

Abstract
Outbreaks of cyclosporiasis, a food-borne illness caused by the coccidian parasite Cyclospora cayetanensis have increased in the USA in recent years, with approximately 2300 laboratory-confirmed cases reported in 2018. Genotyping tools are needed to inform epidemiological investigations, yet genotyping Cyclospora has proven challenging due to its sexual reproductive cycle which produces complex infections characterized by high genetic heterogeneity. We used targeted amplicon deep sequencing and a recently described ensemble-based distance statistic that accommodates heterogeneous (mixed) genotypes and specimens with partial genotyping data, to genotype and cluster 648 C. cayetanensis samples submitted to CDC in 2018. The performance of the ensemble was assessed by comparing ensemble-identified genetic clusters to analogous clusters identified independently based on common food exposures. Using these epidemiologic clusters as a gold standard, the ensemble facilitated genetic clustering with 93.8% sensitivity and 99.7% specificity. Hence, we anticipate that this procedure will greatly complement epidemiologic investigations of cyclosporiasis.
Key words: Cyclospora cayetanensis, clustering, cyclosporiasis, deep sequencing, distance-statistic, genotype, genotyping, machine learning, MLST
Introduction
Cyclosporiasis, a food-borne illness characterised by watery diarrhoea, nausea, abdominal cramps and weight loss, is caused by the coccidian parasite Cyclospora cayetanensis [1]. In the USA, 2299 laboratory-confirmed cases were reported in 2018 [2]. Prior to 2019, this was the largest number of annually reported US cases of cyclosporiasis since the disease became nationally notifiable in 1999, which has been attributed to the increasing use of sensitive molecular diagnostic methods. In 2018, approximately one-third of domestically acquired cases were epidemiologically linked to one of two large multi-state outbreaks associated with produce supplied by two commercial vendors, referred to henceforth as Vendor A (511 confirmed cases) and Vendor B (250 confirmed cases). Additional epidemiologically-defined clusters associated with basil (two clusters, 16 confirmed cases) and cilantro (three clusters, 53 confirmed cases) were identified in 2018 [2]. Despite the identification of multiple clusters including the two large 2018 outbreaks, most domestically acquired cyclosporiasis cases could not be linked to another cluster using available epidemiologic data.
In response to multiple large outbreaks that have occurred in the USA, dating back several years [3–5], the Parasitic Diseases Branch (PDB) at the Centers for Disease Control and Prevention (CDC) prioritised development of a genotyping tool to complement epidemiologic investigations of cyclosporiasis. The first C. cayetanensis genotyping tool required nested polymerase chain reaction (PCR) amplification and Sanger sequencing of five microsatellite repeats from stool DNA extracts [6]. A later appraisal of this method concluded that it had several characteristics that were not amenable to routine molecular surveillance including the low sequencing success rate of the markers, attributed to their repetitive nature [6, 7].
Due to these limitations [6, 7] draft C. cayetanensis genomes were screened for additional markers suitable for genotyping. These loci were PCR amplified from DNA extracts of feces containing C. cayetanensis and amplicons were Sanger sequenced [8–11]. Heterozygosity was encountered at most nuclear markers, evidenced by dual peaks in Sanger chromatograms [9, 11]. Some of these markers were selected based on genomic evidence that they were single-copy genes, suggesting that C. cayetanensis infections often comprise mixtures of genotypes [9, 11]. Nuclear loci sequenced from specimens associated with the same epidemiologic clusters often possessed similar mixtures of haplotypes though were typically not identical, which was attributed to the intrinsic sexual mode of reproduction employed by C. cayetanensis [9]. As a consequence of this heterogeneity, phylogenetic methods are inappropriate analytic approaches for these data.
Instead, statistics such as Jaccard distances or Bray–Curtis dissimilarity, sometimes used to assess similarity based on gene content [12, 13], represent potential analytic solutions and Bray–Curtis dissimilarity has been applied to C. cayetanensis previously [11]. However, these statistics do not consider several informative aspects of genetic data, such as loci entropy, allelic frequencies and nuclear vs. extranuclear inheritance. Furthermore, the ability to generate data for all markers in a MLST panel is subject to physical limitations, such as the volume of specimen submitted for genotyping, or the parasite load in a specimen. Statistics like Bray–Curtis dissimilarity and Jaccard distances treat missing data as being truly absent which is inappropriate given that alleles are present at loci with missing data, but were simply not sampled.
