WT ECT2 or TK, Basic Surface 3A-1, or Basic Surface 3E-1 mutant ECT-2 from siRNA-resistant transgenes were transfected with ECT-2 siRNA and imaged with brightfield microscopy from 24 to 48 hours after transfection. Cells were stained with SiR-DNA (red) to visualize DNA. Time stamp indicates time elapsed after metaphase in minutes. Maximum intensity projections were generated for the 5 x 2.5 μm z-stacks acquired every 5 minutes and are shown at a playback rate of 2 frames per second.
Video S6. Autoinhibition prevents ECT-2 in the cytoplasm from binding to phosphorylated CYK-4 on the central spindle, Related to Figures 7B and 7C. Spinning disk confocal optics were used to image C. elegans one-cell embryos expressing (from left to right) NG::ECT-2 full length, NG::ECT-2 1–320, NG::ECT-2 1–320 Basic Surface 3A mutant, NG::ECT-2 1–320 K166M mutant (related to Figure 7B), NG::ECT-2 1–320 or NG::ECT-2 1–320 (Related to Figure 7C) along with the central spindle marker ZEN-4::Scarlet. In the case of videos related to Figure 7B, embryos were depleted of endogenous CYK-4 and reconstituted with transgenes expressing untagged WT CYK-4 or 4A CYK-4. For all conditions embryos were filmed beginning just before anaphase onset at 20°C. Time stamp indicates time elapsed after anaphase onset in seconds. 8x 2 μm z-stacks were acquired every 20 seconds and maximal intensity projections were generated for each time frame. Playback rate is 7 frames per second.
Highlights.
RhoA regulator screen identifies a single essential PLK1 target site cluster in CYK4
Phosphorylation by PLK1 of CYK4 enables binding to a conserved basic surface in ECT2
ECT2 basic surface is distinct from the canonical BRCT phosphopeptide binding site
Autoinhibition prevents C. elegans ECT2 from binding to P-CYK4 at the central spindle
ACKNOWLEDGEMENTS
We thank Daniel Gerlich for the HeLa Kyoto cell line and Mark Petronczki for providing WT and TK mutant ECT2 constructs. We thank the Oegema and Desai labs for helpful discussions and Rebecca Green for help with the model figure. J.S.G-C was supported by the University of California, San Diego Cancer Cell Biology Training Program (T32 CA067754) and Ruth L. Kirschstein Postdoctoral Individual National Research Service Award (F32GM125347). A.K.S, A.D. and K.O. received salary and other support from the Ludwig Institute for Cancer Research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.D’Avino PP, Giansanti MG, and Petronczki M (2015). Cytokinesis in animal cells. Cold Spring Harb Perspect Biol 7, a015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fededa JP, and Gerlich DW (2012). Molecular control of animal cell cytokinesis. Nat Cell Biol 14, 440–447. [DOI] [PubMed] [Google Scholar]
- 3.Green RA, Paluch E, and Oegema K (2012). Cytokinesis in animal cells. Annu Rev Cell Dev Biol 28, 29–58. [DOI] [PubMed] [Google Scholar]
- 4.Bement WM, Miller AL, and von Dassow G (2006). Rho GTPase activity zones and transient contractile arrays. Bioessays 28, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordan SN, and Canman JC (2012). Rho GTPases in animal cell cytokinesis: an occupation by the one percent. Cytoskeleton (Hoboken) 69, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piekny A, Werner M, and Glotzer M (2005). Cytokinesis: welcome to the Rho zone. Trends Cell Biol 15, 651–658. [DOI] [PubMed] [Google Scholar]
- 7.von Dassow G (2009). Concurrent cues for cytokinetic furrow induction in animal cells. Trends Cell Biol 19, 165–173. [DOI] [PubMed] [Google Scholar]
- 8.Basant A, and Glotzer M (2018). Spatiotemporal Regulation of RhoA during Cytokinesis. Curr Biol 28, R570–R580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishima M (2016). Centralspindlin in Rappaport’s cleavage signaling. Semin Cell Dev Biol 53, 45–56. [DOI] [PubMed] [Google Scholar]
