

Abstract
The human radiolabeled absorption, distribution, metabolism, and excretion (ADME) study offers a quantitative and comprehensive overall picture of the disposition of a drug, including excretion pattern and metabolite profiles in circulation and excreta. The data gathered from the ADME study are highly informative for developing a cohesive strategy for clinical pharmacology studies. Elements of standard ADME study designs are described. An exciting new development in human ADME studies is the application of accelerator mass spectrometry (AMS) as the detection technique for carbon‐14, in replacement of radioactivity measurements. This technology permits administration of 100‐fold to 1,000‐fold lower amounts of carbon‐14, and thus opens the door to the application of new study designs. A new ADME study design, termed the AMS‐Enabled Human ADME study, is described. In this design, both oral and intravenous administration are assessed in a single clinical study with a two‐period crossover. In addition to all of the standard ADME study end points (e.g., mass balance and quantitative metabolite profiles), the AMS‐Enabled ADME study can provide the fundamental pharmacokinetic parameters of clearance, volume of distribution, absolute oral bioavailability, and even estimates of the fraction of the dose absorbed. Thus, we have entered a new era of human ADME study design that can yield vastly more informative and complete data sets enabling a superior understanding of overall drug disposition.
IMPORTANCE OF THE HUMAN ADME STUDY
A quantitative description of the ultimate fate of a new molecule in the human body is required for new drug approval. However, of the many clinical pharmacology studies conducted during the development of new medicines, the radiolabeled absorption‐distribution‐metabolism‐excretion (ADME) study offers the richest dataset to understand how the human body handles the drug. 1 ADME studies are generally conducted for drugs under development that are organic xenobiotics, whereas for drug candidates that are biological macromolecules containing no xenobiotic elements, radiolabeled ADME studies are often not done. An exception to a required ADME study for small organic molecule drug candidates is when the entire dose can be accounted for as unchanged drug in the urine, and, in such a case, the need for the radiolabeled ADME study is obviated. However, this is rare and for most new drugs a radiolabeled ADME study is an essential component of the suite of clinical pharmacology studies included in registration dossiers.
In brief, the human radiolabeled ADME study is typically conducted by administration of a single dose of drug containing a radioactive nuclide followed by collection of plasma and excreta samples. Samples are analyzed for both total radioactivity and the profile of drug‐related material in urine, feces, and plasma. 2 , 3 The use of radiolabeled material permits quantitative accounting for all drug‐related material in biological samples by providing a common analytical response factor. Note that in commonly used analytical methods, such as mass spectrometric, fluorometric, or spectrophotometric detection, each analyte (i.e., parent drug and individual metabolites), possesses a different inherent response factor that is dependent on its chemical structure. This necessitates calibration of instrument response using authentic standards of each analyte. In contrast, quantitative radiometric detection is dependent only on the specific activity of the analytes, which will be constant between parent drug and all metabolites containing the raduionuclide. This is especially important for metabolites where synthetic standards may not exist. In fact, when using radiometric high‐performance liquid chromatography (HPLC) analysis, the structural identity of the metabolites is not even required to characterize their disposition.
There is no specific regulatory requirement as to when the ADME study should be conducted during clinical development other than prior to the initiation of large scale and long duration clinical trials. In the 2012 European Medicines Agency’s guidelines on drug interactions, it is stated that “the results of the mass‐balance study should generally be available before starting phase III.” 4 It is often understood that having this information will enable a robust discussion with regulators prior to phase III regarding the clinical pharmacology studies needed to be conducted before the new drug can be registered as a commercially available therapy. These can include drug‐drug interaction studies and studies in various special populations (e.g., studies evaluating the impact of pharmacogenetic variants of drug disposition proteins, studies in subjects with organ impairment, and others). Knowing the complete human metabolite profile can also be important when planning 2‐year carcinogenicity studies in laboratory animals (typically mice and rats) that are done in parallel with phase III clinical trials so that assurance can be provided that the animals undergoing this evaluation will have been exposed to the metabolites to which humans are exposed. Because of the power of the knowledge gained in the human ADME (especially using a study design described later in this paper), it is advantageous to conduct the study early enough that such knowledge can be leveraged for decision making and planning of other clinical studies. Qualitative analysis of plasma samples from first‐in‐human studies for circulating metabolites (a.k.a. metabolite “scouting”) can yield data that may trigger planning of an earlier human ADME study. Nevertheless, most drug development programs conduct this study only after observing positive signs of efficacy with a proof of concept study in a targeted patient population—either a disease‐modifying outcome or a surrogate marker. This timing balances clinical enablement for candidates that have a greater likelihood of success vs. not wasting efforts conducting ADME studies for those candidates that will ultimately fail.