An ensemble method based on two similarity-based classification algorithms was developed to calculate a dissimilarity statistic using the haplotype composition of genotyped specimens while accounting for informative aspects of MLST data not considered by simpler statistics [9]. This ensemble-based statistic has appeared twice in the published literature to date, once during its first description [9] and a second time where it was applied to MLST datasets generated for the parasitic nematodes Strongyloides fuelleborni and Strongyloides stercoralis [14]. The first description of this ensemble was accompanied by a performance evaluation based on 88 specimens, sequenced at three MLST markers using Sanger technology, which cannot accurately represent all haplotypes in an amplicon if multiple haplotypes are present concomitantly. While this first assessment was encouraging, these limitations – related to data size and sequencing chemistry – meant that a rigorous performance evaluation was still required.
Taking advantage of the large scale of the 2018 US cyclosporiasis outbreaks, this study describes a rigorous performance assessment of this unique ensemble method. We increased the number of markers sequenced from three to eight by combining genotyping markers from multiple studies [9–11] and used targeted amplicon deep sequencing (TADS) to capture the complete haplotype diversity at these markers, amplified from C. cayetanensis DNA in fecal DNA extracts. This TADS approach was applied to hundreds of fecal specimens representing laboratory-confirmed cases of cyclosporiasis acquired in 2018, including specimens from 264 case-patients whose illnesses had been linked to cyclosporiasis outbreaks. This ensemble-based distance statistic was used to identify genetic clusters that were assessed for concordance with analogous epidemiologically-defined clusters. Using the two largest epidemiologic clusters from 2018 as the gold standard for defining clusters, we assessed the performance of the ensemble in terms of its sensitivity, specificity, precision, negative predictive value, and accuracy.
Materials and methods
Epidemiologic investigations
Specimens with epidemiological links (n = 264) were assigned to epidemiologically-defined outbreaks, linked with suspected food vehicles where possible, using methods previously described [2, 9, 10]. An outbreak was defined as at least two cases of cyclosporiasis epidemiologically-linked to a common source or exposure. A temporospatial cluster was defined as cases of cyclosporiasis that occurred in the same geographic area (e.g. same community or town) and had illness onset dates within approximately 15 days of each other. Epidemiologic evidence for linking cases in persons with common exposures (e.g. restaurant, grocery store and/or social events) was considered stronger than for temporospatial clusters.
Human fecal specimens
Fecal samples were received by the Diagnostic Reference Laboratory at CDC in 2018 either frozen without additives, in transport media (Cary-Blair or enteric plus), or in other preservatives compatible with DNA amplification (Total Fix, Zinc Polyvinyl Alcohol; Zinc-PVA, or low-viscosity PVA; LV PVA). These samples were laboratory confirmed as positive for C. cayetanensis infection by either brightfield microscopy of modified acid-fast (MAF) stained stool, UV epifluorescence microscopy, real-time PCR and/or the BioFire FilmArray Gastrointestinal (GI) Panel. Three state health agencies performed C. cayetanensis genotyping at their respective molecular laboratories and provided CDC with sequence data from the eight selected markers, ready for downstream analysis. These included the Texas (TX) Health Molecular diagnostic laboratory, the Parasitology Laboratory at the Wadsworth Center, New York (NY) and the Infectious Disease Laboratory at the Minnesota (MN) Department of Health. These laboratories optimised their DNA extraction, PCR and sequencing protocols at their respective facilities.
DNA extraction
At the CDC and TX laboratories, 2 ml of stool was transferred to a 2 ml tube and washed with Phosphate Buffered Saline (PBS) at pH 7.4 (Gibco, Life Technologies). DNA was extracted from ~0.5 ml aliquots of washed stool using the UNEX-based method [15]. The DNA was eluted in 80 μl of elution buffer and stored at 4 °C. The DNA extraction protocols employed at other participating laboratories differed subtly based on available resources and these differences were controlled for using proficiency specimens tested by each laboratory (see Supplementary File S1).
PCR and targeted amplicon deep sequencing
PCR primers (Table 1) were synthesized at CDC and sent to the TX laboratory, while MN and NY used primers synthesised by LGC Biosearch Technologies, Petaluma, CA and Integrated DNA technologies, Coralville, Iowa, respectively. Due to differences in available equipment at each laboratory (i.e. thermocyclers, centrifuges, etc.), the optimised PCR and TADS protocols differed slightly between laboratories. At the CDC, NY and TX laboratories, sequencing was attempted on all PCR products, irrespective of whether an amplicon was visible following agarose gel electrophoresis. At the MN laboratory, PCR products were sequenced after excision of correctly sized bands from agarose gels. Reactions were accompanied by appropriate positive and negative controls and amplicons were sequenced on the Illumina MiSeq platform. Complete details of these protocols are found in Supplementary File S1.
