

A metal cluster stuck in a protein acts as a synthetic metalloenzyme for cancer-specific catalytic therapy.
Abstract
Metalloenzymes are promising anticancer candidates to overcome chemoresistance by involving unique mechanisms. To date, it is still a great challenge to obtain synthetic metalloenzymes with persistent catalytic performance for cancer-specific DNA cleavage and operando imaging. Here, an artificial metalloenzyme, copper cluster firmly anchored in bovine serum albumin conjugated with tumor-targeting peptide, is exquisitely constructed. It is capable of persistently transforming hydrogen peroxide in tumor microenvironment to hydroxyl radical and oxygen in a catalytic manner. The stable catalysis recycling stems from the electron transfer between copper cluster and substrate with well-matched energy levels. Notably, their high biocompatibility, tumor-specific recognition, and persistent catalytic performance ensure the substantial anticancer efficacy by triggering DNA damage. Meanwhile, by coupling with enzyme-like reactions, the operando therapy effect is expediently traced by chemiluminescence signal with high sensitivity and sustainability. It provides new insights into synthesizing biocompatible metalloenzymes on demand to visually monitor and efficiently combat specific cancers.
INTRODUCTION
DNA-damaging agents that target the integrity of cellular DNA play a profound role in nonsurgical cancer treatment. However, an elevated DNA repair capacity in tumor cells leads to drug or radiation resistance and severely limits the efficacy of these agents (1–3). Anticancer agent with persistent and high-efficient intracellular DNA cleavage property is anticipated to overcome the major obstacle of chemoresistance and radioresistance. Recently, catalytic anticancer architectures that target biochemical traits unique to tumor microenvironment have aroused multidiscipline interest. They are anticipated to enhance the therapeutic effect and alleviate drug tolerance (4, 5). However, there have been few reports on optimal anticancer catalyst that has both high DNA strand scission ability and maintain long-term catalytic efficiency. Meanwhile, the past few decades have witnessed multifunctional and multispatiotemporal probes, which are developed and applied to molecular imaging, for greatly improving the understanding of physiological and pathological processes (6). Among these techniques, catalysis-triggered chemiluminescence imaging without external irradiation is a highly sensitive, facile, and low-cost approach. It enables real-time detection of a wide variety of molecular or cellular events during a therapeutic response and facilitates visible evaluation of the disease therapy effect (7–9). To meet the demand of practical application of in vivo chemiluminescence imaging, it remains a worthy task to design and construct biomimetic catalysts for prolonging chemiluminescence lifetime.
The native metalloenzymes containing metal clusters as enzyme cofactor give a crucial indication of the devisable exquisite biomimetic catalyst paradigm. Commonly, these metal clusters as active centers temporarily react with the substrate, form intermediate to provide lower energy transition state, and consequently accelerate the chemical reaction. During the catalytic process, the metal valence state is efficiently recycled to ensure that the catalytic active centers are not consumed (10–12). Mimicking these metalloenzymes comprising a valence state recycled metal cluster core bound in the cavity of protein shell, from both a structural and functional viewpoint, provides a guiding principle for rationally designing catalytic anticancer metalloenzymes.
Metal clusters have unique catalytic properties because of their special electronic and geometric structures. On one hand, they are deemed to be molecular semiconductors, exhibiting discrete energy level caused by quantum-size effect. The relative position of clusters’ conduction band (CB) and valence band (VB), the inherent electronic property, is facilely tuned in terms of metal atoms number and composition to catalyze the specific reactions. On the other hand, clusters’ geometrical and electronic structures are readily changed and recycled during the catalytic reaction accompanying the activation of substrate and releasing the product. These distinctive dynamics and stability will render new reaction pathways having lower activation barriers (13–15). Among these metal clusters, copper clusters represent a wide range of accessible oxidation states and have been documented as advanced catalysts in various fields such as selective carbon cross-coupling reaction (16), hydrogenated reaction (17), photocatalytic degradation reaction (18), and CO2 reduction reaction (19). Nevertheless, exploring copper clusters as promising artificial metalloenzyme in emerging biomedicine and bioimaging field has yet to be fully achieved.
Tumor cell deregulation is frequently associated with enhanced cellular oxidative stress (20), for instance, the over expression of hydrogen peroxide (H2O2 level, 50 to 100 μM) in both intra- and extracellular tumor microenviroment. Adaptation to this stress is required for cancer cells to survive, while do not exert a vital function on normal cells (H2O2 level, 0.05 to 7 μM) (21, 22). Here, we take one-step biomimetic synthetic strategy to prepare protein protected copper clusters mimicking the natural metalloenzyme, as shown in Fig. 1. Biomimetic catalytic ability of the well-designed catalyst is systematically explored in aqueous model systems, human lung cancer cells, and live mice xenograft lung tumor model. The optimal reusable copper clusters with appropriate CB position and flexible geometrical structure bring about hydroxyl radical (●OH) and oxygen through persistently catalyzing H2O2 decomposition reaction in tumor microenvironment. A closed catalytic cycle as the mechanistic origin is probed by experimental investigation and computational chemistry verification. For therapeutic purpose, copper clusters induce tumor cell DNA strand scission, impair the cellular viability, and significantly suppress tumor growth by evoking catalytic chemical reactions in a tumor-specific manner. Moreover, a sustainable, sensitive chemiluminescent operando imaging mediated by the biomimetic catalysis is used to trace the tumor therapeutic intervention vividly.
Fig. 1. Schematic diagram of protein-cloaked copper cluster as an artificial metalloenzyme with persistent catalytic activity for high-efficient DNA cleavage and operando chemiluminescent imaging in tumor microenvironment.
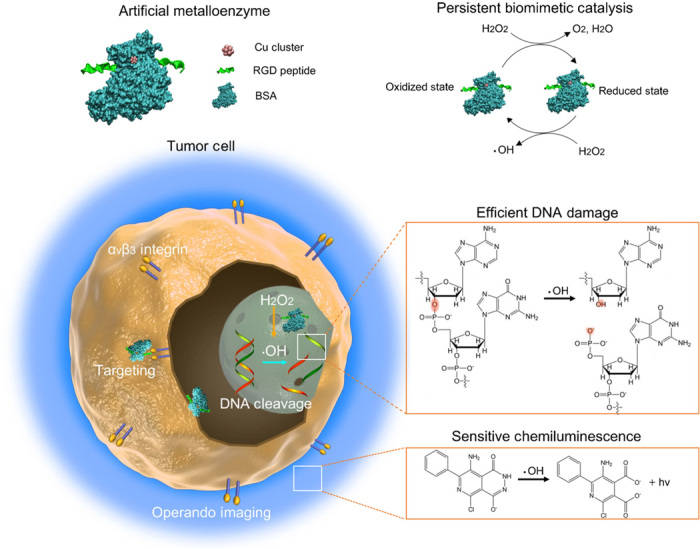
Copper clusters preliminarily recognize αVβ3 overexpressed on vascular endothelial cells and lung tumor cells, then they in situ activate endogenous H2O2 (50 to 100 μM) to give rise to persistent ●OH and O2 generation in both extracellular (pH 6.5) and intracellular (pH 7.4) tumor microenviroment. The energetically favored, regenerated copper clusters as artificial metalloenzyme maintain long-term satisfactory catalytic performance and lead to significant DNA breakage and genotoxicity in tumor cells and tumor xenograft mice models, achieving high-efficient anticancer effect. The metalloenzyme also facilitates the sensitive tracking of tumor therapeutic intervention in vivo through catalyzing persistent chemiluminescence imaging.
