

Abstract
Objective:
Neointima (NI) formation is a primary cause of intermediate to late vein graft (VG) failure. However, the precise source of NI cells in VGs remains unclear.
Approach and Results:
Herein we clarify the relative contributions of mature vascular smooth muscle cells (SMCs) and endothelial cells (ECs) to NI formation in a mouse model of VG remodeling via the genetic inducible fate mapping approaches. Regardless of the magnitude of NI formation, the recipient arterial and the donor venous SMCs contributed ~55% of the NI cells at the anastomotic regions; whereas only donor venous SMCs donated ~68% of the NI cells at the middle bodies. A small portion of the SMC-derived cells became non-SMC cells, mostly likely vascular stem cells and constituted 2–11% of the cells in each major layer of VGs. In addition, the recipient arterial ECs were the major cellular source of re-endothelialization but did not contribute to NI formation. The donor venous ECs donated ~17% NI cells in the VGs with mild NI formation and conditional media from ECs after endothelial mesenchymal transition (EndMT) suppressed vascular SMC dedifferentiation.
Conclusions:
The recipient arterial and donor venous mature SMCs dominate but contribute distinctly to intimal hyperplasia at the anastomosis and the middle body regions of VGs. The recipient arterial ECs are the major cellular source of re-endothelialization but do not donate NI formation in VGs. Only the donor venous ECs undergo EndMT. EndMT is marginal for generating NI cells, but is likely required for controlling the quality of VG remodeling.
Keywords: Vascular smooth muscle cells, Phenotype modulation, Endothelial mesenchymal transition (EndMT), Dedifferentiation, Neointima formation, Vein graft failure
Graphic Abstract
The recipient arterial and donor venous mature SMCs, but not ECs, dominate and contribute distinctly to intimal hyperplasia at the anastomosis and the middle body regions of VGs. A small portion of the SMC-derived cells become non-SMC cells, most likely vascular stem cells and constitute 2–11% of the cells in each major layer of VGs.
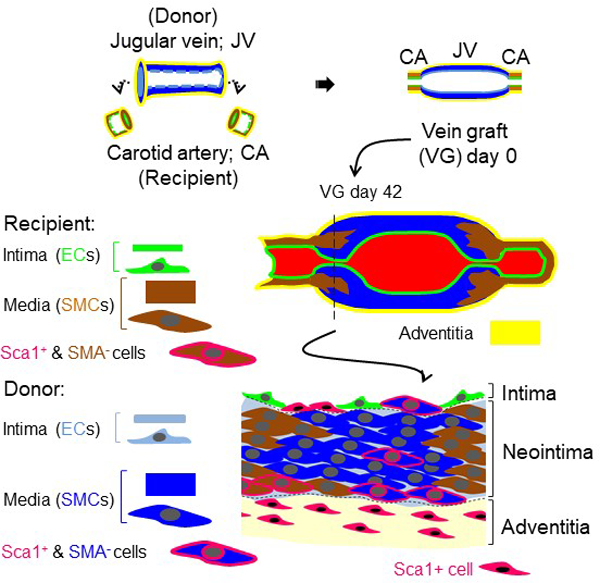
Introduction
Vein bypass graft remains the gold standard revascularization treatment for occlusive arterial diseases.1–3 However, the surgical injury and arterial environment initiate a sophisticated remodeling process in vein grafts (VGs): Within 24 h to 3 days after implantation, the VG exhibits significant endothelial cell (EC) loss, subendothelial and medial edema, and extensive smooth muscle cell (SMC) necrosis associated with inflammation and release of growth factors. One week to one month post-implant, there is re-endothelialization of the luminal surface as well as medial thickening and intimal hyperplasia or neointima (NI) formation, a complicated process involving diverse types of cells including vascular SMCs. One month to three years post-implant, there is atheromatous lesion formation and beyond three years, progressive intimal thickening and superimposed atheromas cause lumenal narrowing. Consequently, as many as 18% of VGs may fail (≥70% stenosis) within 30 days to years after surgery (acute vein graft failure, VGF); 20–50% of VGs fail between 2–5 years after surgery (intermediate VGF); and by 10 years after surgery, 40% of VGs fail and a further 30% have compromised flow.1–3 Acute VGF is predominantly triggered by thrombosis due to surgical trauma to the graft and altered hemodynamics leading to endothelial damage and dysfunction; whereas intermediate VGF is most likely due to the NI formation, particularly at the proximal and distal anastomoses, and late VGF is ascribed to not only the NI formation but also the accelerated atherosclerosis. Of note, most of the VG lesions are focalized.2, 4 In lower extremity VGs, approximately 50% of lesions responsible for VGF are juxta-anastomotic and 30% are located within the body of VGs.4 In the coronary artery bypass VG, the majority of focal stenotic lesions often occur in the perianastomotic regions leading to the intermediate VGF, whereas the diffused stenosis within the graft body contributes to late VGF.2 Continuous improvements in surgical techniques have improved clinical outcome and short-term graft patency; but the overall rate of intermediate and later VGF has largely unchanged for several decades.
Although the major contributors to both intermediate and late VGF remain unclear, over exuberant intimal hyperplasia in VGs has been considered as one potential explanation.1–4 Since the major type of NI cells in human VGs is highly proliferating SMCs, i.e., synthetic SMCs, the role of vascular SMCs in VGF has been a research focus.1–4 Because arterialized vein conduit tissue is rarely retrieved from patients, and the contemporary cell and tissue culture approaches to mimic the multifactorial events of human VG remodeling are still unavailable, animal models serve as an essential tool to explore the mechanisms of VGF. Several studies using different animal models have demonstrated that majority of NI SMCs in VGs are not derived from the bone marrow of the recipient but originated from the SMCs of both the donor VGs and the recipient adjacent arteries.5–9 A more rigorous study using genetic inducible fate mapping (GIFM) technique, i.e., smooth muscle 22 alpha (SM22α) knockin (Ki)-CreERT2::Rosa26Floxed-StopeYFP mice showed that ~16% of the NI cells in VGs are derived from the donor venous SMCs.10 However, this conclusion may need to be revised as the downregulation of SM22α expression due to SM22α-Ki per se in this model may promote SMC dedifferentiation and NI formation in the pathological setting.11–15 In addition, this study did not determine the contribution of recipient mature SMCs to NI formation in VGs.10 On the other hand, a separate study utilizing Stem Cell Leukemia (Scl)-CreERT2::Rosa26Floxed-StopeYFP mice showed that the donor ECs may give rise to approximately 28% of the NI cells and roughly 85% of these EC-derived NI cells are synthetic SMCs via endothelial to mesenchymal transition (EndMT).10 However, this conclusion may also need to be revised because of the weakness associated with the fate mapping approach per se: 1) SCL which is also known as TAL1 (T-cell acute lymphocytic leukemia 1) is expressed not only in adult hematopoietic stem cells and ECs but also in vascular SMCs.16, 17 2) Importantly, the enhancer which is used to control CreERT2 for tracking the fate of adult ECs18 drives a reporter gene expression in adult vascular SMCs.17 Therefore, it is likely that the observed Scl-CreERT2-labeled YFP+ cells in the NI are actually derived from both donor venous SMCs and ECs. Again, the contribution of recipient arterial ECs to the NI formation was not determined in this study. Another study using Cdh5-CreERT2::mT/mG mice demonstrated that only 3–7% of NI SMCs are derived from the recipient arterial ECs in the inferior vena cava graft transplanted into the aorta.19 However, the contribution of donor venous ECs to the NI formation has not been studied. Taken together, the precise contributions of the recipient arterial vs. the donor venous SMCs or ECs to NI formation in VGs still remain unclear.