Briefly, the knowledge gained from an oral ADME study includes:
Percentages of the total drug‐related material in circulation comprised by the drug and each metabolite;
Percentages of the drug‐related material excreted in urine and feces, and the percentages that parent drug and each metabolite comprise of the total dose excreted;
Chemical structures of major metabolites in plasma and excreta; and
A lower limit estimate of the amount of drug absorbed by measurement of total radioactivity recovered in urine.
The quantitative profile of metabolites in the excreta is used to develop an understanding of the mechanism(s) of clearance. By determining the structures of metabolites, a metabolic scheme can be constructed. This scheme can be coupled with in vitro metabolism data wherein each metabolic pathway is attributed to one or more drug‐metabolizing enzymes. This knowledge of the fraction metabolized by various enzymes offers insight used to design the most appropriate drug‐drug interaction (with the new drug as “victim”), pharmacogenetic, and special population studies, as well as used in conjunction with other clinical pharmacology and in vitro studies in construction of a full physiologically‐based pharmacokinetic model of the drug. The knowledge gained from the ADME study builds on previous knowledge obtained in earlier clinical pharmacology and in vitro metabolism and transporter studies, either by refining that knowledge with greater granularity or, in some cases, uncovering new and unanticipated aspects of drug disposition, such as the identification of a previously unknown metabolic pathway.
The quantitative profile of metabolites in circulation addresses two potential issues: (i) the potential for the presence of pharmacologically active metabolites, and (ii) the need for ascertaining that laboratory animal species used in toxicological evaluation of the drug were adequately exposed to human metabolites. To the first point, metabolites in circulation can possibly possess similar activity to the parent compound. 5 The radiometric profile will yield relative abundances of metabolites to parent drug and mass spectral data will offer an indication of the degree of similarity of chemical structure between metabolites and the parent drug. Although structural similarity does not mean that a metabolite will be pharmacologically active, the likelihood is greater and it is advisable that circulating metabolites of structure similar to the parent drug (e.g., metabolites arising by hydroxylation, heteroatom demethylation, dehydrogenation, and others) be evaluated for activity at the target receptor or enzyme. If a circulating metabolite is active, contributes to ≥ 50% of the overall pharmacologic activity, 6 it will need to be monitored in clinical and toxicity studies and be evaluated for drug‐drug interactions. Regarding the second issue, metabolites in safety testing guidance dictates that metabolites reaching a threshold abundance (identified as ≥ 10% of the total drug‐related material in human plasma by area under the curve (AUC), irrespective of activity) must also be present in the plasma of laboratory animal species used in toxicology studies at equal or greater exposure levels to be adequately assessed for safety risk. 7 If they did not achieve such levels in animals, follow‐up is required to gather data for a risk assessment for the specific metabolite in question. It should be noted that the boundary of 10% of total drug‐related material is offered as a general guidance and that for some metabolites, such as ether or quaternary ammonium glucuronides, there can be greater tolerance. For other metabolite types that have been associated with toxicity, such as acyl glucuronides, the tolerance could be stricter. As with any safety risk assessment, the dose level and absolute exposures should be considered.
STANDARD RADIOLABEL ORAL ADME STUDY DESIGN AND EXECUTION
There are three main steps of the standard (i.e., doses of radioactivity in the range of 20–100 µCi) human ADME study:
Pre‐study activities: including synthesis and manufacture of the radioactive drug, and the conduct of a quantitative whole‐body autoradiography (QWBA) study in a rodent species;
Clinical portion: dosing and sample collection with real‐time quantitation of carbon‐14 in excretory and plasma samples;
Analysis: quantitative HPLC profiling of metabolites and metabolite structure elucidation in excretory and plasma samples.