- 10.Golsteyn RM, Mundt KE, Fry AM, and Nigg EA (1995). Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 129, 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brennan IM, Peters U, Kapoor TM, and Straight AF (2007). Polo-like kinase controls vertebrate spindle elongation and cytokinesis. PLoS One 2, e409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, and Jallepalli PV (2007). Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A 104, 4383–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petronczki M, Glotzer M, Kraut N, and Peters JM (2007). Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell 12, 713–725. [DOI] [PubMed] [Google Scholar]
- 14.Burkard ME, Maciejowski J, Rodriguez-Bravo V, Repka M, Lowery DM, Clauser KR, Zhang C, Shokat KM, Carr SA, Yaffe MB, et al. (2009). Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol 7, e1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su KC, Takaki T, and Petronczki M (2011). Targeting of the RhoGEF Ect2 to the equatorial membrane controls cleavage furrow formation during cytokinesis. Dev Cell 21, 1104–1115. [DOI] [PubMed] [Google Scholar]
- 16.Wolfe BA, Takaki T, Petronczki M, and Glotzer M (2009). Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol 7, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalamalasetty RB, Hummer S, Nigg EA, and Sillje HH (2006). Influence of human Ect2 depletion and overexpression on cleavage furrow formation and abscission. J Cell Sci 119, 3008–3019. [DOI] [PubMed] [Google Scholar]
- 18.Kotynkova K, Su KC, West SC, and Petronczki M (2016). Plasma Membrane Association but Not Midzone Recruitment of RhoGEF ECT2 Is Essential for Cytokinesis. Cell Rep 17, 2672–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung CC, and Glover JN (2011). BRCT domains: easy as one, two, three. Cell Cycle 10, 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martino L, Morchoisne-Bolhy S, Cheerambathur DK, Van Hove L, Dumont J, Joly N, Desai A, Doye V, and Pintard L (2017). Channel Nucleoporins Recruit PLK-1 to Nuclear Pore Complexes to Direct Nuclear Envelope Breakdown in C. elegans. Dev Cell 43, 157–171 e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodruff JB, Wueseke O, Viscardi V, Mahamid J, Ochoa SD, Bunkenborg J, Widlund PO, Pozniakovsky A, Zanin E, Bahmanyar S, et al. (2015). Centrosomes. Regulated assembly of a supramolecular centrosome scaffold in vitro. Science 348, 808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho A, Olson SK, Gutierrez E, Zhang K, Noble LB, Zanin E, Desai A, Groisman A, and Oegema K (2011). Acute drug treatment in the early C. elegans embryo. PLoS One 6, e24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose LS, Lamb ML, Hird SN, and Kemphues KJ (1995). Pseudocleavage is dispensable for polarity and development in C. elegans embryos. Dev Biol 168, 479–489. [DOI] [PubMed] [Google Scholar]
- 24.Tse YC, Werner M, Longhini KM, Labbe JC, Goldstein B, and Glotzer M (2012). RhoA activation during polarization and cytokinesis of the early Caenorhabditis elegans embryo is differentially dependent on NOP-1 and CYK-4. Mol Biol Cell 23, 4020–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H, Guo F, Brahma S, Xing Y, and Burkard ME (2014). Centralspindlin assembly and 2 phosphorylations on MgcRacGAP by Polo-like kinase 1 initiate Ect2 binding in early cytokinesis. Cell Cycle 13, 2952–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuce O, Piekny A, and Glotzer M (2005). An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol 170, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou Y, Shao Z, Peng J, Li F, Gong D, Wang C, Zuo X, Zhang Z, Wu J, Shi Y, et al. (2014). Crystal structure of triple-BRCT-domain of ECT2 and insights into the binding characteristics to CYK-4. FEBS Lett 588, 2911–2920. [DOI] [PubMed] [Google Scholar]
- 28.Kim JE, Billadeau DD, and Chen J (2005). The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem 280, 5733–5739. [DOI] [PubMed] [Google Scholar]