RESULTS AND DISCUSSION
Biomimetic synthesis and characterization
Optimizing the trade-off between the stability and activity is the prerequisite for obtaining ideal artificial metalloenzyme (23). Devising bovine serum albumin (BSA) as the template to stabilize copper cluster, on one hand, protects catalytic active center within cavity of protein that simulates natural metalloenzyme to avoid exposed catalyst deactivation. On the other hand, the relative weakly ligated microenvironment allows clusters to activate the reactants and obtain high catalytic capability. Before achieving the effective growth of copper ions into clusters in albumin, a peptide with sequence H2N-CCGPDGRDGRDGRDGR-COOH (naming RGD for short), which recognizes integrin αVβ3 overexpressed on vascular endothelial cells and lung tumor cells (24, 25), was conjugated to the surface of BSA by a heterobifunctional cross-linker sulfosuccinimidyl-4-[N-maleimidomethyl]cyclohexane-1-carboxylate (Sulfo-SMCC; fig. S1A). Under the optimized synthetic condition including precursor concentration and basic pH environment, albumin mineralized copper clusters with the suitable core size were prepared in a controllable manner. The constitutions of the clusters were preliminarily investigated by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF MS; Fig. 2, A and B). The peak of monocation BSA appears at around mass/charge ratio (m/z) of 66,434 (Fig. 2A, black curve), while that of native BSA-mineralized copper clusters (BSA-CuCs) is observed at around m/z 67,261 (Fig. 2A, red curve). After comparison, it clearly indicates that one 13-atom Cu core is cloaked in a single BSA molecule. After bioconjugation of RGD peptide on native BSA, the prominent peak of RGD-BSA has shifted approximately to m/z 70,482, suggesting that one BSA is decorated with two RGD peptides (Fig. 2B, black curve). Subsequently, the unicharged peak of RGD-modified BSA-CuCs (RGD-BSA-CuCs) is observed at around m/z 71,299 (Fig. 2B, red curve). Together, the constitution of this ultimate product as a potential metalloenzyme can be ascribed to (RGD)2-BSA-Cu13.
Fig. 2. Molecular constitution of synthetic copper clusters and energy diagram of RGD-BSA-CuCs to the redox potential of H2O2/·OH and O2/H2O2.
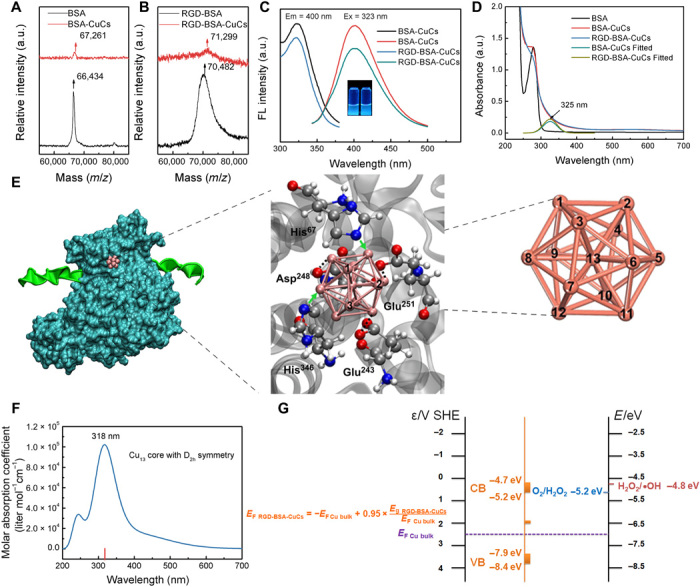
MALDI-TOF MS of (A) BSA and BSA-CuCs as well as (B) RGD-BSA and RGD-BSA-CuCs. a.u., arbitrary units. (C) Excitation and emission spectra of BSA-CuCs and RGD-BSA-CuCs. Inset is digital photograph of BSA-CuCs (left) and RGD-BSA-CuCs (right) under 365-nm hand-held ultraviolet lamp excitation. (D) Ultraviolet-visible (UV-Vis) absorption spectra of BSA, BSA-CuCs, and RGD-BSA-CuCs. (E) Cascade image illustrating the representative conformation of RGD-BSA-CuCs (left) and the orientation of Cu13 core stuck in BSA calculated by molecular docking (middle), as well as the optimized Cu13 core configuration with all copper atomic identities calculated by density functional theory (DFT) (right). Cu13 core is stably stuck in BSA through forming coordination bond with His residues (green arrows) or electrostatic interaction with Glu and Asp (black dotted lines). (F) Molar absorption coefficient of Cu13 core with D2h symmetry from time-dependent DFT (TDDFT) calculation. (G) Proposed energy diagram illustrating the CB and VB position of RGD-BSA-CuCs to the redox potential of H2O2/●OH and O2/H2O2, which explains the recycled catalytic property of biomimetic catalyst. Clusters’ bandgap energy is determined by Tauc’s plot while the Fermi energy is calculated according to an empirical formula. The cluster bandgap and Fermi level determine the position of the VB and CB energies. The electronic level of clusters and redox couples are ranked by standard hydrogen electrode (SHE) level (left vertical line) and vacuum level (right vertical line).
To further clarify the constitution of the artificial metalloenzyme, we investigated the fluorescent excitation and emission spectra of BSA-CuCs and RGD-BSA-CuCs. As shown in Fig. 2C, the maximum excitation and emission peaks of both BSA-CuCs and RGD-BSA-CuCs are located around 323 and 400 nm, respectively, similar to the previously reported characteristic photoluminescence of clusters with Cu13 core (16, 26). This finding can be further demonstrated by their ultraviolet-visible (UV-Vis) absorption spectra (Fig. 2D). Both copper clusters present a newly generated absorption band at around 325 nm, compared with that of BSA. This result is consistent with the reported study (26).
Next, to verify the spatial configuration of Cu13 core, its geometry optimization was achieved by density functional theory (DFT) calculation (27–30). As illustrated in Fig. 2E (right), the calculated Cu13 core with the smallest three-dimensional closed-shell structure is characterized as D2h symmetrical icosahedron with central copper atom coordinated by 12-neighbored copper atoms. Through calculation of molar absorption coefficient of Cu13 core with D2h symmetry by time-dependent DFT (TDDFT; Fig. 2F) and comparison with the corresponding spectrum of RGD-BSA-CuCs from the experiment (Fig. 2D), the coincidence of the maximum peak confirms the characteristic configuration of Cu13 core. It implies that the configuration of initial Cu13 core could be maintained in RGD-BSA-CuCs. Furthermore, we used molecular docking calculation to figure out the orientation of Cu13 core stuck in BSA (Fig. 2E, left and middle). The detailed calculation is present in fig. S1 (B to D) and section S2. The Cu13 core is prone to be stably stuck in a cavity microenvironment consisted of histidine with imidazolyl group and charged aspartic/glutamic acid with carboxyl group. The copper atoms with ID as 1, 2, and 3 (Cu-1, Cu-2, and Cu-3) are exposed to solvation environment in its stuck orientation in BSA. Collectively, all the above findings lead up to that RGD-BSA-CuCs consist of one albumin grafted by two RGD peptides and one Cu13 core mineralized within the protein cavity.