Table 1.
List of PCR primers used to amplify eight Cyclospora cayetanensis genotyping targets
| Marker # | Genome | Target alias | Primer name | Primer Sequence (5′-3′) | Target | Amplicon length (bp) | Reference |
|---|---|---|---|---|---|---|---|
| 1 | Nuclear | CDC-1 | GT1-F | CTCCTTGCTGCTCAGAACGA | ATP synthase | 175 | [11] |
| GT1-R | CAAGAGAGGAGCAGTGGCAA | ||||||
| 2 | Nuclear | CDC-2 | GT2-F | TGCAAACTACTAAGGGCGCA | U3 small nucleolar RNA-associated protein 11 | 246 | |
| GT2N-R | CGCCTTCTCTTGAGCCTTGA | ||||||
| 3 | Nuclear | CDC-3 | GT3-F | AATCGAATCGGTGCAGTGCTTA | uncharacterised | 220 | |
| GT3N-R | GACTGAACGTGTGAGAGGGG | ||||||
| 4 | Nuclear | CDC-4 | GT4-F | GTAGATGGGTCCTTGAAGGCT | ATP-dependent RNA helicase rrp3 | 179 | |
| GT4N-R | CAGACGCCTAAGGAACCGAA | ||||||
| 5 | Nuclear | HC378 | HC378F | CCCCTGCCTTGTTCTTGGTGAA | Sec14 family protein | 650 | [9] |
| HC378R | CCGGCGACACAGAGGTACC | ||||||
| 6 | Nuclear | HC360i2 | HC360i2F | CCCATTACGCCGCATAGAGT | uncharacterised | 469 | |
| HC360i2R | GCATTGCAAAGCCAGTCAGC | ||||||
| 7 | Mitochondrial | Mt-Junction | cyclo_mit-100F | TACCAAAGCATCCATCTACAGC | Mitochondrial junction repeat | 109 to 214 | [10] |
| cyclo_mit-54R | CCCAAGCAATCGGATCGTGTT | ||||||
| 8 | Mitochondrial | MSR | 15F | GGACATGCAGTAACCTTTCCG | Mitochondrial rRNA | 674 | [9] |
| 688R | AGGAAAGGTTAACCGCTGTCA |
Assignment of haplotypes
Illumina data were trimmed and quality-filtered using Geneious Prime (Build 2018-11-06 02:41) (www.geneious.com). Briefly, using BBDuk (v 37.64), reads were trimmed using a minimum Phred quality score of 20 and removal of adapter sequence was performed. Reads less than 50 bases were discarded and paired reads were merged using BBMerge (v 37.64). Haplotypes were identified using customized Geneious workflows that mapped remaining reads to a database of known C. cayetanensis haplotypes compiled at CDC from domestically and internationally (China, Indonesia and Guatemala) acquired infections over several years and validated by Sanger sequencing (Supplementary File S1, Appendix 1) [8–11, 16, 17].
As some markers differ significantly in terms of length, SNP composition and repeat composition, the Geneious workflows used varied between markers. Briefly, each marker-specific workflow used a map-to-reference strategy to generate a haplotype list for markers 1–8 and these marker-specific lists were combined to produce a complete list for each specimen. These lists comprise every haplotype detected in a specimen, including when two or more haplotypes were identified for one locus (i.e. mixed/heterozygous). For specific details on each workflow refer to Supplementary File S1. Each specimens' list was exported to a text file and these files were used to generate a haplotype data sheet (see Supplementary File S2, Tab A). This sheet is saved as a text file or csv file and used as the input for our ensemble procedure as described here: https://github.com/Joel-Barratt/Eukaryotyping.
Clustering and data visualisation
A pairwise matrix was generated from our haplotype data sheet using the R scripts and directions provided here: https://github.com/Joel-Barratt/Eukaryotyping. A value of 0.3072 was calculated for the variable epsilon (ε) which is required before running the scripts. Supplementary File S1 provides complete descriptions of the algorithms underpinning the ensemble (9), including complete equations, methods for calculating epsilon and the importance of selecting appropriate minimum MLST data requirements (also see [14]). The resulting matrix was clustered using hierarchical agglomerative nesting (AGNES), in the R package ‘cluster’. AGNES was performed using Wards clustering method [18]. The resulting hierarchical tree was visualised using the R package ‘ggtree’ [19] and the un-clustered matrix was visualised using MicrobeTrace (https://github.com/CDCgov/MicrobeTrace).
