

A peptidoglycan editing mechanism confers protection against T6SS assaults without relying on immunity proteins.
Abstract
Peptidoglycan (PG) is essential in most bacteria. Thus, it is often targeted by various assaults, including interbacterial attacks via the type VI secretion system (T6SS). Here, we report that the Gram-negative bacterium Acinetobacter baumannii strain ATCC 17978 produces, secretes, and incorporates the noncanonical d-amino acid d-lysine into its PG during stationary phase. We show that PG editing increases the competitiveness of A. baumannii during bacterial warfare by providing immunity against peptidoglycan-targeting T6SS effectors from various bacterial competitors. In contrast, we found that d-Lys production is detrimental to pathogenesis due, at least in part, to the activity of the human enzyme d-amino acid oxidase (DAO), which degrades d-Lys producing H2O2 toxic to bacteria. Phylogenetic analyses indicate that the last common ancestor of A. baumannii had the ability to produce d-Lys. However, this trait was independently lost multiple times, likely reflecting the evolution of A. baumannii as a human pathogen.
INTRODUCTION
Peptidoglycan (PG) is a major component of the bacterial cell envelope. Layers of PG surround the cytoplasmic membrane, maintaining cell shape and providing resistance to osmotic stress. PG is composed of glycan chains of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) that are connected through MurNAc-attached peptides (1). Despite its rigidity, PG is necessarily a dynamic structure, requiring constant turnover to enable fundamental processes such as growth and cell division (2). PG synthesis is a multistep process. The PG precursor lipid II, which consists of a GlcNAc-MurNAc pentapeptide (l-Ala-d-iGlu-mDAP-d-Ala-d-Ala in Gram-negative bacteria) linked to a lipid carrier, is synthesized at the cytoplasmic membrane and subsequently translocated into the outer leaflet of the cytoplasmic membrane. Glycan chains are then polymerized by glycosyltransferases, and peptide cross-links are formed through transpeptidation reactions catalyzed by dd-transpeptidases and ld-transpeptidases.
Because of its essentiality, the bacterial cell wall is targeted by various potentially fatal threats, including attacks from bacterial competitors. Competition among Gram-negative bacteria is largely mediated by the type VI secretion system (T6SS) (3). The T6SS is a dynamic nanomachine structurally related to contractile phage tails, and it is used to deliver toxic effector proteins, including PG hydrolases, from an attacking cell (predator) to nearby competitors (prey) in a contact-dependent manner (4–6). Immunity to T6SS-dependent killing is accomplished by the expression of immunity proteins, which specifically bind and inactivate their cognate effector. The limited repertoire of immunity proteins encoded by one bacterium is insufficient to protect against the large diversity of T6SS effectors encoded by bacterial predators. Therefore, the primary role of immunity proteins is to avoid lethal interactions between sister cells (7–9). General resistance mechanisms to prevent T6SS-dependent killing by nonkin competitors remain poorly characterized (10).
Additional threats to the integrity of the bacterial cell wall include the host immune system, antibiotics, and osmotic stress (11–13). Constant exposure to these threats has exerted selective pressure favoring bacterial species capable of modifying their PG to evade or withstand fatal threats. These modifications can occur during PG precursor synthesis or after PG precursors are incorporated into the growing cell wall meshwork (14). PG modifications enabling immune evasion and resistance to antibiotics have been extensively studied and generally include chemical modifications to the glycan backbone or changes in pentapeptide composition or degree of cross-linking (12, 14–18). In addition, PG editing with noncanonical d-amino acids (NCDAAs) has been shown to make bacteria more resistant to osmotic stress (19). NCDAAs have been best characterized in Vibrio cholerae, which modifies up to ~5% of its PG subunits with d-Met during stationary phase. In addition to incorporating d-Met into its PG, this organism produces and secretes a wide variety of NCDAAs to millimolar concentrations (19). Some of the secreted NCDAAs inhibit the growth of diverse bacteria at physiological concentrations, making NCDAAs important mediators of interbacterial competition (20). It is currently unknown whether PG editing with NCDAAs plays an important role during bacterial warfare.
The Gram-negative bacterial pathogen Acinetobacter baumannii has a remarkable ability to withstand antibiotic treatment and persist in health care settings. As such, it is categorized by the World Health Organization and the U.S. Centers for Disease Control and Prevention as a critical priority for the research and development of novel antimicrobial therapies. Given the remarkable ability of A. baumannii to withstand a wide variety of environmental stressors (21), we investigated whether this organism modifies its PG during stationary phase, as has been reported in other bacterial species (1, 19, 22). To this end, we purified PG from Ab17978 grown to exponential phase (PGexp) or stationary phase (PGstat), digested it with muramidases, and analyzed the resulting muropeptides by high-performance liquid chromatography (HPLC). The muropeptide profile obtained from PGexp is similar to that of Escherichia coli (Fig. 1A and figs. S1 and S2). However, the HPLC chromatogram derived from PGstat showed five unique peaks (Fig. 1A). Mass spectrometry (MS) analysis identified these products as monomeric or cross-linked muropeptides containing a lysine residue (Lys) in the fourth (l-Ala-d-iGlu-mDAP-lysine, hereafter referred to as “Tri-Lys”) or fifth (l-Ala-d-iGlu-mDAP-d-Ala-lysine, hereafter referred to as “Tetra-Lys”) position (Fig. 1B, fig. S8, and table S1). Quantification of HPLC profiles showed that the Tri-Lys muropeptide and its cross-linked forms constitute ~24% of the total muropeptides of PGstat (Fig. 1A and table S2). Lys-containing muropeptides were also present in PGexp but in a considerably lower amount (~6% of total muropeptides). In contrast, Tetra-Lys muropeptides were unique to PGstat and accounted for ~10% of the total muropeptides. Together, our results indicate that during stationary phase, Ab17978 is capable of modifying about one-third of its PG with Lys.
Fig. 1. Ab17978 incorporates d-Lys into its PG at stationary phase.