In the present study, we applied the GIFM approach to track down the fate of both recipient arterial and donor venous SMCs and ECs in murine VG remodeling and demonstrated that both recipient arterial and donor venous mature SMCs dominate but contribute distinctly to intimal hyperplasia at the anastomosis region and the middle body of VGs. In addition, the recipient arterial ECs are the major cellular source of re-endothelialization but do not contribute to NI formation in VGs. Also, only the donor venous ECs undergo EndMT, but the direct contribution of donor venous ECs via EndMT to overall NI formation in VGs is marginal. EndMT is not critical for generating NI cells but likely required for controlling the quality of VG remodeling.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Wild type (WT) C57BL/6J and Rosa26Floxed-StopeYFP mice were purchased from the Jackson Laboratory. SM22α knockin (Ki)-CreERT2 mice were kindly provided by Dr. Robert Feil at Technische Universität München, Germany. Myh11-CreERT2::Rosa26Floxed-StopeYFP mice, Cdh5-Cre CreERT2::Rosa26Floxed-StopeYFP mice and SM22α knockin (Ki)-CreERT2::Rosa26Floxed-StopeYFP mice were generated as previously reported.20–22 Since myh11-CreERT2 transgene is integrated on the Y chromosome, only male could carry the myh11-CreERT2 transgene. Due to the technique limitation, only male mice were used in this study. All animals were housed at the AAALAC-accredited animal facility of University of South Carolina School of Medicine. All animals were treated in compliance with the USA National Institute of Health Guideline for Care and Use of Laboratory Animals. The use of animals and all animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at University of South Carolina.
Induction and Quantification of Cre-loxP Recombination by Tamoxifen
Tamoxifen (Cat#: T5648, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in warm sunflower seed oil at a concentration of 10 mg/ml and injected intraperitoneally (i.p.) into the reporter mice, i.e., Myh11-CreERT2::Rosa26Floxed-StopeYFP mice, Cdh5-Cre CreERT2::Rosa26Floxed-StopeYFP mice and SM22α knockin (Ki)-CreERT2::Rosa26Floxed-StopeYFP mice, at age of 8 weeks at 1 mg/day/per mouse for 5 consecutive days as previously reported.23 After at least 20 days of washout time period, the tamoxifen-induced YFP labeling of mature SMCs and ECs as well as their progeny were first quantified prior to the vein graft transplantation. Moreover, after additional 6 weeks, at the time of the experimental end point for lineage tracking, the tamoxifen-induced YFP-labeling efficiencies in the reporter mice without receiving operation were assessed again. The labeling efficiencies were assessed by quantifying the YFP positive SMCs or ECs in 5 consecutive cross-sections with an interval of 100 µm were selected from each vessel (n=5). The expression patterns of YFP in SMCs and ECs in the other organs were analyzed via the staining of a few tissue sections (2–3 sections) randomly selected from these organs.
Venous Bypass Graft Procedure
Vein graft transplantation in mice was carried out as previously described.24 Briefly, in anesthetized recipient mice, the right common carotid artery was mobilized and divided. A 1-mm cuff with a 1-mm handle (0.65 mm in diameter outside and 0.5 mm inside) (REF 800/200/100/200, Portex LTD, United Kingdom) was placed on both ends of the artery, and the ends were reverted over the cuff and ligated with an 8–0 silk ligature. The vein segment which was isolated from donor mice was grafted between the 2 ends of the carotid artery by sleeving the ends of the vein over the artery cuff and ligating them together with the 8–0 suture without changing the direction of blood flow.
Tissue Harvest, Immunohistochemical Staining and Morphological Analyses
Mice were euthanized with an overdose of pentobarbital (50 mg/kg, i.p., Cat#: 2821, Vortech, USA) or CO2 inhalation. After washing out blood, blood vessels and the other organs were dissected after perfusion fixation with 4% paraformaldehyde (Cat#: J19943-K2, Thermo Fisher Scientific, USA) for 5 min and further fixed with 4% paraformaldehyde in 4°C overnight, and then embedded using Optimal cutting temperature (O.C.T.) compound (Cat#: 4585, Fisher Health Care, USA) prior to cryosection. The frozen tissue sections with a 5-µm thickness were subject to immunochemical staining and morphological analyses as described elsewhere.25 The antibodies used are listed in Online supplementary Tables S1 and S2.
Western Blot Analysis and Polymerase Chain Reaction (PCR)
Harvested tissues were snap-frozen in liquid nitrogen and stored in −80°C. The cultured cells were washed with ice-cold 1x PBS and snap-frozen in liquid nitrogen and stored in −80°C. Genomic DNAs were also extracted from mouse tails and subjected to PCR for genotyping of transgene mice. PCR primers and reaction conditions for genotyping are listed in Online Table III. Western blot and PCR analyses were performed as described elsewhere.25
Cell Culture and EndMT
Mouse aortic SMCs were isolated and cultured as described elsewhere.26 EndMT of human umbilical vein endothelial cells (HUVECs) was induced by transforming growth factor beta 1 (TGF-β1) or bone morphogenetic protein 4 (BMP4) as previously reported.27
Statistics
Data are shown as mean ± SEM if they are not specified. Differences between 2 groups were evaluated for statistical significance using the Student t test. When differences among > 3 groups were evaluated, results were compared by one-way ANOVA with Bonferroni test for multiple comparisons. Differences were considered significant at p < 0.05.
Details of “Materials and Methods” can be found in the online-only Data Supplement.
Results
There are two types of VG remodeling in a mouse model of VG intimal hyperplasia
Several animal models of VG intimal hyperplasia have been established by engrafting veins into arteries using either sutures or cuffs.28, 29 Notably, a murine model of VG intimal hyperplasia using a polyethylene cuff, which was originally described by Zou, et. al. appears to be most feasible to quantify and obtain reproducible results.28, 29 Nevertheless, the intimal hyperplasia along VGs in this mouse model has not been completely characterized. Thus, we studied the intimal hyperplasia of entire VGs in this model. At each experimental end point, the entire isologous jugular VGs (n=30) with an average of 4.5-mm length were consecutively and completely sectioned from the proximal to the distal end, resulting in ~900 5 µm-thick tissue cross-sections per graft (Figure 1A and Online Figure IIIA–B). Because of the inherently ill-defined nature of the lamina in venous tissues, delineation of the boundary of the media layer in veins is often difficult.30 Thus, we carried out morphological analyses in 90 consecutive cross-sections from the proximal to distal end with a 50 µm interval per graft at 3 weeks (wks) and 6 wks after transplantation (n=3), which were stained with hematoxylin and eosin (HE) or anti-SMA antibodies and Dapi. Accordingly, we established a new method by SMA and Dapi staining to quantify VG remodeling and named it C-method (Online Figures IV–VII). C-method revealed that overall NI area and thickness per VG were time-dependently increased; however, overall lumen area per VG was time-dependently decreased (Figure 1B). In addition, the lumen area near both the proximal and distal ends was usually smaller than that at the middle portion; whereas, the NI thickness near either the proximal or distal ends was usually larger than that at the middle portion (Figure 1B). These results indicate that like the clinical setting,2, 4 mouse jugular vein remodeling results in both focal stenotic lesions at the perianastomotic regions and the diffused stenosis within the bodies of VGs towards VGF.
Figure 1.

Characterization of vein graft (VG) remodeling in mice. (A) A schematic presentation of consecutive cross-sectioning of mouse jugular VGs. S, cross-section. (B) Neointima (NI) areas and thicknesses, lumen areas, Neointimal hyperplasia (Nh) and lumen patency (Lp) indexes of VGs measured by C-method as described in “Methods”. The left panel is the overall averages and the right panel shows the average values at each anatomical location along the VG. (C) The representative HE and SMA staining of normal jugular veins (JVs) and carotid arteries (CAs), and JV isografts with severe and mild NI formation of adult male WT C57BL/6J mice at age of 12 wks. (D) Nh index and Lp index of normal JVs and JV isografts at 3, 6 and 12 wks after transplantation. Male WT C57BL/6J mice at age of 12 wks were used for these experiments. VG-All, all VGs; VG-S, VGs with severe NI formation; VG-M, VGs with mild NI formation.