- 29.Bigot N, Day M, Baldock RA, Watts FZ, Oliver AW, and Pearl LH (2019). Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9–1-1 to control the G1 DNA damage checkpoint. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Day M, Rappas M, Ptasinska K, Boos D, Oliver AW, and Pearl LH (2018). BRCT domains of the DNA damage checkpoint proteins TOPBP1/Rad4 display distinct specificities for phosphopeptide ligands. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M, Pan H, Sun L, Shi P, Zhang Y, Li L, Huang Y, Chen J, Jiang P, Fang X, et al. (2020). Structure and regulation of human epithelial cell transforming 2 protein. Proc Natl Acad Sci U S A 117, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lekomtsev S, Su KC, Pye VE, Blight K, Sundaramoorthy S, Takaki T, Collinson LM, Cherepanov P, Divecha N, and Petronczki M (2012). Centralspindlin links the mitotic spindle to the plasma membrane during cytokinesis. Nature 492, 276–279. [DOI] [PubMed] [Google Scholar]
- 33.Zhang D, and Glotzer M (2015). The RhoGAP activity of CYK-4/MgcRacGAP functions noncanonically by promoting RhoA activation during cytokinesis. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Avino PP, Savoian MS, Capalbo L, and Glover DM (2006). RacGAP50C is sufficient to signal cleavage furrow formation during cytokinesis. J Cell Sci 119, 4402–4408. [DOI] [PubMed] [Google Scholar]
- 35.Wagner E, and Glotzer M (2016). Local RhoA activation induces cytokinetic furrows independent of spindle position and cell cycle stage. J Cell Biol 213, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14, 53–56. [DOI] [PubMed] [Google Scholar]
- 37.Prokopenko SN, Brumby A, O’Keefe L, Prior L, He Y, Saint R, and Bellen HJ (1999). A putative exchange factor for Rho1 GTPase is required for initiation of cytokinesis in Drosophila. Genes Dev 13, 2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su KC, Bement WM, Petronczki M, and von Dassow G (2014). An astral simulacrum of the central spindle accounts for normal, spindle-less, and anucleate cytokinesis in echinoderm embryos. Mol Biol Cell 25, 4049–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, and Jorgensen EM (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. (2000). A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401. [DOI] [PubMed] [Google Scholar]
- 41.Hattersley N, Lara-Gonzalez P, Cheerambathur D, Gomez-Cavazos JS, Kim T, Prevo B, Khaliullin R, Lee KY, Ohta M, Green R, et al. (2018). Employing the one-cell C. elegans embryo to study cell division processes. Methods Cell Biol 144, 185–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee KY, Green RA, Gutierrez E, Gomez-Cavazos JS, Kolotuev I, Wang S, Desai A, Groisman A, and Oegema K (2018). CYK-4 functions independently of its centralspindlin partner ZEN-4 to cellularize oocytes in germline syncytia. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, and Baker NA (2007). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35, W522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. PLK-1 localizes to the central spindle but C. elegans ECT-2 does not, Related to Figure 1A and 2A. Spinning disk confocal optics were used to image one-cell stage C. elegans embryos expressing either in situ-tagged PLK-1::GFP (top) or NeonGreen::ECT-2 (bottom) along with the central spindle marker ZEN-4::mScarlet. In the lower set of panels, endogenous ECT-2 and ZEN-4 were depleted to maximize the detected signal of the probes. Time stamp indicates time elapsed after anaphase onset in seconds. For PLK-1::GFP, images are maximum intensity projections of 8 x 2 μm z-stacks acquired every 40 seconds. For NG::ECT-2 images are single central sections. Playback rate is 7 frames per second. Embryos were filmed at 20°C.
Video S2. PLK-1 is essential for RhoA activation during cytokinesis in C. elegans, Related to Figure 1B. Spinning disk confocal optics were used to image permeabilized C. elegans one-cell embryos expressing H2B::mCherry and a GFP-tagged Plasma Membrane marker along with untagged wild-type (WT; left) or analog sensitive (AS; right) PLK-1. The analog-sensitive kinase inhibitor 1-NM-PP1 was added at metaphase. Embryos were imaged without (top) or with (bottom) depletion of NOP-1. Time stamp indicates time elapsed after anaphase onset in seconds. 8 x 2 μm z-stacks were acquired every 20 seconds and a single slice was selected for each time frame. Playback rate is 7 frames per second. Embryos were filmed at 20°C.