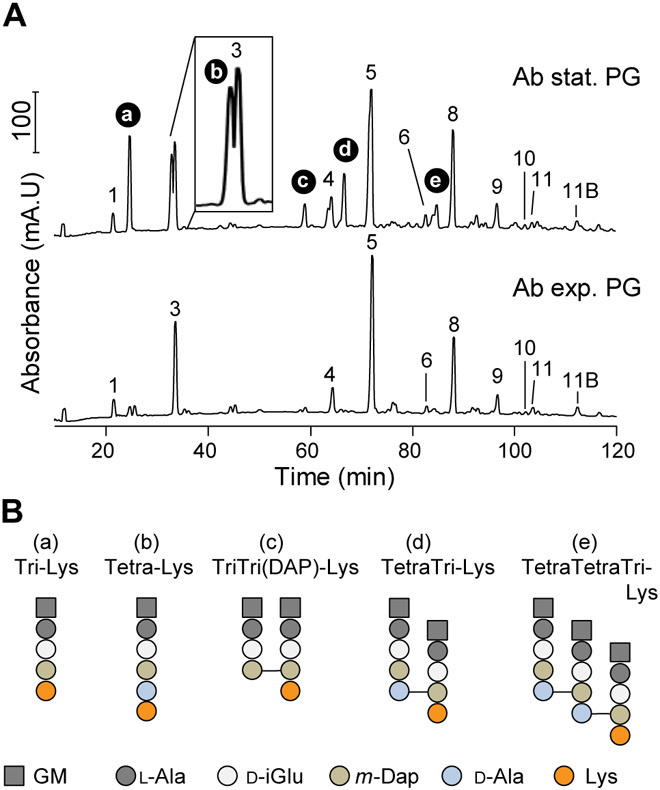
(A) Chromatograms of purified muropeptides from Ab17978 during stationary (stat.) and exponential (exp.) phase. Main muropeptides (no. 1-11B) are numbered according to Boll et al., 2016 (43). d-Lys–containing muropeptides (a to e) were identified by MS. Proposed muropeptide structures are shown in fig. S2. mA.U, milli absorbance units.(B) Proposed structures of d-Lys–modified muropeptides a to e. GM, N-acetylglucosamine-N-acetylmuramitol; l-Ala, l-alanine; d-iGlu, d-isoglutamate; m-Dap, meso-diaminopimelic acid; Lys, lysine; d-Ala, d-alanine. “Tri” or “Tetra” indicates the number of amino acids in the peptides chain preceding Lys.
MS is unable to differentiate l-Lys from d-Lys. Previous work showed that bacteria that produce and incorporate NCDAAs into their PG meshwork also secrete NCDAAs into the culture supernatant at millimolar concentrations (19, 22). Thus, d-Lys secretion by Ab17978 would suggest editing with the d-enantiomer. We developed a colorimetric assay using the d-Lys–specific oxidase AmaD from Pseudomonas putida to detect and quantify the concentration of d-Lys in culture supernatants of Ab17978 (Fig. 2A) (23). Using our AmaD assay, we determined that Ab17978 produces and secretes d-Lys into culture supernatants to a concentration of ~0.3 mM (Fig. 2B), which is comparable to levels of NCDAA secreted by other bacterial species (19, 24). d-Lys secretion has not been reported in Acinetobacter, leading us to investigate how d-Lys is produced. Amino acid racemases are enzymes that catalyze the conversion between l-amino acids and d-amino acids. Ab17978 encodes five putative amino acid racemases, only one of which is predicted to have a signal peptide (ACX60_11360, hereafter referred to as “RacK”). To identify the racemase involved in d-Lys production, we constructed mutant strains of Ab17978 lacking each racemase and determined the levels of d-Lys in their culture supernatants. Deletion of only one racemase, RacK, completely abrogated d-Lys secretion (fig. S3). Complementation of the racK gene (RacK+) restored d-Lys secretion to levels comparable with wild-type (WT) Ab17978 (Fig. 2B). Having identified RacK as responsible for d-Lys production, we determined whether d-Lys was the Lys enantiomer incorporated into the PG of Ab17978 during stationary phase. We analyzed muropeptides from Ab17978 WT, ΔRacK, and RacK+ by HPLC. PGstat from ΔRacK lacked the peaks corresponding to Tri-Lys and Tetra-Lys subunits, all of which were present in the WT and RacK+ strains (Fig. 2C). Together, our results demonstrate that Ab17978 secretes d-Lys to millimolar concentrations and that PG editing is dependent on the production of d-Lys by RacK. Because RacK is predicted to localize to the periplasm, we propose that PG editing with NCDAAs in Ab17978 occurs in the periplasm, as has been suggested for V. cholerae (19, 22). Previous transcriptomics study in Ab17978 showed that racK was also up-regulated in biofilms compared to cells in exponential phase (25).
Fig. 2. Racemase RacK is responsible for d-Lys secretion and PG editing.
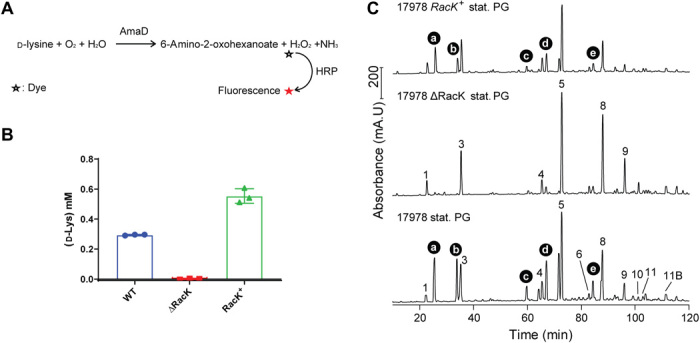
(A) Overview of the colorimetric assay used to quantify d-Lys secretion. Purified d-Lys oxidase AmaD from P. putida catalyzes the oxidative deamination of d-Lys to release 6-amino-2-oxohexanoate, hydrogen peroxide (H2O2), and ammonium (NH3). H2O2 produced was then quantified by treatment with horseradish peroxidase (HRP) and a fluorophore dye. The amount of fluorescence is converted to d-Lys concentration through a standard curve. (B) Quantification of d-Lys present in supernatant fractions of Ab17978 WT, 17978ΔRacK, and the complemented strain (17978 RacK+). Data represent means ± SD of three biological replicates. (C) Chromatograms of purified, stationary phase muropeptides from the indicated strains. Muropeptide structures are shown in fig. S2.
Virtually all bacteria have the ability to incorporate NCDAAs, either produced by their own racemases or present in the environment, into their PG meshwork (14, 26, 27). This can be accomplished by the activity of dd- or ld-transpeptidases (22, 27–29). Previous work has demonstrated that PG editing with NCDAAs affects cell physiology and decreases both dd– and ld–cross-links, likely by competing with transpeptidase activity (19, 22, 28–30). Consistently, PG of Ab17978 WT and RacK+ showed a lower percentage of muropeptides in cross-linking compared with Ab17978 ΔRacK, indicating that d-Lys interferes with the PG cross-linking activity of transpeptidases (table S4). Previous studies have shown that PG cross-linking enhances a bacterium’s ability to withstand stress (31, 32). Thus, we next tested whether PG editing with d-Lys affected the ability of Ab17978 to withstand a variety of environmental stressors. We found that Ab17978 ΔRacK was nearly indistinguishable from WT in growth under pH and osmotic stress as well as cell morphology (fig. S4), which suggest an alternative role for d-Lys in Acinetobacter. NCDAAs are considered part of the warfare between bacterial species, as certain secreted d-amino acids can inhibit the growth of diverse bacteria (20). However, we found that the concentration of d-Lys present in the spent media of Ab17978 was not sufficient to inhibit the growth of Pseudomonas aeruginosa PAO1, Acinetobacter nosocomialis strain M2 (M2), or A. baumannii strain 19606 (Ab19606) (fig. S5).