Moreover, the ratio of the NI area to the lumen area that we named as the neointimal hyperplasia (Nh) index near both the proximal and distal ends, especially the nearest two segments (#1 & #2 or #8 & #9) at both ends were higher than that at the middle portion (#3 to #7); whereas the ratio of the lumen area to the NI area that we named as the lumen patency (Lp) index exhibited the opposite results (Figure 1B). Nh or Lp index values of VGs were similar at 3 wks (n=13), but split into approximately two groups with either bigger values or smaller values after 6 wks (Figure 1D and Online Figure VIII). While Nh index values of both groups increased up to 12 wks, the magnitude of Nh index increase was more dramatic in the group with a larger Nh index compared as to the group with a smaller Nh index (Figure 1D). In contrast, the Lp indexes of these groups were opposite (Figure 1D). To find a model which well fit the data, we compared the goodness of fit of two models: (1) a unimodal fit, which assume data follows a single normal distribution and (2) a bimodal fit, which assumes data follows a mixture of two normal distributions. We found that the mixture model fit the data significantly better than the unimodal model (p-value+0.19E-4, log likelihood ratio test) (Online Figure IX). Therefore, there are two subpopulations with means at 0.25 and 0.58 separately (Online Figure IX). We empirically set a cutoff at 0.4 to split the data into two subgroups. As expected, we found these two subgroups have significantly different means (p-value=0.14E-12, two sample t-test). There results indicate that the transplanted veins undergo at least two major types of vascular remodeling; one is characterized by exuberant or severe NI formation with aggressive loss of the lumen patency and another is characterized by modest or mild NI formation with less loss of the lumen patency. We referred the first one as to VG remodeling severe type (VG-S) and the other as to mild type (VG-M). We found that approximately 79% of VGs are the severe type and the other 21% are the mild type.
Mature SMCs of both donor veins and recipient arteries dominate but differentially contribute to intimal hyperplasia in the arteriovenous anastomosis and the middle portion of VGs
We tracked down the fate of mature SMCs in VG remodeling using tamoxifen-inducible Myh11-CreERT2::Rosa26Floxed-StopeYFP mice in which mature SMCs and their progeny could be permanently labeled with YFP by tamoxifen injection.31 Prior to transplantation, the efficiencies of tamoxifen-induced YFP labeling of SMCs in the aorta, carotid arteries, jugular veins and other organs were measured. Almost all of SMA+ cells in these vessels and the other organs were positive for YFP (Online Figure XA and XD) and quantified analyses revealed that around 97% of SMA+ cells in the media of the aorta were positive for YFP; 98% of SMA+ cells in the media of the carotid artery were positive for YFP; and 95% of SMA+ cells in the media of the jugular vein were positive for YFP (Online Figure XA). Then, we performed reciprocal jugular vein transplantation between WT and Myh11-CreERT2::Rosa26Floxed-StopeYFP after the tamoxifen injection for 6 wks. Prior to the assessment of SMC fate in these VGs, we checked again the genetic labeling efficiency in the aorta and carotid artery as well as the jugular vein and vena cava of reporter mice to make sure the labeling was maintained in these reporter mice during the experimental time period. We confirmed that YFP labeling efficiency was maintained in each reporter mouse (Online Figure XI).
Firstly, we analyzed the VGs of WT mice (n=12) transplanted into the carotid arteries of the reporter mice (arterial SMC-YFP). In agreement with the results of Figure 1C and D, 9 of the VGs developed severe NI and 3 of them had mild NI (Figure 2). Regardless of the magnitude of NI formation, SMCs (SMA+ cells) are the major cell type in the NI across the VGs, constituting approximately ~62%, ~81% and ~75% of NI cells at the proximal end, the middle body and the distal end, respectively (Figure 2, Online Table IV and Online Figures XII, XIII, and XIV). In addition, the recipient mature arterial SMCs gave rise to ~13% or ~27% (YFP+ cells) of NI cells at the proximal or distal anastomotic segments, respectively; but to none of NI cells at the middle body (Figure 2C–D, Online Table V and Online Figures XII, XIII, and XIV). The majority of these arterial SMC-derived cells are SMCs (YFP and SMA double positive; ~62% at the proximal portion and ~82% at the distal part) (Figure 2C–D, Online Tables V and VI, and Online Figures XII, XIII, and XIV). Secondly, we analyzed the VGs of the reporter mice (venous SMC-YFP) (n=13) transplanted into the carotid arteries of WT mice. While the incident rate of VG remodeling types in the VGs (10 VG-S and 3 VG-M) (Figure 3A) was very consistent with the other experiments (Figures 1D and 2A), the percentages of SMCs (SMA+ cells) and donor venous SMC-derived cells (YFP+ cells) at the proximal, body and distal portions are similar independent of the VG remodeling types (Figure 3C–D, Online Tables IV and V, and Online Figures XV, XVI and XVII). In addition, while the donor mature venous SMCs gave rise to ~22% or ~47% of NI cells at the proximal or distal anastomotic regions, respectively; they donated to ~68% of NI cells at the body (Figure 3C–D, Online Tables IV and V, and Online Figures XV, XVI and XVII). The majority of donor venous SMC-derived cells (YFP+SMA+; ~82% at the proximal end, ~94% at the body and ~69% at the distal end) were also SMCs (Figure 3C–D, Online Tables V and VI, and Online Figures XV, XVI and XVII).
Figure 2.

Characterization of vein graft (VG) remodeling of wild type (WT) jugular veins (JVs) transplanted to the carotid arteries (CAs) in which mature SMCs were genetically labeled with YFP. (A) Schematic presentation of WT JVs grafted into CAs of the reporter mice. Total number of VGs is 12. (B) The Nh index and Lp index of the VGs. VG, all VGs; VG-S, VGs with severe NI formation; VG-M, VGs with mild NI formation. (C) The percentages of SMA, YFP and SMA and YFP double positive cells in each major layer of VGs with two type of VG-remodeling (n=3). Data are means ± SEM. (D) The representative images of co-staining of YFP and SMA as well as co-staining of YFP and CD31 in the cross-sections of #1 segment at the proximal end (Pro), #3–#7 segments at the middle part (Mid), and #9 segment at the distal end (Dis) of VGs. YFP is green; SMA and CD31 are red. L, the lumen; IN, the intima; NI, the neointima; Ad, the adventitia. White dotted lines separate the NI and the Ad layers.
Figure 3.

Characterization of VG remodeling of the JVs in which mature SMCs were genetically labeled with YFP transplanted to the CAs of WT mice. (A) Schematic presentation of reporter JVs grafted into the CAs of WT mice. (B) The Nh index and Lp index of the VGs. (C) The percentages of SMA, YFP and SMA and YFP double positive cells in each major layer of VGs with two type of VG-remodeling (n=3). (D) The representative images of co-staining of YFP and SMA as well as co-staining of YFP and CD31 in the cross-sections of #1 segment at the proximal end, #3–#7 segments at the middle part, and #9 segment at the distal end of VGs. YFP is green; SMA and CD31 are red. White dotted lines separate the NI and the Ad layers. The abbreviations are described in Figures 1 and 2.
In summary, we have demonstrated that the focal nature of vein graft remodeling is linked with distinct contribution of recipient arterial and donor venous SMCs to the arteriovenous anastomotic regions and the middle body. Both the recipient arterial and donor venous mature SMCs give rise to approximately ~55% of the NI cells at the arteriovenous anastomosis (~35% at the proximal end and ~74% at the distal end). However, only donor venous mature SMCs contribute up to 68% of the NI cells at the middle body and more than 94% of them are synthetic SMCs.