Video S3. The PLK-1 target sites in the CYK-4 N-terminus and a conserved basic surface on the ECT-2 BRCT repeat module are essential for cytokinesis, Related to Figures 3C and 5E. A comparison of the phenotypes resulting from preventing phosphorylation of the CYK-4 N-terminus (top row) and mutating the conserved basic surface on the ECT-2 BRCT repeat module that binds phosphorylated CYK-4 (bottom row). Spinning disk confocal optics were used to image C. elegans one-cell embryos expressing an mCherry::plasma membrane marker. Embryos were depleted of endogenous NOP-1 and CYK-4 (top) or NOP-1 and ECT-2 (bottom). (top row) Representative embryos with no transgene or reconstituted with transgenes expressing untagged WT, 4A or Δ163–180 CYK-4. (bottom row) Representative embryos with no transgene or reconstituted with transgenes expressing untagged WT or Basic Surface 3A or Basic Surface 3E mutant ECT-2. Time stamp indicates time elapsed after anaphase onset in seconds. 8 x 2 μm z-stacks were acquired every 20 seconds and a single central slice was selected for each time point. Embryos were filmed at 20°C. Playback rate is 7 frames per second.
Video S4. ECT2 Basic Surface Mutations compromise targeting to the spindle midzone and enrichment at the equatorial cell membrane during cytokinesis in human cells, Related to Figure 6G. HeLa Kyoto cells stably expressing GFP fusions with WT ECT2 or TK, Basic Surface 3A-1, or Basic Surface 3E-1 mutant ECT-2 from siRNA-resistant transgenes were transfected with ECT-2 siRNA and imaged from 24 to 48 hours after transfection. Time stamp indicates time elapsed after anaphase onset in minutes. Maximum intensity projections were generated for the 5 x 2.5 μm z-stacks acquired every 5 minutes and are shown at a playback rate of 2 frames per second.
Video S5. ECT2 Basic Surface Mutations lead to cytokinesis failure in human cells, Related to Figure 6H. HeLa Kyoto cells stably expressing GFP alone or GFP fusions with WT ECT2 or TK, Basic Surface 3A-1, or Basic Surface 3E-1 mutant ECT-2 from siRNA-resistant transgenes were transfected with ECT-2 siRNA and imaged with brightfield microscopy from 24 to 48 hours after transfection. Cells were stained with SiR-DNA (red) to visualize DNA. Time stamp indicates time elapsed after metaphase in minutes. Maximum intensity projections were generated for the 5 x 2.5 μm z-stacks acquired every 5 minutes and are shown at a playback rate of 2 frames per second.
Video S6. Autoinhibition prevents ECT-2 in the cytoplasm from binding to phosphorylated CYK-4 on the central spindle, Related to Figures 7B and 7C. Spinning disk confocal optics were used to image C. elegans one-cell embryos expressing (from left to right) NG::ECT-2 full length, NG::ECT-2 1–320, NG::ECT-2 1–320 Basic Surface 3A mutant, NG::ECT-2 1–320 K166M mutant (related to Figure 7B), NG::ECT-2 1–320 or NG::ECT-2 1–320 (Related to Figure 7C) along with the central spindle marker ZEN-4::Scarlet. In the case of videos related to Figure 7B, embryos were depleted of endogenous CYK-4 and reconstituted with transgenes expressing untagged WT CYK-4 or 4A CYK-4. For all conditions embryos were filmed beginning just before anaphase onset at 20°C. Time stamp indicates time elapsed after anaphase onset in seconds. 8x 2 μm z-stacks were acquired every 20 seconds and maximal intensity projections were generated for each time frame. Playback rate is 7 frames per second.
Data Availability Statement
All primary data associated with the paper is available upon request.