Metal clusters exhibit semiconductor or molecule-like behavior, and their inherent electronic properties mainly rely on the atom number and constitution (13–15). The electronic structures are quantized with energy gap, which gives a clear judgment on whether substrates can be activated via electron transfer from or to the electronic orbital of metal clusters (26, 31). Before investigating the catalytic performance of copper clusters, it is imperative to identify their bandgaps. The optical bandgaps (Eg) of both clusters are calculated by Tauc’s approach applied to semiconductors (fig. S2 and section S3) (26). As a result, the Eg values of BSA-CuCs and RGD-BSA-CuCs are evaluated in the range of 3.01 to 3.74 eV and 2.89 to 3.64 eV, respectively. Moreover, through a theoretical prediction of Fermi level of copper clusters
| (1) |
the approximate CB position of RGD-BSA-CuCs is estimated to locate at −4.7 to −5.2 eV, while that of VB is at around −7.9 to −8.4 eV (with respect to the vacuum level). Compared to VB, free moving electrons are occupied in CB of clusters at ambient temperature through thermal activation. Therefore, the electron transfer occurs in CB where clusters could accept or donor an electron between redox couples in the biological aqueous environment that of approximately the same level (32). As a major thrust, we mainly focus on H2O2 relating redox pairs because the overexpressed level of H2O2 in both extracellular biological fluid and the cellular interior of tumor microenvironment. As illustrated in Fig. 2G, the redox potentials of H2O2/●OH (−4.8 eV) (30) and O2/H2O2 (−5.2 eV) (33) are close to CB of RGD-BSA-CuCs (−4.7 to −5.2 eV). It suggests that RGD-BSA-CuCs could serve as potential conduits to decompose H2O2 to produce ●OH and O2 by electron transfer.
Intrinsic enzyme-like activities
With the aim of elucidating whether RGD-BSA-CuCs evoke sustainable ●OH and O2 generation, electron spin resonance (ESR) technique and polarographic analysis were adopted. Initially, as depicted in Fig. 3 (A and E) using 5,5-dimethyl-1-pyrroline n-oxide (DMPO) as trapping agent under both weak acidic [(pH 6.5),100 μM H2O2, mimicking extracellular tumor environment] and weak alkaline [(pH 7.4), 100 μM H2O2, mimicking intracellular tumor environment] conditions, RGD-BSA-CuCs persistently catalyze formation of a large amount of ●OH for a long time, even up to 24 hours. It is evidenced by the observed DMPO-OH spin adducts distinguished by four-line spectrum with intensity and hyperfine splitting features. The spectroscopic signature indicates RGD-BSA-CuCs have peroxidase (POD)-mimic activity. Furthermore, the steady-state kinetics in the initial catalytic period was examined to validate the kinetic parameters for comparing clusters’ POD activity under two pH conditions. Practically, ●OH generation rate is expediently followed by the oxidation of Amplex UltraRed, a commercial reactive oxygen species (ROS) sensitive fluorescence probe (34) with the maximum fluorescence emission rising in a time-dependent manner at both pH conditions (Fig. 3, B and F). After setting up typical Michaelis-Menten curves (Fig. 3, C and G), apparent Michaelis-Menten constant (Km) and catalytic constant (Kcat) are assessed by Lineweaver-Burk plot (Fig. 3, D and H). The ultimate comparative results are listed in table S1 that Kcat value of RGD-BSA-CuCs under pH 7.4 (6.38 × 10−4 s−1) is higher than that under pH 6.5 (1.01 × 10−4 s−1), implying that clusters have much evident POD-like activity under weak alkaline condition. Next, to support RGD-BSA-CuCs’ intrinsic catalase (CAT)-like activity (33, 35), oxygen production was thereby measured in two pH buffers by a dissolved oxygen electrode. As expected, in the catalytic system, oxygen production lasts for at least 24 hours under both conditions (Fig. 3, I to L). CAT-like activity of metalloenzyme is more distinct at higher pH level. As discussed above, RGD-BSA-CuCs’ CB is energetically matching the redox potential of H2O2/●OH and O2/H2O2. Given that H2O2 is an active molecule displaying oxidant and reductant features (33, 35), the recycled catalytic mechanism is tentatively proposed. When H2O2 behaves as oxidant and is reduced into ●OH, clusters exhibit POD-like activity. In contrast, when H2O2 acts as reductant and is oxidized into O2, clusters present CAT-like activity. Coupled with oxidation/reduction cycle of RGD-BSA-CuCs as catalyst, H2O2 is ultimately decomposed into ●OH and O2. It is safe to draw a conclusion hereto that through electron transfer, RGD-BSA-CuCs with dual enzyme-mimetic activities can oxidize and reduce H2O2 into ●OH and O2 continually in a long term.
Fig. 3. RGD-BSA-CuCs catalyze hydrogen peroxide decomposition to continually produce hydroxyl radical and oxygen under mimetic physiologic condition.

Time-dependent catalytic •OH generation of mixture containing 100 μM H2O2 and 12 μM RGD-BSA-CuCs within 24 hours under (A) pH 6.5 and (E) pH 7.4 condition at 37°C. Time-dependent fluorescence change of oxidized Amplex UltraRed at 585 nm in mixture containing 25 μM Amplex UltraRed, 100 μM H2O2, and 12 μM RGD-BSA-CuCs under (B) pH 6.5 and (F) pH 7.4 condition at 37°C. Representative steady-state kinetic analyses of mimetic enzyme by varying the concentrations of H2O2 (0 to 500 μM) with fixed 25 μM Amplex UltraRed and 12 μM RGD-BSA-CuCs under (C) pH 6.5 and (G) pH 7.4 condition at 37°C. Fitted curves (D and H) are relevant double-reciprocal plots reflecting POD activity of RGD-BSA-CuCs using Michaelis-Menten and Lineweaver-Burk models at double pH conditions. Oxygen production traced by mixing 12 μM RGD-BSA-CuCs and 100 μM H2O2 immediately (I) and 24 hours after initiating the catalysis (J) under pH 6.5 condition at 37°C, measured by a Clark-type oxygen electrode. The similar assays (K and L) were conducted under pH 7.4 condition.
Recycled catalytic property
To verify RGD-BSA-CuCs remain their intrinsic geometrical structure during the entire catalytic process, we carried out multiple steady-state measurements. Notwithstanding high-resolution transmission electron microscope (HRTEM) is not suitable to characterize clusters’ geometrical structure ascribing to their low melting point, the average size of formed nanostructures will reflect clusters’ monodispersity to an extent. As shown in Fig. 4A, under the high energy electron beam irradiation, agglomerated nanosized structures larger than individual cluster were formed. By virtue of statistical analysis of 50 representative crystalline nanoparticles, no substantial distinction in the size is detected before and after catalysis under two pH conditions (Fig. 4B). Simultaneously, dynamic light scattering tests reveal that the average hydrated diameter of RGD-BSA-CuCs fluctuates between 4.9 and 6.6 nm (fig. S3A). On the basis of the knowledge that the average hydrated diameter of BSA is around 5.0 nm (36), the comparative results disclose that Cu13 core as catalytic active center is fairly stable within the cavity of the protein, rather than dissociates into copper ions to leaches or be further mineralized into large nanoparticles to complicate the catalysis. The relative photoluminescence intensity (Ft/F0) evolution of clusters, the essential character closely correlated with their structure parameter, is found to remain constant during the catalysis (Fig. 4C). It is a coupling evidence to support the stability of RGD-BSA-CuCs.
Fig. 4. Intrinsic geometrical and electronic properties of biomimetic catalyst remain unaltered during the catalytic process studied by the experimental and DFT methods.