We then hypothesized that PG editing with d-Lys could constitute a protective mechanism against T6SS-mediated attacks from nonkin bacteria. In this model, PG editing masks the target of T6SS PG hydrolase effectors, similar to how bacteria become resistant to antibiotics by modifying the structure of the antibiotic target. The H1-T6SS of P. aeruginosa PAO1 delivers at least two PG hydrolase effectors into target cells: the amidase (or d,l-endopeptidase) Tse1 and the muramidase Tse3 (4). Thus, to test our hypothesis, we performed bacterial competition assays with P. aeruginosa PAO1 as the predator and Ab17978 WT or ∆RacK as the prey. We found that ∆RacK was ~15-fold more susceptible to T6SS killing compared with WT Ab17978 (Fig. 3A). Consistently, UPAB1 RacK+ showed ~25-fold higher survival than WT UPAB1 when co-incubated with PAO1 (Fig. 3A and fig. S6). Similarly, we observed that PG editing by d-Lys increased the survival of A. baumannii against Serratia marcescens and A. nosocomialis M2 (Fig. 3A). Collectively, these results indicate that PG editing with d-Lys provides A. baumannii immunity against T6SS-dependent attacks from diverse bacteria.
Fig. 3. PG editing protects A. baumannii from T6SS amidase effector.
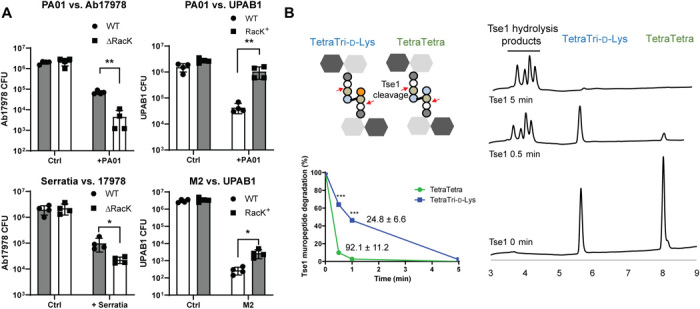
(A) PG editing with d-Lys provides protection to T6SS-dependent attack from P. aeruginosa PAO1. Competition assay between PAO1, S. marcescens (Serratia), or A. nosocomialis M2 as predator and A. baumannii strains expressing (Ab17978 WT or UPAB1 RacK+) or not expressing (Ab17978ΔRacK or UPAB1 WT) RacK as prey. Bar graphs represent the means ± SD from four biological replicates. (B) Nonedited (TetraTetra) and edited muropeptides (TetraTri-d-Lys) were treated together with purified PAO1 amidase effector Tse1. Reaction products were analyzed by HPLC at time t = 0 (Ctrl), t = 0.5 min, and t = 5 min. Undigested muropeptides were quantified, and specific activity of Tse1 against TetraTetra and TetraTri-d-Lys was calculated. Specific activity values shown on graph are expressed in milligrams of muropeptide degraded/min/Tse1 concentration. Data present means ± SD of two biological replicates. Statistical analyses were performed using the Student’s unpaired t test, *P < 0.05, **P < 0.01, and ***P < 0.001.
To gain better insight into the mechanism of PG editing–based immunity to T6SS-mediated attacks, we compared the activity of P. aeruginosa PAO1 Tse1 toward nonedited (TetraTetra) and d-Lys–edited (TetraTri-d-Lys) dimeric muropeptides. A mix of both muropeptides was incubated with purified Tse1, and samples were taken at different time points and analyzed by HPLC (Fig. 3B). At 0.5 min, we found that most of the TetraTetra muropeptides were hydrolyzed by Tse1, whereas nearly 40% of TetraTri-d-Lys remained uncleaved. We observed a ~4-fold decrease in the specific activity of Tse1 toward the d-Lys–modified muropeptide compared with the nonedited muropeptide, consistent with the results of our bacterial competition assays (Fig. 3A). Together, these results support the hypothesis that PG editing in Ab17978 provides an immunity protein–independent mechanism to prevent T6SS-dependent killing by masking the target PG from the injected effector.
Some bacterial species modify their PG to counteract the host innate immune response (12, 33). We examined whether the presence of RacK would affect A. baumannii pathogenesis in the murine acute pneumonia model of infection (34). Ab17978 or UPAB1 strains either encoding or lacking RacK were introduced by intranasal inoculation, and bacterial burdens were enumerated 36 hours after infection. Unexpectedly, strains unable to produce d-Lys showed substantially higher bacterial burdens [1 to 2 log colony-forming units (CFU)] in the kidney, heart, and liver, suggesting that d-Lys production is disadvantageous for bacterial dissemination (Fig. 4A). Recent findings have suggested that the d-amino acid oxidase (DAO), expressed by human neutrophils, macrophages, and epithelial cells, contributes to host defense against various pathogens through the production of H2O2, a by-product of the DAO-dependent oxidative deamination of d-amino acids (24, 35, 36). Thus, we hypothesized that d-Lys–producing bacteria would be more susceptible to DAO-dependent killing. To test this, we incubated stationary phase Ab17978 or UPAB1 strains either encoding or lacking RacK with purified DAO and enumerated surviving CFU after 4.5 hours. DAO showed significant antibacterial activity against A. baumannii strains harboring RacK, resulting in a 103-fold reduction in CFU compared with strains not producing d-Lys (Fig. 4B). The addition of purified catalase, an enzyme that eliminates H2O2, provided full protection against DAO, indicating that A. baumannii killing by DAO is H2O2-dependent. Together, our results demonstrate that d-Lys production is detrimental to pathogenesis, at least in part, due to DAO activity.
Fig. 4. d-Lys secretion impairs A. baumannii virulence.
