


Abstract
Recent large-scale multi-omics studies resulted in quick accumulation of an overwhelming amount of cancer-related data, which provides an unprecedented resource to interrogate diverse questions. While certain existing web servers are valuable and widely used, analysis and visualization functions with regard to re-investigation of these data at cohort level are not adequately addressed. Here, we present CVCDAP, a web-based platform to deliver an interactive and customizable toolbox off the shelf for cohort-level analysis of TCGA and CPTAC public datasets, as well as user uploaded datasets. CVCDAP allows flexible selection of patients sharing common molecular and/or clinical characteristics across multiple studies as a virtual cohort, and provides dozens of built-in customizable tools for seamless genomic, transcriptomic, proteomic and clinical analysis of a single virtual cohort, as well as, to compare two virtual cohorts with relevance. The flexibility and analytic competence of CVCDAP empower experimental and clinical researchers to identify new molecular mechanisms and develop potential therapeutic approaches, by building and analyzing virtual cohorts for their subject of interests. We demonstrate that CVCDAP can conveniently reproduce published findings and reveal novel insights by two applications. The CVCDAP web server is freely available at https://omics.bjcancer.org/cvcdap/.
INTRODUCTION
A massive amount of cancer sequencing and molecular profiling data from recent international consortiums (1–3) creates unparalleled opportunities for data mining, which could significantly improve our understanding of molecular mechanisms underlying tumorigenesis and help develop new therapeutic approaches. Substantial efforts have also been made to make these data capable for pan-cancer analysis. For example, The Cancer Genomics Atlas (TCGA) generated a re-calling of uniform files by applying an ensemble of seven mutation-calling algorithms with scoring and artifact filtering (4), International Cancer Genome Consortium (ICGC) uniformed data processing and merged mutation calling results from three established pipelines in The PanCancer Analysis of Whole Genomes (PCAWG) project (5), and Clinical Proteomic Tumor Analysis Consortium (CTPAC) applied a Common Data Analysis Pipeline (CDAP) to reduce the variability introduced by disparate data analysis platforms and ensure uniformly formatted results with consistent identification thresholds (6). However, exploration and re-analysis of these high-quality data for a specific research question often entails intensive programming in data processing and calling statistical and analytical software tools needing heterogeneous running environments, which creates a huge obstacle for experimental and clinical researchers to investigate these data.
The community has endeavored to facilitate access of these large amounts of data, and several web portals have been developed and widely used. Genomic Data Commons (GDC) data portal (7), ICGC data portal (3) and CPTAC data portal (8) are main repositories to browse, query and download data for their corresponding consortium members projects. cBioPortal provides advanced gene-centered query and visualization functions across multiple studies (9). Oncoscape offers numerous interactive analysis and visualization tools for individual TCGA study (10). Xena provides an interactive sample-level visualization of multi-omics datasets for individual integrated study (11). COSMIC provides the most comprehensive catalog of somatic mutations in cancer (12). LinkedOmics (13) provides analysis and visualization of associations between different types of molecular features including proteomic datasets from CPTAC. OASIS integrates both tumor tissue and Cancer Cell Line Encyclopedia (CCLE) (14) data to enable identifying cell lines carrying a mutation of interest for further functional and therapeutic study (15).
Within these extremely valuable tools, GDC portal, ICGC portal and cBioPortal (beta version) recently enabled selection of patients across multiple studies, as a virtual cohort for exploratory analysis and comparison. However, many analytical and visualization functions essential for discovery analysis of single cohort or comparison of two relevant cohorts are not adequately addressed. For example, multivariate Cox regression analysis is often needed to control confounding variables, which is not available in the existing tools. In addition, none of these tools provides genomic analysis functions, including tumor mutational burden (TMB) estimation, mutational signature analysis and driver gene analysis, which are essential to investigate genomic landscape of a user-defined cohort. Furthermore, cohort analysis functions (e.g. dimensional reduction, Gene Set Enrichment Analysis, unsupervised clustering) are lacking for the rapidly growing proteomic datasets from CPTAC.
To address the above challenges, we developed the Cancer Virtual Cohort Discovery Analysis Platform (CVCDAP), an interactive and customizable toolbox for molecular and clinical analysis of user-defined cancer virtual cohorts, to complement with the existing data portals.