(A) HRTEM images of RGD-BSA-CuCs before and after catalysis for 24 hours under pH 6.5 (top) and pH 7.4 (bottom) conditions at 37°C. (B) Statistical size analysis of the representative 50 crystalline nanoparticles characterized by HRTEM at pH 6.5 (top) and pH 7.4 (bottom) before and after the catalysis. (C) The relative photoluminescence intensity (Ft/F0) evolution of RGD-BSA-CuCs at both pH conditions at 37°C. F0 and Ft represent the fluorescent intensity of the catalyst before and after initiation of the catalysis, respectively. Error bars represent variation between three measurements. (D) Representative normalized x-ray absorption near-edge structure (XANES) spectra at the Cu K edge of RGD-BSA-CuCs before and after catalysis for 24 hours under two pH conditions at 37°C. The spectra of Cu foil, Cu2O, and CuSO4 compounds are nominated as standards. (E) The reaction pathway of the recycled catalytic reaction from the initial Cu13 core to the intermediate compound Cu13═O. Int1, Int2, and Int3 are the intermediate products, while TS1 and TS2 are the transition states. The reaction rate determining step is highlighted in the blue dash frame. (F) The reaction pathway of the recycled catalytic reaction from the intermediate compound Cu13═O to the initial Cu13 core. Int4, Int5, and Int6 are the intermediate products, while TS3 and TS4 are the transition states. The reaction rate determining step is highlighted in the blue dash frame. Copper, oxygen, and hydrogen atoms are depicted in pink, red, and white, respectively.
X-ray photoelectron spectroscopy (XPS) and auger electron spectroscopy (AES) analysis were simultaneously carried out to delineate the oxidation state of Cu before and after catalysis, crucial to interpret electronic recyclability of RGD-BSA-CuCs (fig. S3, B and C). In deconvoluted Cu 2p XPS spectra, the peak centered around 943.6 eV is definitely ascribed to Cu (II) 2p3/2 signal, whereas the peaks at 933.7 and 953.7 eV are assigned to Cu 2p3/2 and Cu 2p1/2 signals, which are approximately attributed to either Cu (I) or Cu (0). Because of the proximity of Cu (I) and Cu (0) binding energy, the oxidation state of Cu was further explored by AES. In x-ray excited Cu LMM Auger spectra, a strong peak depicted at 572.3 eV unambiguously indicates the existence of Cu (I). In contrast, Cu (0) signal expected around 568.0 eV is indistinguishable (37, 38). Considering XPS and AES parameters comprehensively, Cu (0), Cu (I), and Cu (II) atomic ratios of RGD-BSA-CuCs were roughly quantified from constituent peak area (table S2). Although unnoticeable valent changes were discerned under both catalytic pH conditions, XPS and AES characterization may be overstated because x-ray beam-induced photoreaction is inevitable.
Because photoreduction does not occur with synchrotron radiation technique (39), we thereupon exploited x-ray absorption near-edge structure (XANES) spectroscopy as a powerful tool to quantify Cu oxidation state before or after catalysis. As shown in Fig. 4D, compared to the standard Cu metal or compounds, we note a broadened feature in the normalized XANES spectra of copper clusters due to the quantum size effect (40). In addition, the peak position and shape of RGD-BSA-CuCs before or after catalysis under both pH conditions are similar to those of Cu2O (red arrow points) and CuSO4 (black arrow points). It means that samples exhibit mixed Cu (I) and Cu (II) states. The result is in good consistence with previous report that because the surface/center Cu atom ratio of Cu13 core is 12 to 1, the surface Cu (I) atom contribution dominates the XANES spectra of the clusters and center Cu (0) signature is obscure (41). Moreover, note that RGD-BSA-CuCs have been thoroughly purified to remove free unmineralized CuSO4 prior further characterization or initiation catalytic reaction. Therefore, we deduce that the minor Cu (II) components might stem from the firmly adsorbed ions on RGD-BSA-CuCs’ surface even after repeatedly purification (18, 42). In terms of quantitative evaluation, principal components analysis (PCA) and linear combination fit (LCF) of copper clusters’ XANES spectra were performed using Cu2O and CuSO4 as standards because clusters do not feature any Cu (0) character. Table S3 reports the steady-state Cu fraction obtained by XANES fitting. We notice that clusters have a similar Cu (I)/Cu (II) ratio, irrespective of the catalyst before or after catalysis under two pH conditions, ascertaining clusters’ recycling nature. Furthermore, compared to Cu (I)/Cu (II) ratios summarized by XPS and AES results (table S2), that obtained through XANES fitting is a little bit lower. It might be attributed to the photoreduction behavior of x-ray beam concomitant with XPS and AES characterization (39).
It is widely accepted that Cu (II) ions can catalyze the depletion of H2O2 through Haber-Weiss and Fenton-like reactions to produce ●OH (43). The subsequent comparative ESR experiments were conducted to exclude the possibility that radical generation is mainly caused by the adsorbed Cu (II). As shown in fig. S3D, the radical level catalyzed by RGD-BSA-CuCs comprising 50 μM Cu (II) was compared with that triggered by equivalent-free Cu (II) ions. Obviously, RGD-BSA-CuCs behave fourfold more efficiency in ●OH production at pH 6.5 and twofold more efficiency at pH 7.4 than that induced by the dissolved Cu (II) ions. It is thereupon safe to presume that ●OH generation is attributed primarily to the valence state recycled catalytic reactions involving the skeleton components of RGD-BSA-CuCs, rather than merely caused by the adsorbed Cu (II). Until recently, the benign H2O2 has been reported in triggering in situ Fenton reaction to liberate ●OH by several Fe-based nanocatalysts. The homogeneous Fenton reaction is initiated by iron ions released from the nanocatalysts (44), while the heterogeneous catalysis directly takes place on the nanocatalysts’ surface (45). Compared with the retrospective homogeneous and heterogeneous catalysis, copper clusters maintaining long-term catalytic efficiency is plausibly premised on their unique electronic and geometrical structures. We envision that clusters, as intermediate state of matter between isolated atoms and nanoparticles, are anticipated to comprise a critical complement to bridge the gap between homogeneous and heterogeneous catalysis in biomedical field.
Theoretical study of the catalytic mechanism
To disclose the beneath catalytic molecular mechanism, we used DFT to explore the recycling catalytic reaction pathways by considering the cluster’s configurational and electronic structures. The catalytic reaction pathways are revealed with reasonable reaction efficiency by taking account into the intermediate products and transition states (46–48). Specifically, the xth intermediate product is denoted as Intx, and the yth transition state is denoted as TSy. As illustrated in Fig. 4E, first, in the structural calculation of Int1, we test all possible reaction sites of Cu13 core to H2O2 and find the most active copper atoms with ID as 1, 2, and 3 (Cu-1, Cu-2, and Cu-3). This reaction interface is consistent to the exposed interface of Cu13 core stuck in BSA (Fig. 2E). In the first reaction step, H2O2 homogeneously splits into double ●OH. Then, the first ●OH bonds to two Cu atoms (noted as Cu-1 and Cu-3), and the second ●OH bonds to one Cu atom (noted as Cu-2) sequentially to construct Int1 complex. After H2O2 is activated by Cu13 core, Int1 overcomes 0.27 eV energy barrier via TS1 and evolves into Int2 with the reaction energy as −0.12 eV in the first reaction step. The moderate reaction barrier and the minus reaction energy ensure that the reaction occurs in physiological environment. In Int2, the active hydrogen favors to approach the adjacent OH and bonds OH to produce H2O molecule in Int3. The second step of the reaction has the reaction barrier as 0.50 eV and the reaction energy as 0.26 eV, which is the rate determining step in this half-recycle reaction (highlighted in the blue dash frame in Fig. 4E). The activation energy as 0.50 eV supports an appreciable reaction rate at room temperature. The products of the half-recycle reaction are the oxidized compound Cu13═O and H2O molecule separated from the cluster structure in Int3. The detailed structure information is shown in fig. S4 (A to E) including the key bond lengths for both Int1, Int2, Int3, TS1, and TS2.