Pre‐study activities
Preparation of the dose
The human ADME study activities begin long before any dosing is performed. 2 The first endeavor is the preparation of radiolabeled drug, which typically takes several months. The identity and site of incorporation of the radionuclide must be carefully selected so that the label is not lost as a small portion of the drug through metabolism. Thus, carbon‐14 is almost always selected over hydrogen‐3 (the latter label can be lost though tritium exchange or simple oxidation reactions), and the position of the carbon‐14 must be part of the core structure of the molecule. For drugs that are cleaved into two substantial portions, it may be necessary to make two labeled materials with carbon‐14 on each portion and conduct two ADME studies so that each portion of the drug can be monitored. Precursor reagents and chemistries for radiosynthesis are much more limited than standard synthesis, thus the synthetic route for preparation of the radiolabeled material will likely need to be designed anew and cannot merely mirror the process route that has already been established for making the drug. Furthermore, although not stated as a regulatory requirement, many human ADME studies utilize radiolabeled drug prepared under (good manufacturing practices). This adds considerably to the time needed, documentation, and qualification of synthetic reagents, and hence cost, and it is a point of debate (beyond the scope of this paper) as to whether such stringency is of any scientific value for a single‐dose study conducted in a well‐controlled clinical environment. Furthermore, this is especially notable when considering doses of < 1 µCi used in the accelerator mass spectrometry (AMS)‐Enabled Human ADME study design described later. Most important is establishing the radiopurity and specific activity of the material. In most cases, the selection of the total dose (radiolabeled drug + unlabeled drug) is typically similar to the pharmacologic dose and is based on the clinical safety profile of the administered drug. For most ADME studies, the radiolabeled material must be diluted with unlabeled material to match the correct total dose level to be administered with an appropriate amount of radioactivity. It is critical to ensure that this combination of labeled and unlabeled material is homogeneous, because the quantitation will be done using radiometric measurements. Mixing radiolabeled material with unlabeled material to the desired specific activity needs to be done in a solution step during the radiosynthetic process (either at some point in the synthesis or as the final crystallization step). Mixing solid radiolabeled material with solid unlabeled material and dosing as a nonhomogeneous solid or suspension can yield aberrant results and can invalidate a study. If the material is dosed in a solution, then the issue of mixing unlabeled and labeled is solved. Additionally, the material must be formulated as necessary, based on the pharmaceutical characteristics of the drug.
Conduct of a QWBA study in rodents
In order to dose humans with typical 20–100 µCi amounts of carbon‐14, a tissue distribution study must be conducted in an animal species (usually rats) to make estimates of exposure to radioactivity in tissues of humans (a.k.a. dosimetry). For example, if the drug of interest binds and is retained in a specific tissue (e.g., melanin binding for basic drugs in ocular microstructures is commonly observed), then the amount of radioactivity that may be administered may need to be correspondingly decreased. In extreme cases, if the tissue residence of radioactivity is extensive and prolonged, the amount of radioactivity that can be safely dosed to humans may be too small to practically carry out the study. Limits on human exposure to radiolabeled materials are described and vary with the organ being exposed (most are 3 rem). 8
Clinical portion
Successful human radiolabeled ADME studies begin with meticulous laboratory and clinical practices when carrying out the dosing and sample collection. Calculations done regarding mass balance, excretion, and the percentage that each metabolite comprises of the dose all derive from an accurate determination of the dose delivered to each study subject. The nominal (i.e., intended) dose is less important than knowing exactly the amount of radioactivity that was actually administered. After dosing, excreta are collected as quantitatively as possible, as any missed excreta will compromise assessment of mass balance. Blood is also sampled from study subjects using a pharmacokinetic sampling scheme that may be more extended than typical in anticipation of the possibility of metabolites with elimination half‐lives longer than the parent drug. Routine safety monitoring is also done mostly in a precautionary measure because by the time the ADME study is performed the safety profile of the single dose level used in the study is well‐established. To ensure proper and complete collection of excreta, study subjects need to remain in the clinical facility for the duration of the sample collection intervals. However, to enable timely subject release, measurements of radioactivity in excreta need to be done on an ongoing basis to determine when discharge criteria are met (i.e., total excretion exceeds a pre‐agreed value; e.g., 90–95%) or the rate of excretion decreases to be under a pre‐agreed threshold (e.g., 1% per day). There can be instances in which excretion occurs at an extremely slow rate and this precludes keeping study subjects in the clinical facility until acceptable mass balance is achieved. In such instances, subjects can be dismissed from the clinical facility and recalled at future dates when excreta can be collected over a predefined period (i.e., 1 day). Data gathered in this fashion can be modeled to estimate the rate of excretion and projection of mass balance. Mass balance in and of itself is not a parameter that offers much toward the understanding of the drug and is mostly a value used in making other calculations (such as amount of each metabolite as % of dose), nevertheless it can be disconcerting when a good mass balance is not achieved. There are practical limitations to the thoroughness that excreta can be collected and an analysis of past studies suggests that when > 85% recovery is obtained, the study can be considered successful. 9 Values marginally below that will require scrutinization of the dosing and sample collection procedures and development of a plausible explanation of why higher balance was not achieved. However, a well‐executed study with recoveries below 85% will raise questions as to whether a substantial portion of the dose is sequestered in the body and whether there are any safety considerations of such a phenomenon.