MATERIALS AND METHODS
Data sources
Somatic mutations and transcriptomic data were downloaded from PanCanAtlas (version 20190101). The TCGA PanCancer Atlas MC3 set consists of uniform re-calling results by the Multi-Center Mutation Calling in Multiple Cancers project to remove batch effects, and was imported to enable robust cross-tumor-type genomic analyses, and batch-corrected mRNA expression levels (FPKM) was imported for unbiased gene expression analysis. Copy number data (thresholded GISTIC2 focal-level score) were downloaded from the Genomic Data Commons (GDC) hub. Proteomics data were download from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) data portal to import relative abundance of proteins generated by the Common Data Analysis Pipeline (6). Clinical data (survival time, tumor site, age, ethnicity and grade) were downloaded from both PanCanAtlas and CPTAC for corresponding samples with molecular data, and only primary tumors were included in CVCDAP. Details will be updated online for each release.
Pre-processing of expression data
We used R package impute to perform KNN imputation within individual cancer type for both mRNA and proteins expression data. Regarding multiple samples from the same patient, median values were used for patient-level analysis. For RNA-seq data, FPKM values was converted to TPM and log2 transformed before importing into the database. For proteomic data, downloaded expression matrix was subjected to quantile normalization using normalizeQuantiles function implemented in R package limma (16) v3.36.1. All CVCDAP data were stored in a MySQL relationship database (version 5.7). Details will be updated online for each release.
Integrated analysis tools and job running
CVCDAP developed dozens of tools to enable customized cohort-level data analysis with a uniform web user interface, by in-house pipeline scripts to utilize multiple R packages (maftools (17) v1.8.0, deconstructSigs v1.8.0, limma (16) v3.36.1, Rtsne v0.15, survival v2.43.3, and survminer v0.4.4) and Gene Set Enrichment Analysis (GSEA) software v3.0 (18,19). In addition to result files, CVCDAP allows experienced users to download input files (in Rdata format) for their further analysis running locally. After user submission of an analysis request, the records will be available in the analysis history for future access. A detailed documentation introducing parameters details of each analysis tool and tutorial slides illustrating how to perform the case studies using CVCDAP are available online for new users.
Website development
The website is free and open to all users. There is no login requirement to access any feature; however, the logged-in user can reuse and share the results of their cohorts, which are stored and managed in a MySQL relational database. The server-side interface is implemented using JSP (Java Server Pages) running on an Apache Tomcat web server, with Spring (https://spring.io/) and MyBatis (https://github.com/mybatis/mybatis-3/) frameworks employed to improve the efficiency and stability of the web services. The client-side interface is developed using HTML5, JavaScript native and third-party libraries including Bootstrap (http://getbootstrap.com/) and jQuery (http://jquery.com). The interactive charts and tables that summarize a cohort are based on DataTables.js (https://www.datatables.net/) and ECharts.js (https://www.echartsjs.com/zh/index.html) JavaScript libraries.
RESULTS
CVCDAP imported and pre-processed the molecular and clinical data from PanCanAtlas and CPTAC that includes a total of 11 263 patients across 33 cancer types in the release 2019. Moreover, it allows users to upload their own datasets to combine with the integrated public data in CVCDAP for query and analysis. The schematic overview of CVCDAP is illustrated in Figure 1; key components and features are listed in Supplementary Table S1 and elucidated in the following sections.
Figure 1.

Schematic of CVCDAP. Top panel describes the available molecular and clinical data in CVCDAP. Middle panel shows the typical workflow of CVCDAP starting from cohort creation using single or a combination of tissue, clinical and molecular filters. Bottom panel presents example visualization of analyses results using CVCDAP built-in functions. Example outputs of single cohort analysis include oncoplot, mutual exclusive analysis, mutation signature analysis, TMB plot, survival analysis, rainfall plot, t-SNE, clustering and prevalence plot. Example outputs of two cohorts analysis include differentially mutated genes analysis, co-oncoplot, lollipopplot2 plot, cox regression analysis, GSEA analysis, differential expression analysis, volcano plot and box plot.