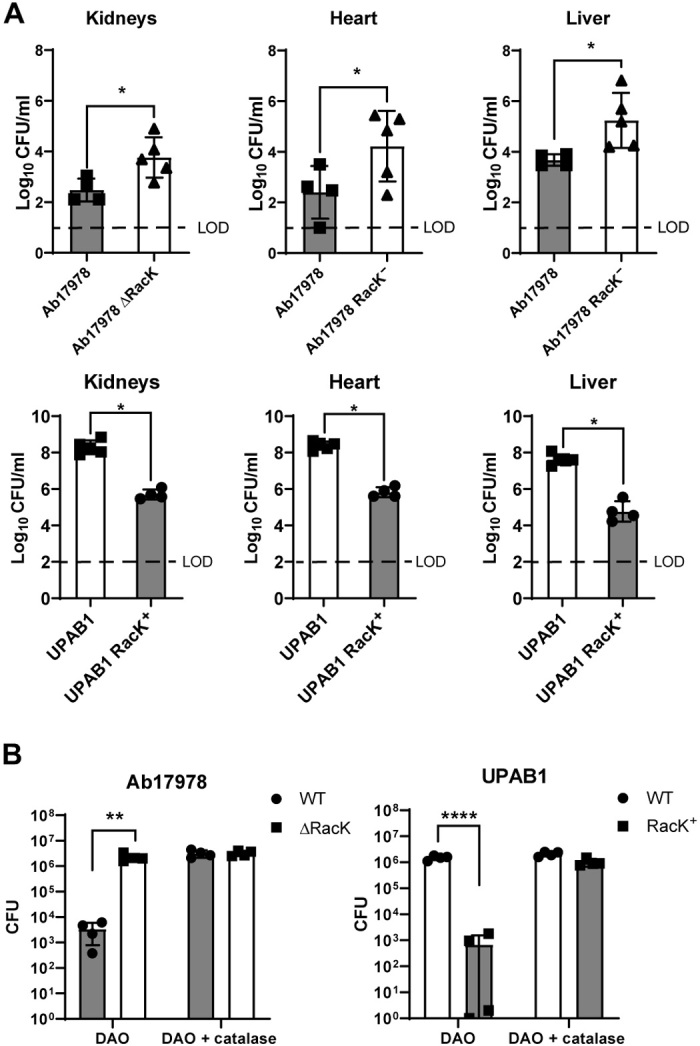
(A) RacK is detrimental in a murine model of acute pneumonia. Mice were intranasally inoculated with either the Ab17978 WT or ΔRacK (left) or the UPAB1 WT or RacK+ (right). Following 36 hours of infection, the bacterial burdens of the kidneys, heart, liver, spleen, lungs, and blood were determined. Each symbol represents an individual mouse. Statistical analyses were performed using the Mann-Whitney U test, and LOD represents the limit of detection. (B) DAO has increased antibacterial activity on A. baumannii strains having RacK. WT and ΔRacK Ab17978 (left) or WT and RacK+ UPAB1 (right) were incubated with DAO or DAO + catalase for 4.5 hours, and CFU were enumerated. Bar graphs represent the means ± SD of four biological replicates. Statistical analyses were performed using the unpaired Student’s t test, *P < 0.05, **P < 0.01, and ****P < 0.0001.
The benefit of PG editing in bacterial competition and detriment to pathogenesis present an interesting evolutionary paradox, making it difficult to predict whether natural selection would favor Acinetobacter strains capable or incapable of modifying their PG. To this end, we performed a phylogenetic profiling analysis of RacK across 3052 Acinetobacter strains representing 53 species. We found that RacK is largely confined to the Acinetobacter calcoaceticus/baumannii (Acb)–complex, occurring only sporadically in other species (fig. S7). Within the Acb-complex, RacK is prevalent in the two earliest branching lineages, suggesting its presence in the last common ancestor of the Acb-complex (Fig. 5). Furthermore, RacK is always flanked by the same four genes, and this gene order is maintained irrespective of the presence of RacK (Fig. 5). A phylogenetic analysis provided no evidence that the evolutionary history differs between the four flanking genes and RacK, indicating that the five genes were likely present in the last common ancestor of A. baumannii. We found that only 7% of A. baumannii strains encode RacK, and these strains are distributed across different clades in the A. baumannii phylogeny (Fig. 5 and fig. S6). Thus, our results indicate that RacK was encoded by the last common ancestor of the Acb-complex and was lost multiple times independently during A. baumannii evolution.
Fig. 5. Evolutionary history of A. baumannii RacK.
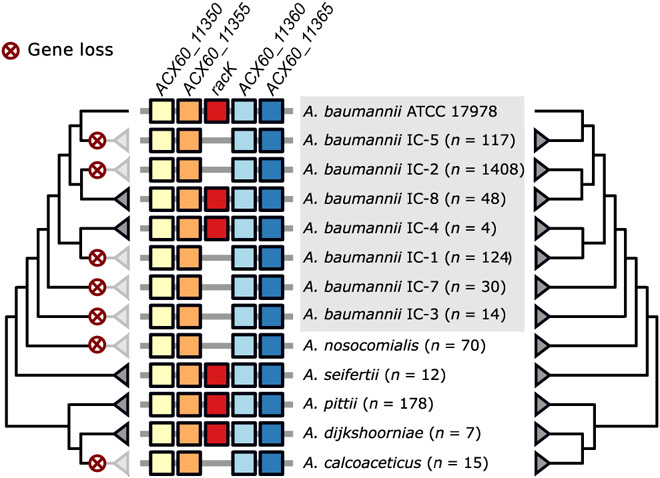
The presence of RacK (red boxes) is limited to individual lineages within A. baumannii, while it is ubiquitously present in the closely related species Acinetobacter seifertii, Acinetobacter pitti, and Acinetobacter dijkshoorniae. Whenever present, RacK is embedded within the same four flanking genes (colored boxes) throughout the Acb complex. This gene order is maintained in the species and strains where RacK is missing. The phylogenetic relationships of RacK (left tree) and of the four flanking genes (right tree) are congruent, indicating that the contemporary patchy distribution of RacK is a consequence of recurrent and lineage specific gene loss (marked by the red crosses). ATCC, American Type Culture Collection.
Our results are summarized in Fig. 6. During stationary phase, the periplasmic racemase RacK catalyzes the conversion of l-Lys to d-Lys. Once produced, d-Lys is incorporated into the PG of Ab17978 by Ldt (l,d-transpeptidase) and penicillin-binding proteins (PBPs) (22, 28–30). PG editing with d-Lys confers immunity to T6SS-dependent attacks by reducing the specific activity of PG hydrolase effectors, such as Tse1. Because T6SS effectors recognize and cleave conserved amide bonds in PG, we propose that certain types of PG editing constitute an immunity mechanism against T6SS-dependent killing. Previous work reported that l-DAP amidation in the PG of the environmental bacterium Gluconobacter frateurii resulted in reduced in vitro digestion by T6SS endopeptidases compared with nonmodified PG (18). This mechanism of T6SS immunity is independent of immunity proteins, which bind with high specificity to their cognate T6SS effectors. In contrast, PG editing–mediated immunity might protect against various T6SS effectors that recognize canonical PG structures. We therefore propose that PG editing provides A. baumannii with a type of “innate immunity” against diverse bacterial competitors. It is tempting to speculate that additional common targets for T6SS effectors (or T7SS effectors in Gram-positive bacteria) are also modified to evade the toxicity of these effectors. However, PG modification may not protect against all bacteria, as the degree of protection may depend on the specific effectors secreted by different species.
Fig. 6. Proposed model of d-Lys production role in Acinetobacter spp.
