

This study reveals the contribution of gut bacteria to Alzheimer’s disease.
Abstract
The gut-brain axis is bidirectional, and gut microbiota influence brain disorders including Alzheimer’s disease (AD). CCAAT/enhancer binding protein β/asparagine endopeptidase (C/EBPβ/AEP) signaling spatiotemporally mediates AD pathologies in the brain via cleaving both β-amyloid precursor protein and Tau. We show that gut dysbiosis occurs in 5xFAD mice, and is associated with escalation of the C/EBPβ/AEP pathway in the gut with age. Unlike that of aged wild-type mice, the microbiota of aged 3xTg mice accelerate AD pathology in young 3xTg mice, accompanied by active C/EBPβ/AEP signaling in the brain. Antibiotic treatment diminishes this signaling and attenuates amyloidogenic processes in 5xFAD, improving cognitive functions. The prebiotic R13 inhibits this pathway and suppresses amyloid aggregates in the gut. R13-induced Lactobacillus salivarius antagonizes the C/EBPβ/AEP axis, mitigating gut leakage and oxidative stress. Our findings support the hypothesis that C/EBPβ/AEP signaling is activated by gut dysbiosis, implicated in AD pathologies in the gut.
INTRODUCTION
Alzheimer’s disease (AD) is the most common progressive neurodegenerative disease with aging as the major risk factor. The pathological hallmarks include extracellular senile plaques that are mainly composed of aggregated polypeptide β-Amyloid 42 (Aβ42) and intraneuronal neurofibrillary tangles (NFTs), principally consisting of hyperphosphorylated Tau. Although intensive efforts have been made in this field, the key molecular mechanism dictating AD etiopathogenesis still remains unclear. The interaction between genetic and environmental factors contributes to AD pathogenesis (1). Gut microbiota regulate host brain functions and behavior via the microbiota-gut-brain axis. GF (germ-free) animals, antibiotics interference, probiotics intervention, pathogen infection, and dietary habits not only affect the composition of gut microbiota and physiological function of the gut but also impinge on host cognitive behavior and alter the risk of AD (2). Emerging evidence suggests that gut microbiota may play some role in mediating AD pathology. For instance 16S ribosomal RNA (16S rRNA) sequences from fecal samples reveal that the bacterial gut microbiome of AD patients differs taxonomically from that of controls (3). The gut microbiota of patients with AD show reduced diversity and a distinct taxonomic composition, with decreased Firmicutes, increased Bacteroidetes, and decreased Bifidobacterium compared to the microbiota of healthy controls (4). Most recently, Haran et al. (5) demonstrated that the microbiome of AD patients is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. The microbiome of aged AD patients shows a lower proportion and prevalence of bacteria with the potential to synthesize butyrate, as well as higher abundances of taxa that are known to cause proinflammatory conditions.
Many diseases have been correlated with variations in numbers and composition of gut microbiota, including obesity, diabetes, hypertension, autism, depression, and Parkinson’s disease (PD) (6, 7). Irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) are two common gastrointestinal (GI) diseases. IBS is a functional GI disease related to changes in the microbiota-gut-brain axis, usually manifested in the case of stress or enteric infection. IBD is characteristic of increased intestinal permeability, disrupted gut microbiota, and inflammation. Patients with IBS and IBD show a decline in cognitive ability, suggesting the potential relationship between gut and cognitive functions (8). A consistent association of brain amyloidosis with proinflammatory gut bacteria of cognitively impaired patients has been reported (9). Furthermore, antibiotic-mediated perturbation in the gut microbiome modulates amyloid deposition in an AD mouse model (10). The microbiota diversity of APP/PS1 mice (containing mutations in both APP 670/671NL (Swedish) and PSEN L166P) decreases with increased age. The association of bacterial taxa with cerebral Aβ pathology observed in conventionally raised APP/PS1 mice indicates that specific microbes may be involved in the progression of cerebral Aβ amyloidosis (11).
Mammalian asparagine endopeptidase (AEP; also called legumain; gene name, LGMN) is a cysteine protease that specifically cleaves the substrates after asparagine (N) in a pH-dependent manner. AEP usually resides in the endolysosomes, and it is activated under acidosis. AEP is activated in human AD brains (12). It simultaneously cleaves both APP and Tau, promoting amyloid and NFT pathologies in AD mouse models. In addition, it cleaves both APP N585 and Tau N368 in human AD brains. Knockout of AEP from 5xFAD or Tau P301S mice mitigates senile plaques or NFT pathologies, restoring the cognitive functions (13, 14). Notably, Tau N368 is demonstrable in the cerebrospinal fluid of patient with AD, correlating with Tau positron emission tomography signals (15). Recently, we reported that LGMN mRNA transcription is predominantly regulated by CCAAT/enhancer binding protein β (C/EBPβ) in the brain in an age-dependent way (16). In addition to promoting the production of inflammatory mediators, C/EBP family members are themselves induced by the classical proinflammatory triad of interleukin-1β (IL-1β), IL-6, and tumor necrosis factor–α (TNF-α) (17, 18), all of which are substantially increased in pathologically affected regions of AD brain (19). Hence, there is a feedback loop between amyloid and neuroinflammation through activating C/EBPβ in microglia or astrocytes. Most recently, we reported that the C/EBPβ/AEP axis is spatiotemporally activated in 3xTg AD mouse brain, mediating AD pathologies (20).
In the current report, we provide extensive evidence demonstrating that gut dysbiosis occurs in 5xFAD mice in an age-dependent manner, associated with temporal activation of C/EBPβ/AEP pathway and amyloid pathology in the gut. Microbiota from the aged 3xTg but not aged wild-type (WT) mice accelerates AD pathology in young 3xTg mice, accompanied with active C/EBPβ/AEP signaling in the brain. Chronic antibiotic treatment represses this pathway, alleviates AD pathology, and rescues cognitive dysfunctions in 5xFAD. R13, a small molecular prodrug of 7,8-dihydroxyflavone (7,8-DHF) that is a tropomyosin receptor kinase B (TrkB) agonist, acts as a prebiotic, blocks the C/EBPβ/AEP pathway in the gut, and reduces amyloidogenesis. R13-enriched probiotic decreases gut leakage and oxidative stress in 5xFAD mice.
RESULTS
Gut dysbiosis occurs in 5xFAD mice in an age-dependent manner
Previous studies have indicated an association between alterations in the intestinal microbial community and the development of AD pathologies in murine models. However, there is a scarcity of mechanistic knowledge on how dysbiotic gut microbiota affect amyloid pathology in the gut. Therefore, we first characterized the gut microbial community composition in the AD mouse model in an age-dependent manner, where we used a murine AD model, specifically 5xFAD mice and their WT littermates. For this purpose, the total DNA was purified from mouse fecal samples, which were collected from 3- or 6-month-old 5xFAD mice and their WT littermates. Bacterial 16S rRNA genes (V4 region) were polymerase chain reaction (PCR)–amplified, and the amplicons were sequenced using the Illumina MiSeq high-throughput sequencing (HTS) platform. The microbiome analysis showed a remarkable alteration of the gut microbiota composition in 5xFAD mice, when we compared the microbial community structure with the 6-month-old WT mice. On the other hand, at 3 months of age, the microbial community structures remained mostly similar in both 5xFAD and WT mice (Fig. 1A). At the phyla level, microbiota analysis revealed that the gut microbiota of both 3-month-old 5xFAD and WT mice consisted of a lower abundance of Firmicutes and a higher abundance of Bacteroidetes. However, microbiome analysis demonstrated a marked temporal shift in the taxonomic distribution of the gut microbiota at the age of 6 months in the 5xFAD and WT littermate mice, as evident from the dynamically altered abundance of the two dominant phyla Firmicutes and Bacteroidetes (Fig. 1A). The microbiota community in the 6-month-old 5xFAD mice is characterized by the dynamic increase in the abundance of proinflammatory phyla, including Proteobacteria, Bacteroidetes, and Deferribacteres (fig. S1A). At the genus level, 6-month-old 5xFAD mice also demonstrated the increase in the numbers of Helicobacter, Prevotella, and Sutterella compared with WT littermate mice (fig. S1B). In addition, the genus Helicobacter, which is a member of the phylum Proteobacteria, particularly showed more than a10-fold increase in the abundance in the stool of the 6-month-old 5xFAD mice in comparison with WT littermates (fig. S1C). Our analysis further indicated that Helicobacter bilis was significantly increased in the relative abundance in 6-month-old versus 3-month-old 5xFAD mice (fig. S1D). Furthermore, the dynamic alteration in the microbial community composition was also evident from the β-diversity analysis (Fig. 1B), which demonstrated a marked temporal shift in the taxonomic distribution of the gut microbiota at the age of 6 months in the 5xFAD and WT mice. Moreover, the phylogenetic richness (α-diversity) at the operational taxonomic unit (OTU) level significantly decreased in the 6-month-old 5xFAD mouse intestine (Fig. 1, C to E). Hence, these findings indicate age-dependent dynamic alterations in the gut microbiota of 5xFAD mice, which are characterized by a diminished microbial diversity, followed by a marked depletion of anti-inflammatory Firmicutes and temporal enrichment of proinflammatory Proteobacteria.