Convenient construction and management of virtual cohorts
CVCDAP provides the flexibility to allow users to create a virtual cohort consisting of samples from single or multiple studies sharing common molecular and/or clinical characteristics. A virtual cohort can be defined by applying any of tissue features (tissue of origin, disease type), clinical features (stage, grade, gender, ethnicity and age), molecular features (somatic mutations, tumor mutational burden, copy number estimate, level of mRNA or protein expression aberration) or a combination of features at discretion. It can also be directly created based on a list of patient/sample IDs provided by users from previous analysis or knowledge.
Created cohorts can be saved for operations with another saved cohort, including union, intersection or subtraction. Registered users can share a saved cohort to the community (Figure 2A) by enabling the sharing function and providing the URL for open access. Together, these features enable CVCDAP to conveniently and effectively create virtual cohorts of relevance for a wide range of biological and clinical questions.
Figure 2.
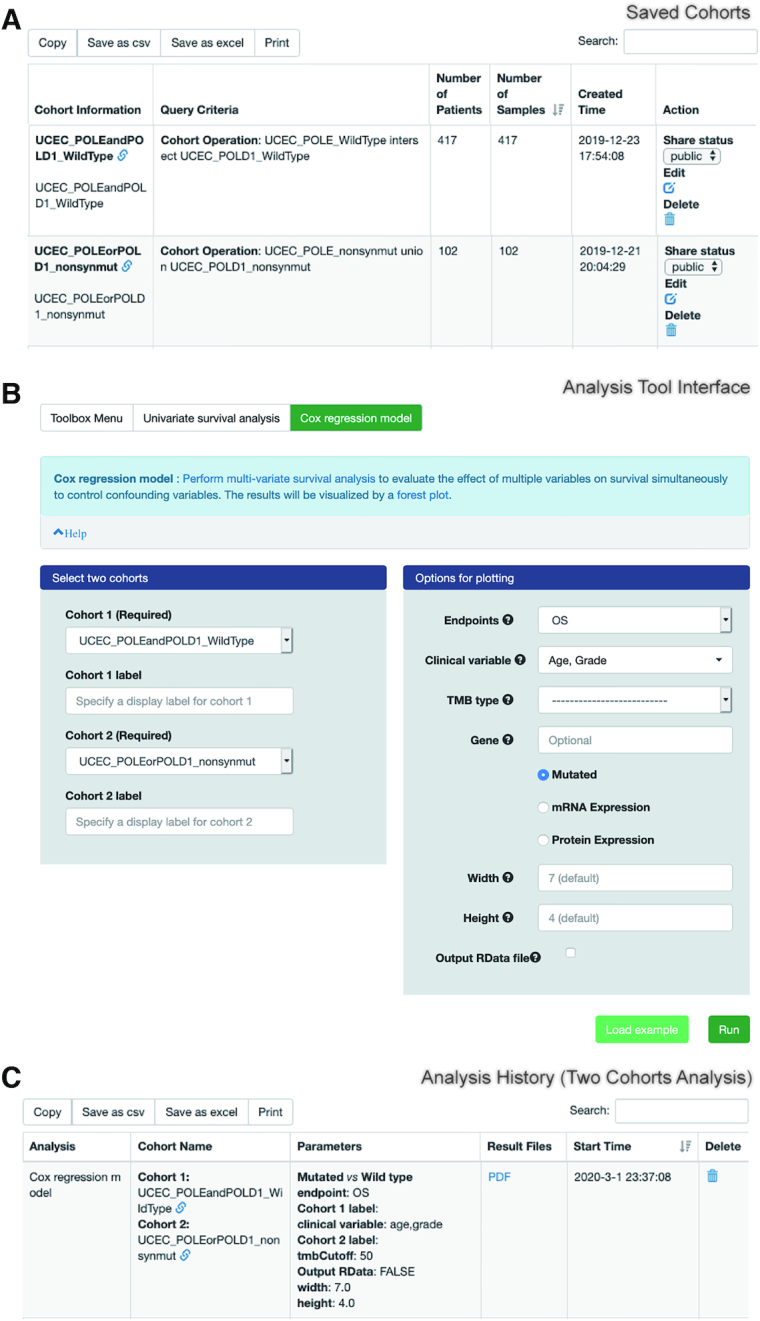
Key web interfaces of CVCDAP. (A) ‘Saved Cohorts’ shows the name, query criteria, numbers of patients and samples of each created cohort, and provides links to rename, share and remove a selected cohort. (B) Each analysis tool provides a uniform interface for flexible customization of analysis and plotting parameters. (C) ‘Analysis History’ enables users to check the status of analysis task, recall the analysis parameters and download the result files.