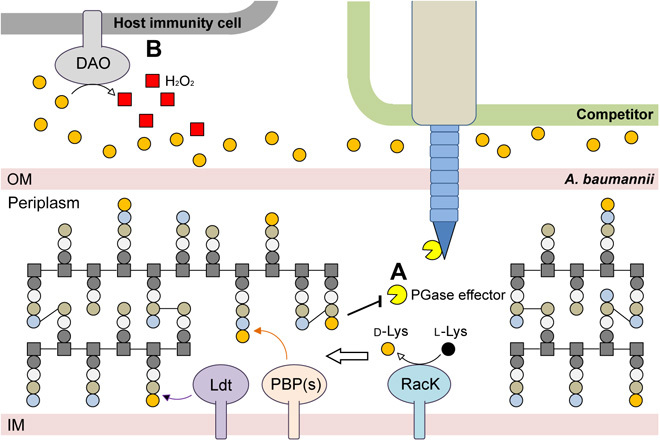
(A) The lysine racemase RacK converts l-Lys to d-Lys in the periplasm. The presence of d-Lys in the periplasmic space leads to PG editing by Ldts and PBPs in competition with their canonical cross-link activity. This PG editing mechanism provides a form of innate immunity against foreign PG targeting T6SS. PGase, Peptidoglycanase (B) As a trade-off, secretion of d-Lys during infection increases host DAO activity and thus decreases A. baumannii virulence. PGase, peptidoglycanase; OM, outer-membrane; IM, inner-membrane.
Our data show that d-Lys production is unfavorable for A. baumannii pathogenesis. We have shown that, in vitro, purified DAO is able to recognize the d-Lys secreted by A. baumannii and produce enough H2O2 to kill bacteria expressing RacK. DAO is known to be produced by key players in the initial immune response against A. baumannii infections (36–39). Despite the competitive advantages conferred by the expression of RacK during interbacterial competition, modern clinical Ab (Acinetobacter baumannii) strains and other Acinetobacter species of the Acb-complex have relinquished their copy of the racK gene. Strains from the most common lineages of clinical A. baumannii strains, such as international clone 1 (IC1), IC2, and IC3, do not carry the racK gene. This is reminiscent of the loss of the T6SS via genetic disruptions to the T6SS gene locus or the accumulation of inactivating point mutations in ~40% of A. baumannii clinical isolates (40, 41). Both PG editing and a functional T6SS are crucial in the environment but appear to impose a fitness cost during infection. These examples illustrate the multiple adaptations that A. baumannii has undertaken to become a pathogen capable of successfully infecting the human host.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in table S5. Unless otherwise noted, strains were grown in lysogeny broth (LB) liquid medium at 37°C with shaking (200 rpm). The antibiotics rifampicin (150 μg/ml), irgasan (25 μg/ml), kanamycin (7.5 or 50 μg/ml), gentamicin (20 μg/ml), chloramphenicol (15 μg/ml), carbenicillin (100 or 200 μg/ml), and zeocin (50 μg/ml) were added when necessary. Spontaneous rifampicin-resistant mutant strains were obtained by plating an overnight culture on LB agar with rifampicin.
PG isolation and analysis
PG from exponential and stationary cells from A. baumannii strains was isolated and analyzed by HPLC as previously described (41, 42). Briefly, A. baumannii strains were grown at 37°C in 500 ml of LB media to a final OD600 (optical density at 600 nm) of 0.6 for exponential and grown for 24 hours for stationary phase samples. Cells were collected by centrifugation for 15 min at 4°C and 7000g. Cell pellets were resuspended in 50 ml of cold water and lysed by dropwise addition to 50 ml of boiling 8% SDS under vigorous stirring. Samples were boiled for further 30 min to ensure complete solubilization of the membranes and degradation of the high–molecular weight DNA. Crude PG samples were collected by ultracentrifugation for 60 min at 110,000g at 25°C. Pellets were washed several times with phosphate buffer (pH 6.0) to remove SDS. Crude PG samples were treated with α-amylase (1 mg/ml) for 1 hour at 37°C and then with Pronase (2 mg/ml) for overnight at 60°C to remove trapped high–molecular weight glycogen and PG-associated proteins, respectively. Further PG preparation steps were performed as previously described (41). Briefly, muropeptides were released from PG by the muramidase cellosyl (Hoechst, Frankfurt am Main, Germany), reduced by sodium borohydride, and separated on a 250 mm by 4.6 mm 3-μm ProntoSIL 120-3-6C18 AQ reversed-phase column (Bischoff, Leonberg, Germany). The eluted muropeptides were detected by their absorbance at 205 nm. The PG composition from exponentially and stationary growing cells was analyzed in two biological replicates. Muropeptides were assigned according to their retention times of the known muropeptides from E. coli and A. baumannii (42) and quantified using the Laura software (LabLogic Systems).
MS/MS analysis
New muropeptide fractions with retention times other than standard muropeptides were collected and analyzed by tandem MS (MS/MS) as previously described (43). Linear trap quadrupole-Fourier transform MS analyses revealed the presence of d-Lys in the collected muropeptide fractions c to e (fig. S7). Table S1 shows the proposed structures and the theoretical neutral and measured neutral atomic mass units. Muropeptide fractions a and b were also acetylated using an established protocol (17) prior MS/MS analysis to confirm the number of amino groups.
Construction of A. baumannii mutant and complement strains
The primers used in this study are listed in table S5. Mutants were constructed as described previously (44). Briefly, an antibiotic resistance cassette was amplified with primers pair P1_kan Fwd-Rev (Integrated DNA Technologies) with homology to the flanking regions of the racK gene with additional 3′ 18 to 25 nucleotides of homology to the FRT (FLP recognition target) site–flanked kanamycin resistance cassette from plasmid pKD4. This polymerase chain reaction (PCR) product was electroporated into competent Ab17978 carrying pAT04, which expresses the RecAB recombinase (44). Mutants were selected on kanamycin (10 μg/ml), and integration of the resistance marker was confirmed by PCR. To remove the kanamycin resistance cassette, electrocompetent mutants were transformed with pAT03 plasmid, which expresses the FLP recombinase. To create Ab17978 RacK+ strain, the racK gene was cloned into pSH vector and under an arabinose inducible promotor, and then the pSH_RacK plasmid was electroporated into Ab17978 ΔRacK strain. The racK gene was introduced into UPAB1 via a four-parent conjugal strategy as described by Kumar et al. (45). Briefly, 100 μl of stationary cultures were normalized to an OD600 of 2.0 of each recipient strain, and HB101(pRK2013), EC100D(pTNS2), and EC100D containing the pUC-miniTn7-racK plasmid were added to 600 μl of warm LB. Each suspension was washed twice by centrifugation at 7000g, followed by resuspension of the bacterial pellet in 1 ml of warm LB. On the final wash, the bacterial pellet was resuspended in 25 μl of LB, and the suspension was spotted on a prewarmed low-salt LB agar plate and incubated overnight at 37°C. The bacteria were scraped from the plate and resuspended in 1 ml of LB and vortexed, and serial dilutions were plated on L agar plates supplemented with chloramphenicol to select against E. coli strains and kanamycin or zeocin to select for A. baumannii strains that had received the mini-Tn7 constructs. To verify that mini-Tn7 had successfully transposed downstream of the glmS2 gene, it is amplified by PCR using primers pair UPAB1_check Fwd-Rev, and the products were verified by sequencing. The strains were saved as Ab17978 RacK+ and UPAB1 RacK+.