In the following half-recycle reaction (shown in Fig. 4F), one hydrogen atom of H2O2 molecule interacts with the oxygen atom of the compound Cu13═O, which produces Int4 complex consisting of Cu13═O and deformed H2O2 molecule during the process of H2O2 attacking Cu13═O. In the first step of the reaction, oxygen atom of Cu13═O in Int4 catches one hydrogen atom to form ●OOH free radical, which generates Int5 complex. TS3 is the transition state between Int4 and Int5 in the first step of the reaction. The energy barrier is 0.21 eV, and the reaction energy is −0.55 eV in this step, signifying that this step is a high rate reaction. In Int5, OH catches the hydrogen of ●OOH to form one H2O molecule. Meanwhile, two oxygen atoms form one O2 molecule. Thus, the resulting Int6 includes one Cu13, H2O, and O2. The second step of this reaction has the reaction barrier as 0.34 eV and the reaction energy as −0.01 eV, which is the rate determining step in this half-recycle reaction (highlighted in the blue dash frame in Fig. 4F). The detailed structure information is shown in fig. S4 (F to J), including the key bond lengths for both Int4, Int5, Int6, TS3, and TS4. Obviously, computational approaches lend support to energetically favorable recycled catalysis with desired reaction efficiency.
In vitro therapeutic effect
After corroborate catalysis by RGD-BSA-CuCs in the model aqueous system and computational simulation with high efficacy, satisfied sustainability, and stability, in vitro/in vivo activity of the artificial metalloenzyme warrants fundamental study. Their targeting specificity and distribution in lung tumor cells were first vividly delineated through microscopic observation. As shown in fig. S5A, after culturing RGD-BSA-CuCs with A549 cells (human tumorigenic adenocarcinomic alveolar basal epithelial cells overexpressing integrin αVβ3) (25), apparent blue photoluminescence in cytoplasm was observed, where endogenously generated H2O2 is abundant (20, 49). In notable contrast, vacant cells do not emit noticeable blue photoluminescence, when images were collected at the same setting. The uptake of clusters induced by selective recognition of RGD peptide to integrin is further demonstrated by a peptide blocking study. After blocked by 2.0 mM free RGD peptide, the blue fluorescence signal in cytoplasm has almost vanished. As a corollary test, dimmer photoluminescence is observed when BSA-CuCs was cultured with cells, implying the marking specificity of RGD-BSA-CuCs.
To gain an insight into the antiproliferative activities of RGD-BSA-CuCs on hyperoxidative stress tumor type, we incubated A549 cells with clusters over a wide dosage range (4.8 to 77 μM) for 48 hours and evaluated cytotoxicity via a cell counting kit-8 (CCK-8) assay (Fig. 5A). In contrast to RGD-BSA with no significant effect on cell viability, the half-maximum inhibitory concentration (IC50) value of RGD-BSA-CuCs against A549 cells proliferation is around 18 μM, whereas that of BSA-CuCs is about 53 μM. We evaluated the intracellular average Cu atoms accumulation after 24 hours clusters exposure and found that the value is around 123 ± 3.1 μM and 162 ± 4.0 μM Cu atoms per A549 cell for incubation with BSA-CuCs and RGD-BSA-CuCs, respectively, much higher than that of endogenous Cu ions content in single cell (see fig. S5B and section S7). The much evident cytotoxicity is thereupon accounted for the higher internalization efficiency of RGD-BSA-CuCs. In particular, in the above catalytic tests in tube, the concentration of RGD-BSA-CuCs (12 μM) in model aqueous system is deliberately selected by simulating the copper atom content ingested by single cancer cell (162 ± 4.0 μM), considering that one cluster comprises 13 Cu atoms. Moreover, as shown in Fig. 5A, the cytotoxicity toward A549 cells induced by 77 μM RGD-BSA-CuCs or BSA-CuCs is completely rescued by coculturing mannitol, a typical ●OH quencher (50). It indicates that cell mortality does originate from the oxidative damages evoked by catalytically produced ●OH. Meanwhile, we examined the cytotoxicity of equimolar concentration clusters on noncancerous lung primary cells. As depicted in fig. S5C, proliferation of human embryonic lung diploid cell line CCC-HPF-1 (human embryonic lung diploid cell line CCC-HPF-1) cells is not significantly affected and with around 90% confidence level when administrated with 19 μM RGD-BSA-CuCs. It means that targeting H2O2 dependency and biomimetic catalysis is a reliable strategy to selectively kill hyperoxidative stress lung cancer cells compared to healthy lung cells (44).
Fig. 5. Copper clusters as anticancer biomimetic catalysts exert therapeutic effect in vitro and in vivo.

(A) In vitro viability of A549 lung tumor cells after incubation of RGD-BSA-CuCs or BSA-CuCs with the identical clusters concentration for 48 hours. Viability of A549 cells pretreated by RGD-BSA is set as control group (means ± SD, n = 3). DIC, differential interference contrast. (B) Fluorescence microscopy images showing ROS burst through fluorogenic reaction with CM-H2DCFDA in cells pretreated with a series concentration of RGD-BSA-CuCs for 48 hours. Cells treated with 39 μM RGD-BSA-CuCs and 250 μM ROS quencher mannitol are set as control. (C) Quantification the proportion of apoptotic cells in series dosages of RGD-BSA-CuCs measured by flow cytometry. DIC, differential interference contrast. (D) Agarose gel electrophoretic patterns of supercoiled pUC19 plasmid DNA (20 ng ml−1) in the presence of a series concentration of RGD-BSA-CuCs and 100 μM H2O2 (left). The mixture was preincubated in pH 7.4 buffer solution at 37°C for 12 hours. The persistent DNA cleavage property of 9.6 μM RGD-BSA-CuCs in the presence of 100 μM H2O2 (right). The mixture was preincubated in pH 7.4 buffer solution at 37°C for 0, 3, 6, and 12 hours. DNA treated by 100 μM H2O2 for 12 hours is set as control. (E) DNA lesions induced by the increased concentration of RGD-BSA-CuCs measured by the basic comet assay. DNA content in the comet tail represents the extent of DNA cleavage, analyzed by 50 cells selected randomly by CASP software. ***P < 0.001. (F) Scheme of establishing subcutaneous (s.c.) xenografted A549 cells tumor model and catalytic mediated therapeutic protocol. i.p., intraperitoneal. (G) The relative tumor volumes of mice treated with control group and therapeutic groups (means ± SD, n = 5). *P < 0.05 and ***P < 0.001. (H) Tumor tissues were dissected and compared visually showing inhibition of tumor growth after 21 days of therapy (n = 5). (I) Representative hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate biotin nick end labeling (TUNEL) staining images of dissected tumor tissues. Tumor sections present obvious apoptosis and prominent necrosis. In TUNEL assay, positive apoptotic cells are observed to emit green luminescence [(fluorescein isothiocyanate (FITC)–labeled], while the cell nuclei are stained with 4, 6-diamidino-2-phenylindole (DAPI, blue) (Photo credit: X.G., Beijing University of Technology).
For the sake of proving that ●OH catalyzed by clusters could exhaust the cellular antioxidant capacity and push ROS level beyond a “threshold” that enables the sequential cell apoptosis, microscopic observation and flow cytometry analysis were conducted. Accompanying the increased clusters exposure dosage, the cells stained by ROS commercial kit behave substantial fluorescence enhancement (Fig. 5B). The signal is distributed in the whole A549 cell, including the nucleus and cytoplasm, signifying overwhelming oxidative stress. To further confirm the existence of ●OH, cells were incubated with the highest dosage of clusters and mannitol. In this case, green fluorescence has almost vanished, implying that ●OH generation is completely compromised by the internalized quencher. Next, the cell death induced by the biomimetic catalysis was examined with the dual fluorescence annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining, applied to differentiate viable cells from dead cells. As shown in Fig. 5C, the population of apoptosis cells also presents a dose-dependent manner. It is highly plausible that the cell impairment is ascribed to the catalysis generated ●OH, which is prone to react with various intracellular molecules including nucleic acids, proteins, and lipids.