A built-in interactive and customizable toolbox for cohort-level molecular and clinical analysis
Virtual cohorts from query results can be directly used as input for built-in analysis and visualization tools. For a single cohort, we provided: (i) 12 analysis tools to characterize genomic aberrations, including plotting a sample-level overview of genomic landscape, and identification of potential driver genes, mutational spectrum and signature, co-occurring or exclusive patterns, etc.; (ii) tools for dimensionality reduction and unsupervised clustering analysis based on gene expression and protein abundance respectively; (iii) univariate and multi-variate analysis tools to evaluate and visualize statistical associations of specific molecular aberration (mutation or overexpression/downregulation), and/or clinical variables with patient outcome.
For cohort comparison, what we offered includes: (i) analysis tools to compare genomic landscapes and mutation hotspots of two cohorts side-by-side, as well as to identify and visualize differentially mutated genes; (ii) analysis tools for differential expression analysis and GSEA analysis of transcriptomic and proteomic profiles between two cohorts, as well as common visualization functions including box plot, heatmap and volcano plot; (iii) univariate and multi-variate analysis tools to evaluate and visualize statistical associations of specific molecular aberration and/or clinical parameters with patient outcome.
A uniform user interface (Figure 2B) is provided for each CVCDAP analysis tool to hide the complexity of running the underlying software tools from end users. Submitted analysis tasks are listed at the ‘Analysis History’ page (Figure 2C), which lists input cohort(s), parameters and results files of each analysis task for reproducibility. Together, this interactive and customizable toolbox allows users to re-analyze their defined cohorts of interests on the fly, with elegant visualization of analysis results.
Given these advanced features, CVCDAP could enable rapid discovery of molecular mechanisms underlying biological/clinical subjects of interest. We demonstrated the advantages of CVCDAP by two case studies, which conveniently reproduced published findings, as well as revealed novel insights.
Investigation of molecular features associated with tumor recurrence difference in breast cancer
African Americans have higher breast cancer mortality rate compared to white patients. Conventionally, this survival gap has been attributed to variability in financial level, disease stage, subtype and clinical management (20,21), notwithstanding recent studies indicated biological difference may also account for the racial/ethnic inequity in breast cancer outcome (22,23). By analyzing a total of 793 TCGA stage I-III breast cancer patients (148 African Americans versus 645 white patients, Figure 3A) in CVCDAP, we identified that three genes with ethnicity-specific expression pattern are significantly associated with tumor recurrence. First, we compared gene expression profiles between two ethnic groups of patients and identified 27 genes with significant difference (Benjamini and Hochberg adjusted P < 0.01 and log2 fold change > 1.5, Figure 3B), in which expression of three genes (Figure 3C) are also associated with disease-free interval (DFI) (Log-rank test; IL6ST, P = 0.001; SCUBE2, P = 0.0031; MSLN, P = 0.0033) (Figure 3D). After adjusting for the clinical variables (age and stage), high expression of IL6ST and SCUBE2, and low expression of MSLN are still associated with better prognosis, respectively. Interestingly, separate adjustment for each of these three genes decreased the hazard ratio and statistical significance of the ethnic association with tumor recurrence (Figure 3E; Supplementary Figure S1A–C). We also compared genomic profiles of two groups of patients and revealed African Americans harbored more TP53 mutations and fewer PIK3CA mutations (P < 0.001) (Supplementary Figure S1D and E), which agrees with previous studies (22,23). However, mutations in neither TP53 nor PIK3CA were associated with patient prognosis. Taken together, our analysis revealed three candidate genes for further investigation. Although the number of African Americans is relatively small and socioeconomic factors have not been taken into account due to the lack in the original TCGA study (24), it highlights the needs for further studies to investigate the molecular landscape of cancer in minority and under-represented populations.