Metabolite profiling and identification
Plasma and excreta samples are analyzed using HPLC in line with high‐resolution mass spectrometry and radiometric detection, or the latter is done off‐line by collecting fractions from the HPLC and analyzing these fractions for radioactivity. The radioactivity measurements yield the fraction that each metabolite comprises of the total drug‐related material. Mass spectrometry (MS) offers some informative insight regarding the structure of a metabolite, but alone does not unambiguously prove the structure in most instances. From the MS data, one can determine the type of chemical modification(s) that have occurred, and fragmentation data can point to the region of the drug molecule that has been modified. Proof of the chemical structure requires other information, such as comparison of chromatographic and spectral properties with a synthesized authentic standard of the metabolite, or isolation of the metabolite for analysis by 1D and 2D nuclear magnetic resonance spectroscopy. Drug metabolism scientists who carry out the metabolite profiling portion of the human ADME study will strive to account for as much of the drug‐related material and identify as many of the metabolites as possible, but there are practical limitations. Metabolites present at > 10% of dose in excreta or drug‐related material in plasma should always have structures definitively identified. When the metabolite profile is highly complex with numerous metabolites (e.g., > 20) at low levels, it can be challenging to determine structures for much of the drug‐related material because there will not be enough mass of each metabolite to carry out the needed spectral characterization.
TECHNOLOGY ADVANCE: THE “ACCELERATOR MASS SPECTROMETRY ENABLED HUMAN ADME STUDY”
AMS is a technique that has been available for decades to sensitively measure specific isotopes. In the past, AMS instruments were very large, complex, expensive, and were only located at large national laboratories where operators were highly trained experts. However, over the past decade, instrumentation has been modified to be smaller and specialized to measure carbon‐14. More facile combustion methods to prepare samples for AMS analysis can replace tedious sample graphitization to reduce cost and time. 10 , 11 For use in human ADME studies, AMS can be leveraged and its exquisite sensitivity permits the administration of much lower quantities of carbon‐14. This is because AMS measures carbon‐14 by MS and not radioactivity. Instead of typical doses of 20–100 µCi in a human ADME study, when using AMS as the detection and quantitation technique, doses of 0.1–1 µCi (3.7–37 kBq) can be used and still yield reliable measurements. Although there can be advantages of using less radioactivity (lower radioactive exposure to study subjects, and reduced cost of waste) the greatest advantage offered using AMS is in the design of more informative ADME studies. Additionally, the very low amounts of radioactivity used in this study reduces the exposure to humans so significantly that it obviates the need for prerequisite QWBA animal studies to estimate dosimetry. Many standard oral‐dose human carbon‐14 ADME studies have now been reported, wherein AMS is used as the measurement approach in place of liquid scintillation counting, and it can be concluded that AMS as applied to ADME has evolved from a niche technology used in special circumstances to a mainstay approach. A discussion of the technical aspects of AMS analysis are beyond the scope of this paper and the interested reader is referred to other papers on this topic. 11 , 12 , 13
The AMS‐Enabled Human ADME study consists of a two‐period, fixed sequence, crossover design (Figure 1 ). Period 1 consists of a standard ADME study protocol. A pharmacologically relevant oral dose is administered, albeit with material of a much lower carbon‐14 content suitable for analysis by AMS (0.1–1 µCi instead of 20–100 µCi). Excreta are collected with an intent to assess mass balance. Blood is collected and plasma harvested to determine the pharmacokinetics of total carbon‐14 and parent drug. These matrices are analyzed as above to determine the identities and chemical structures of metabolites. However, the HPLC eluent is also fractionated and the fractions are analyzed by AMS to generate a carbon‐14 chromatogram from which quantities of each metabolite can be determined. The complexity of the AMS technique, as currently done, precludes evaluating the metabolite profile of every individual sample. Thus, pools of each matrix are constructed across sampling times and across the individual subjects yielding three injections: one plasma, one urine, and one fecal homogenate. Thus, although the differences in metabolite profiles among individual subjects in the study are no longer captured, ADME studies, in general, are not powered to robustly evaluate interindividual variability. Instead, variability in exposure to and/or excretion of specific metabolites (i.e., pharmacologically active or metabolites exceeding the metabolites in safety testing guidance threshold) can be followed up in larger numbers of individuals in other clinical studies using standard bioanalytical methods, as needed.