Ensemble performance assessment
Genotyping strategies are assessed on their discriminatory power, reproducibility and epidemiologic concordance [20]. Algorithms performing classification tasks are routinely assessed against a gold standard comprising a set of specimens falling into known classification categories. The rate at which the algorithm assigns specimens of a known classification to the correct category is then used to assess performance by calculating metrics such as sensitivity, specificity, precision and negative predictive value [21–23]. Assessment of our ensemble against these criteria first required identification of specimens belonging to known classification categories (i.e. a gold standard). In this study, specimens from case-patients whose illnesses were linked to specific outbreaks represented specimens of known categories (e.g. Vendor A, Vendor B, etc) and were used as our gold standard.
Next, a method for delineating genetic clusters from the hierarchical tree was required to assess whether specimens of a known category had been assigned to the appropriate genetic cluster, considering that the level at which a hierarchical tree is dissected impacts which specimens are interpreted as being related. For routine molecular surveillance of bacterial pathogens, cluster delineation typically involves selecting a cut-off based on the number of single nucleotide polymorphism (SNP) differences observed in the core genomes of related isolates [24, 25]. Cutoffs vary drastically amongst taxa and even serotypes of the same bacterial species may require different cutoffs [25–27]. Consequently, the selection of an appropriate SNP-cutoff is non-trivial and is usually based on a priori knowledge of the pathogen's genetic diversity.
This a priori knowledge is unavailable for C. cayetanensis and cutoffs based on SNP-differences are not applicable in the present context. Therefore, we devised a simple bootstrapping procedure to determine an appropriate way to divide the hierarchical tree. Firstly, the hierarchical tree was dissected empirically at different levels resulting in 5, 10 or 20 distinct clusters, each representing a distinct model. The most parsimonious of these three models was the one that: (1) minimised assigning specimens associated with different epidemiologic clusters to the same genetic cluster and (2) minimised separation of specimens from epidemiologically-linked case-patients across multiple genetic clusters. After selection of the most parsimonious model, the rate at which specimens of the same category were assigned to the same genetic cluster (e.g. sensitivity) could be calculated, along with other performance metrics.
Human ethics
Human fecal specimens were used in accordance with the protocol entitled ‘Application of genetic typing to Investigations of cases/clusters of cyclosporiasis’, Human Research Protection Office at the CDC Center for Global Health (Determination Number: 2018-123).
Results
Genotyping success and concordance with epidemiology
A total of 686 fecal specimens were received by the molecular diagnostic laboratory in the Parasitic Diseases Branch at CDC from 14 US State health departments between April and September of 2018. As two case-patients submitted two specimens each, these 686 specimens represented 684 case-patients. Genotyping was attempted on 118 specimens received by TX, 91 by NY and 32 by MN. Overall, genotyping was attempted on 927 fecal specimens. Of these 927 specimens, 869 (93.7%) had at least one marker successfully sequenced and 648 (70%) met the minimum data criteria for inclusion in the ensemble analysis (see Supplementary File S1). Of these 648 genotyped specimens, 264 were from case-patients whose illnesses were epidemiologically linked to large outbreaks or smaller clusters of cyclosporiasis, while the remaining 384 cases could not be confidently linked to an epidemiologic cluster using the available data.
The pairwise distance matrix was hierarchically clustered and visualised as a dendrogram (Fig. 1). Of the three models assessed (5, 10 and 20 clusters), the epidemiologic data best supported 10 clusters, revealing six relatively small clusters and four larger clusters. Specimens assigned to the four large clusters were associated with multiple outbreaks: one contained most specimens associated with Vendor A, a second contained most specimens associated with Vendor B, third contained specimens associated with three restaurant-associated clusters (restaurants A, B and F) and a fourth genetic cluster contained most specimens associated with temporospatial cluster A. Visualisation of pairwise distances using MicrobeTrace reaffirmed this observation, supporting the existence of four major clusters (Fig. 2).
Fig. 1.

Cluster dendrogram generated from the ensemble matrix of pairwise distances. The ensemble matrix was clustered using Wards clustering method to generate the dendrogram shown. A 10-cluster model was considered the most parsimonious and branches are colour-coded according to the clusters identified using this model. Peripheral bar colours indicate specimens from case-patients epidemiologically linked to outbreaks of cyclosporiasis identified in the USA in our study, where at least one specimen was genotyped; colours of these bars indicate identified epidemiologic linkages per the legend. To determine the specific location of a given specimen in this dendrogram refer to Supplementary File S1, Appendix 2, which is a searchable pdf of the same dendrogram that includes all specimen names. The number of specimens assigned to each of the 10 genetic clusters is as follows: genetic cluster 1 (34 cases), cluster 2 (92 cases), cluster 3 (93 cases), cluster 4 (144 cases), cluster 5 (10 cases), cluster 6 (40 cases), cluster 7 (150 cases), cluster 8 (35 cases), cluster 9 (28 cases), cluster 10 (40 cases).