Figure 3.

Molecular profiles associated with ethnic difference in breast cancer recurrence. (A) Kaplan–Meier curve of DFI for 793 stage I-III breast cancer patients in CVCDAP. The analysis was performed using all follow-up events, though we plotted the curves for the first 10 years only. (B) Volcano plot of significance of gene expression difference between African American and white women with breast cancer. A gene is considered to be significantly differentially expressed if |log2(FC)| > 1.5 and adjusted P-value < 0.05. (C) Boxplot of significance of gene expression levels of three candidate genes. IL6ST and SCUBE2 are significantly upregulated, while MSLN is significantly downregulated in white patients compared with African Americans. (D) Kaplan–Meier curve of DFI of three candidate genes. Expression levels of IL6ST, SCUBE2 and MSLN are significantly associated with DFI (P < 0.05). (E) Forest plot shows multivariate HR for tumor recurrence adjusted for ethnicity, age, stage and SCUBE2 expression level. Adjustment for SCUBE2 level decreased the magnitude and significance of the ethnic association with tumor recurrence. Adjusted HRs and their corresponding P values are presented (∗∗∗ indicates P < 0.001; ∗∗ indicates P < 0.01; * indicates P < 0.05; Cox regression). Square data markers indicate estimated HR. Error bars represent 95% CIs. Abbreviations: CI, confidence interval; DFI, disease-free interval; HR, hazard ratios.
Evaluation of clinical utilities of POLE and POLD1 mutations in endometrial cancer
DNA polymerase epsilon (POLE) and delta 1 (POLD1) are essential for proofreading and faithful replication of DNA, and mutations in POLE or POLD1 are widely distributed across multiple cancer types. CVCDAP pan-cancer analysis identified that uterine corpus endometrial carcinoma (UCEC) (n = 530) has the highest mutation frequency for POLE (15%) and POLD1 (8%) (Figure 4A) among 19 cancer types with patients harboring POLE/POLD1 mutations (Supplementary Figure S2). Although clinical implications of POLE mutations have been suggested in UCEC (25), the clinical significance of POLD1 mutations has not been investigated yet, to our knowledge. Thus, we utilized CVCDAP to conduct a comprehensive analysis of UCEC patients with POLE or POLD1 mutations. First, the univariate analysis identified that patients with POLE/POLD1 mutation were significantly associated with better overall survival (OS), disease-specific survival (DSS), progression-free interval (PFI) and DFI (Log-rank test, OS, P = 0.0019; DSS, P = 0.0024; PFI, P < 0.0001; DFI, P = 0.028) (Supplementary Figure S3A). The associations remain statistically significant after adjusting for age and grade (Figure 4B and Supplementary Figure S3B). UCEC patients with POLE/POLD1 mutations exhibited significantly greater TMB than those without mutations (Wilcoxon rank sum test, P < 2.2e-16) (Figure 4C). Additionally, we confirmed POLE codons 286 and 411 in exonuclease domain as mutation hotspots (Figure 4D) and no hotpot mutation was seen in POLD1 (Supplementary Figure S3C) (26). Furthermore, GSEA analysis revealed a significant up-regulation of pathways (FDR < 0.25) in POLE/POLD1 mutant tumors, which are related to immune response activity including natural killer cell–mediated cytotoxicity and T-cell receptor signaling (Figure 4E). These results (increased TMB and upregulation of immune response activity) imply UCEC samples with POLE/POLD1 mutations could benefit from immune-checkpoint inhibitors as indicated in the previous pan-cancer studies (27,28). Taken together, our results suggest combined mutation status of POLE and POLD1 has potential clinical utilities for endometrial carcinoma patients, although further clinical investigation is needed for metastatic UCEC patients treated with immunotherapy.
Figure 4.