A novel aspect of mature vascular SMCs in VG remodeling
Intriguingly, we noticed that there were a few scattered cells positive for YFP but negative for SMA in the reciprocally transplanted VGs independent of the magnitude of intimal hyperplasia (Figure 4, Online Tables VII and VIII, and Online Figures XVIII and XIX), revealing that mature SMCs give rise to a novel type of non-SMC cells in VGs. Both the arterial and venous SMC-derived non-SMC cells constitute around 3–6% of the intimal cells, 6–14% of the NI cells and 9–11% of Ad cells at the arteriovenous anastomotic regions; whereas the donor venous SMC-derived non-SMC cells predominantly account for around 0.5% of the intimal cells, 2% of the NI cells and 2% of the Ad cells at the middle bodies (Figure 4A and IVC, Online Tables VII and VIII, and Online Figures XVIII and XIX). These novel findings indicate that mature SMCs may not only serve as the major cellular source of NI formation via the well-established mechanism of SMC dedifferentiation,32 but also are a source of the intimal and the adventitial (Ad) cells contributing to re-endothelialization and adventitial reconstruction via a yet unknown mechanism in VG remodeling.
Figure 4.

Characterization of non-SMC cells derived from mature SMCs in VGs. (A, B) The non-SMC cells derived from the recipient arterial SMCs. (A) The top panel is a scheme of wild WT JVs grafted into reporter CAs. Total number of vein graft (VG) is 12. The lower panels are quantified percentages of YFP positive but SMA negative cells and percentages of YFP positive but SMA negative in YFP positive cells. (B) The representative images of YFP (green) and SMA (red) staining in VG cross-sections at the anatomical location as indicated. (C, D) The non-SMC cells derived from the donor venous SMCs. (C) The top panel is a scheme of reporter JVs grafted into WT CAs. Total number of vein graft (VG) is 13. The lower panels are quantified percentages of YFP positive but SMA negative cells and percentages of YFP positive but SMA negative in YFP positive cells. (D) The representative images of YFP (green) and SMA (red) staining in VG cross-sections at the anatomical location as indicated. Green arrows indicate YFP+SMA− cells. White dotted lines separate the NI and the IN or Ad. The abbreviations are described in Figures 1 and 2.
It has been reported that mature vascular SMCs may lose the SMC identity while gaining stemness, thereby transforming or reprogramming themselves into stem cell-like cells.33–36 Thus, we questioned whether or not VG transplantation may drive mature SMCs to gain stemness, i.e., SMC reprogramming into stem or stem cell-like cells. We carried out triple staining of stem cell markers (Sca1, CD44, CD45, and Gli1)37, 38 with YFP and SMA in the tissue cross-sections of 42-day VGs (n=13) in which mature SMCs were genetically labeled with YFP. We found that Sca1, CD44 or CD45, but not Gli1 are expressing in the YFP+SMA− cells and these unique cells reside most frequently at the intima and the boundary areas between the NI and the adventitia (Ad) (Figure 5, Online Figures XX and XXI, and Online Table IX). Of note, up to 35% of YFP+SMA− cells in the intima are positive for Sca1, ~35% are positive for CD44, and ~23% are positive for CD45; whereas up to 78% of YFP+SMA− cells in the Ad are positive for Sca1, ~45% are positive for CD44, and ~25% are positive for CD45 (Figure 5 and Online Table IX). These findings reveal that mature SMCs could be transformed into stem or stem cell-like cells in VGs.
Figure 5.

The expression of Sca1, a vascular stem cell marker in non-SMC cells derived from donor venous mature SMCs in VGs. Tissue sections of VGs described in Figure 4 were used for the staining. (A, B) Representative images of Sca1, YFP and SMA co-staining in different segments of VGs. EC areas (intimal areas) are circled with yellow lines and NI/Ad areas (the boundary area between NI and Ad are circled with white lines. (B) The enlarged images of marked areas in (A). (C) The percentages of YFP+Sca1+SMA− cells in different layers of VGs. Two or three cross-sections of each segment (Pro end: #1+2; Middle portion: #3–#7; Dis end: #8+9) of each vein were randomly chosen for the staining (20–30 sections from 13 JVs in total for each location).
SM22α (Ki)-CreERT2::Rosa26Floxed-StopeYFP mice are not the appropriate tool for SMC lineage tracking in vein graft remodeling
The homozygotes of SM22α (Ki)−/−-CreERT2 mice in which the CreERT2 gene is inserted into the endogenous SM22α locus39 do not express SM22α. The Ki approach per se actually results in global SM22α KO in mice. Thus, the reporter mice carrying SM22α (Ki)+/−-CreERT2 heterozygous alleles are usually used for SMC lineage tracking. Notably, SM22α is not merely a SMC marker; instead it plays a critical role in the regulation of SMC phenotype modulation and vascular remodeling.11–15 Therefore, we questioned whether the SM22α (Ki)+/−-CreERT2 approach affects the VG remodeling of the reporter mice. We found that knockdown of SM22α dramatically enhanced H2O2-induced cell death in cultured mouse aortic SMCs (Figure 6A and Online Figure XXII). Then we performed vein graft transplantation using sex and age matched WT C57BL/6J mice as the recipient and littermates of WT (SM22α+/+), SM22α(Ki)+/−-CreERT2 (SM22α+/−) and SM22α(Ki)−/−-CreERT2 (SM22α−/−) mice in C57BL/6J genetic background as the donor. At 4 h after transplantation, a substantial number of cells including SMCs were positive for Tunel staining in WT vein grafts, and the Tunel positive numbers were increased in both SM22α(Ki)+/−-CreERT2 and SM22α(Ki)−/−-CreERT2 vein grafts (Figure 6B and Online Figure XXIII). Importantly, the magnitude of cell death in SM22α (Ki)+/−-CreERT2 and SM22α(Ki)−/−-CreERT2 vein grafts were comparable (Figure 6B and Online Figure XXIII). These results reveal that knockdown of SM22α expression in SMCs could sufficiently enhance SMC death in VGs, presumably leading to adverse outcome. Indeed, loss of SM22α in SMCs exaggerated the NI formation in VGs at 6 wks after transplantation (Figure 6C).
Figure 6.

Loss of SM22α sensitizes SMCs to stress-induced cell death. (A) H2O2-induced cell death in cultured mouse aortic SMCs. SMCs transfected with scramble control sequences (si-Ctl) and SM22 α siRNA (si-SM22 α) were treated with or without H2O2 for 2h and then subject to propidium iodide (PI) uptake assay as described in “Method”. The results are means ± SD (n=4). *p<0.01 between indicated groups. The SM22α knockdown efficiency was confirmed by Western blot analysis (inserted immunoblots). (B) SMC death in transplanted jugular veins (JVs) of WT, SM22α (Ki)-CreERT2+/− (SM22α(Ki)+/−) and SM22α (Ki)-CreERT2−/− (SM22α(Ki)−/−) mice. JVs of WT, SM22α(Ki)+/−, and SM22α(Ki)−/− mice at age of 3 months were transplanted into WT CAs (n=4) for 4 hours. Cell death in the media of these VGs was determined by Tunel staining. The results are means ± SD. *p<0.01 between indicated groups. (C) Loss of SM22α promotes NI formation in VGs. JVs of WT and SM22α(Ki)−/− mice at age of 3 months were transplanted into WT carotid arteries (CAs) for 6 wks (n=6). The overall averages of NI areas in these VGs were measured as described in “Methods”. The results are means ± SD. *p<0.01 between indicated groups.