Fig. 2.

Ensemble pairwise distance matrix visualised using MicrobeTrace. To generate this network the same ensemble matrix used to construct Figure 1 (Supplementary File S2, Tab E) was filtered to a value of 0.15 using MicrobeTrace (https://github.com/CDCgov/MicrobeTrace/wiki). Nodes were colour-coded according to their epidemiological linkage, using the same colours used to denote epidemiologically-defined clusters in Figure 1.
Algorithm performance
Taking advantage of the large number of genotyped specimens associated with Vendor A (n = 116) and Vendor B (n = 126), we established that a true positive result required the presence of an epidemiologic link to Vendor A or B only, where specimens from case-patients associated with these outbreaks were assigned to genetic clusters 7 and 4, respectively. Specimens associated with other epidemiologic clusters were not considered when defining true positive results because the small number of genotyped fecal specimens associated with these clusters (Table 2) could drastically bias **this performance assessment. However, specimens associated with epidemiologic clusters other than Vendor A and B were used to define true negative results (see Table 3).
Table 2.
Concordance of epidemiologic information and genetic clustering.
| Epi-cluster (genetic cluster) | Food item or suspected food vehicle | Number of genetically linked specimens for epi-cluster | Number of case-patients in epi-cluster | Concordance |
|---|---|---|---|---|
| Vendor A (cluster 7)α | Salad | 106 | 116 | 91% |
| Vendor B (cluster 4)β | Vegetables | 121 | 126 | 96% |
| Restaurant A (cluster 2)* | Herb 1 | 3 | 3 | 100% |
| Restaurant B (cluster 2)* | Herb 1 | 3 | 3 | 100% |
| Restaurant C (cluster 6) | Herb 2 | 2 | 2 | 100% |
| Restaurant D (cluster 3) | Unknown | 1 | 1 | NA |
| Restaurant E (cluster 1) | Herb 1 | 1 | 1 | NA |
| Restaurant F (cluster 2) | Unknown | 1 | 1 | NA |
| Temporospatial cluster A (cluster 3) | Unknown | 8 | 10 | 80% |
| Cruise associated cluster (cluster 10) | Unknown | 1 | 1 | NA |
| TOTALS | NA | 247 | 264 | Overall Concordance: 94% |
Note: Epidemiologically-defined clusters with genotyping data available for one case are shaded grey. These were not included in the concordance calculations. Specimens with unknown epidemiologic linkage = 384.
α Of the 384 specimens with unknown epidemiologic linkage, 44 were assigned to genetic cluster 7.
β Of the 384 specimens with unknown epidemiologic linkage, 22 were assigned to genetic cluster 4.
* These restaurants shared the same supplier of herb 1.
Table 3.
Assessment of the ensemble performance against an epidemiologic gold standard
| Metric | Result | Definition |
|---|---|---|
| True Positives (TP) | 227 | Specimens associated with Vendor A and correctly assigned to genetic cluster 7 and those associated with Vendor B and assigned to genetic cluster 4. |
| True Negatives (TN) | 321 | Specimens not associated with Vendor A (i.e. associated with any other epidemiologically defined cluster) and not placed in genetic cluster 7 and those not associated with Vendor B and not assigned to genetic cluster 4. |
| False Positives (FP) | 1 | Specimens not associated with Vendor A but incorrectly assigned to genetic cluster 7 and specimens not associated with Vendor B but incorrectly assigned to genetic cluster 4. |
| False Negatives (FN) | 15 | Specimens associated with Vendor A but not assigned to genetic cluster 7 and those associated with Vendor B but not assigned to genetic cluster 4. |
| Sensitivity | 93.8% | 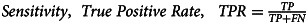 |
| Specificity | 99.7% | 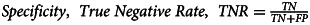 |
| Precision/Positive Predictive Value | 99.6% | 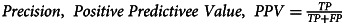 |
| Negative Predictive Value | 95.5% | 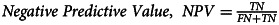 |
| Accuracy | 97.2% | 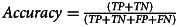 |
Note: Cyclosporiasis case-patients with unknown epidemiologic linkage were not included in these calculations. Supplementary File S2, Tab B shows which specimens were considered TPs, TNs, FPs and FNs based on the definitions in this table.