Protein expression and purification
The amaD gene was PCR-amplified from P. putida and was cloned in pET22b+ vector void of the pelB sequence using Hi-Fi DNA Assembly mix (New England Biolabs). The pET22b+_amaD vector was electroporated into E. coli Rosetta II (Invitrogen) and selected on carbenicillin. The bacteria were grown in LB to an OD600 of 0.6, and AmaD protein expression was induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside for 3 hours at 30°C. Cells were harvested and lysed with two rounds of a cell disruptor using 35 k.p.s.i. (Constant Systems Ltd., Kennesaw, GA). Cell lysates were clarified at 10,000 rpm for 10 min and then were passed over a nickel-nitrilotriacetic acid-agarose column Ni-NTA (GoldBio, St. Louis, MO). Protein was eluted with 300 mM imidazole, buffer-exchanged using Sephadex G-25 PD10 column, and stored in 50 mM tris-HCl (pH 8.0) and 150 mM NaCl at −80°C.
P. aeruginosa Tse1 protein was obtained as described (18). LdtA from V. cholerae was obtained as described (22).
AmaD enzymatic assay
Bacteria cultures (3 ml) grown for 20 hours were centrifuged at 10,000g, and supernatants (0.5 ml) were collected. The supernatants were deproteinized using Amicon Ultra 0.5-ml Centrifugal Filters with 3000 MWCO (molecular weight cut-off). Samples were incubated with AmaD (0.4 mg/ml) for 1 hour at 37°C. Hydrogen peroxide coproduct was then measured using Hydrogen Peroxide Assay Kit (Abcam) as instructed by the manufacturer.
Muropeptide isolation
TetraTetra and TetraTri-d-Lys were purified by HPLC on an Aeris peptide column (250 mm by 4.6 mm; 3.6-μm particle size; Phenomenex, USA), concentrated, and desalted using a water-methanol gradient on the same column before MS analysis. TetraTetra was obtained from muramidase treatment of E. coli sacculi (46). Chromatographic analyses of muropeptides were performed on ACQUITY Ultra Performance Liquid Chromatography (UPLC) BEH C18 column (130 Å, 1.7 μm, 2.1 mm by 150 mm; Waters), and peptides were detected at Abs. (absorbance) 204 nm using ACQUITY UPLC UV-Visible Detector. Muropeptides were separated using a linear gradient from buffer A (0.1% of formic acid in water) to buffer B (0.1% of formic acid in acetonitrile) for 18 min and flow (0.25 ml/min). Muropeptide identity was confirmed by MS/MS analysis using a Xevo G2-XS QTof system (Waters Corporation, USA). Quantification of muropeptides was based on their relative abundances (relative area of the corresponding peak).
In vitro reaction
TetraTri-d-Lys was obtained by mixing 10 μg of TetraTetra with 20 mM d-Lys and 1 μM LdtA for 4 hours in 20 mM tris (pH 8.0). PG digestion was analyzed over time (0.5, 1, and 5 min) using a final concentration of purified Tse1 enzyme (either 0.01 or 0.02 mg/ml) from P. aeruginosa at 37°C in 20 mM tris-HCl (pH 8.0). The specific activity was calculated from two independent experiments in duplicates (n = 4). The dataset resulting from the lower enzyme concentration (0.01 mg/ml; n = 2) was plotted to illustrate the differential substrate consumption by Tse1 on canonical versus noncanonical dimers. Individual muropeptides were quantified from their integrated areas using samples of known concentration as standards. Reactions were terminated by inactivation at 100°C for 10 min and centrifuged at 14,000g for 30 min to discard the coagulated Tse1. Next, Tse1-treated muropeptides were analyzed by UPLC. Determination of the extent of Tse1-dependent degradation was performed by comparing the integration areas with respect to a nontreated sample.
T6SS competition assay
Competition assays were performed as previously described (47). Briefly, predator and prey overnight cultures were pelleted, washed three times in fresh LB, and resuspended at an OD600 of 1.0. The cultures were mixed at a predator:prey ratio of 5:1 (PAO1:Ab or M2:Ab) or 10:1 (Serratia:Ab), and 10 μl of drops was spotted on a LB 3% agar plate. After 4 hours at 37°C, the spots were harvested, resuspended in 0.7 ml of LB, and serially diluted and plated on rifampicin or gentamicin LB agar plates to determine the number of surviving prey cells. In parallel, the number of surviving PAO1 predator was also enumerated on irgasan LB agar plates.
Murine model of A. baumannii acute pneumonia
All infection experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee and are in compliance with the guidelines set by the Animal Welfare Act, the National Institutes of Health, and the American Veterinary Medical Association. Lung infection experiments were performed essentially as previously described (32). Briefly, before infection, overnight cultures were subcultured 1:100 in 30 ml of liquid LB media and incubated at 37°C with shaking. Bacteria in mid-exponential phase growth were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), and resuspended in PBS to the same concentration. Anesthetized 7- to 9-week-old C57BL/6 mice (the Jackson laboratory) were inoculated intranasally with 4 × 108 to 6 × 108 CFU of Ab17978, UPAB1, or their respective isogenic mutants in 35 μl of PBS. Lungs, livers, spleens, kidneys, hearts, and blood were aseptically harvested from mice euthanized at 36 hours after infection. Organs were homogenized and then serially diluted and plated to LB agar to determine bacterial burdens.
DAO antimicrobial in vitro assay
Bacteria cultures (3 ml) grown for 20 hours were centrifuged at 10,000g, and supernatants (900 μl) were collected. One hundred microliters of fresh LB was added as well as purified porcine kidney DAO and catalase (Millipore Sigma) to 50 and 25 μg/ml, respectively. The bacterial pellets were resuspended and added to the supernatant-LB-DAO or DAO/catalase mixtures to an OD600 of 0.005. After 4.5 hours of incubation at 37°C, bacteria were serially diluted and spotted on LB agar plates for enumeration.
Genome data
Genomic data used in this study were obtained from the National Center for Biotechnology Information reference sequencing database (version 87, accessed 13 March 2018). All 3052 Acinetobacter spp. genome assemblies together with the corresponding coding and protein sequences were downloaded into a local database and integrated in this analysis. To reduce the computational burden for tree reconstruction, we compiled a core set of 232 Acinetobacter spp. encompassing all described IC types of A. baumannii as well as type and reference strains of 53 validly named species.
Phylogenetic profiling and synteny analysis
For all protein sequences, orthologous groups (OGs) across the 3052 genomes were inferred using a tandem of OMA (orthologous matrix) standalone v. 2.2.0 (https://doi.org/10.1093/nar/gkx1019) and HaMStR v. 13.2.9 (https://github.com/BIONF/hamstr, https://doi.org/10.1186/1471-2148-9-157) as previously described (https://doi.org/10.1080/21505594.2018.1558693). The synteny of racK and its two flanking genes was inferred with the genomic loci of the corresponding orthologs using the gene order and orientation of the strain American Type Culture Collection (ATCC) 17978 as a reference.