Fig. 1. Microbiome analysis in 5xFAD mouse stool reveals an alteration in the microbial community.
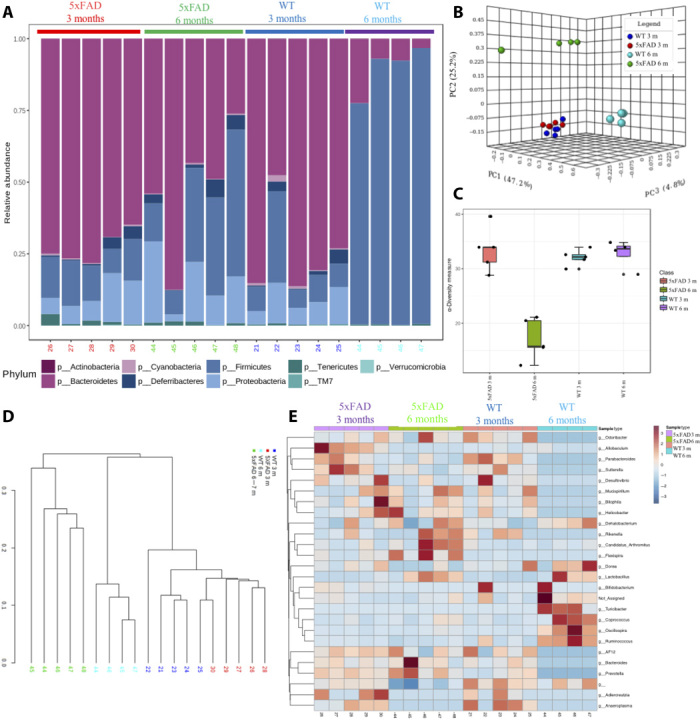
(A) Relative abundance of bacterial phyla determined by HTS analysis (n = 5 per group). (B) Principal coordinate plot (PCoA) (Bray-Curtis index) of microbial community structure in an age-dependent manner in 5xFAD and WT mouse stool. m, months. (C) Boxplot of α-diversity pattern (Chao1) of the microbiota across different age groups in the 5xFAD and WT mouse stool. (D and E) Hierarchical clustering of the core microbial taxa shows alterations of abundances at the genus level across different age groups of 5xFAD and WT mouse. (D) A dendrogram showing clustering of samples across age and genetic backgrounds. (E) Heatmap analysis using taxonomic abundances. Hierarchical clustering was performed using a Spearman’s correlation–based dissimilarity matrices and agglomeration method.
Gut microbiota modulate C/EBPβ/AEP pathway in the brain, regulating cognitive functions in 3xTg AD mice
To further assess the contribution of gut microbiome to AD pathologies in the brain, we cohoused 6-week-old 3xTg mice with either aged 3xTg mice (12 months old) or aged WT mice (12 months old) for 5 months. The microbiome analysis showed an alteration of the gut microbiota components in young 3xTg mice cohoused with aged 3xTg mice, as compared with those cohoused with aged WT mice. At 6 months of age, the microbial community structures displayed distinct differences between two groups of young recipient mice. Moreover, some similarities in microbial community structures were observed between the recipient 3xTg mice and their donor mice, respectively (Fig. 2, A and B). Notably, fluorescein isothiocyanate (FITC)–dextran assay indicated that receiving gut microbiome from aged 3xTg mice enhanced the gut leakage in young 3xTg mice (Fig. 2C). Immunoblotting revealed that introducing the gut microbiome from aged 3xTg mice increased both p-C/EBPβ and C/EBPβ levels in young 3xTg mice brains, resulting in higher activation of AEP. Subsequently, the downstream APP and Tau were pronouncedly augmented, leading to evident escalation of both APP N373/N585 and Tau N368 fragments. Notably, AT8 signals [phospho-Tau (Ser 202, Thr205)] were increased in young 3xTg mice cohoused with aged 3xTg, as compared to aged WT mice or to the control group (Fig. 2D). AEP enzymatic assays validated that gut microbiome from aged 3xTg mice triggered AEP activation in the brains of young 3xTg, while gut microbiome from healthy aged mice blocked the protease activity (Fig. 2E). Moreover, electron microscopy (EM) analysis revealed that the synapses were strongly reduced in young 3xTg mice brains upon receiving gut microbiome from aged 3xTg mice (Fig. 2, F and G). Morris water maze (MWM) behavioral tests revealed significant cognitive defects in young 3xTg mice receiving the gut microbiome from aged 3xTg mice, compared to microbiome transplants from aged WT mice (Fig. 2, H to K). Therefore, gut microbiome alteration in young 3xTg mice due to cohousing with aged 3xTg mice activates C/EBPβ/AEP signaling in the brains of young recipient mice, accelerating the onset of cognitive dysfunction.
Fig. 2. Gut microbiomes from aged 3xTg mice activate C/EBPβ/AEP in young 3xTg mice brain, facilitating cognitive dysfunction.

(A) Relative abundance of bacterial phyla determined by HTS analysis (n = 5 per group). (B) PCoA of microbiota community structure in young 3xTg mice cohoused with either aged 3xTg mice or aged WT mice, aged 3xTg mice, and aged WT mice. (C) GI permeability barrier defect, as determined by FITC-dextran translocation in 3xTg mice. 3xTg mice cohoused with aged 3xTg mice developed much severe leaky gut than control 3xTg mice (n = 3 per group). Data represent the means ± SEM; *P < 0.05 compared with control, one-way analysis of variance (ANOVA). (D) Immunoblot showing p-C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and processing in the mouse brains. (E) AEP activity assay in the brain lysates (n = 3 per group). Data represent the means ± SEM; **P < 0.01 and ***P < 0.001 compared with control, one-way ANOVA. RFU, relative fluorescence units. (F) Representative EM of the synaptic structures. Red circles indicate the synapses. Scale bars, 1 μm. (G) Quantitative analysis of the synaptic densities in 3xTg mice brains. 3xTg mice cohoused with aged 3xTg mice showed decreased synaptic densities. (n = 5 per group). Data are shown as means ± SEM; **P < 0.01. (H to K) MWM cognitive assays. 3xTg control mice, 3xTg mice cohoused with aged 3xTg mice, or aged WT mice were trained in the water maze more than 5 days. Shown are the means ± SEM latency to platform and the area under curve of latency (AUC latency) (H), means ± SEM swim path distance and the area under curve of distance (AUC distance) (I), the percentage of time spent in the target quadrant in the probe trail (J), and swim speed (K); *P < 0.05, **P < 0.01 compared to control 3xTg mice.
C/EBPβ/AEP pathway is activated in the gut of 5xFAD mice in an age-dependent way
The C/EBPβ/AEP axis is activated in an age-dependent manner in different brain regions of the 3xTg AD mouse model, elevating AEP-truncated APP and Tau fragments and promoting senile plaques and NFT formation in the brain, associated with gradual neuronal loss and chronic neuroinflammation (20). To examine whether this pathway is also activated in the gut, as implicated in AD pathologies, we conducted immunofluorescent (IF) costaining on the colon sections from different ages of 5xFAD and their WT littermates. p-C/EBPβ/microtubule-associated protein 2 (MAP2) costaining indicated that C/EBPβ phosphorylation in MAP2-positive ENS (enteric nerve system) neurons was slightly escalated in the WT gut in an age-dependent manner. Notably, MAP2 neuronal signals were reduced, suggesting enteric neuronal loss during aging. Notably, p-C/EBPβ was highly augmented in 5xFAD mice, while MAP2-positive neurons were evidently declined in the gut (Fig. 3A). To validate the specificity of p-C/EBPβ staining signals, we incubated the brain and colon slides from 6-month-old 5xFAD mice with control or anti–p-C/EBPβ, a well-characterized specific antibody for active C/EBPβ; we only observed the fluorescent signals from the samples incubated with p-C/EBPβ antibody (fig. S2A), indicating that the positive signals are from active p-C/EBPβ cells. Consistent with these findings, the downstream target AEP was strongly escalated during aging, as were the total levels of C/EBPβ in both WT and 5xFAD gut with the latter stronger than the former (Fig. 3B). These findings suggest that extensive inflammation occurs in the gut during aging with 5xFAD mice more strongly than with WT mice. Consequently, Aβ abundance was highly elevated in 5xFAD mice during aging, as was AEP-truncated Tau N368. Although Aβ signals were demonstrable in WT gut tissues, Tau N368 was barely detectable (Fig. 3C). On the other hand, AEP-truncated APP C586 was also temporally increased in 5xFAD gut tissues, accompanied by escalating p-Tau AT8 activities. By contrast, APP C586 signals were much weaker in WT gut, and AT8 was not demonstrable (Fig. 3D).
Fig. 3. C/EBPβ/AEP pathway is escalated in an age-dependent way in 5xFAD mice colon.