Figure 1.
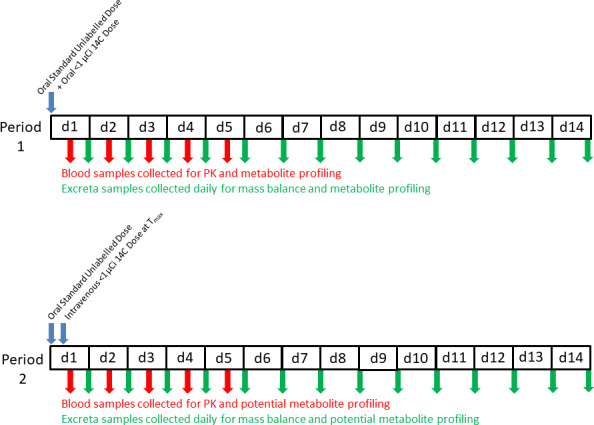
Study Design of the accelerator mass spectrometry (AMS)‐Enabled Human absorption, distribution, metabolism, and excretion (ADME) study. This is an open‐label fixed sequence crossover study in which subjects receive a single oral administration of a clinically relevant dose level of a drug with a total amount of carbon‐14 of 0.2–1 µCi in period 1. Blood samples are collected to obtain the pharmacokinetics of unchanged drug and total carbon‐14. Excreta samples are collected daily and total carbon‐14 is measured to determine mass balance. After a suitable washout period, the second period is conducted with the same subjects. An oral dose of unlabeled drug is administered at the same dose level as in period 1 but with no carbon‐14. This is followed by intravenous administration of labeled drug at time of maximum plasma concentration using the same amount of carbon‐14 as was given orally in period 1. Blood and excreta samples are collected and analyzed similarly to period 1.
The greatest advantage of using AMS in the human ADME study is that it enables the addition of a second period wherein the same subjects are administered an intravenous microdose of carbon‐14 labeled material. With intravenous administration, the fundamental and important pharmacokinetic parameters clearance and volume of distribution can be calculated. Combining these data with the data from period 1 permits estimation of fraction of dose absorbed. Importantly, in period 2, the same dose is administered orally (no carbon‐14) and at approximately the time of the oral time of maximum plasma concentration (Tmax) an intravenous microdose of carbon‐14 labeled material is administered. This is done because the total amount of material in the intravenous dose is very low (in the range of 1 µg or less). However, unlike other microdose studies in which pharmacokinetic data at the low doses may not scale to those measured following higher doses, 14 this study design is not prone to that problem because there is still the appropriate total mass of material in the body from the nonlabeled oral dose. Calculation of clearance, volume of distribution, and absolute bioavailability requires the measurement of the drug in plasma following the intravenous doses by first separating it from its metabolites by HPLC and then quantitating it using AMS. For oral bioavailability, the parent drug in plasma measured by AMS is used to calculate the AUCIV and the parent drug measured in the same plasma samples from period 2 by a standard analytical method (e.g., HPLC‐MS) is used to calculate AUCPO. The ratios of these two AUC values, corrected for the differences in intravenous and oral doses, is used to calculate the oral bioavailability. Although the use of AMS to make estimates of oral bioavailability 15 and absorption 16 have occurred, examples of the use of this approach for a combined study that truly evaluates all four of the letters in the ADME acronym have only recently appeared in the literature. 17 , 18
Estimation of the fraction absorbed (F a) can be made by comparing the total carbon‐14 excreted in urine following the two dose routes using the following ratio 3 , 16 , 17 :
It should be noted that some assumptions and approximations require acceptance to make this estimate (listed in Table 1 ). In addition, it assumes that overall mass balance is the same following each route of administration and, if this is not the case, the ratio will need to be corrected for any difference. If urinary total radioactivity is low, the estimate made in this manner may be prone to greater experimental variability. Alternately, comparison of unchanged parent drug in feces after intravenous and oral administration can be leveraged to make an estimate of F a. When unchanged drug is observed in feces following oral administration, it could be due to either unabsorbed material or drug that is absorbed but then secreted back into the gut via bile or intestinal secretion. Unchanged drug in feces following intravenous administration can help demonstrate whether biliary clearance and/or intestinal secretion mechanisms contribute to the observation of drug in feces