Phylogenetic tree reconstruction
The protein sequences of racK and its flanking genes in the core set were aligned for each OG individually with MAFFT-linsi v7.407 (https://doi.org/10.1093/nar/gkt389). The resulting multiple sequence alignment together with the corresponding coding sequences served then as input for PAL2NAL v.14 (https://doi.org/10.1093/nar/gkl315) (with option “-codontable 11”) to generate corresponding codon alignments. Individual or concatenated MSAs (multiple sequence alignment) were then used for a maximum likelihood tree reconstruction with IQ-Tree v1.6.12 (https://doi.org/10.1093/nar/gkw256) performing 1000 nonparametric bootstrap replicates (https://doi.org/10.1093/molbev/msx281). The best-fitting substitution model was determined with the model-testing routines implemented into IQ-Tree (https://doi.org/10.1038/nmeth.4285). Topology tests were performed using the weighted and unweighted Shimodaira-Hasegawa test (https://doi.org/10.1093/oxfordjournals.molbev.a026201) as implemented in IQ-Tree.
MLST classification and IC assignments
We predicted for each of the 3052 Acinetobacter spp. strains the sequence type with two different multilocus sequence typing (MLST) schemes, Oxford (https://doi.org/10.1128/JCM.43.9.4382-4390.2005) and Pasteur (https://doi.org/10.1371/journal.pone.0010034), that we obtained from the PubMLST website (http://pubmlst.org/abaumannii/) using MLSTcheck v2.1.17 (https://doi.org/10.21105/joss.00118). Strains of A. baumannii were assigned to an IC type if predicted sequence types and IC were unambiguously linked in the literature (https://doi.org/10.1016/j.ijantimicag.2019.03.019, https://doi.org/10.1016/j.meegid.2019.103986, https://doi.org/10.1016/j.watres.2018.04.057, https://doi.org/10.1038/s41426-018-0127-9, https://doi.org/10.1111/1462-2920.13931, https://doi.org/10.1186/1471-2180-13-234, https://doi.org/10.1371/journal.pone.0010034, https://doi.org/10.1371/journal.pone.0153014, https://doi.org/10.1371/journal.pone.0179228, https://doi.org/10.3389/fmicb.2019.00930, and https://doi.org/10.1186/s13059-015-0701-6).
Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., La Jolla, CA). For the murine acute pneumonia model, data were log-transformed and analyzed for Gaussian distribution using the D’Agostino-Pearson omnibus normality test. For normally distributed datasets, Student’s unpaired t tests were used. For datasets with unknown distribution type, nonparametric Mann-Whitney U tests were used.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) grant R21 AI137188. Work in the Vollmer lab was supported by the Wellcome Trust grant 101824/Z/13/Z and the U.K. Medical Research Council within the Antimicrobial Resistance Cross-Council Initiative collaborative grant MR/N002679/1. Work in the Cava lab was supported by the Swedish Research Council (VR), the Knut and Alice Wallenberg Foundation (KAW), the Laboratory of Molecular Infection Medicine Sweden (MIMS), and the Kempe Foundation. Work in the Skaar lab was supported by the NIAID grant R01 AI101171. Work in the Levin lab was supported by the NIH grant GM127331 and the NSF grant DGE-1745038. This study was supported by a grant by the German Research Foundation (DFG) in the scope of the Research Group FOR2251 “Adaptation and persistence of A. baumannii.” Grant ID EB-285-2/2 (“The evolutionary origins of genomic innovation in the human pathogen A. baumannii”). Author contributions: Conceived and designed the experiments: N.-H.L., K.P., A.E., J.R.S., G.D.V., B.D., E.A.M., S.W.H., J.G., I.E., E.P.S., F.C., W.V., and M.F.F. Performed the experiments: N.-H.L., K.P., A.E., J.R.S., G.D.V., J.L., B.D., E.A.M., J.G., and S.W.H. Analyzed the data: N.-H.L., K.P., A.E., J.R.S., G.D.V., B.D., E.A.M., J.G., P.A.L., I.E., E.P.S., F.C., W.V., and M.F.F. Wrote the paper: N.-H.L., J.L., and M.F.F. All authors commented on the manuscript. Competing interests: E.P.S. is a consultant for Shionogi. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eabb5614/DC1
REFERENCES AND NOTES
- 1.Vollmer W., Blanot D., De Pedro M. A., Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Typas A., Banzhaf M., Gross C. A., Vollmer W., From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell A. B., Peterson S. B., Mougous J. D., Type VI secretion system effectors: Poisons with a purpose. Nat. Rev. Microbiol. 12, 137–148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell A. B., Hood R. D., Bui N. K., LeRoux M., Vollmer W., Mougous J. D., Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou S., Bui N. K., Russell A. B., Lexa K. W., Gardiner T. E., LeRoux M., Vollmer W., Mougous J. D., Structure of a peptidoglycan amidase effector targeted to gram-negative bacteria by the type VI secretion system. Cell Rep. 1, 656–664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basler M., Pilhofer M., Henderson G. P., Jensen G. J., Mekalanos J. J., Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483, 182–186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitney J. C., Chou S., Russell A. B., Biboy J., Gardiner T. E., Ferrin M. A., Brittnacher M., Vollmer W., Mougous J. D., Identification, structure, and function of a novel type VI secretion peptidoglycan glycoside hydrolase effector-immunity pair. J. Biol. Chem. 288, 26616–26624 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srikannathasan V., English G., Bui N. K., Trunk K., O’Rourke P. E. F., Rao V. A., Vollmer W., Coulthurst S. J., Hunter W. N., Structural basis for type VI secreted peptidoglycan dl-endopeptidase function, specificity and neutralization in Serratia marcescens. Acta Crystallogr. D Biol. Crystallogr. 69, 2468–2482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulthurst S., The Type VI secretion system: A versatile bacterial weapon. Microbiology 165, 503–515 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Toska J., Ho B. T., Mekalanos J. J., Exopolysaccharide protects Vibrio cholerae from exogenous attacks by the type 6 secretion system. Proc. Natl. Acad. Sci. U.S.A. 115, 7997–8002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Höltje J.-V., Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62, 181–203 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis K. M., Weiser J. N., Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect. Immun. 79, 562–570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohanski M. A., Dwyer D. J., Collins J. J., How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 8, 423–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yadav A. K., Espaillat A., Cava F., Bacterial strategies to preserve cell wall integrity against environmental threats. Front. Microbiol. 9, 2064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melnyk J. E., Mohanan V., Schaefer A. K., Hou C.