(A) IF staining of p-C/EBPβ and MAP2 in colon of 5xFAD mice. Quantitative analysis of p-C/EBPβ–positive cells and MAP2-positive cells. Data represent the means ± SEM; representative data of five samples; ****P < 0.0001. (B) IF staining of C/EBPβ and AEP in the colon of 5xFAD mice. Quantitative analysis of C/EBPβ-positive cells and AEP-positive cells. Data represent the means ± SEM; representative data of five samples; *P < 0.05, ***P < 0.001, and ****P < 0.0001. (C) IF staining of Aβ and cleaved Tau N368 in colon of 5xFAD mice. Quantitative analysis of Aβ-positive cells and cleaved Tau N368–positive cells. Data represent the means ± SEM; representative data of five samples; *P < 0.05 and ****P < 0.0001. (D) IF staining of AT8 and cleaved APP C586 in the colon of 5xFAD mice. Quantitative analysis of AT8-positive cells and cleaved APP C586–positive cells. Data represent the means ± SEM; representative data of five samples. ****P < 0.0001. Scale bars, 20 μm (A to D).
The neurotrophic factors BDNF (brain-derived neurotrophic factor) and netrin-1 both promote neuronal survival and regulate gut functions. BDNF, the cognate ligand for TrkB receptors, is distributed abundantly in the intestinal tract, including ENS neurons and intestinal mucosa epithelia and interstitial, mediating the gut motility (21). In peripheral organs, netrin-1 is expressed in intestinal epithelium and modulates inflammation (22). We found that both BDNF and netrin-1 were reduced in the gut from WT mice age-dependently and these effects were much more prominent in 5xFAD mice (fig. S2B), which is consistent with previous reports that BDNF is reduced in patients with AD (23). As expected, C/EBPβ and AEP were demonstrable and temporally augmented in 5xFAD mouse brains (fig. S2C). Again, Aβ aggregates were abundantly accompanied by detectable AEP-truncated Tau N368 activities in both 6- and 9-month-old 5xFAD brains. In contrast, both signals were barely detectable in 6-month-old WT brains (fig. S2D). Hence, C/EBPβ/AEP signaling is temporally activated in 5xFAD mice, inversely correlated with trophic factors reduction.
Antibiotic treatment represses C/EBPβ/AEP signaling in 5xFAD mice, alleviating AD pathologies
To assess the impact of the gut microbes on amyloid deposition, we treated 6-week-old 5xFAD mice with a chronic dose of antibiotics cocktail (ABX; composition in methods) in the drinking water for 5 months. We used the regimens of antibiotics (a mixture of ampicillin, vancomycin, neomycin, gentamycin, and erythromycin) that are commonly used to disrupt the microbiota in mouse models of enteric infection and inflammation and evaluated their effects on Aβ deposition in the brains of 5xFAD mice and prepared the brain lysates and serial sections from each hemibrain from ABX- and vehicle-treated animals at 6.5 months of age. Immunoblotting revealed that chronic ABX treatment repressed both C/EBPβ and p-C/EBPβ levels, resulting in the reduction of AEP. Subsequently, the downstream substrates of APP and Tau proteolytic cleavage was pronouncedly attenuated upon ABX treatment, leading to APP N585 and N373, two truncated fragments from AEP cleavage of APP, and Tau N368 evidently decreased. Conversely, the full-length APP recognized by N-terminal (APP NT) antibody was escalated in ABX-treated 5xFAD brains. Notably, AT8 and AT100, two Tau phosphorylation antibodies, showed that Tau hyperphosphorylation was robustly mitigated in ABX-treated 5xFAD brains (Fig. 4A). AEP enzymatic assay with brain lysates validated that ABX significantly blocked its protease activity (Fig. 4B). Accordingly, Aβ42 concentrations were substantially reduced upon ABX treatment (Fig. 4C). IF costaining with anti-Aβ showed that senile plaques deposited in the 5xFAD hippocampus were largely removed by ABX treatment (Fig. 4, D and E), consistent with quantitative Aβ42 enzyme-linked immunosorbent assay (ELISA) data.
Fig. 4. Antibiotic treatment represses C/EBPβ/AEP signaling in 5xFAD mice and rescues the cognitive dysfunctions.

(A) Immunoblot showing p-C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and processing in the mouse brains. MW, molecular weight. (B) AEP activity assay in the brain lysates from 5xFAD treated with antibiotics or vehicle. Data represent the means ± SEM; representative data of three samples; *P < 0.05 compared with control, one-way ANOVA. (C) Aβ1-42 concentrations in the cortex of 5xFAD treated with antibiotics or vehicle. Data represent the means ± SEM; representative data of four samples; *P < 0.05 compared with control, one-way ANOVA. (D) Anti-Aβ staining of amyloid plaques in the hippocampus of 5xFAD mice brain sections. Scale bar, 200 μm. (E) Quantitative analysis of Aβ-positive plaques. The density of plaques in male 5xFAD mice brain was significantly decreased by antibiotic treatment. Data represent the means ± SEM; representative data of three samples; ***P < 0.001 compared with vehicle, one-way ANOVA. (F) The percentage of time spent in the target quadrant in the probe trail. Mice treated with antibiotics spent more time in the target quadrant than the control mice. Data represent the means ± SEM; representative data of 10 samples; *P < 0.05 compared with control, one-way ANOVA. (G) Area under curve of latency. Data represent the means ± SEM; representative data of 10 samples; **P < 0.01 compared with control, one-way ANOVA. (H) Fear-conditioning tests. Data represent the means ± SEM of n = 10 mice per group; *P < 0.05, one-way ANOVA. CS, conditional stimulus.
In accordance with immunoblotting analysis, IF costaining revealed that both AEP and C/EBPβ were reduced in 5xFAD brains after ABX treatment (fig. S3A). In alignment with immunoblotting observations, both APP C586 and Tau N386 activities were attenuated; consistently, Aβ and p-Tau AT8 activities were strongly blocked in 5xFAD brains (fig. S3, B and C). Aβ and thioflavin S (ThS) costaining demonstrated that aggregated Aβ fibrils were almost completed eradicated by ABX (fig. S3D). Positive signals from Iba-1, a microglia/macrophage-specific calcium-binding protein as microglia marker, were reduced upon ABX treatment (fig. S3E). Golgi staining and EM analysis indicated that the dendritic spines and the synapses were strongly elevated (fig. S3, F and I).
Immunoblotting analysis with colon lysates from 5xFAD mice demonstrated that both C/EBPβ and p-C/EBPβ were reduced by ABX, as was active AEP. Consequently, APP N373, N585, and Tau N368 fragmentation in the gut by AEP was evidently antagonized by ABX treatment (fig. S4A). IF costaining on the gut sections revealed that AEP and C/EBPβ were decreased by ABX, as were AEP-truncated APP C586 and Aβ costaining, and the deposited Aβ aggregates, as indicated by ThS staining (fig. S4, B to D). Notably, FITC-dextran assay showed that the gut leakage was significantly diminished by ABX (fig. S4E). MWM behavioral test showed that the cognitive memory in 5xFAD mice was clearly augmented by the ABX (Fig. 4, F and G). Fear conditioning assay indicated that precontextual freezing time was significantly increased by ABX, as compared to vehicle control, indicating that the learning functions were improved (Fig. 4H). Therefore, these studies support that chronic antibiotic treatment antagonizes C/EBPβ/AEP signaling pathway and inhibits amyloid pathology in 5xFAD mice, leading to restoration of cognitive functions.
R13, a 7,8-DHF prodrug, blunts C/EBPβ/AEP signaling in the gut of 5xFAD mice
Our previous report shows that the BDNF mimetic compound 7,8-DHF, a potent small molecular TrkB agonist, displays prominent therapeutic efficacy against AD (24). To improve 7,8-DHF oral bioavailability and in vivo pharmacokinetics profiles, we optimized 7,8-DHF into a carbamate prodrug R13 that displays ~3-fold higher oral bioavailability and longer in vivo half-life, resulting in more promising therapeutic efficacy (25). We found that R13 decreased C/EBPβ in the gut of 5xFAD in a dose-dependent manner; subsequently, AEP was oscillated in the similar format. As a result, both APP N373 and Tau N368 cleavage by AEP was blunted in the gut of 5xFAD mice in a concentration-dependent manner by R13. Notably, full-length (APP FL) was also dose-dependently decreased by R13 (Fig. 5A). IF staining revealed that C/EBPβ and AEP signals in the gut were increasingly attenuated by R13 (Fig. 5, B, C, E, and F). Aβ signals in the gut also prominently decreased (Fig. 5, D and G). Consistent with decreased active AEP levels in the brain lysates, AEP enzymatic assay indicated that the protease activities in the gut were significantly reduced at doses of 21.8 and 43.6 mg/kg (Fig. 5H). Furthermore, R13 mitigated the gut leakage in 5xFAD mice in a dose-dependent manner (Fig. 5I). As expected, the levels of IL-6, the downstream target of C/EBPβ, were progressively decreased in the gut, as the doses of R13 were gradually escalated. By contrast, IL-1β and TNF-α remained unchanged (Fig. 5J).