DNA cleavage in test tube and living cancerous cells
Of the types of biomacromolecule damage that exist within cells, DNA breakage is considered as one of the most hazardous lesions that results in genomic instability and cell death. Before investigating the cytogenotoxicity caused by RGD-BSA-CuCs, we examined DNA cleavage activity of the metalloenzyme using plasmid DNA strand scission assay (51). In specific, to study DNA damage in vitro, we mixed RGD-BSA-CuCs, H2O2, and pUC19 plasmid DNA with 2686 base pairs (bp) in pH 7.4 buffer solution under mimic physiological condition for 12 hours. Figure 5D (left) shows the result of gel electrophoretic separation of pUC19 DNA cleaved by the increasing concentration of RGD-BSA-CuCs in the presence of 100 μM H2O2. When clusters concentration varies from 0 to 4.8 μM, the amount of supercoiled form DNA (form I) obviously decreases and even disappears, while the nicked form DNA (form II) with single-strand break substantially increases. This finding persuasively indicates a nuclease-like activity. As the concentration of RGD-BSA-CuCs increases from 9.6 to 39 μM, the treatment leads to a marked DNA degradation, characterized by the smearing of DNA fragments (52). It suggests that copper clusters have impressing DNA degradation activity in the presence of H2O2, most likely occurs through an oxidative mechanism. The radical ●OH is considered as an initiator of DNA breakdown by the abstraction of a hydrogen atom from deoxyriboses along the DNA backbone. Subsequently, the phosphodiester bonds between bases are cut down, and DNA is efficiently degraded (53). Because of its small size, ●OH diffuses rapidly and then cleaves DNA molecules at any position (54). In contrast, under similar condition, RGD-BSA-CuCs behave mild DNA cleavage activity in the absence of H2O2 (fig. S5D). In this case, the transformation of DNA from form I to form II is positively related to the concentration of clusters. When the catalysts’ concentration is as high as 77 μM, a finite amount of supercoiled DNA can still be observed (fig. S5D). The limited DNA damage activity without H2O2 is probably ascribed to the adsorbed Cu (II) ions on the protein. The activity is reminiscent of the Cu (II)–mediated DNA damage involving a classic metal center reduction (55). Furthermore, to confirm clusters with persistent cleavage property, DNA cutting efficiency by 9.6 μM RGD-BSA-CuCs in the presence of H2O2 was studied in a time-dependent manner (Fig. 5D, right). The results disclose that, with catalytic time proceeds from 0 to 12 hours, form I–type DNA is gradually converted to form II–type DNA and is lastly degraded into small fragments or even nucleotides that hardly observed in lanes.
Oxidative DNA damage in living cells includes single-strand or double-strand breaks, alkali-labile sites, and base damage. Single cell gel electrophoresis assay (comet assay) under the alkaline condition was next performed to detect all these forms of lesion (56). Images in Fig. 5E (left) show that RGD-BSA-CuCs induce typical concentration-dependent DNA damage in tumor cells, demonstrated by the migration of cleaved DNA fragments (comet tail) from the nucleoid (comet head) under an electric field. DNA content in the comet tail, which is positively correlated with the extent of DNA breakage in a cell, was considered as an index of DNA damage (57). Statistically, exposure to RGD-BSA-CuCs and intracellular H2O2 triggers severe fragmentation of DNA in A549 cells (Fig. 5E, right). The strongest genotoxic effect, with DNA content in the comet tail up to 64 ± 8.5%, is induced when 39 μM clusters was delivered. The lesioned DNA declines at higher concentrations of clusters (58 and 77 μM). The variation in concentration-dependent catalysis is consistent with the result present in Fig. 5D (left). It could be ascribed to the reason that along with the increasing concentration of clusters, generated ●OH preferably attacks amino acid residues of nearby biomolecules including BSA shell or intracellular proteins, leading to the decrease in cleaved DNA by ●OH. In addition, ●OH triggers the release of metal ions that are liberated from metal storage proteins as reactive ions. These ions, in turn, generate more ROS, which could also contribute to oxidative DNA lesions and have deleterious consequences for cells, including apoptosis or necrosis.
In vivo therapeutic effect and mechanism study
Before testifying RGD-BSA-CuCs as potential anticancer metalloenzymes, in vivo behaviors of clusters, including their targeting ability, pharmacokinetics, and biodistribution profile, were initially assessed. Active targeting capacity was evaluated through intraperitoneally injection RGD-BSA-CuCs and BSA-CuCs to A549 lung tumor cells xenograft BALB/c nude mice. The result suggests that, as predicted, RGD-BSA-CuCs behave enhanced accumulation in tumor microenvironment, compared to BSA-CuCs (fig. S5E). Then, we investigated the pharmacokinetic character of RGD-BSA-CuCs by examining the Cu concentration in the blood through inductively coupled plasma MS measurements. Given that the blood clearance half-life (t1/2β) of clusters is fitted as 69 hours (fig. S5F), the following in vivo anticancer administration frequency is therefore tentatively adopted as once every 2 days. It ensures that the next dosage of RGD-BSA-CuCs is administrated before one-half of clusters in the blood are thoroughly cleared. Subsequently, the biodistribution of the metalloenzyme in major organs via a single intraperitoneal injection was studied (fig. S5G). It is found that shortly (1 hour) after injection, RGD-BSA-CuCs accumulation in tumor passes through a maximum, presumably via enhanced permeability and retention and active targeting effects. In time, RGD-BSA-CuCs are heavily distributed in the liver arising from the capture capacity of the mononuclear phagocyte system. Accompanied a longer observation period, the accumulation of clusters in the liver declines, suggesting that RGD-BSA-CuCs, which not entrapped by tumor, may be degraded by following phagocytosis in the liver.
The intriguing in vitro catalysis–evoked DNA strand scission and tumor cell damage by RGD-BSA-CuCs imply their potential in vivo therapeutic outcome. The antitumor performance of RGD-BSA-CuCs was then examined on established lung tumor xenograft mice. Saline (control group) and RGD-BSA-CuCs with two doses (1.0 and 2.5 mg kg−1, therapeutic groups) were administrated intraperitoneally every 2 days to investigate the therapeutic effects, respectively (Fig. 5F). After 21 days of therapeutic intervention, tumors grew substantially more slowly in mice administrated with clusters, indicating a significant anticancer effect (fig. S5H). To be more specific, according to the variation of the relative tumor volume, we calculate the averaged suppression rate to be 45 ± 15% and 67 ± 6%, respectively, at Cu doses (1.0 and 2.5 mg kg−1) (Fig. 5, G and H). We suppose that the satisfactory tumor suppression effect is contributed to the highly toxic ●OH, which is derived from catalytic decomposition of endogenous H2O2 by RGD-BSA-CuCs.
To evaluate the pathological changes and DNA damages to tumors treated by RGD-BSA-CuCs, we observed the histopathology images of the dissected lung tumor tissue. As described in Fig. 5I, the apparent destruction of tumor cells is noticed in hematoxylin and eosin (H&E) staining images in therapeutic groups. In tissue section from the group treated by low-dose clusters (RGD-BSA-CuCs, 1.0 mg kg−1), tumor cells present marked apoptosis with featured cell shrinkage and chromatic agglutination. Apart from apoptosis, prominent necrosis is also imaged from the group administrated by RGD-BSA-CuCs (2.5 mg kg−1) with amount of cell nuclei disappear markedly. In contrast, no evident apoptosis or necrosis in tumor tissue from control group is observed. Besides, as a corollary observation, in the fluorescent images of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate biotin nick end labeling (TUNEL) assays on tumor sections, several green TUNEL–positive apoptotic cells in therapeutic groups are captured compared with the control group. It indicates that substantial detected tumor cells undergo extensive DNA degradation during the late stages of apoptosis. All the evidence confirms that the developed metalloenzymes as abiotic nucleases can be successfully applied for in vitro/in vivo cleaving oncogenic DNA.