-W., Grimes C. L., Peptidoglycan modifications tune the stability and function of the innate immune receptor Nod2. J. Am. Chem. Soc. 137, 6987–6990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bera A., Herbert S., Jakob A., Vollmer W., Götz F., Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 55, 778–787 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Vollmer W., Tomasz A., The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 275, 20496–20501 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Espaillat A., Forsmo O., El Biari K., Björk R., Lemaitre B., Trygg J., Cañada F. J., de Pedro M. A., Cava F., Chemometric analysis of bacterial peptidoglycan reveals atypical modifications that empower the cell wall against predatory enzymes and fly innate immunity. J. Am. Chem. Soc. 138, 9193–9204 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Lam H., Oh D.-C., Cava F., Takacs C. N., Clardt J., de Pedro M. A., Waldor M. K., d-amino acids govern stationary phase cell wall remodeling in bacteria. Science 325, 1552–1555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez L., Aliashkevich A., De Pedro M. A., Cava F., Bacterial secretion of D-arginine controls environmental microbial biodiversity. ISME J. 12, 438–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding C. M., Hennon S. W., Feldman M. F., Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 16, 91–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cava F., de Pedro M. A., Lam H., Davis B. M., Waldor M. K., Distinct pathways for modification of the bacterial cell wall by non-canonical d-amino acids. EMBO J. 30, 3442–3453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Revelles O., Wittich R.-M., Ramos J. L., Identification of the initial steps in d-lysine catabolism in Pseudomonas putida. J. Bacteriol. 189, 2787–2792 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasabe J., Miyoshi Y., Rakoff-Nahoum S., Zhang T., Mita M., Davis B. M., Hamase K., Waldor M. K., Interplay between microbial d-amino acids and host d-amino acid oxidase modifies murine mucosal defence and gut microbiota. Nat. Microbiol. 1, 16125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rumbo-Feal S., Gómez M. J., Gayoso C., Álvarez-Fraga L., Cabral M. P., Aransay A. M., Rodríguez-Ezpeleta N., Fullaondo A., Valle J., Tomás M., Bou G., Poza M., Whole transcriptome analysis of Acinetobacter baumannii assessed by RNA-sequencing reveals different mRNA expression profiles in biofilm compared to planktonic cells. PLOS ONE 8, e72968 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Pedro M. A., Quintela J. C., Höltje J. V., Schwarz H., Murein segregation in Escherichia coli. J. Bacteriol. 179, 2823–2834 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuru E., Radkov A., Meng X., Egan A., Alvarez L., Dowson A., Booher G., Breukink E., Roper D. I., Cava F., Vollmer W., Brun Y., VanNieuwenhze M. S., Mechanisms of incorporation for d-amino acid probes that target peptidoglycan biosynthesis. ACS Chem. Biol. 14, 2745–2756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caparrós M., Pisabarro A. G., de Pedro M. A., Effect of d-amino acids on structure and synthesis of peptidoglycan in Escherichia coli. J. Bacteriol. 174, 5549–5559 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lupoli T. J., Tsukamoto H., Doud E. H., Wang T.-S. A., Walker S., Kahne D., Transpeptidase-mediated incorporation of d-amino acids into bacterial peptidoglycan. J. Am. Chem. Soc. 133, 10748–10751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caparrós M., Torrecuadrada J. L., de Pedro M. A., Effect of d-amino acids on Escherichia coli strains with impaired penicillin-binding proteins. Res. Microbiol. 142, 345–350 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Morè N., Martorana A. M., Biboy J., Otten C., Winkle M., Serrano C. K. G., Montón Silva A., Atkinson L., Yau H., Breukink E., den Blaauwen T., Vollmer W., Polissi A., Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. MBio 10, e02729-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hugonnet J.-E., Mengin-Lecreulx D., Monton A., den Blaauwen T., Carbonnelle E., Veckerlé C., Brun Y. V., van Nieuwenhze M., Bouchier C., Tu K., Rice L. B., Arthur M., Factors essential for L,D-transpeptidase-mediated peptidoglycan cross-linking and β-lactam resistance in Escherichia coli. eLife 5, e19469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf A. J., Underhill D. M., Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 18, 243–254 (2018). [DOI] [PubMed] [Google Scholar]
- 34.L. D. Palmer, E. R. Green, J. R. Sheldon, E. P. Skaar, Methods in Molecular Biology (Humana Press Inc., 2019), vol. 1946, pp. 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura H., Fang J., Maeda H., Protective role of d-amino acid oxidase against Staphylococcus aureus infection. Infect. Immun. 80, 1546–1553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuinema B. R., Reid-Yu S. A., Coombes B. K., Salmonella evades d-amino acid oxidase to promote infection in neutrophils. MBio 5, e01886 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson J. M., Briggs R. T., Karnovsky M. J., Localization of d-amino acid oxidase on the cell surface of human polymorphonuclear leukocytes. J. Cell Biol. 77, 59–71 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Faassen H., KuoLee R., Harris G., Zhao X., Conlan J. W., Chen W., Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect. Immun. 75, 5597–5608 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gee J. B., Vassallo C. L., Vogt M. T., Thomas C., Basford R. E., Peroxidative metabolism in alveolar macrophages. Arch. Intern. Med. 127, 1046–1049 (1971). [PubMed] [Google Scholar]
- 40.Lewis J. M., Deveson Lucas D., Harper M., Boyce J. D., Systematic identification and analysis of Acinetobacter baumannii Type VI secretion system effector and immunity components. Front. Microbiol. 10, 2440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez J., Ly P. M., Feldman M. F., The tip of the VgrG spike is essential to functional type VI secretion system assembly in Acinetobacter baumannii. MBio 11, e03761-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glauner B., Höltje J. V., Schwarz U., The composition of the murein of Escherichia coli. J. Biol. Chem. 263, 10088–10095 (1988). [PubMed] [Google Scholar]
- 43.Boll J. M., Crofts A. A., Peters K., Cattoir V., Vollmer W., Davies B. W., Trent M. S., A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc. Natl. Acad. Sci. U. S. A. 113, E6228–E6237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tucker A. T., Nowicki E. M., Boll J. M., Knauf G. A., Burdis N. C., Trent M. S., Davies B. W., Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. MBio. 5, e01313–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A., Dalton C., Cortez-Cordova J., Schweizer H. P., Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J. Microbiol. Methods. 82, 296–300 (2010). [DOI] [PubMed] [Google Scholar]
- 46.L. Alvarez, S. B. Hernandez, M. A. De Pedro, F. Cava, in Methods in Molecular Biology (Humana Press Inc., 2016), vol. 1440, pp. 11–27. [DOI] [PubMed] [Google Scholar]
- 47.Weber B. S., Ly P. M., Irwin J. N., Pukatzki S., Feldman M. F., A multidrug resistance plasmid contains the molecular switch for type VI secretion in Acinetobacter baumannii. Proc. Natl. Acad. Sci. U. S. A. 112, 9442–9447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eabb5614/DC1