Fig. 5. R13 administration blunts C/EBPβ/AEP signaling in the gut of 5xFAD mice.

(A) Immunoblot showing p-C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and processing in the mouse colon. (B) IF staining of C/EBPβ. Scale bar, 20 μm. (C to G) IF staining of AEP and Aβ in colon of 5xFAD mice and the quantification. Scale bars, 200 μm. (H) AEP activity assay of the colon lysates from 5xFAD treated with antibiotics or vehicle. Data represent the means ± SEM; representative data of six samples; *P < 0.05 compared with control, one-way ANOVA. (I) GI permeability barrier defect as determined by FITC-dextran translocation in R13-treated 5xFAD mice and vehicle mice. Data represent the means ± SEM; representative data of four samples; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control, one-way ANOVA. (J) R13 treatment decreased IL-6 but not TNF-α or IL-1β expression. Data represent the means ± SEM; representative data of four samples; ***P < 0.001 compared with control, one-way ANOVA.
IF analysis showed that Aβ and AEP-truncated APP C586 activities were steadily reduced in the gut of 5xFAD mice by R13 (fig. S5A). Moreover, the aggregated Aβ fibrils in the gut tissues, revealed by ThS costaining, were diminished in a dose-dependent manner (fig. S5B). To investigate whether R13 activates TrkB receptors in the brain involves any other mechanisms, in addition to direct agonistic activation by 7,8-DHF that is released after R13 hydrolysis, we conducted IF staining with anti-BDNF and ELISA quantification. We found that R13 up-regulated BDNF levels in the hippocampus of 5xFAD dose-dependently (fig. S5, C and D). Immunohistochemistry (IHC) staining with 4-HNE (4-hydroxynonenal), a biomarker for lipid peroxidation and oxidative stress, showed that R13 progressively inhibits the oxidative stress in both the 5xFAD brain and gut (fig. S5E). Hence, R13 exerts its anti-AD therapeutic efficacy via multiple mechanisms including direct TrkB agonistic effect, BDNF up-regulation in the central nervous system (CNS), and C/EBPβ/AEP signaling inhibition in the gut.
R13 prebiotic mitigates AD pathologies in the gut of 5xFAD mice
To assess whether chronic R13 treatment alleviates the gut dysbiosis, we analyzed the fecal samples from 5xFAD mice 3 months after treatment and compared them with vehicle controls. The microbiome analysis indicated a slight reduction of the Bacteroidetes in the gut of vehicle-treated mice at 6 months of age. Besides, there is a moderate escalation in the abundance of Firmicutes and Tenericutes and a modest reduction in certain Proteobacteria following R13 treatment in 5xFAD mice (Fig. 6A). Notably, the relative abundance of proteobacterial Helicobacter hepaticus increased by 3.15-fold (P < 0.05) in the stool of the 6-month-old vehicle-treated 5xFAD mice in comparison with the 3-month-old 5xFAD mouse stool. R13 treatment significantly inhibited this expansion (2.2-fold; P < 0.05) in the abundance of H. hepaticus species (Fig. 6B). On the other hand, the abundance of Barnesiella intestinihominis, Lactobacillus salivarius, and Bacteroides ovatus increased by approximately twofold (P < 0.05) in the stool of the 6-month-old R13-administered 5xFAD in comparison with the vehicle-treated 5xFAD mouse stool (Fig. 6, C to E). The principal coordinate plot (PCoA) demonstrated that the microbiota of the R13-treated mice clustered distinctly but closely from that of the vehicle-treated mice (Fig. 6F, circled cluster). Hence, our data indicate that R13 treatment reduced the abundance of these proinflammatory Proteobacteria, especially Helicobacter in the mouse gut. This finding was supported by an in vitro analysis, which demonstrated that it is 7,8-DHF not T1, which is an intermediate metabolite from R13, and decreased the number of H. hepaticus in a dose-dependent manner. Our in vitro analysis also indicated that 7,8-DHF significantly increased the number of L. salivarius in a dose-dependent manner (Fig. 6G). Thus, 16S-dependent microbiota analysis revealed a progressive decrease in the flavonoid using Firmicutes in 5xFAD control mice in an age-dependent manner. However, the administration of R13 moderately increased the abundance of this microbial population. Further microbiome analysis demonstrated that the R13 treatment stimulated the enrichment of specific anti-inflammatory bacterium L. salivarius and also reduced the abundance of certain proinflammatory pathobionts of the intestine.
Fig. 6. Gut microbiota analysis by 16S rRNA from the fecal samples of 5xFAD mice, chronically treated with R13.
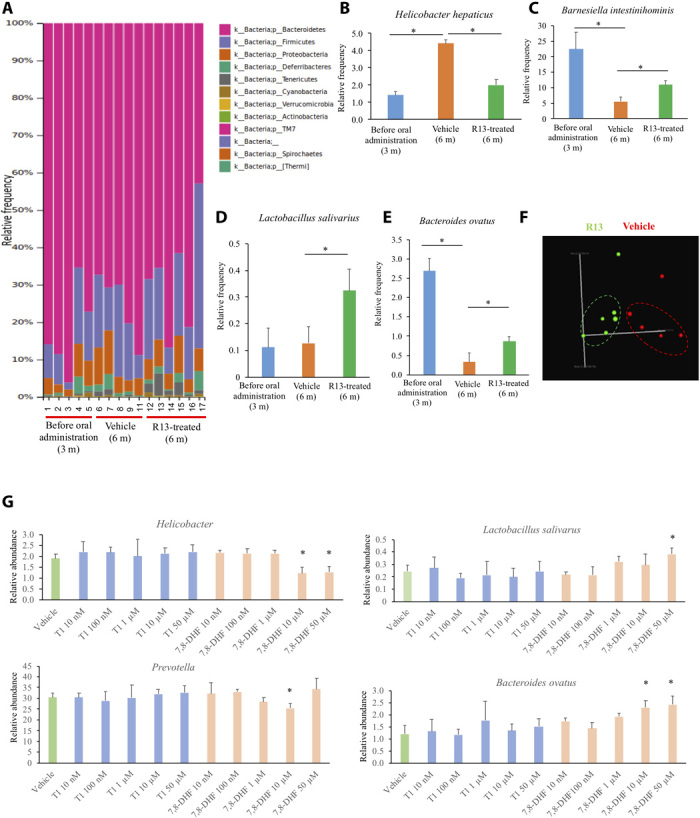
(A) Relative abundance of bacteria phyla determined by HTS (n ≥ 5 mice per group). (B to E) Mean bacteria abundance of bacterial species. Data represent the means ± SEM; representative data five samples; *P < 0.05 compared with control, one-way ANOVA. (F) PCoA of microbiota community structure in vehicle- and R13-treated 5xFAD mice. (G) In vitro analysis of T1 and 7,8-DHF on bacteria growth. Mean bacteria abundance of bacteria species and phyla was determined. Data represent the means ± SEM; representative data of five samples; *P < 0.05 compared with control; one-way ANOVA.
R13-induced probiotic suppresses C/EBPβ/AEP signaling in 5xFAD mouse brain, decreasing gut leakage and oxidative stress
Next, we evaluated the effect of chronic antibiotic treatment on specific intestinal bacterial groups identified in Fig. 1 and analyzed R13-induced probiotic’s impact on gut microbiota. As expected, the administration of ABX for 5 months significantly reduced the relative frequency of the proinflammatory Helicobacter and Prevotella (fig. S6A, top). The relative abundance of Lactobacillus salivarus and B. ovatus remained similar (fig. S6A, bottom). Thus, in corroboration with previous reports, our data suggest that the application of antibiotic regimens diminishes the number of proteobacteria, which are predominantly associated with the inflammatory immune responses. On the other hand, chronic R13 treatment selectively escalated and enriched L. salivarus in 5xFAD mice (Fig. 6D). We hypothesized that this L. salivarus may act as probiotic and partially account for the R13’s therapeutic effect. To test this possibility, we chronically fed 5xFAD mice with live or boiled L. salivarus for 3 months and compared them with phosphate-buffered saline (PBS) vehicle. The microbiome analysis indicated a significant reduction of the phyla inflammatory Proteobacteria and robust increase in the class Mollecutes in the gut of the live L. salivarus–administered mice in comparison with the vehicle-treated control mice at 6 months of age (fig. S6C). However, as evidenced by the PCoA analysis, the overall community composition remained essentially unchanged and clustered closely together across the treatment groups (fig. S6B). In addition, live L. salivarus treatment also decreased the relative abundance of Helicobacter to some extent.