following oral administration. For any of these methods estimating F a, there are assumptions needed regarding specific phenomena that could undermine the estimate, such as the presence of enterohepatic recirculation, presystemic metabolism in gut lumen, and others. However, if both estimates are made and offer agreement, then there may be greater confidence in the estimate of F a. Thus, using this design, the AMS‐Enabled Human ADME study offers a comprehensive picture of the overall pharmacokinetics and disposition of the drug. It should be noted that the oral dosage form used in ADME studies is frequently a solution or suspension. Thus, the estimates for F a made using this approach may be different from the absorption of the drug in its final product form (e.g., tablets with various excipients) for which dissolution characteristics can be different.
Table 1.
Assumptions and considerations required when using the ratio of urinary excreted carbon‐14 after oral and intravenous administration to estimate fraction absorbed
| Assumption | Reason |
|---|---|
| No metabolism in GI lumen with the metabolites absorbed and renally cleared. | This could yield an overestimate of F a because metabolites generated in the lumen would be included as absorbed drug. |
| No first pass intestinal enterocyte metabolism of drug with instantaneous secretion of metabolites into the lumen. | This could yield an underestimate of F a because some of the parent drug that was absorbed orally into enterocytes was metabolized and the metabolite secreted. (Thus, the estimate of F a is truly an estimate of the composite F a · F g). |
| No first pass intestinal enterocyte metabolism of drug coupled with first pass biliary secretion of these metabolites that are ultimately excreted in feces (and not reversibly converted back into parent drug in the lumen). | This could yield an underestimate of F a because some of the parent drug that was absorbed orally into enterocytes was metabolized and the metabolite was extracted by the liver and secreted in bile before reaching the kidney. |
| There is enough excretion of drug‐related material in the urine that reliable and accurate measurements of total urinary carbon‐14 can be obtained. | If 14C is very low in urine, variability could be greater and F a estimates will be less precise. |
F a, fraction absorbed; GI, gastrointestinal.
CONSTRUCTION OF THE MASS BALANCE MODEL
The data from the human ADME study, when combined with in vitro metabolism and transport data, can be used to develop a total mass balance model of drug disposition (Figure 2 ). The elements of this model begin with an estimate of the fraction of the oral dose that is absorbed. First‐pass hepatic extraction is estimated from the hepatic clearance as compared with hepatic blood flow, whereas the difference among the absorption, oral bioavailability, and hepatic extraction is attributed to intestinal first‐pass extraction (which itself is not directly measurable). Using in vitro drug metabolism data (as well as hepatic uptake transport data when necessary) contributions to first‐pass extraction and systemic hepatic clearance can be attributed to individual enzymes. Finally, the amount of the dose as unchanged drug in urine and feces following intravenous administration can be used to estimate renal clearance and biliary clearance. However, for the latter, it cannot be truly discerned whether the drug is cleared through the bile or by active transport secretion by the intestine. Direct measurement of biliary clearance in humans would require the quantitative collection of bile and measurement of drug and this cannot be readily done in humans. It has been reported that sampling of human bile during an ADME study can be done using the Enterotest device, 19 however, this will only offer a “snapshot” of drug‐related material in human bile and does not represent a quantitative collection of bile. There may also be cases in which parent drug is glucuronidated, metabolites are secreted into the bile and hydrolyzed back to parent drug in the gastrointestinal tract. Substantial extrahepatic metabolism, if not accounted for, will confound construction of a mass balance model.