We also carefully quantified the genetic labeling efficiency of SMCs in adult SM22α (Ki)+/−-CreERT2::Rosa26Floxed-StopeYFP mice. In aorta and carotid arteries, we found that ~26% of SMA+ cells were positive for YFP in the tunica media (Online Figure XXIV). In jugular veins, we found that ~27% of SMA+ cells were positive for YFP in the media (Online Figure XXIV). Considering such a low labeling efficiency and the adverse impact on NI formation in VGs, SM22α (Ki)+/−-CreERT2::Rosa26Floxed-StopeYFP mice may not be a reliable tool for tracking down the fate of SMCs in VG remodeling.
Recipient arterial, but not the donor venous ECs, repair damaged endothelium in VGs. Only donor venous ECs undergo EndMT contributing to the mild type, but not the severe type NI formation in VGs.
To verify whether ECs via EndMT serve as a major source of NI SMCs in VGs, we mapped the fate of both recipient arterial and donor venous ECs in the murine cuff model of intimal hyperplasia using Cdh5-CreERT2::Rosa26Floxed-StopeYFP mice as Cdh5-CreERT2 is the most established tamoxifen-inducible EC-specific Cre.40 Prior to transplantation, the efficiencies of tamoxifen-induced YFP labeling of ECs in the aorta, carotid arteries, jugular veins and other organs were measured. Almost all of intimal CD31+ cells in these vessels and the other organs were positive for YFP (Online Figure XXV) and quantified analyses revealed that ~98% of intimal CD31+ cells of the aorta were positive for YFP; ~94% of intimal CD31+ cells of the carotid artery were positive for YFP; and ~88% of intimal CD31+ cells of the jugular vein were positive for YFP (Online Figure XXV). Then, we performed reciprocal jugular vein transplantation between WT and Cdh5-CreERT2::RosaFloxed-StopeYFP after the tamoxifen injection for 6 wks. Prior to the assessment of EC fate in these VGs, we confirmed that the YFP labeling efficiency has been maintained in the reporter mice during the experimental time periods (Online Figure XXVI).
Firstly, we analyzed the VGs of WT mice (n=11) transplanted into the carotid arteries of the reporter mice (arterial EC-YFP). Again, there were two types of VG remodeling with a similar incident rates (Figure 7A) to other experiments (Figures ID, 2A, and 3A). Also, independent of the magnitude of NI formation, SMCs were the major cellular component of the NI with the percentages of ~63%, ~85%, and ~83% at the proximal end, the middle body and the distal end, respectively (Figure 7C–D, Online Table X and Online Figures XXVII, XXVIII and XXIX), which were similar to the other experiments above mentioned. Regardless of the VG remodeling types, the majority of intimal cells at both perianastomotic regions were positive for YFP with the positive percentages of ~96% and ~69% at the proximal and distal segments, respectively; whereas near half of the intimal cells (~47%) were positive for YFP in the middle body (Figure 7C–D, Online Tables XI–XIV and Online Figures XXVII, XXVIII and XXIX). Most of the intimal YFP+ cells at the perianastomotic regions were also positive for vWF; whereas around 80% of the intimal YFP+ cells at the bodies were positive for vWF (Figure 7D and Online Figures XXVII and XXIX). However, none of the NI cells were positive for YFP (Figure 7C–D, Online Tables XI–XIV and Online Figures XXVII, XXVIII and XXIX). These results reveal that recipient arterial mature ECs are not the cellular source of intimal hyperplasia; but they are the major cellular source of the re-endothelialization in VGs. In addition, the re-endothelialization begins at the perianastomotic ends and extends into the middle part of VGs over time.
Figure 7.

Characterization of VG remodeling of WT JVs transplanted to the CAs in which mature ECs were genetically labeled with YFP. (A) Schematic presentation of WT JVs grafted into reporter CAs. Total number of VGs is 11. (B) The Nh index and Lp index of the VGs. (C) The percentages of SMA, YFP and SMA and YFP double positive cells in each major layer of VGs with two type of VG-remodeling (n=3). Data are means ± SEM. (D) The representative images of co-staining of YFP and SMA as well as co-staining of YFP and vWF in the cross-sections of #1 segment at the proximal end, #3–#7 segments at the middle part, and #9 segment at the distal end of VGs. YFP is green; SMA and vWF are red. White dotted lines separate the NI and the Ad layers. The abbreviations are described in Figures 1 and 2.
Secondly, we analyzed the VGs of reporter mice (venous EC-YFP) (n=15) transplanted into the carotid arteries of WT mice. Compared to the other experiments, the differences observed were that the contributions of donor mature ECs to the re-endothelialization and the NI formation were distinct depending on the magnitude of NI formation in VGs. In the VG-S group (n=11), none of the intimal or the NI cells were positive for YFP (Figure 8B–C, Online Tables XI–XIV and Online Figures XXX, XXXI and XXXII), indicating a negligible contribution of donor venous ECs to VG remodeling towards the severe NI formation. In the VG-M group (n=4), only a small portion of the intimal cells at the proximal part (8.3±11.8%) and at the body (1.7±2.3%), but none of the intimal cells at the distal part were positive for YFP (Figure 9A–B, Online Tables XI–XIV and Online Figures XXXIII and XXXIV), suggesting a minor contribution of donor venous ECs to the re-endothelialization. In addition, 28.2±9.3% of the NI cells at the proximal portion and 13.4±11.1% at the middle body were positive for YFP, whereas the number of YFP+ cells at the distal end was negligible (Figure 9A–B, Online Table X and Online Figures XXXIII and XXXIV). Of interest, the majority of these YFP+ cells are SMCs (YFP+SMA+vWF−) (Figure 8A–B, Online Table X and Online Figures XXX), supporting that the survived donor venous ECs undergo EndMT in VGs with mild intimal hyperplasia as previously reported.41, 42 However, the EndMT occurred only in the sites of NI with less hyperplastic growth of the same VGs (Figure 9B).
Figure 8.

Characterization of VG remodeling of the JVs in which mature ECs were genetically labeled with YFP transplanted to the CAs of WT mice. (A) Schematic presentation of the reporter JVs grafted into the CAs of WT mice. Total number of VGs is 15 and 11 of them are VGs with severe NI formation (VG-S). (B) The percentages of SMA, YFP and SMA and YFP double positive cells in each major layer of VGs of VG-S (n=3). (C) The representative images of co-staining of YFP and SMA as well as co-staining of YFP and vWF in the cross-sections of #1 segment at the proximal end, #3–#7 segments at the middle part, and #9 segment at the distal end of VGs. YFP is green; SMA and vWF are red. White dotted lines separate the NI and the Ad layers. The abbreviations are described in Figures 1 and 2.
Figure 9.

Characterization of VG remodeling of the JVs in which mature ECs were genetically labeled with YFP transplanted to the CAs of WT mice. (A) The percentages of SMA, YFP and SMA and YFP double positive cells in each major layer of VGs with mild NI formation (VG-M) (n=3). (B) The representative images of co-staining of YFP and SMA as well as co-staining of YFP and vWF in the cross-sections of VGs of VG-M. YFP is green; SMA and vWF are red. White dotted lines separate the NI and the Ad layers, green dotted lines indicate less hyperplasia side of VG and red dotted lines indicate more hyperplasia side of VG (C) Schematic presentation of transplantation of the reporter JVs grafted into the CAs of WT mice. Total number of VGs is 15 and 4 of them are VG-M. The abbreviations are described in Figures 1 and 2.