Note that live L. salivarus treatment robustly repressed C/EBPβ in the brain and inhibited AEP activation, as compared to dead L. salivarus or vehicle. Accordingly, APP N585 and Tau N368 proteolytic cleavage by active AEP was blunted by live L. salivarus treatment. Consequently, p-Tau AT8 activities were antagonized (Fig. 7A). Consistently, AEP enzymatic activity was significantly suppressed by live L. salivarus treatment compared to dead L. salivarus or vehicle (Fig. 7B). Moreover, proinflammatory cytokine IL-6 concentration in the brain decreased as well (Fig. 7C). However, BDNF in the brain was not significantly changed regardless of live or dead L. salivarus treatment (Fig. 7D). Although Aβ40 and Aβ42 concentrations in the brain were not statistically significantly reduced by live L. salivarus versus vehicle or dead L. salivarus, they displayed the decrease trend (Fig. 7E). FITC-dextran assay indicated that the gut leakage in 5xFAD mice was strongly reduced by the live probiotic L. salivarus and dead L. salivarus also decreased the gut leakage weakly yet significantly, as compared to vehicle (Fig. 7F), indicating that mice treated with boiled L. salivarus might also benefit from some components or metabolites from L. salivarus, which alleviate the gut leakage. Notably, live probiotic L. salivarus evidently mitigated both C/EBPβ and AEP levels, associated with Aβ and APP C586 reduction in 5xFAD brains (Fig. 7, G and H). Consequently, ThS-positive Aβ aggregates in the brains were attenuated by live L. salivarus versus vehicle or boiled L. salivarus (Fig. 7I). IHC staining with 4-HNE on both the brain and gut sections from 5xFAD mice showed that live L. salivarus decreased oxidative stress in the cortex and the hippocampus versus vehicle or boiled L. salivarus (fig. S7A). IF staining showed that live L. salivarus treatment decreased both C/EBPβ and AEP in the gut (fig. S7B). As a result, APPC586 proteolytic cleavage by active AEP was also decreased, and Aβ deposition in the gut of 5xFAD mice was mitigated by live L. salivarus (fig. S7C).
Fig. 7. R13-induced probiotic L. salivarius suppresses C/EBPβ/AEP signaling in the brain of 5xFAD mice, decreasing gut leakage.

(A) Immunoblot showing p-C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and processing in the mouse brains. LS, L. salivarius. (B) AEP activity assay in the brain lysates from 5xFAD treated with live L. salivarius, boiled L. salivarius, or control. Data represent the means ± SEM; representative data of three samples; *P < 0.05 compared with control, one-way ANOVA. (C) Live L. salivarius treatment decreased IL-6 concentrations in the mouse brains. Data represent the means ± SEM; representative data of three samples; *P < 0.05 compared with control, one-way ANOVA. (D) BDNF concentrations in the brains from 5xFAD treated with live L. salivarius, boiled L. salivarius, or control. Data represent the means ± SEM; representative data of three samples. (E) Aβ1-40 and Aβ1-42 concentrations in 5xFAD mice treated with live L. salivarius, boiled L. salivarius, or control, respectively. Data represent the means ± SEM; representative data of three samples. (F) GI permeability barrier defect, as determined by FITC-dextran translocation in 5xFAD mice treated with live L. salivarius, boiled L. salivarius, or control. Data represent the means ± SEM; representative data of six samples; **P < 0.01 and ***P < 0.001, compared with control, one-way ANOVA. (G to I) IF staining of C/EBPβ and AEP; Aβ/APP C586 and Aβ/ThS on the brain sections from 5xFAD mice treated with live L. salivarius, boiled L. salivarius, or control. Scale bars, 20 μm.
DISCUSSION
Human symbiotic microbes are very important environmental factors influencing host health. Approximately 95% of the symbiotic microbes are located in the gut, and they play a major role in human nutrition, digestion, neurotrophy, inflammation, growth, immunity, and protection against pathogen infections (26). Dysbiosis and alterations of gut microbiome composition contribute to the development of several diseases in humans, including IBS, type 2 diabetes, metabolic syndrome, obesity, and AD (6, 7, 27). These changes are integral to gut homeostasis and to programming of the hypothalamic-pituitary-adrenal axis, and thus play an important role in stress responses (28). In this study, we show that gut dysbiosis takes place in 5xFAD mice versus WT littermates in an age-dependent manner. Note that the gut microbial dysbiosis is intricately associated with a reduction in phylogenetic richness, especially in diseases that are characterized by intestinal inflammation and deregulated GI barrier integrity. For instance, Helicobacter is highly enriched in 5xFAD mice, compared to WT littermates at 6 months of age (fig. S1B). In particular, H. bilis showed a significant increase in both 3-month-old and 6-month-old 5xFAD mice (fig. S1D). Moreover, microbiota analysis also revealed that Prevotella species increased (~100-fold) at 6 months in 5xFAD mice (fig. S1B). Prevotella is the predominant genus in human colonic microbiota, and is genetically equipped to function as an efficient mucin degrader in the intestine. Prevotella has been reported to promote GI dysfunction in diabetes and autism (29). Similarly, the genus Sutterella has been frequently associated with human diseases, such as autism, Down syndrome, and IBD (30).
Gut microbiota may contribute to aging and influence brain disorders. In particular, a new connection between gut microbiota and PD has been reported in humans (31, 32). The Aβ and NFT pathologies and associated neuroinflammation in AD brain cross-talk with the gut via the microbiota-gut-brain axis. Conceivably, gut leakage due to gut dysbiosis in patients with AD may allow the endotoxins from bacterial metabolites such as lipopolysaccharides (LPS) to penetrate into the ENS system in the colon, where they may stimulate C/EBPβ activation and AEP up-regulation, resulting in the augmentation of Aβ and NFT pathologies in myenteric and submucosal nervous system. The aggregated Aβ or Tau fibrils might spread into the brain along the vagus nerve. For instance, previous studies show that α-Syn fibrils derived from patients with PD can transport into the brain along the vagus nerve after injection into the colon of the rat (33). Most recently, we have reported that AEP-truncated α-Syn N103 and Tau N368 form compact fibrils, have higher neurotoxicity, and propagate from the colon into the brain, triggering PD pathologies and motor disorders (34). A healthy gut microbiota is very important for maintaining normal brain development and function. Mounting evidence supports that the gut-brain axis is a bidirectional communication that simultaneously applies to both the brain and the gut. Kim et al. (35) reported that the transfer of healthy microbiota would reduce amyloid and Tau pathology in a different AD animal model, providing that a proof of concept of gut microbiota affects the brain. To further test this notion, we conducted a cohousing experiment using 3xTg mice, in which young 3xTg mice were either cohousing with aged 3xTg mice or aged WT mice from 1.5 to 6.5 months of age. This is an alternative for fecal microbiota transplant experiment. Our data suggest that microbiota from the aged 3xTg but not WT mice accelerate AD pathology in young 3xTg mice, associated with active C/EBPβ/AEP signaling in the brain (Fig. 2).
The C/EBPβ/AEP pathway is also temporally activated in the gut of 5xFAD mice, concurrent with the onset of amyloid pathology in the gut. Chronic antibiotic treatment represses C/EBPβ/AEP signaling and Aβ pathology, leading to the restoration of cognitive functions in 5xFAD mice. Notably, oral administration of a small molecular TrkB receptor agonist prodrug R13 that is metabolized into T1 and then into 7,8-DHF, which blunts C/EBPβ/AEP axis in the gut and antagonizes Aβ pathology (fig. S5, A and B). Thus, R13 acts as a prebiotic, in addition to the activation of BDNF/TrkB neurotrophic signaling via 7,8-DHF in the brain. H. hepaticus preferentially colonizes mouse small intestine, cecum, and colon and induces intestinal inflammation and colitis in the murine model of IBD (36), and R13 treatment significantly repressed its expansion (Fig. 6B). Notably, fecal sample analysis reveals that the beneficial bacterium L. salivarius is selectively enriched after chronic R13 treatment in 5xFAD mice. Accordingly, long-term feeding of 5xFAD mice with L. salivarius probiotic attenuates gut leakage and inflammation and oxidative stress. However, probiotic treatment alone is not sufficient to reverse the cognitive deficits in 5xFAD mice, indicating that R13-induced L. salivarius probiotic contributes to a portion of the therapeutic efficacy of R13 in treating AD. Previous studies demonstrated that the probiotic L. salivarius ameliorates colonic damage and reduces mucosal inflammation (37).
Previously, we have reported that the parent compound 7,8-DHF penetrates into the brain,binds TrkB receptors, and stimulates the neurotrophic signaling in the CNS (25, 38). Notably, we also found that R13 dose-dependently elevates BDNF production in the brains (fig. S5, C and D). Conceivably, R13 or 7,8-DHF exerts the therapeutic efficacy in AD via multiple mechanisms from the TrkB agonistic functions to the augmentation of BDNF production in the brains to the prebiotic actions in the gut microbiota. Note that treatment with antibiotics, R13, or probiotic in 5xFAD mice, C/EBPβ, a crucial proinflammatory cytokines transcription factor, is robustly diminished. Consequently, its downstream target AEP is repressed in the gut and the brain, leading to APP and Tau proteolytic fragmentation reduction and Aβ pathology inhibition (Figs. 4, 5, and 7). Our previous study demonstrated that BDNF and C/EBPβ inversely regulate each other, providing a mechanistic link between BDNF/TrkB reduction, C/EBPβ up-regulation, δ-secretase (AEP) activation, and Aβ and Tau alterations in the murine brains (39). In the current study, we found that R13 (7,8-DHF) also functions as antioxidant in the murine colon, leading to proinflammatory cytokine IL-6 decrease and suppression of C/EBPβ.