Association of POLE/POLD1 mutation with tumor mutation load and prognosis in UCEC. (A) Oncoplot shows mutation landscape of POLE and POLD1 in UCEC. Top panel presents the number (log 10 transformed) of mutations of each classification in individual samples, bottom panel presents mutation classification in each sample (left) and the frequency of mutations in the cohort (right). (B) Forest plot illustrates a multivariate HR for OS fitted with POLE/POLD1 mutation status and clinical factors (age and grade). Adjusted HRs and their corresponding P values are presented (∗∗∗ indicates P < 0.001; ∗∗ indicates P < 0.01; * indicates P < 0.05; Cox regression). Square data markers indicate estimated HR. Error bars represent 95% CIs. (C) Comparison of mutation rates of UCEC patients with and without mutations in POLE or POLD1. (D) Lollipop plot shows the distribution of somatic mutations in protein domains of POLD1. (E) Bar plot shows the up-regulated KEGG pathways identified in POLE/POLD1 mutated patients by GSEA analysis. Pathways are ordered by the NES, and FDR < 0.25 is considered as statistically significant. Abbreviations: CI, confidence interval; FDR, false discovery rate; HR, hazard ratios; MT, mutant; NES, normalized enrichment score; OS, overall survival; UCEC, uterine corpus endometrial carcinoma; WT, wild-type.
DISCUSSION
CVCDAP is a web-based platform to allow flexible selection of cancer patients of relevance as virtual cohorts for specific research and clinical questions, with a toolbox of analytic and visualization functions empowered for discovering and comparing molecular and clinical profiles of user-defined cohorts. We illustrated the value of cohort-level re-analysis of published data in generating novel hypothesis, which is essential for a number of reasons: (i) The original published study usually includes a smaller dataset, thus statistical power is underestimated to detect low-frequency mutations or low expression variations. (ii) Overlaying additional features, such as patient outcome, will help rank candidate genes for further investigation. (iii) Extending previous signatures with additional genes included from latest relevant research findings, which could be easily validated using the same cohort. There are more scenarios than we can enumerate here, the community could conveniently and effectively reveal all possibilities for their questions of interest via CVCDAP.
CVCDAP will be updated quarterly, with more analytical functions and interactive plots developed, as well as additional valuable cohort-based public datasets integrated in the future, such as the recently launched ICGC-ARGO project (https://www.icgc-argo.org/), which will collect molecular and clinical data from about 100,000 cancer patients participating in clinical trials. With these further developments, CVCDAP will complement other available tools to help biological and clinical researchers reveal the molecular mechanisms and indicate novel therapeutic approaches from the massive amount of public cancer-related data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lihua Cao for suggestions on data normalization method, Huan Yu for comments on the POLE/POLD1 case study. We also thank users who provided valuable feedback, bug reports, and feature requests during the development of the website.
Contributor Information
Xiaoqing Guan, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Center for Cancer Bioinformatics, Peking University Cancer Hospital and Institute, Beijing 100142, China; Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Gastrointestinal Cancer Center, Peking University Cancer Hospital and Institute, Beijing 100142, China.
Meng Cai, Institute of Systems Biomedicine, Department of Medical Bioinformatics, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China.
Yang Du, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Center for Cancer Bioinformatics, Peking University Cancer Hospital and Institute, Beijing 100142, China.
Ence Yang, Institute of Systems Biomedicine, Department of Medical Bioinformatics, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China.
Jiafu Ji, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Gastrointestinal Cancer Center, Peking University Cancer Hospital and Institute, Beijing 100142, China.