Taken together, our results demonstrate that the recipient arterial ECs are the major cellular source of re-endothelialization in VGs and do not contribute to NI formation in VGs independent of the magnitude of intimal hyperplasia. The donor venous ECs do not directly contribute to the re-endothelialization and NI formation in VG-S. Although the donor venous ECs do serve as a cellular source of 21% (13–28%) NI cells at the proximal perianastomotic region and the middle portion in VG-M, they do not directly contribute to the intimal hyperplasia at the distal perianastomotic region. Given that the incidence rate of VG-M is around 20% and only 17% (9~25%) of NI cells of the proximal perianastomotic region and a portion of the middle part in VG-M are derived from the donor venous ECs most likely via EndMT, the overall contribution of ECs via EndMT (~5%) to the intimal hyperplasia is marginal. Thus, the major role of ECs, and mostly the recipient arterial ECs, is to act as a cellular source of endothelium reconstruction in VG remodeling.
EndMT is likely an adaptive response in VG remodeling.
Because EndMT occurred only VG-M group, we postulated there is yet unappreciated mechanism responsible for the low incidence rate of EndMT in vein graft remodeling. We also questioned whether there is a link between the EndMT and onset of the mild type of vein graft remodeling. To find clues, we performed a detailed time course study of cellular dynamics with a focus on SMCs and ECs in VG remodeling. We found that ~99% of ECs and ~97% of SMCs die within 3 days after transplantation; however, the death rate of ECs is higher than that of SMCs (Figure 10 and Online Figures XXXA–C). The re-endothelialization did not occur within 2 weeks after transplantation, whereas a substantial number of rapidly proliferating SMCs appeared, forming NI at 2 weeks (Online Figures XXXA–B). Thus, it is unlikely that EndMT of the survived ECs occurs and contributes to the generation of synthetic SMCs at an early stage of VG remodeling. Instead, EndMT may happen at a late stage of VG remodeling and play a regulatory role in VG arterialization or occlusion. To test this hypothesis, we studied the paracrine regulation of vascular SMC dedifferentiation by native ECs and the ECs after EndMT in vitro. Utilizing an established system of EndMT in cultured human umbilical vein endothelial cells (HUVECs),27 we prepared conditional media (CM) from cultured native HUVECs (EC CM) and HUVECs after EndMT induced by transforming growth factor beta 1 (TGF-β1) (EndMT-T CM) or bone morphogenetic protein 4 (BMP4) (EndMT-B CM) (Figure 11A and B). We then determined the impact of these CM on mouse aortic SMC dedifferentiation. Compared to the SMCs cultured in SMC growth medium, the SMCs cultured in EndMT-T CM and EndMT-B CM, but not in EC culture base medium (EC baseM) or EC-CM, underwent dramatic morphological changes characterized by decreasing the horizontal width while increasing the longitudinal length of the cells, resulting in an elongated and spindlleshaped morphology, a typical feature of mature vascular SMCs (Figure 11C). Like the SMC growth medium, EC CM was capable of stimulating SMC proliferation (Figure 11D) and migration (Online Figure 36). Compared to EC CM, however, EndMT-T CM and EndMT-B CM were less effective for promoting SMC proliferation (Figure 11D) and migration (Online Figure 36). Interestingly, EC CM downregulated the expression of SMC genes including SMA, CNN1 and SM22α; however, EndMT-T CM was less effective for downregulating the SMC gene expression, and EndMT-B CM even upregulated the SMC gene expression (Figure 11E). These results demonstrate that in the conventional 2 dimensional culture system, native ECs are activated thereby releasing substances to promote SMC dedifferentiation, and EndMT prevents the activation of ECs as well as the subsequent release of pro-SMC dedifferentiating factors. Of note, the potential of EC EndMT CM in suppressing SMC dedifferentiation is proportional to the magnitude of EC EndMT (Figure 11). Collectively, our findings indicate that EndMT may be an adaptive response, at least in VG remodeling, towards VG arterialization.
Figure 10. The cellular dynamics of SMCs and ECs during VG remodeling in mice.

(A) The percentages of SMA+ SMCs and CD31+ ECs in the intima (I) and the neointima (NI). (B) The cell numbers per field are stained positive for EC markers CD31 and vWF, SMC markers SMA, SM22α, CNN1 and MYH11, and Tunel in the intima (I) and the neointima (NI). Normal jugular veins were used as the control (0 day). Three randomly selected tissue cross-sections per vein or VG of 4 mice were stained and analyzed as described in Online Supplement “Methods”.
Figure 11. The effects of conditional media from HUVECs with or without EndMT on vascular SMC dedifferentiation in vitro.

(A) Schematic diagram of CM preparation from HUVECs with or without EndMT. (B) TGF-β1- or BMP4-induced EndMT in HUVECs. Western blot analyses of EC markers Tie2, eNOS and Cdh5; mesenchymal stem cell markers CD44 and CD90; and mesenchymal marker SMA. Left panel is the quantified results (n=4). (C) Morphological changes, (D) proliferation and (E) SMC gene expression of mouse aortic SMCs cultured in different CMs, i.e., ① EC baseM: EC basal media; ② EC CM: CM from ECs without EndMT; ③EndMT-T CM: CM from ECs with EndoMT induced by TGF-β1; and ④ EndMT-B CM: CM from ECs with EndoMT induced by BMP4 as described in Online Supplement :Methods”. *p<0.05 between the indicated groups.
Discussion
In the present study, we have clarified several controversial issues regarding VG remodeling as follows: (1) Mature SMCs are the major cellular source of intimal hyperplasia. However, the recipient arterial and the donor venous SMCs differentially contribute to NI formation at different portions of VGs. While both the recipient arterial and donor venous mature SMCs give rise up to 55% of the NI cells at the arteriovenous anastomosis, only donor venous mature SMCs contribute up to 68% of the NI cells at the middle body and more than 90% of them are synthetic SMCs. (2) SM22α (Ki)+/−-CreERT2 mice may not be a reliable tool for tracking down the fate of SMCs in VG remodeling since the Ki approach per se has great impact on VG remodeling. (3) Mature ECs, particularly the recipient arterial ECs are not the major cellular source of intimal hyperplasia; instead they are the cellular source of re-endothelialization. The direct contribution of ECs via EndMT to NI formation is marginal; however, EndMT may play an important role in VG adaptation. In addition, we have found that there are two types of VG remodeling, i.e., up to 80% of VGs develop severe NI formation, termed VG remodeling severe type (VG-S); and around 20% of VGs develop mild NI formation, termed VG remodeling mild type (VG-M). Also, we have uncovered a novel aspect of mature SMCs in VG remodeling; i.e., the recipient arterial and/or the donor venous SMCs are transformed into the scattered non-SMC cells, most likely vascular stem cells, constituting ~11% of the intimal or adventitial cells at the arteriovenous anastomosis and~2% of the intimal or adventitial cells at the middle body. Our findings not only underscore a predominant role of mature SMCs in intimal hyperplasia but also reveal a potential role of mature SMCs in repopulating the intimal and the adventitial cells in VGs, providing new insight into the pathogenesis of occlusive VG remodeling towards VGF.