It has been shown that populating the gut microbiota can release significant amounts of amyloids and LPS, which might play a role in the modulation of signaling pathways and the production of proinflammatory cytokines related to the pathogenesis of AD. Nutrients have been shown to affect the composition of the gut microbiota and the formation and aggregation of cerebral Aβ, suggesting that modulating the gut microbiome and amyloidogenesis through specific nutritional interventions might be an effective strategy to prevent or reduce the risk of AD (40).
Antibiotics seriously disrupt the gut microbiota. Long-term ABX administration in male mice leads to reduced Aβ deposition and associated gliosis and reduces Aβ deposition selectively in males but not in females (10). Transplants of fecal microbiota from age-matched APP/PS1-21 male mice into ABX-treated APP/PS1-21 male restore the gut microbiome and partially restore Aβ pathology and microglial morphology, thus demonstrating a causal role of the microbiome in the modulation of Aβ amyloidosis and microglial physiology in mouse models of Aβ amyloidosis (41). Accordingly, we showed that ABX treatment inhibits C/EBPβ/AEP pathway in the gut and the brain of 5xFAD mice and mitigates Aβ pathologies, attenuating AD cognitive disorders (Fig. 4, and figs. S3, S4, and S6).
Host gut microbiota also constantly control maturation and function of microglia in the CNS. Modulating the gut microbiome and restoring its diversity in the elderly through diet and nutrition intervention might improve the physical and mental health of the elderly. Dietary intake of some other antioxidant nutrients, such as vitamin C, vitamin E, and flavonoids, is also considered to be related to decreased risk of AD. Chronic treatment with R13 also results in alleviation of gut dysbiosis in 5xFAD mice by increasing the beneficial bacteria and attenuating inflammatory bacteria, which is associated with AD pathology reduction in the gut (Fig. 5 and fig. S5). Previously, we have reported that R13 dose-dependently activates TrkB signaling via its small molecular agonist 7,8-DHF in the brain and inhibits AEP via activated Akt-mediated phosphorylation, leading to escalation of synaptic plasticity and suppression AD pathologies in 5xFAD mice (25). The metabolite 7,8-DHF is a flavonoid with multiple functions including TrkB agonistic activity (38), antioxidative effect (fig. S5), and prebiotic activity (Fig. 5), whose effects on gut microbiota might be independent of the effect in the brain as a TrkB agonist in alleviating AD pathogenesis. Accordingly, chronic treatment with R13-induced probiotic L. salivarius bacteria suppresses C/EBPβ/AEP signaling in the gut and the brain. Notably, it also significantly mitigates the gut leakage and displays the antioxidative effect in both the brain and the gut of 5xFAD mice (Fig. 7 and fig. S7). Note that both ABX and R13 treatments reduce the proinflammatory bacteria such as Helicobacter. The abundance of anti-inflammatory L. salivarus remained unchanged or even increased after ABX or R13 treatment, respectively. Thus, our findings indicate that L. salivarus abundancy after both treatments remained beyond a certain level, which is associated with the regulation of signaling pathways. C/EBPβ/AEP signaling in both treatments was repressed compared with control (vehicle). However, R13 itself exerted the direct effect on the host as many flavonoids, in addition to physically activating TrkB receptors. It remains unclear exactly how gut microbiota alteration mediates brain BDNF levels. Presumably, R13 treatment decreases the inflammation in the brain and inactivates C/EBPβ (Fig. 5), alleviating the repression by C/EBPβ on BDNF transcription.
After consumption, polyphenols, which are characterized by low bioavailability, need biotransformation by gut microbiota into bioactive metabolites, recognized as xenobiotics, and become readily absorbable. Therefore, to sufficiently obtain the health benefits and increase the bioavailability and activity of polyphenols, one may need healthy gut microbiota. The protective role of other antioxidants and nutrients may also depend on the balance of gut microbiota to some extent. We have reported that both 7,8-DHF and R13 are orally bioactive, and R13 improves 7,8-DHF oral bioavailability from 4.6 to 11% (25). Currently, R13 is under phase 1 clinical trial for treating AD indication. Therefore, external environmental factors induce gut microbiota disturbance, and the influence of host genes on gut microbiota may interact together to determine disease susceptibility, including the risk of AD. Conceivably, R13 exerts its therapeutic efficacy toward AD via both direct TrkB receptor agonistic effect and beneficial modulation of gut microbiota.
MATERIALS AND METHODS
Mice
5xFAD (APP SwFlLon, PSEN1*M146L*L286V) mice were from the Jackson laboratory (stock number, 008169) and were bred in a pathogen-free environment in accordance with the Emory Medical School guidelines. The animals were randomly allocated to experimental groups. Investigators were blinded to group allocation during the animal experiments. The protocol was reviewed and approved by the Emory Institutional Animal Care and Use Committee. In the antibiotic treatment experiment, the male 5xFAD mice were provided ampicillin (1 g/liter; Sigma-Aldrich, St. Louis, MO), vancomycin (0.5 g/liter; Sagent Pharmaceuticals, Schaumburg), neomycin (0.5 g/liter; Thermo Fisher Scientific), gentamycin (100 mg/liter; Sigma-Aldrich), and erythromycin (10 mg/liter; Sigma-Aldrich) in drinking water beginning at 8 to 9 weeks of age through 24 to 25 weeks of age. In the R13 treatment experiment, the 5xFAD mice received vehicle or R13 dissolved in 5% dimethyl sulfoxide/0.5% methylcellulose at doses of 7.25, 21.8, or 43.6 mg/kg per day at 12 weeks of age through 24 to 25 weeks of age. In probiotic treatment experiment, GF animals treated with either live bacteria or heat-killed bacteria were provided with 5 × 108 colony-forming units/ml of L. salivarius (ATCC11741). Treatments were given through oral gavage twice a week starting from 8 weeks of age through 24 to 25 weeks of age. 3xTg (APP Swe, MAPT P301L, PSEN1 M146V) mice were ordered from the Jackson laboratory (stock number, 34830) and were bred in a pathogen-free environment in accordance with the Emory Medical School guidelines. The animals were randomly allocated to experimental groups. Investigators were blinded to group allocation during the animal experiments. The protocol was reviewed and approved by the Emory Institutional Animal Care and Use Committee. In the cohousing experiment, the young 3xTg mice at the age of 6 weeks were either receiving dirty bedding from the cages of 14-month-old 3xTg mice or 14-month-old WT mice for 5 months on a weekly basis.
Antibodies and reagents
δ-Secretase antibody (clone 6E3 and 11B7) was a present from C. Watts, University of Dundee. TrkB antibody was from BioVision (Milpitas, CA). Phospho-TrkB Y816 antibody was raised against [H]-CKLQNLAKASPV-pY-LDILG-[OH] (amino acids 806 to 822) (EM437 and EM438) as rabbit polyclonal antibody. 4-HNE antibody was from Abcam (San Francisco, CA). Aβ, AT8, AT100, Tau 5, tubulin, and β-actin antibody were from Sigma-Aldrich (St. Louis, MO). C/EBPβ (H-7) antibody was bought from Santa Cruz Biotechnology (Dallas, TX). Legumain (D6S4H) and p-C/EBPβ antibodies were bought from Cell Signaling Technology (Boston, MA). Histostain-SP kit, Aβ1-42, and Aβ1-40 ELISA kit were from Invitrogen (Grand Island, NY). TNF-α, IL-1β, and IL-6 ELISAs were from eBioscience (San Diego, CA). All chemicals not included above were purchased from Sigma-Aldrich.
Protocol for microbiota analysis
DNA was extracted from stool samples (n = 5 mice per group) using a PowerSoil kit from MO BIO Laboratories (Carlsbad, CA). 16S rRNA genes were PCR-amplified from each sample using a composite forward primer and a reverse primer containing a unique 12-base barcode, designed using the Golay error-correcting scheme, which was used to tag PCR products from respective samples. We used primers for paired-end 16S community sequencing on the Illumina platform using bacteria/archaeal primer 515F/806R. Primers were specific for the V4 region of the 16S rRNA gene. The forward PCR primer sequence contained the sequence for the 5′ Illumina adapter, the forward primer pad, the forward primer linker, and the forward primer sequence. Each reverse PCR primer sequence contained the reverse complement of the 3′ Illumina adapter, the Golay barcode (each sequence contained a different barcode), the reverse primer pad, the reverse primer linker, and the reverse primer (42). Three independent PCR reactions were performed for each sample, combined, and purified with AMPure magnetic purification beads (Agencourt). The products were quantified, and a master DNA pool was generated from the purified products in equimolar ratios. The pooled products were sequenced using an Illumina MiSeq sequencing platform. Bioinformatics analysis was performed using QIIME (Quantitative Insights into Microbial Ecology). Sequences were assigned to OTUs with UPARSE using 97% pairwise identity and were classified taxonomically using the RDP classifier retrained with Greengenes. After chimera removal, the average number of reads per sample was 21,511. A single representative sequence for each OTU was aligned using PyNAST, and a phylogenetic tree was then built using FastTree. The phylogenetic tree was used to compute the UniFrac distances. The PCoA analysis shown is unweighted.