Figure 2.
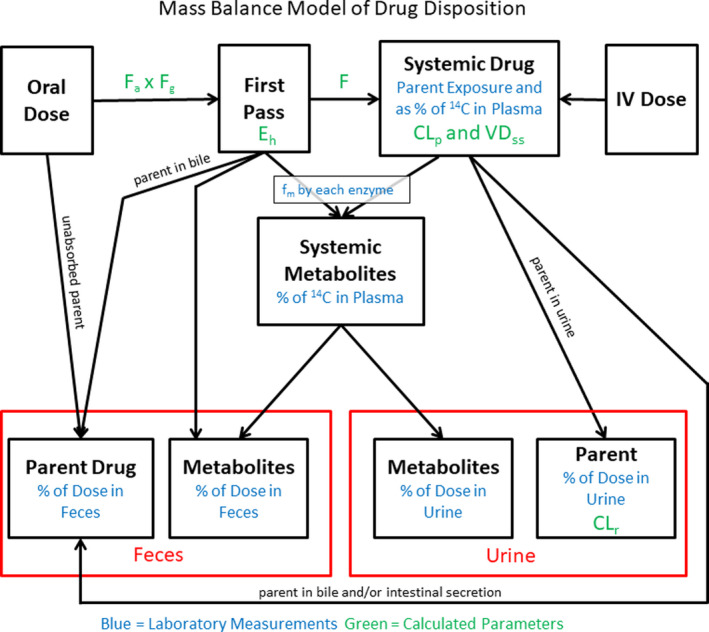
Mass balance model of total drug disposition. From the accelerator mass spectrometry (AMS)‐Enabled Human absorption, distribution, metabolism, and excretion (ADME) study, a picture of the total disposition of the drug can be constructed. This is coupled with in vitro metabolism and transport data, along with other clinical pharmacology studies that quantitatively describe specific clearance routes (e.g., drug interaction studies with specific probe inhibitors).
Once constructed, the mass balance model can be leveraged in developing full physiologically‐based pharmacokinetic models of the drug, which, in turn, can inform a clinical pharmacology study strategy with a quantitative description of overall drug disposition. 20 Appropriate clinical pharmacology studies can be planned to evaluate the effect of other drugs on the pharmacokinetics of the drug of interest, impact of genetic polymorphisms of drug metabolizing enzymes and transporters, changes in drug disposition under various disease states (e.g., hepatic impairment, renal impairment, etc.), effects of age, and possibly others. Overall, the mass balance model is essential in “telling the complete story” of what happens to the drug in the human body. This knowledge enhances the drug development process and ultimately the safe use of the drug in the clinic.
CONCLUSIONS
Among clinical pharmacology studies, the human ADME study offers the most complete picture of how a new drug is handled by the human body. Using carbon‐14 labeled drug, all of the drug‐related material can be quantitatively monitored. AMS offers an exciting new tool that has opened the possibility of more comprehensive ADME evaluations because it facilitates the use of intravenous microdosing of carbon‐14 labeled drugs. Pharmacokinetic parameters that are unobtainable if only oral administration is done are now readily obtained in the same study. With a complete and quantitative understanding of the metabolite profile a total mass balance model of a new drug can be constructed.
Funding
No funding was received for this work.
Conflict of Interest
All authors are employees and shareholders of Pfizer, Inc.
References
- 1. Coppola, P. , Andersson, A. & Cole, S. The importance of the human mass balance study in regulatory submissions. CPT Pharmacometrics Syst. Pharmacol. 8, 792–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Penner, N. , Klunk, L.J. & Prakash, C. Human radiolabeled mass balance studies: objectives, utilities and limitations. Biopharm. Drug Dispos. 30, 185–203 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Tse, F.L.S. Pharmacokinetics in drug discovery and development: nonclinical studies In: Welling P.G. & Tse F.L.S. eds. Pharmacokinetics. Regulatory, Industrial, Academic Perspectives. New York: Marcel Dekker, Inc., 1995, pp. 281–334. [Google Scholar]
- 4. European Medicines Agency . Guideline on the investigation of drug interactions. <https://www.google.com/search?q=ema+drug+interaction+guidance&rlz=1C1GCEA_enUS837US837&oq=EMA+Drug+&aqs=chrome.2.69i57j0j69i59j0l5.6200j0j7&sourceid=chrome&ie=UTF‐8#> (2012).