Given that overwhelming literatures have firmly established a central role of vascular SMCs in various forms of vascular lesion formation32 including the intimal hyperplasia in VGs,1–3 it wasn’t surprising to find that mature SMCs are the major cellular source of intimal hyperplasia in VGs independent of the magnitude of NI formation. Considering the important role of SM22α per se in vascular remodeling,11–15 the limitation of SM22α (Ki)+/−-CreERT2 approach in tracking down the fate of SMCs in VG remodeling could also be postulated. However, the clarification of relative contributions of the recipient arterial and the donor venous SMCs to the NI formation in arteriovenous anastomoses and bodies of VGs may provide a research guideline to understand the focal stenotic nature of VGs towards the intermediate and late failure observed in patients.2, 4 In this regard, our findings that the recipient arterial and donor venous mature SMCs contributed to up to 55% of the NI cells at the arteriovenous anastomosis have emphasized the necessity to understand the mechanism associated with the recipient arterial and/or donor venous mature SMC-mediated intimal hyperplasia at these specific locations which are associated with to the intermediate VGF.2, 4 On the other hand, the findings that only donor venous mature SMCs contributed up to 68% of the NI cells at the middle body and more than 90% of them are synthetic SMCs have highlighted the focus to explore essential determinants of the donor venous SMC-mediated NI formation in the body which leads to the late VGF.2, 4
The minimal contribution of the cells derived from mature ECs via EndMT to intimal hyperplasia in VGs is intriguing and has challenged the emerging concept that mature ECs via EndMT serve as a major cellular source of vascular lesion formation.41, 42 In fact, previous studies have shown that in transplant settings, over 80% of luminal ECs express mesenchymal markers while the frequency of EndMT elsewhere is much less and varies in a range of 3% to 50% depending on the type of transplantation and the fate mapping tools used. It is worthy to note that the observed high incident rate of EndMT is clearly associated with the lineage tracing mouse tools41 that are not specific for tracking EC fate in vivo as previously mentioned. In agreement with our findings, the studies using Cdh5-CreERT2 mice, the most commonly used inducible and EC-specific genetic labelling approach43 have consistently demonstrated that less than 7% of NI cells are derived from mature ECs in VGs (inferior vena cava to aorta).41 Therefore, EndMT may not be a critical process to produce NI cells in VGs. However, the strong inhibitory effect of conditional medium from ECs after EndMT on vascular SMC dedifferentiation suggests that it may be an adaptive response in VG remodeling. Given that EndMT has the potential in paracrine control of SMC dedifferentiation while giving rise to a smaller portion of NI cells in VGs, although not conclusive at this point, a plausible explanation is likely that it may serve as a regulatory mechanism in the pathogenesis of VG diseases, which needs to be further investigated.
The observation that both the recipient arterial and the donor venous mature SMCs could be transformed into the scattered non-SMCs, most likely vascular stem cells, constituting a small but noticed portion of the intima, the NI and the Ad of VGs is also very interesting. Although the fates of these cells have not been uncovered, several options could be postulated from the known mechanisms of vascular SMC phenotypic transition.33–36 Accordingly, VG transplantation may drive mature SMCs to gain stemness, i.e., SMC reprogramming into stem cell-like cells, and then differentiate into the other vascular cells, such as the intimal ECs, the neointimal SMCs and the adventitial cells as previously proposed.33, 34, 36 Indeed, we found that some of ECs are actually derived from mature SMCs in VGs (data not shown), indicating a process of mesenchymal to endothelial transition. Alternatively, they are probably the progeny of the resident adventitial stem cell antigen 1 (Sca1)+ stem cells which are derived from the mature SMCs.35, 36 Our findings uncover that mature SMCs could be reprogrammed into stem cells in VGs. However, the molecular mechanism of mature SMC reprogramming in VG remodeling and the contribution of mature SMC reprogramming to VG remodeling need to be further investigated.
The pathophysiological significance of VG-S and VG-M has not been fully determined in the present study. However, a few possibilities may be assumed. Given that VG-S was the major type of VG remodeling and characterized by progressive intimal hyperplasia and loss of lumen patency, it is conceivable that VGs of VG-S may eventually fail. In addition, because the contribution of mature vascular SMCs to these two types of VG remodeling was similar, it is not likely that vascular SMCs determine the consequences of specific types of VG remodeling. On the other hand, EndMT observed only in VGs of VG-M was capable of suppressing vascular SMC dedifferentiation, indicating a potential link between EndMT and the quality of VG remodeling. Nevertheless, whether EndMT plays a key role in the quality control of VG remodeling or a merely disease associated epiphenomenon remain to be elucidated.
It should be noted that like most mammalian models of vascular disease, mouse studies hold many confounding factors that contribute to the observed variations in VG remodeling, which include, but are not limited to mouse genetic backgrounds, surgical technical skill, differences in the grafting protocol, and tissue processing.29 Although our rigorous approaches cleared the genetic background and technical issues for a mouse interposition graft model of VG intimal hyperplasia, whether our findings are applicable to other VG models such as vein end to artery side anastomosis remains to be verified. In addition, the significance of our findings needs to be interrogated by the experiments which integrate the inherent anatomical and physiological differences between mice and human, as well as the factors such as aging and comorbidities associated with human vein graft remodeling and failure.
In conclusion, our present study demonstrates that it is mature vascular SMCs, but not ECs, are the major cellular source of intimal hyperplasia in VGs. In addition, a small portion of mature vascular SMCs are dedifferentiated into non-SMCs, most likely vascular stem cells residing at all different layers of VGs. EndMT does not play a major role in driving mature ECs into NI cells, but may be linked to the quality control of VG remodeling. Further investigations of the identities and roles of mature vascular SMC-derived non-SMCs as well as the potential role of EndMT in the quality control of VG remodeling will provide not only novel insights into VG remodeling but also new perspectives for the rational design and development of a novel therapeutic approach for the treatment of VGF.
Supplementary Material
Highlights.
Both the recipient arterial and donor venous mature SMCs give rise to approximately ~55% of the neoinitima cells at the arteriovenous anastomosis of vein grafts.
Only donor venous mature SMCs contribute up to 68% of the neoinitima cells at the middle body of vein grafts.
A small portion of the SMC-derived cells become non-SMC cells, most likely stem cell-like cells and constitute 2–11% of the cells in each major layer of vein grafts.
The major role of ECs, and mostly the recipient arterial ECs, is to act as a cellular source of endothelium reconstruction in vein graft remodeling.
EndMT is marginal for generating neointima cells but is likely required for controlling the quality of vein graft remodeling.
Acknowledgments
Sources of Funding
This work as partly supported by grants from the National Institute of Biomedical Imaging and Bioengineering of the National Institute of Health (NIH) to T. Cui (1R21EB022131-01A1) and from the National Heart, Lung, and Blood Institute of NIH to D.C. Porter and T. Cui (1R43HL137525-01, 1R01 HL131667-01A1) and M. Azhar (R01HL126705, R01CA218578, R01HL145064), and from the National Institute of General Medical Science of NIH to I.B. Roninson and T. Cui (P20GM109091), and from the National Center for Complementary and Integrative Health of NIH to P. Nagarkatti and T. Cui (2P01 AT003961-06A1) and from American Diabetes Association to T. Cui (1-16-IBS-059) and from American Heart Association to M. Azhar (17GRNT33650018).