Enteric commensal-specific quantitative PCR
For quantification, total microbial DNA and universal bacterial primer sets 515F and 806R were used. To quantify the abundance of specific bacterial species or genus, we used the validated forward and reverse primers specific for the 16S rRNA genes. Detection was achieved using a Fast SYBR Green Master mix according to the manufacturer’s instructions. The following reverse transcription PCR (RT-PCR) protocol was followed: 95°C for 15 min, 40 cycles of 95°C for 15 s, 66°C (bacteria-specific) and 50°C (universal) for 40 s, and 72°C for 30 s. A melting curve was performed after amplification to distinguish between the targeted and nontargeted PCR products. All reactions were performed in duplicate. Bacterial abundance was analyzed as genome equivalents. For quantitative PCR (qPCR) analyses, the sample-specific relative abundance of bacteria was determined as genome equivalents amplified by bacteria-specific primers divided by genome equivalents amplified by universal primers. Absolute abundance per gram of stools was determined by adjusting for the dilutions performed during DNA extraction, normalization, and qPCR setup and dividing this concentration by the total grams of stools used for the original DNA extraction. Last, data were expressed as a fold change in stools collected from different mouse backgrounds compared to stool of the WT mice.
In vitro bacteria culture experiment
Stool samples were collected from 6-month-old 5xFAD and mice incubated with suspension of vehicle, T1, or 7,8-DHF at different concentrations for 4 hours. Subsequently, DNA was extracted and commensal-specific quantitative RT-PCR was performed following methods described above.
Western blot analysis
The mice brain tissue was lysed in lysis buffer [50 mM tris (pH 7.4), 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, and 10 mM sodium β-glycerophosphate, supplemented with protease inhibitor cocktail] and centrifuged for 15 min at 16,000g. The supernatant was boiled in SDS loading buffer. After SDS–polyacrymide gel electrophoresis, the samples were transferred to a nitrocellulose membrane. Western blotting analysis was performed with a variety of antibodies.
Immunostaining
Paraffin-embedded mouse brain and colon sections went through dewaxing and rehydration process by incubating the slides in xylene first and then immerse them into a decreasing percentage of ethanol. The sections were boiled in 10 mM citric acid for 20 min, followed by room temperature cooling down for antigen retrieval. Then, sections were treated with 3% H2O2 for 10 min, followed by three times washing in PBS and 30-min blocking in 1% radioimmunoassay–bovine serum albumin (BSA) and 0.3% Triton X-100, as well as the overnight incubation with p-TrkB (1:300; Y816, homemade), 4-HNE antibody (1:50; Abcam), and Aβ antibody (1:500; Sigma-Aldrich) at 4°C. The signal was developed using Histostain-SP kit (Invitrogen). To detect the localization of C/EBPβ and p-C/EBPβ, the slides were incubated with C/EBPβ antibody (H-7) (1:100) and p-C/EBPβ antibody (1:200) at 4°C. To detect the localization of AEP, AEP-derived Tau fragment, and phosphorylated Tau in mouse brain section, the slides were incubated with AEP antibody (11B7) (1:500; from C. Watts, University of Dundee), Tau N368 (1:1000; homemade), AT-8 (1:500; MN1020, Thermo Fisher Scientific) at 4°C. After overnight incubation, the slides were washed three times in PBS and incubated with Texas Red–conjugated anti-rabbit immunoglobulin G (IgG) or FITC-conjugated anti-mouse IgG for 1 hour at room temperature. The slides were washed three times in PBS, then covered with a glass cover using a mounting solution, and examined under a fluorescence microscope (Olympus).
Golgi staining
Mice brains were fixed in 10% formalin for 24 hours and then immersed in 3% potassium bichromate for 3 days in the dark. The solution was changed each day. Then, the brains were transferred into 2% silver nitrate solution and incubated for 24 hours in the dark. Vibratome sections were cut at 60 μm, air-dried for 10 min, dehydrated through 95 and 100% ethanol, cleared in xylene, and coverslipped. For measurement of spine density, only spines that emerged perpendicular to the dendritic shaft were counted.
Aβ plaque staining
Amyloid plaques were stained with ThS. The deparaffinized and hydrated sections were incubated in 0.25% potassium permanganate solution for 20 min, rinsed in distilled water, and incubated in a bleaching solution containing 2% oxalic acid and 1% potassium metabisulfite for 2 min. After rinsed in distilled water, the sections were transferred to a blocking solution containing 1% sodium hydroxide and 0.9% hydrogen peroxide for 20 min. The sections were incubated for 5 s in 0.25% acidic acid, then washed in distilled water, and stained for 5 min with 0.0125% ThS in 50% ethanol. The sections were washed with 50% ethanol and placed in distilled water. Then, the sections were covered with a glass cover using a mounting solution.
Aβ ELISA
The mice brains were homogenized in 8× mass of 5 M guanidine HCl/50 mM tris-HCl (pH 8.0) and incubated at room temperature for 3 hours. Then, the samples were diluted with cold reaction buffer (PBS with 5% BSA and 0.03% Tween 20, supplemented with protease inhibitor cocktail) and centrifuged at 16,000g for 20 min at 4°C. The supernatant was analyzed by human Aβ40 and Aβ42 ELISA kit according to the manufacturer’s instructions (KHB3481 and KHB3441, respectively, Invitrogen). The Aβ concentrations were determined by comparison with the standard curve.
Morris water maze
5xFAD mice maintained on administration were trained in a round, water-filled tub (1.32 meters in diameter) in an environment rich with extramaze cues. An invisible escape platform was located in a fixed spatial location 1 cm below the water surface independent of a subject start position on a particular trial. In this manner, experimental subjects needed to use extramaze cues to determine the platform’s location. At the beginning of each trial, the mouse was placed in the water maze with their paws touching the wall from one of four different starting positions (North, South, East, and West). Each subject was given 4 trials/day for five consecutive days with a 15-min intertrial interval. The maximum trial length was 60 s, and if subjects did not reach the platform in the allotted time, then they were manually guided to it. Upon reaching the invisible escape platform, subjects were left on it for an additional 5 s to allow for survey of the spatial cues in the environment to guide future navigation to the platform. After each trial, subjects were dried and kept in a dry plastic holding cage filled with paper towels to allow the subjects to dry off. The temperature of the water was monitored every hour so that the mice were tested in water that was between 22° and 25°C. Following the 5 days of task acquisition, a probe trial was presented during which time the platform was removed, and the percentage of time spent in the quadrant that previously contained the escape platform during task acquisition was measured more than 60 s. All trials were analyzed for latency, swim path length, and swim speed by means of MazeScan (CleverSys Inc.).
Statistical analysis
All data are expressed as means ± SEM from three or more independent experiments, and the level of significance between two groups was assessed with Student’s t test. For more than two groups, one-way analysis of variance (ANOVA) followed by least significant difference post hoc test was applied. A value of P < 0.05 was considered to be statistically significant.
Ethical approval
Experimental protocol was approved by the Emory University Institutional Animal Care and Ethical Committee.