Using the 10-cluster model, 121 of the 126 specimens associated with Vendor B were assigned to genetic cluster 4. For Vendor A, 106 of the 116 associated specimens were assigned to genetic cluster 7 (Table 2). In accordance with the definitions in Table 3, the sensitivity, specificity and precision of the ensemble were 93.8%, 99.7% and 99.6%, respectively. Its negative predictive value is 95.5% and its accuracy is 97.2% (Table 3). Construction of an epidemiologic curve for each genetic cluster confirmed temporal clustering of specimens assigned to genetic cluster 4, with the majority of illness onset dates occurring between 26 May and 7 June (Fig. 3). For specimens assigned to genetic cluster 7, temporal clustering of cases was observed between 24 June and 6 July (Fig. 3).
Fig. 3.

Epidemiologic curve for cyclosporiasis cases (cases over time) plotted for each genetic cluster. Onset of illness dates for cases of cyclosporiasis is plotted as a separate histogram for each genetic cluster. Temporal clustering of specimens from cluster 4 and cluster 7 is apparent. Some temporal clustering seems apparent for cluster 2, which may possess a bimodal distribution. Colours used to denote each genetic cluster here corresponds to those used to denote genetic clusters in Figure 1.
Discussion
The ensemble-based distance statistic facilitated genetic clustering of specimens at 94% concordance with analogous epidemiologic clusters and possesses analytical sensitivity, specificity, precision, accuracy, and negative predictive values of at least 93.8%, 99.7%, 99.6%, 97.2% and 95.5%, respectively (Table 3). The utility of our TADS-MLST genotyping methodology is also supported, as it resolved the C. cayetanensis population examined here into 10 distinct genetic clusters. In spite of an earlier report describing C. cayetanensis as possessing a clonal population structure [6], these data unequivocally indicate that this is not the case. Genotypes observed among specimens from epidemiologically linked case-patients were usually very similar but often not identical (Supplementary File S2), which may be attributable to the sexual reproductive cycle or gametogony of C. cayetanensis. This sexual cycle is ongoing throughout infection and likely drives the diversity observed, even among cases with strong epidemiologic links. Indeed, Cinar and colleagues [28] expressed concerns surrounding the use of nuclear markers for genotyping C. cayetanensis, stating that while these loci might possess greater discriminatory power, they ‘may have limited use in typing due to recombination events during meiosis that could introduce substantial genetic variations within related populations’. The results of the present study show that these concerns no longer have any real basis, as our unique analysis procedure utilises this nuclear diversity to provide excellent resolving power while achieving high epidemiologic concordance. In fact, our data support that the addition of more high-entropy nuclear markers to our current MLST panel would provide improved resolution of genetic clusters.
Whole-genome sequencing is impracticable for routine molecular surveillance of C. cayetanensis outbreaks due to the current inability to culture it, difficulties in obtaining a sufficient mass of parasite DNA from feces, the absence of an animal model and its 44 megabase genome [9]. Instead, MLST protocols based on TADS are more appropriate. However, generating data for all markers in a MLST panel is often subject to physical limitations, such as the volume of specimen submitted or the parasite load in a specimen, which can vary drastically. This variation is compounded by differences in amplification efficiency for different markers. In this study, sequencing success varied from 53.2% to 97.9% depending on the marker, so the occurrence of missing data was common. Only 34.4% of specimens retained in this analysis had data for all markers and 13.1% of retained specimens were genotyped at only four markers (Supplementary File S1). Despite this, the ensemble performed very well. This also gives credence to a recent report where the same ensemble-based distance statistic was applied to MLST datasets generated from the human-infecting nematodes Strongyloides stercoralis and Strongyloides fuelleborni, that also suffered from an abundance of missing data [14]. In that report, the ensemble method was used to identify novel trends among populations of these worms. Importantly, that study also confirms that the ensemble approach is applicable beyond Cyclospora and will likely perform robustly when applied to MLST datasets generated for a range of other eukaryotes [14].
While genetic clustering of specimens was 94% concordant with analogous epidemiologic clustering, in some cases the genotype and/or the illness onset dates of case-patients associated with these discordantly clustered specimens were inconsistent with their epidemiologic linkage, meaning that the epidemiologic information may have been the source of a discordant result. This could be due to case-patients having multiple exposures or error introduced during recall. Examining the genotypes of discordantly clustered specimens (n = 15) provided additional insights into the cluster assignments made. For 11 of the 15 false-negative results (Table 3), the genotypes strongly suggested that the ensemble had actually made an appropriate cluster assignment (discussed in great detail in Supplementary File S1). Therefore, the performance metrics calculated here probably represent a conservative lower boundary for the ensembles' true performance.