Chemiluminescence imaging in vitro/in vivo to trace therapeutic intervention
To exploit RGD-BSA-CuCs as a theranostic agent to image oxidative damage level in tumor microenvironment, we used 8-amino-5-chloro-7-phenylpyrido[3,4-d]-pyridazine-1,4(2H,3H)dione (L-012) (58), a chemiluminescence substrate, to trace ●OH production in test tube and live mice tumor environment via an in vivo imaging system (IVIS) spectrum imaging system. As illustrated in fig. S6A, in the presence of H2O2 and clusters, the chemiluminescence spectrum of oxidized L-012 presents the maximum emission peak locates at around 470 nm, which is derived from the energy release of an excited-state intermediate (59). In addition, the relevant control groups confirm that ●OH is the predominant reactant for chemical ignition. Next, catalyst or substrate concentration–dependent chemiluminescence properties were determined (Fig. 6, A and B). It implies that the delivered dosage of clusters and the biologically relevant concentration of H2O2 fall within the detection window. The high signal-to-noise ratio promises the following operando imaging in vivo. Through prolonged tracing, the half-life of chemiluminescence is evaluated around 7.9 hours (Fig. 6C). Such a persistent luminescence is not only consistent with the long-term catalytic property of the synthetic metalloenzyme discussed in Fig. 3 but also be beneficial for the following in vivo imaging applications.
Fig. 6. Chemiluminescence imaging in vivo to trace the process of therapeutic intervention.
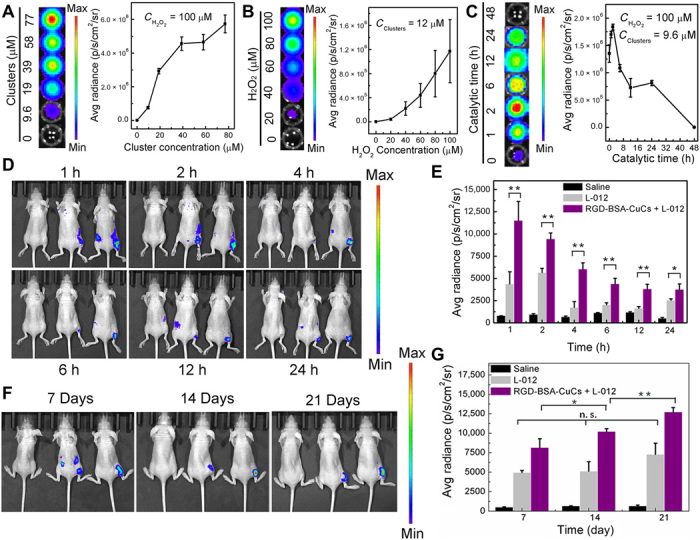
(A) Chemiluminescence images (left) and quantitative signal data (right) of 1.0 mM L-012 in the presence of different concentrations of RGD-BSA-CuCs with fixed 100 μM H2O2 under pH 6.5. (B) Chemiluminescence luminescence images (left) and quantitative signal data (right) of 1.0 mM L-012 in the presence of different concentrations of H2O2 with fixed 12 μM RGD-BSA-CuCs under pH 6.5. (C) Time-dependent luminescence (left) and signal intensity (right) of 1.0 mM L-012 upon incubation with 100 μM H2O2 and 9.6 μM RGD-BSA-CuCs. (D) Chemiluminescence images present a higher intensity signal in the tumor region of mice treated with CuCs and L-012 (the right mice in each group) compared with that mice treated with L-012 only (the middle mice) or saline treated mice (the left mice). (E) The corresponding luminescence intensity of tumor region at indicated time (means ± SD, n = 3). The pseudo-colors represent photons per second per square centimeter per steradian. *P < 0.05 and **P < 0.01. (F) Chemiluminescence images of tumor region of mice treated with CuCs and L-012 (the right mice in each group) compared with that mice treated with L-012 only (the middle mice) or saline treated mice (the left mice) for 7, 14, and 21 days of therapeutic intervention. (G) The corresponding luminescence intensity of tumor region at indicated time (means ± SD, n = 3). The pseudo-colors represent photons per second per square centimeter per steradian. *P < 0.05 and **P < 0.01. n.s., no significance. (Photo credit: X.G., Beijing University of Technology).
Subsequently, to sensitively image ROS generation in complex biological systems, we in vivo assessed chemiluminescence by imaging system without photoexcitation. When mice displayed tumor volumes around 100 mm3, they were intraperitoneally administrated RGD-BSA-CuCs (2.5 mg kg−1). Indicated time intervals later, mice were intratumorally injection of L-012, and in vivo endogenously catalytic ●OH generation was visualized in real time (Fig. 6D). Notably, L-012–mediated luminescence reaches a maximal signal at 1 hour after injection of clusters with declined emission at longer indicated time. Substantial distinction of tumor signal intensities was observed on the clusters and L-012–treated (right mice in each group), sole L-012–treated (middle mice in each group), and saline-treated (left mice in each group) groups. Through quantitative analysis, at 1 hour after injection of clusters, luminescence intensity of clusters treated group is increased by 1.6-fold compared with that of L-012–treated group and 13.2-fold compared with that of saline group, respectively (Fig. 6E). In addition, the luminescence shows a sustainable profile during the examined 24 hours, demonstrating that the artificial metalloenzyme can continually function in tumor microenvironment. The ex vivo imaging analysis further confirms that this ●OH-involved chemiluminescence signal specifically stems from tumor tissue (fig. S6B).
We further used the chemiluminescence to monitor the therapeutic intervention effect in mice at different stages of therapy in vivo. As a comparative group, the intrinsic oxidative stress level of mice tumor environment was evaluated at varied therapeutic time points for real-time imaging by luminescence (Fig. 6F, the middle mice in each group). It finds that the luminescence increases a little bit with tumor volume promotion in 21 days (Fig. 6G, gray columns). In sharp contrast, after 7, 14, and 21 days of catalysis-evoked tumor therapy by RGD-BSA-CuCs, the luminescence of tumor enhances significantly (Fig. 6G, purple columns), signifying that ●OH with high oxidative nature is much more reactive with L-012 than that of endogenous H2O2. The increased signal is a collective consequence of catalysis-induced signal amplification and intrinsic enhanced oxidative stress in situ. The ex vivo imaging analysis provides a supporting evidence (fig. S6C). Collectively, the results above demonstrate that clusters can serve as sustainable metalloenzyme for in vivo sensitive visualization of tumor that is associated with high H2O2 expression, by which the therapeutic process can be vividly traced.
Biosafety assessment
Copper, one of the transition metals, is an essential trace element for life. Although excessive amount of Cu adversely results in serious diseases, low amount of Cu plays a pivotal role in many fundamental physiological processes in organisms (60). To inquire into the biocompatibility and biosafety of RGD-BSA-CuCs, we recorded the body weights of the mice during the treatment. Insignificant body variation indicates that the metalloenzyme has no obvious in vivo toxicity toward the mice growth (fig. S7A). In addition, H&E staining with major organ tissues (e.g., heart, liver, spleen, lung, and kidneys) of mice after therapy was carried out. No abnormalities are found in therapeutic groups compared with those mice treated with saline (fig. S7B), confirming clusters’ high catalytic selectivity to tumor and minimal side effects to organs. Last, unnoticeable distinction on blood indices is observed, including blood biochemistry and complete blood counts, proving the uniform hematology status triggered by RGD-BSA-CuCs during the whole evaluation period (fig. S7C). Since RGD-BSA-CuCs accumulate in the liver for at least 24 hours, we attentively refer to the blood biochemistry concerning liver function. All biochemical indices representing liver function were within the normal range during treatment. These preliminary in vivo evaluations demonstrate the high biocompatibility of RGD-BSA-CuCs holding great potential in vivo therapeutic applications in the future.