Nonstandard Abbreviations and Acronyms
- VG
vein graft
- VGF
vein graft failure
- SMCs
smooth muscle cells
- ECs
endothelial cells
- NI
neointima
- EndMT
endothelial mesenchymal transition
- SM-MHC or Myh11
smooth muscle myosin heavy chain
- SM22α or Tagln
smooth muscle 22 alpha
- CNN1
calponin 1
- SMA or Acta2
smooth muscle actin alpha
- Ki
knockin
- Nh
neointimal hyperplasia
- Lp
lumen patency
- GIFM
genetic inducible fate mapping
- Sca1
stem cell antigen 1
- Gli1
glioma-associated oncogene homolog 1
References
- 1.Shukla N and Jeremy JY. Pathophysiology of saphenous vein graft failure: a brief overview of interventions. Current opinion in pharmacology. 2012;12:114–20. [DOI] [PubMed] [Google Scholar]
- 2.Harskamp RE, Lopes RD, Baisden CE, de Winter RJ and Alexander JH. Saphenous vein graft failure after coronary artery bypass surgery: pathophysiology, management, and future directions. Ann Surg. 2013;257:824–33. [DOI] [PubMed] [Google Scholar]
- 3.Owens CD, Gasper WJ, Rahman AS and Conte MS. Vein graft failure. Journal of vascular surgery. 2015;61:203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owens CD, Ho KJ and Conte MS. Lower extremity vein graft failure: a translational approach. Vascular medicine. 2008;13:63–74. [DOI] [PubMed] [Google Scholar]
- 5.Hu Y, Mayr M, Metzler B, Erdel M, Davison F and Xu Q. Both donor and recipient origins of smooth muscle cells in vein graft atherosclerotic lesions. Circulation research. 2002;91:e13–20. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Freedman NJ, Brian L and Peppel K. Graft-extrinsic cells predominate in vein graft arterialization. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:470–6. [DOI] [PubMed] [Google Scholar]
- 7.Jevon M, Ansari TI, Finch J, Zakkar M, Evans PC, Shurey S, Sibbons PD, Hornick P, Haskard DO and Dorling A. Smooth muscle cells in porcine vein graft intimal hyperplasia are derived from the local vessel wall. Cardiovasc Pathol. 2011;20:e91–4. [DOI] [PubMed] [Google Scholar]
- 8.Liang M, Liang A, Wang Y, Jiang J and Cheng J. Smooth muscle cells from the anastomosed artery are the major precursors for neointima formation in both artery and vein grafts. Basic Res Cardiol. 2014;109:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang M, Wang Y, Liang A, Mitch WE, Roy-Chaudhury P, Han G and Cheng J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires Notch activation to form neointima. Kidney Int. 2015;88:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC and Boehm M. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Science translational medicine. 2014;6:227ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J, Yang M, Ju D, Jiang H, Zheng JP, Xu Z and Li L. Disruption of SM22 promotes inflammation after artery injury via nuclear factor kappaB activation. Circulation research. 2010;106:1351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong LH, Wen JK, Liu G, McNutt MA, Miao SB, Gao R, Zheng B, Zhang H and Han M. Blockade of the Ras-extracellular signal-regulated kinase 1/2 pathway is involved in smooth muscle 22 alpha-mediated suppression of vascular smooth muscle cell proliferation and neointima hyperplasia. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:683–91. [DOI] [PubMed] [Google Scholar]
- 13.Chen R, Zhang F, Song L, Shu Y, Lin Y, Dong L, Nie X, Zhang D, Chen P and Han M. Transcriptome profiling reveals that the SM22alpha-regulated molecular pathways contribute to vascular pathology. Journal of molecular and cellular cardiology. 2014;72:263–72. [DOI] [PubMed] [Google Scholar]
- 14.Lv P, Miao SB, Shu YN, Dong LH, Liu G, Xie XL, Gao M, Wang YC, Yin YJ, Wang XJ and Han M. Phosphorylation of smooth muscle 22alpha facilitates angiotensin II-induced ROS production via activation of the PKCdelta-P47phox axis through release of PKCdelta and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circulation research. 2012;111:697–707. [DOI] [PubMed] [Google Scholar]
- 15.Dong LH, Li L, Song Y, Duan ZL, Sun SG, Lin YL, Miao SB, Yin YJ, Shu YN, Li H, Chen P, Zhao LL and Han M. TRAF6-Mediated SM22alpha K21 Ubiquitination Promotes G6PD Activation and NADPH Production, Contributing to GSH Homeostasis and VSMC Survival In Vitro and In Vivo. Circulation research. 2015;117:684–94. [DOI] [PubMed] [Google Scholar]
- 16.Pulford K, Lecointe N, Leroy-Viard K, Jones M, Mathieu-Mahul D and Mason DY. Expression of TAL-1 proteins in human tissues. Blood. 1995;85:675–84. [PubMed] [Google Scholar]
- 17.Pimanda JE, Silberstein L, Dominici M, Dekel B, Bowen M, Oldham S, Kallianpur A, Brandt SJ, Tannahill D, Gottgens B and Green AR. Transcriptional link between blood and bone: the stem cell leukemia gene and its +19 stem cell enhancer are active in bone cells. Molecular and cellular biology. 2006;26:2615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gothert JR, Gustin SE, Hall MA, Green AR, Gottgens B, Izon DJ and Begley CG. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood. 2005;105:2724–32. [DOI] [PubMed] [Google Scholar]
- 19.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, Anderson DG, Kotelianski V, Wang F, Tellides G and Simons M. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell reports. 2012;2:1684–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS and Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nature medicine. 2008;14:64–8. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD and Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–6. [DOI] [PubMed] [Google Scholar]
- 22.Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F and Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15–22. [DOI] [PubMed] [Google Scholar]
- 23.Herring BP, Hoggatt AM, Burlak C and Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell. 2014;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou Y, Dietrich H, Hu Y, Metzler B, Wick G and Xu Q. Mouse model of venous bypass graft arteriosclerosis. The American journal of pathology. 1998;153:1301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L, Wang X, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL and Cui T. Nrf2-Mediated Cardiac Maladaptive Remodeling and Dysfunction in a Setting of Autophagy Insufficiency. Hypertension. 2016;67:107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, Murgai M, Turner SD, Geng YJ, Bekiranov S, Connelly JJ, Tomilin A and Owens GK. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nature medicine. 2016;22:657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R and Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nature medicine. 2010;16:1400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Q Mouse models of arteriosclerosis: from arterial injuries to vascular grafts. The American journal of pathology. 2004;165:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu P, Nguyen BT, Tao M, Campagna C and Ozaki CK. Rationale and practical techniques for mouse models of early vein graft adaptations. Journal of vascular surgery. 2010;52:444–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terry CM, Blumenthal DK, Sikharam S, Li L, Kuji T, Kern SE and Cheung AK. Evaluation of histological techniques for quantifying haemodialysis arteriovenous (AV) graft hyperplasia. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2006;21:3172–9. [DOI] [PubMed] [Google Scholar]
- 31.Feil S, Krauss J, Thunemann M and Feil R. Genetic inducible fate mapping in adult mice using tamoxifen-dependent Cre recombinases. Methods Mol Biol. 2014;1194:113–39. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, Giachelli CM, Parmacek MS, Raines EW, Rusch NJ, Speer MY, Sturek M, Thyberg J, Towler DA, Weiser-Evans MC, Yan C, Miano JM and Owens GK. Smooth muscle cell plasticity: fact or fiction? Circulation research. 2013;112:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bukovsky A Sex steroid-mediated reprogramming of vascular smooth muscle cells to stem cells and neurons: possible utilization of sex steroid combinations for regenerative treatment without utilization of in vitro developed stem cells. Cell cycle. 2009;8:4079–84. [DOI] [PubMed] [Google Scholar]
- 34.Hong X, Margariti A, Le Bras A, Jacquet L, Kong W, Hu Y and Xu Q. Transdifferentiated Human Vascular Smooth Muscle Cells are a New Potential Cell Source for Endothelial Regeneration. Scientific reports. 2017;7:5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA and Weiser-Evans MC. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4. Circulation research. 2017;120:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M and Jorgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nature communications. 2018;9:4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Psaltis PJ and Simari RD. Vascular wall progenitor cells in health and disease. Circulation research. 2015;116:1392–412. [DOI] [PubMed] [Google Scholar]
- 38.Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, Kaesler N, Chang-Panesso M, Machado FG, Gratwohl S, Madhurima K, Hutcheson JD, Jain S, Aikawa E and Humphreys BD. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell stem cell. 2016;19:628–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M and Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circulation research. 2014;115:662–7. [DOI] [PubMed] [Google Scholar]
- 40.Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF and Iruela-Arispe ML. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn. 2006;235:3413–22. [DOI] [PubMed] [Google Scholar]
- 41.Dejana E, Hirschi KK and Simons M. The molecular basis of endothelial cell plasticity. Nature communications. 2017;8:14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Payne S, De Val S and Neal A. Endothelial-Specific Cre Mouse Models. Arteriosclerosis, thrombosis, and vascular biology. 2018;38:2550–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.