A weakness of this study was the inability to consider many epidemiologic clusters in the same fashion as the Vendor A and B outbreaks when calculating performance metrics, due to the small number of specimens collected from case-patients associated with these clusters. Four epidemiologic clusters had only one associated specimen genotyped, making it impossible to assess if they clustered correctly (Table 2). One small cluster was identified via temporospatial links which are not as strong as links traced back to a specific event or restaurant. Specimens associated with three separate epidemiologic clusters (n = 7) were each assigned to genetic cluster 2 (restaurants A, B and F). For restaurants A and B, herb 1 was the suspected vehicle of infection and it was retrospectively confirmed that these two restaurants shared the same supplier of herb 1. Given the genetic relatedness of specimens associated with the A, B and F clusters, herb 1 obtained from the same supplier could have been the vehicle associated with cluster F as well, but this was not confirmed. Consequently, it is unknown whether the assignment of the F-associated specimen to genetic cluster 2 represents a correct assignment. However, outbreak clusters represented by a small number of genotyped specimens could be used as ‘true negatives’ for the Vendor A and B outbreaks because we knew with high certainty that these were not associated with Vendors A or B. In any case, a reasonable performance assessment executed in the fashion described here should involve at least 10–20 (as an empirically-defined minimum) genotyped specimens from case-patients strongly linked to each of multiple outbreaks, genotyped alongside a large and diverse reference population of ideally 100 specimens or more. However, an assessment based on outbreaks of a similar scale to those observed for Vendors A and B (i.e. more than 100 cases) will obviously provide a stronger indication of performance.
Identifying outbreaks of cyclosporiasis in a timely manner, allowing implementation of strategies that reduce their impact, is a priority for US public health agencies. To be considered useful, molecular surveillance tools must be robust so they can be deployed to various US public health laboratories in a concerted control effort while still producing consistent results. When used in combination with our novel TADS methodology, the ensemble-based distance statistic evaluated here will likely strengthen our ability to detect common sources of C. cayetanensis exposure, increasing the likelihood of detecting food vehicles of cyclosporiasis. These methods also improve our ability to estimate the scope of outbreaks by providing putative genetic links between cases where no epidemiologic information was obtained. Finally, while this ensemble-based statistic was assessed in the context of a C. cayetanensis MLST dataset, we also emphasise that its design does not preclude its application to other eukaryotic pathogens.
Acknowledgements
The authors would like to acknowledge the assistance of the following people for their contribution to this study, either by providing post-diagnostic fecal specimens and/or providing additional epidemiologic meta-data that assisted in the generation of this manuscript: Jacob Garfin, Dave Boxrud, Youngmi Kim, Katherine Hebbeln, Stephen Hendren, Nicholas Desuno, Lori Saathoff-Huber, Selam Tecle and Jeffrey Higa.
Conflict of interest
The authors have no conflicts of interest to disclose.
Author contribution
FSN and JLNB wrote manuscript drafts. KH, FSN, BC, EC, SMA, CS, JZ, RT, AR and JH performed laboratory work, including specimen organisation, DNA extraction, PCR, library preparation and assay optimisation. MP and JLNB designed and implemented the ensemble. JK prepared, ran, and maintained the MiSeq for this study. SC, CB and TR performed epidemiologic investigations. JLNB designed bioinformatic workflows, data analysis procedures, designed figures, designed bioinformatic experiments and performed assessment of the ensemble. RSB, ET and JLNB wrote grant for funding source. MJA, ET, RSB and YQ approved grant and led project. JLNB prepared final manuscript draft. All authors edited and approved the final manuscript draft.
Financial support
This research was supported by a grant from the Centers for Disease Control and Prevention Office of Advanced Molecular Detection.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry.
Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0950268820001697.