Jianmin Wu, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Center for Cancer Bioinformatics, Peking University Cancer Hospital and Institute, Beijing 100142, China; Peking University International Cancer Institute, Peking University, Beijing 100191, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Peking University [PKU2018LCXQ015]; Science Foundation of Peking University Cancer Hospital [16-01]; PKU-Baidu Fund [2019BD012]; Michigan Medicine-PKUHSC Joint Institute for Translational and Clinical Research [BMU2019JI010]; Beijing Municipal Bureau of Health [2019-1]. Funding for open access charge: PKU-Baidu Fund [2019BD012].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cancer Genome Atlas Research Network Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., Townsend R.R., Kinsinger C., Mesri M., Rodriguez H. et al.. Connecting genomic alterations to cancer biology with proteomics: the NCI clinical proteomic tumor analysis consortium. Cancer Discov. 2013; 3:1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang J., Bajari R., Andric D., Gerthoffert F., Lepsa A., Nahal-Bose H., Stein L.D., Ferretti V.. The international cancer genome consortium data portal. Nat. Biotechnol. 2019; 37:367–369. [DOI] [PubMed] [Google Scholar]
- 4. Ellrott K., Bailey M.H., Saksena G., Covington K.R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K.E., McLellan M. et al.. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018; 6:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020; 578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rudnick P.A., Markey S.P., Roth J., Mirokhin Y., Yan X., Tchekhovskoi D.V., Edwards N.J., Thangudu R.R., Ketchum K.A., Kinsinger C.R. et al.. A description of the clinical proteomic tumor analysis consortium (CPTAC) common data analysis pipeline. J. Proteome Res. 2016; 15:1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M.. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016; 375:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edwards N.J., Oberti M., Thangudu R.R., Cai S., McGarvey P.B., Jacob S., Madhavan S., Ketchum K.A.. The CPTAC data portal: a resource for cancer proteomics research. J. Proteome Res. 2015; 14:2707–2713. [DOI] [PubMed] [Google Scholar]
- 9. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McFerrin L.G., Zager M., Zhang J., Krenn G., McDermott R., Horse-Grant D., Silgard E., Colevas K., Shannon P., Bolouri H. et al.. Analysis and visualization of linked molecular and clinical cancer data by using Oncoscape. Nat. Genet. 2018; 50:1203–1204. [DOI] [PubMed] [Google Scholar]
- 11. Goldman M., Craft B., Hastie M., Repečka K., Kamath A., McDade F., Rogers D., Brooks A.N., Zhu J., Haussler D.. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. 2019; bioRxiv doi:26 September 2019, preprint: not peer reviewed 10.1101/326470. [DOI] [PMC free article] [PubMed]
- 12. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E. et al.. COSMIC: the catalogue of somatic mutations in Cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vasaikar S.V., Straub P., Wang J., Zhang B.. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018; 46:D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghandi M., Huang F.W., Jane-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R. 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H. et al.. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019; 569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernandez-Banet J., Esposito A., Coffin S., Horvath I.B., Estrella H., Schefzick S., Deng S., Wang K., K A.C., Ding Y. et al.. OASIS: web-based platform for exploring cancer multi-omics data. Nat. Methods. 2016; 13:9–10. [DOI] [PubMed] [Google Scholar]
- 16. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayakonda A., Lin D.C., Assenov Y., Plass C., Koeffler H.P.. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018; 28:1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. et al.. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34:267–273. [DOI] [PubMed] [Google Scholar]
- 20. Wheeler S.B., Spencer J.C., Pinheiro L.C., Carey L.A., Olshan A.F., Reeder-Hayes K.E.. Financial impact of breast cancer in black versus white women. J. Clin. Oncol. 2018; 36:1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daly B., Olopade O.I.. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J. Clin. 2015; 65:221–238. [DOI] [PubMed] [Google Scholar]
- 22. Keenan T., Moy B., Mroz E.A., Ross K., Niemierko A., Rocco J.W., Isakoff S., Ellisen L.W., Bardia A.. Comparison of the genomic landscape between primary breast cancer in African American versus white women and the association of racial differences with tumor recurrence. J. Clin. Oncol. 2015; 33:3621–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huo D., Hu H., Rhie S.K., Gamazon E.R., Cherniack A.D., Liu J., Yoshimatsu T.F., Pitt J.J., Hoadley K.A., Troester M. et al.. Comparison of breast cancer molecular features and survival by African and European ancestry in the cancer genome atlas. JAMA Oncol. 2017; 3:1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C. et al.. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015; 163:506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McConechy M.K., Talhouk A., Leung S., Chiu D., Yang W., Senz J., Reha-Krantz L.J., Lee C.H., Huntsman D.G., Gilks C.B. et al.. Endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Clin. Cancer Res. 2016; 22:2865–2873. [DOI] [PubMed] [Google Scholar]
- 26. Cancer Genome Atlas Research, N. Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R. et al.. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang F., Zhao Q., Wang Y.-N., Jin Y., He M.-M., Liu Z.-X., Xu R.-H.. Evaluation of POLE and POLD1 Mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019; 5:1504–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samstein R.M., Lee C.H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A. et al.. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019; 51:202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.