Supplementary Material
Acknowledgments
Funding: This work is supported by a grant from the NIH (RF1, AG051538; RO1, NS105982) to K.Y. This study was supported in part by the Rodent Behavioral Core (RBC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical and Translational Science Alliance of the National Institutes of Health under award number UL1TR002378. Author contributions: K.Y. conceived the project, designed the experiments, analyzed the data, and wrote the manuscript. C.C., E.H.A., S.S.K., and A.A. designed and performed most of the experiments and analyzed the data. X.L. conducted genotype and bred the transgenic mice. A.A. assisted with data analysis and interpretation and critically read the manuscript. Competing interests: K.Y. is a shareholder of Wuhan Yuanzheng Pharmaceuticals Inc. and Shanghai Braegen Pharmaceuticals Inc. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba0466/DC1
REFERENCES AND NOTES
- 1.Heneka M. T., Carson M. J., Khoury J. E., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., Kummer M. P., Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu X., Wang T., Jin F., Alzheimer's disease and gut microbiota. Sci. China Life Sci. 59, 1006–1023 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Zhuang Z. Q., Shen L.-L., Li W.-W., Fu X., Zeng F., Gui L., Lü Y., Cai M., Zhu C., Tan Y.-L., Zheng P., Li H.-Y., Zhu J., Zhou H.-D., Bu X.-L., Wang Y.-J., Gut microbiota is altered in patients with Alzheimer’s disease. J. Alzheimers Dis. 63, 1337–1346 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Vogt N. M., Kerby R. L., Dill-McFarland K. A., Harding S. J., Merluzzi A. P., Johnson S. C., Carlsson C. M., Asthana S., Zetterberg H., Blennow K., Bendlin B. B., Rey F. E., Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7, 13537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haran J. P., Bhattarai S. K., Foley S. E., Dutta P., Ward D. V., Bucci V., McCormick B. A., Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. MBio 10, e00632-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stecher B., The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol. Spectr. 3, (2015). [DOI] [PubMed] [Google Scholar]
- 7.Esteve E., Ricart W., Fernandez-Real J. M., Gut microbiota interactions with obesity, insulin resistance and type 2 diabetes: Did gut microbiote co-evolve with insulin resistance? Curr. Opin. Clin. Nutr. Metab. Care 14, 483–490 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Gomborone J. E., Dewsnap P. A., Libby G. W., Farthing M. J., Selective affective biasing in recognition memory in the irritable bowel syndrome. Gut 34, 1230–1233 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cattaneo A., Cattane N., Galluzzi S., Provasi S., Lopizzo N., Festari C., Ferrari C., Guerra U. P., Paghera B., Muscio C., Bianchetti A., Volta G. D., Turla M., Cotelli M. S., Gennuso M., Prelle A., Zanetti O., Lussignoli G., Mirabile D., Bellandi D., Gentile S., Belotti G., Villani D., Harach T., Bolmont T., Padovani A., Boccardi M., Frisoni G. B.; INDIA-FBP Group , Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Minter M. R., Zhang C., Leone V., Ringus D. L., Zhang X., Oyler-Castrillo P., Musch M. W., Liao F., Ward J. F., Holtzman D. M., Chang E. B., Tanzi R. E., Sisodia S. S., Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 6, 30028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen L., Liu L., Ji H. F., Alzheimer's disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J. Alzheimers Dis. 56, 385–390 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Basurto-Islas G., Gu J. H., Tung Y. C., Liu F., Iqbal K., Mechanism of Tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J. Alzheimers Dis. 63, 821–833 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z., Song M., Liu X., Kang S. S., Kwon I.-S., Duong D. M., Seyfried N. T., Hu W. T., Liu Z., Wang J.-Z., Cheng L., Sun Y. E., Yu S. P., Levey A. I., Ye K., Cleavage of Tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 20, 1254–1262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z., Song M., Liu X., Kang S. S., Duong D. M., Seyfried N. T., Cao X., Cheng L., Sun Y. E., Yu S. P., Jia J., Levey A. I., Ye K., Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 6, 8762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foiani M. S., Cicognola C., Ermann N., Woollacott I. O. C., Heller C., Heslegrave A. J., Keshavan A., Paterson R. W., Ye K., Kornhuber J., Fox N. C., Schott J. M., Warren J. D., Lewczuk P., Zetterberg H., Blennow K., Höglund K., Rohrer J. D., Searching for novel cerebrospinal fluid biomarkers of tau pathology in frontotemporal dementia: An elusive quest. J. Neurol. Neurosurg. Psychiatry 90, 740–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z.-H., Gong K., Liu X., Zhang Z., Sun X., Wei Z. Z., Yu S. P., Manfredsson F. P., Sandoval I. M., Johnson P. F., Jia J., Wang J.-Z., Ye K., C/EBPβ regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat. Commun. 9, 1784 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magalini A., Savoldi G., Ferrari F., Garnier M., Ghezzi P., Albertini A., Lorenzo D. D., Role of IL-1β and corticosteroids in the regulation of the C/EBP-α, β and δ genes in vivo. Cytokine 7, 753–758 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Wedel A., Ziegler-Heitbrock H. W., The C/EBP family of transcription factors. Immunobiology 193, 171–185 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G. M., Cooper N. R., Eikelenboom P., Emmerling M., Fiebich B. L., Finch C. E., Frautschy S., Griffin W. S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I. R., McGeer P. L., O'Banion M. K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F. L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., Wyss-Coray T., Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H., Liu X., Chen S., Ye K., Spatiotemporal activation of the C/EBPβ/δ-secretase axis regulates the pathogenesis of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, E12427–E12434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen F., Yu Y., Wang P., Dong Y., Wang T., Zuo X., Li Y., Brain-derived neurotrophic factor accelerates gut motility in slow-transit constipation. Acta Physiol (Oxford) 212, 226–238 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Mirakaj V., Gatidou D., Potzsch C., Konig K., Rosenberger P., Netrin-1 signaling dampens inflammatory peritonitis. J. Immunol. 186, 549–555 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Phillips H. S., Hains J. M., Armanini M., Laramee G. R., Johnson S. A., Winslow J. W., BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7, 695–702 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z., Liu X., Schroeder J. P., Chan C.-B., Song M., Yu S. P., Weinshenker D., Ye K., 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 638–650 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C., Wang Z., Zhang Z., Liu X., Kang S. S., Zhang Y., Ye K., The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, 578–583 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooper L. V., Gordon J. I., Glycans as legislators of host-microbial interactions: Spanning the spectrum from symbiosis to pathogenicity. Glycobiology 11, 1R–10R (2001). [DOI] [PubMed] [Google Scholar]
- 27.Bekkering P., Jafri I., van Overveld F. J., Rijkers G. T., The intricate association between gut microbiota and development of type 1, type 2 and type 3 diabetes. Expert. Rev. Clin. Immunol. 9, 1031–1041 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Sudo N., Chida Y., Aiba Y., Sonoda J., Oyama N., Yu X.-N., Kubo C., Koga Y., Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 558, 263–275 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scher J. U., Sczesnak A., Longman R. S., Segata N., Ubeda C., Bielski C., Rostron T., Cerundolo V., Pamer E. G., Abramson S. B., Huttenhower C., Littman D. R., Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2, e01202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q., Han Y., Dy A. B. C., Hagerman R. J., The gut microbiota and autism spectrum disorders. Front. Cell. Neurosci. 11, 120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheperjans F., Aho V., Pereira P. A. B., Koskinen K., Paulin L., Pekkonen E., Haapaniemi E., Kaakkola S., Eerola-Rautio J., Pohja M., Kinnunen E., Murros K., Auvinen P., Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Sampson T. R., Debelius J. W., Thron T., Janssen S., Shastri G. G., Ilhan Z. E., Challis C., Schretter C. E., Rocha S., Gradinaru V., Chesselet M.-F., Keshavarzian A., Shannon K. M., Krajmalnik-Brown R., Wittung-Stafshede P., Knight R., Mazmanian S. K., Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmqvist S., Chutna O., Bousset L., Aldrin-Kirk P., Li W., Björklund T., Wang Z.-Y., Roybon L., Melki R., Li J.-Y., Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Ahn E. H., Kang S. S., Liu X., Chen G., Zhang Z., Chandrasekharan B., Alam A. M., Neish A. S., Cao X., Ye K., Initiation of Parkinson’s disease from gut to brain by δ-secretase. Cell Res. 30, 70–87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M.-S., Kim Y., Choi H., Kim W., Park S., Lee D., Kim D. K., Kim H. J., Choi H., Hyun D.-W., Lee J.-Y., Choi E. Y., Lee D.-S., Bae J.-W., Mook-Jung I., Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 69, 283–294 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Bain C. C., Oliphant C. J., Thomson C. A., Kullberg M. C., Mowat A. M., Proinflammatory role of monocyte-derived CX3CR1int macrophages in Helicobacter hepaticus-induced colitis. Infect. Immun. 86, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peran L., Camuesco D., Comalada M., Nieto A., Concha A., Diaz-Ropero M. P., Olivares M., Xaus J., Zarzuelo A., Galvez J., Preventative effects of a probiotic, Lactobacillus salivarius ssp. salivarius, in the TNBS model of rat colitis. World J. Gastroenterol. 11, 5185–5192 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang S.-W., Liu X., Yepes M., Shepherd K. R., Miller G. W., Liu Y., Wilson W. D., Xiao G., Blanchi B., Sun Y. E., Ye K., A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z.-H., Xiang J., Liu X., Yu S. P., Manfredsson F. P., Sandoval I. M., Wu S., Wang J.-Z., Ye K., Deficiency in BDNF/TrkB neurotrophic activity stimulates δ-secretase by upregulating C/EBPβ in Alzheimer’s disease. Cell Rep. 28, 655–669.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pistollato F., Cano S. S., Elio I., Vergara M. M., Giampieri F., Battino M., Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 74, 624–634 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Dodiya H. B., Kuntz T., Shaik S. M., Baufeld C., Leibowitz J., Zhang X., Gottel N., Zhang X., Butovsky O., Gilbert J. A., Sisodia S. S., Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp. Med. 216, 1542–1560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alam A., Leoni G., Quiros M., Wu H., Desai C., Nishio H., Jones R. M., Nusrat A., Neish A. S., The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat. Microbiol. 1, 15021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba0466/DC1