click here to view supplementary material
Data availability statement
Raw reads have been made publicly available by submission to NCBI under BioProject accession number PRJNA578931. The R scripts required to run the classifier can be accessed here: https://github.com/Joel-Barratt/Eukaryotyping
References
- 1.Ortega YR et al. (1993) Cyclospora species--a new protozoan pathogen of humans. The New England Journal of Medicine 328, 1308–1312. [DOI] [PubMed] [Google Scholar]
- 2.Casillas SM, Bennett C and Straily A. (2018) Notes from the field: multiple cyclosporiasis outbreaks - United States, 2018. MMWR Morbidity and Mortality Weekly Report 67, 1101–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hedberg CW and Osterholm MT. (2016) Foodborne outbreaks caused by Cyclospora: the message is more important than the messenger. Epidemiology and Infection 144, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abanyie F et al. (2015) 2013 Multistate outbreaks of Cyclospora cayetanensis infections associated with fresh produce: focus on the Texas investigations. Epidemiology and Infection 143, 3451–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herwaldt BL. (2000) Cyclospora cayetanensis: a review, focusing on the outbreaks of cyclosporiasis in the 1990s. Clinical Infectious Diseases 31, 1040–1057. [DOI] [PubMed] [Google Scholar]
- 6.Guo Y et al. (2016) Multilocus sequence typing tool for Cyclospora cayetanensis. Emerging Infectious Diseases 22, 1464–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofstetter JN et al. (2019) Evaluation of multilocus sequence typing of Cyclospora cayetanensis based on microsatellite markers. Parasite 26, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qvarnstrom Y et al. (2015) Draft genome sequences from Cyclospora cayetanensis oocysts purified from a human stool sample. Genome Announcements 3, e01324–e01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barratt JLN et al. (2019) Genotyping genetically heterogeneous Cyclospora cayetanensis infections to complement epidemiological case linkage. Parasitology 146, 1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nascimento FS et al. (2019) Mitochondrial junction region as genotyping marker for Cyclospora cayetanensis. Emerging Infectious Diseases 25, 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houghton KA et al. (2020) Development of a workflow for identification of nuclear genotyping markers for Cyclospora cayetanensis. Parasite 27, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kupczok A et al. (2018) Rates of mutation and recombination in Siphoviridae Phage genome evolution over three decades. Molecular Biology and Evolution 35, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rihova J et al. (2017) Legionella becoming a mutualist: adaptive processes shaping the genome of symbiont in the louse Polyplax serrata. Genome Biology and Evolution 9, 2946–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrratt JLN and Sapp SGH. (2020) Machine learning-based analyses support the existence of species complexes for Strongyloides fuelleborni and Strongyloides stercoralis. Parasitology 1–12. doi: 10.1017/S0031182020000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qvarnstrom Y et al. (2018) Molecular detection of Cyclospora cayetanensis in human stool specimens using UNEX-based DNA extraction and real-time PCR. Parasitology 145, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cinar HN et al. (2015) The complete mitochondrial genome of the foodborne parasitic pathogen Cyclospora cayetanensis. PLoS One 10, e0128645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogedengbe ME et al. (2015) A linear mitochondrial genome of Cyclospora cayetanensis (Eimeriidae, Eucoccidiorida, Coccidiasina, Apicomplexa) suggests the ancestral start position within mitochondrial genomes of eimeriid coccidia. International Journal for Parasitology 45, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strauss T and von Maltitz MJ. (2017) Generalising ward's method for Use with Manhattan distances. PLoS One 12, e0168288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G et al. (2016) Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 8, 28–36. [Google Scholar]
- 20.van Belkum A et al. (2007) Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clinical Microbiology and Infection 13 (Suppl. 3), 1–46. [DOI] [PubMed] [Google Scholar]
- 21.Goodswen SJ, Kennedy PJ and Ellis JT. (2013) A novel strategy for classifying the output from an in silico vaccine discovery pipeline for eukaryotic pathogens using machine learning algorithms. BMC Bioinformatics 14, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajaraman S, Jaeger S and Antani SK. (2019) Performance evaluation of deep neural ensembles toward malaria parasite detection in thin-blood smear images. PeerJ 7, e6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poostchi M et al. (2018) Image analysis and machine learning for detecting malaria. Translational Research 194, 36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meehan CJ et al. (2018) The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine 37, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker TM et al. (2013) Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. The Lancet Infectious Diseases 13, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payne M et al. (2019) Enhancing genomics-based outbreak detection of endemic Salmonella enterica serovar Typhimurium using dynamic thresholds. Microbial Genomics, 1–9. doi: 10.1099/mgen.0.000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coipan CE et al. (2020) Concordance of SNP- and allele-based typing workflows in the context of a large-scale international Salmonella Enteritidis outbreak investigation. Microbial Genomics 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cinar HN et al. (2020) Molecular typing of Cyclospora cayetanensis in produce and clinical samples using targeted enrichment of complete mitochondrial genomes and next-generation sequencing. Parasites & Vectors 13, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0950268820001697.
click here to view supplementary material
Data Availability Statement
Raw reads have been made publicly available by submission to NCBI under BioProject accession number PRJNA578931. The R scripts required to run the classifier can be accessed here: https://github.com/Joel-Barratt/Eukaryotyping