- 5. Obach, R.S. Pharmacologically active drug metabolites: impact on drug discovery and pharmacotherapy. Pharmacol. Rev. 65, 578–640 (2013). [DOI] [PubMed] [Google Scholar]
- 6. US Food and Drug Administration . In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry. <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/vitro‐drug‐interaction‐studies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions> (2020).
- 7. International Conference on Harmonization . Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. <https://www.ich.org/page/multidisciplinary‐guidelines> (2012). [PubMed]
- 8. US Food and Drug Administration . The Radioactive Drug Research Committee: Human Research Without and Investigational New Drug Application (2010).
- 9. Roffey, S.J. , Obach, R.S. , Gedge, J.I. & Smith, D.A. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugs. Drug Metab. Rev. 39, 17–43 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Lozac’h, F. et al Evaluation of cAMS for 14C microtracer ADME studies: opportunities to change the current drug development paradigm. Bioanalysis 10, 321–339 (2018). [DOI] [PubMed] [Google Scholar]
- 11. van Duijn, E. , Sandman, H. , Grossouw, D. , Mocking, J.A. , Coulier, L. & Vaes, W.H. Automated combustion accelerator mass spectrometry for the analysis of biomedical samples in the low attomole range. Anal. Chem. 86, 7635–7641 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Young, G.C. , Seymour, M. , Dueker, S.R. , Timmerman, P. , Arjomand, A. & Nozawa, K. New frontiers‐accelerator mass spectrometry (AMS): recommendation for best practices and harmonization from Global Bioanalysis Consortium Harmonization Team. AAPS J. 16, 357–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vogel, J.S. , Giacomo, J.A. , Schulze‐König, T.S. , Keck, B.A. , Lohstroh, P.N. & Dueker, S.R. AMS best practices for accuracy and precision in bioanalytical 14C measurements. Bioanalysis 2, 455–468 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Smith, D.A. , Johnson, D.E. & Park, B.K. Use of microdosing to probe pharmacokinetics in humans ‐ is it too much for too little? Curr. Op. Drug Disc. Dev. 6, 39–40 (2003). [Google Scholar]
- 15. Sarapa, N. , Hsyu, P.‐H. , Lappin, G. & Garner, R.C. The application of accelator mass spectrometry to absolute bioavailability studies in humans: simultaneous administration of an intravenous microdose of 14C‐nelfinavir mesylate solution and oral nelfinavir to healthy volunteers. J. Clin. Pharmacol. 45, 1198–1205 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Raje, S. et al Novel application of the two‐period microtracer approach to determine absolute oral bioavailability and fraction absorbed of ertugliflozin. Clin. Transl. Sci. 11, 405–411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harrell, A.W. et al An innovative approach to characterize clinical ADME and pharmacokinetics of the inhaled drug nemiralisib using an intravenous microtracer combined with an inhaled dose and an oral radiolabel dose in healthy male subjects. Drug Metab. Dispos. 47, 1457–1468 (2019). [DOI] [PubMed] [Google Scholar]
- 18. de Vries, R. et al Apalutamide absorption, metabolism, and excretion in healthy men, and enzyme reaction in human hepatocytes. Drug Metab. Dispos. 47, 453–464 (2019). [DOI] [PubMed] [Google Scholar]
- 19. Mamaril‐Fishman, D. et al Investigation of metabolism and disposition of GSK1322322, a peptidase deformylase inhibitor, in healthy humans using the entero‐test for biliary sampling. Drug Metab. Dispos. 42, 1314–1325 (2014). [DOI] [PubMed] [Google Scholar]
- 20. Grimstein, M. et al Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration's Office of Clinical Pharmacology. J. Pharm. Sci. 108, 21–25 (2019). [DOI] [PubMed] [Google Scholar]