In addition, some potential limitations of this work should be concerned. First, the synthetic metalloenzyme is composed of copper cluster, BSA, and RGD peptides. In particular, RGD peptides have attributed to metalloenzyme recognition to tumor microenvironment. However, the number of decorated peptides is only two on each BSA. Therefore, the targeting efficiency of metalloenzyme in vivo is limited but may be enhanced by the following optimization. Second, although the synthetic metalloenzyme has distinct advantages, we still need to devise more promising complex systems with multiple or sequential catalytic activities. With these efforts, the synthetic metalloenzymes will be competent to natural enzymes, which are easily denatured and degraded in circulation. Third, in this study, therapeutic intervention with the metalloenzyme via intraperitoneal injection suppresses tumor growth with high efficiency. Future studies are planned to investigate whether other drug administration strategies, such as intravenous injection, can potentiate in vivo efficacy.
In summary, we have developed an artificial metalloenzyme with persistent catalytic activity through biomimetic mineralization. The engineered copper cluster–based metalloenzyme exhibit mimic-POD and mimic-CAT activities simultaneously. That is, they are capable of continually catalyzing decomposition of H2O2 into ●OH and O2, accompanying a stable catalytic cycle process. The copper cluster’ CB level is matched well to redox potentials of H2O2-invovled couples, which enables electron transfers and the reliable catalysts recyclability. Consequently, cancer therapy with high efficacy is achieved by selectively and persistently amplifying ROS in tumor microenvironment. The freely diffusible ●OH is one of the intermediates involved in the DNA scission process and lastly induces tumor cell apoptosis. The artificial metalloenzyme with persistent and high-efficient intracellular DNA cleavage property is anticipated as selective and mechanistic strategy for overcoming tumor resistance in the future. Meanwhile, the sustainable and sensitive chemiluminescence imitated by the metalloenzyme enables real-time chasing of tumor therapeutic evaluation in situ. This proof-of-concept study could open a new route to synthesize metal cluster–based biomimetic catalysts to tune the specific biochemical reactions for cancer theranostics.
MATERIALS AND METHODS
Materials
The RGD peptide H2N-CCGPDGRDGRDGRDGR-COOH was synthesized by a solid phase method (ChinaPeptides Co. Ltd.; purity, 95%). BSA, DMPO, and mannitol were purchased from Sigma-Aldrich. Copper (II) sulfate (CuSO4), cuprous oxide (Cu2O), hydrogen peroxide (H2O2), sodium hydroxide (NaOH), nitric acid (HNO3), and hydrochloric acid (HCl) were obtained from Beijing Chemical Reagent Co., China. Amplex UltraRed, LysoTracker Red DND-99, and CM-H2DCFDA were from Molecular Probes, USA. Sulfo-SMCC, CCK-8 reagent, and annexin V–FITC/PI apoptosis detection kit were purchased from Dojindo Laboratories, Japan. The Supercoiled pUC19 plasmid DNA with 2686 bp was purchased from Thermo Fisher Scientific for the DNA double-strand break assays. Cell culture 1640 medium and fetal bovine serum (FBS) were purchased from HyClone. Chemiluminescence probe L-012 for detection ROS was purchased from Wako Chemicals (Neuss, Germany). All other materials were commercially available and used as received unless otherwise mentioned. The water used in all experiments was Milli-Q grade with a resistivity of 18.2 megohm⋅cm.
Preparation of RGD-BSA-CuCs and BSA-CuCs
In a typical experiment, 10 ml of 80 mg of BSA was mixed with 1.5 ml of 4.0 mg of Sulfo-SMCC at 4°C in phosphate-buffered saline (PBS) (pH 7.4) for 2 hours. After dialyzed for 4 hours, the mixture was concentrated to 10 ml and reacted with 2.0 ml of 20 mg of RGD peptide at 4°C in PBS for 2 hours. The as-obtained mixture containing RGD-BSA was dialyzed and concentrated to 2.0 ml before 0.80 ml of 20 mM CuSO4 was added under vigorous stirring. After 5 min, 0.10 ml of 0.50 mM NaOH aqueous solution was introduced, and the mixture was allowed to react under darkness for another 8 hours at 55°C to produce RGD-BSA-CuCs. Furthermore, the as-prepared clusters dispersion was concentrated by superfilter tube (molecular weight cutoff, 3 kDa) to 1.0 ml to cut off free ions. BSA-CuCs was synthesized by a similar procedure without bioconjugating peptide to BSA as template. The as-prepared clusters are substantially stable in water for a long storage period at 4°C.
Characterization of RGD-BSA-CuCs and BSA-CuCs
MALDI-TOF MS assays were performed using Bruker Autoflex III MS (Germany). The samples were tested in positive ion linear mode using sinapinic acid as the matrix. A UV-1800 spectrophotometer (Shimadzu, Japan) was used to record the UV-Vis spectra of various samples. Fluorescence spectra of the samples were measured on a Hitachi F-7000 fluorescence spectrometer (Japan).
Theoretical study
The DMol3 module in the Material Studio package with the generalized gradient approximation/Perdew-Burke-Ernzerhof functional and the Double Numeric Polarization (DNP) basis set were used. The relativistic effects and spin interactions are fully considered for all electrons in the complex systems. The multiplicity was set as doublet, and the system is neutral. The geometry optimizations of copper cluster and its catalytic intermediate products with the convergence accuracy as customized were completed. In addition, the complete LST/QST (Linear Synchronous Transit/Quadratic Synchronous Transit) protocol to search the transition states with the initial accuracy as medium based on the above geometry optimizations was used. The solvation effects are considered. The other parameters are as the same as those in geometry optimization calculations of catalytic intermediate products. Some transition states from the imaginary frequency results were obtained. After further improving the search accuracy, the configurations of transition states were harvested. The recycled catalytic reaction pathway was constructed of the intermediate products and transition states.
ESR test
The ESR spectra were acquired with a Bruker model ESP 300 spectrometer (Germany) operating at room temperature. To 50 μl of mixture containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 in pH 6.5 or pH 7.4 buffer solution premixed for 0, 6, 12, and 24 hours at 37°C, 10 μl of 1.0 M DMPO in aqueous solution was added. Immediately, 50 μl of aliquot of control or experimental sample was put in glass capillary tube and sealed. The capillary tube was then inserted into the ESR cavity, and the spectra were recorded. Other settings were as follows: microwave power, 10.12 mW; time constant, 40.96 ms; frequency, 9.8 GHz; scan width, 100 G.
RGD-BSA-CuCs catalyze oxidation of Amplex UltraRed
The oxidation of Amplex UltraRed reagent by H2O2 catalyzed via RGD-BSA-CuCs produced a bright yellow fluorescence with a major emission peak at 581 nm, shown in the following equation. First, 200 μl of reaction buffer at pH 6.5 or pH 7.4 containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 together with 25 μM Amplex UltraRed was kept at 37°C water baths for 0, 6, 12, 24, 36, 48, 60, and 72 hours. Then, the fluorescence of oxidized Amplex UltraRed was evaluated at 37°C in 96-well plates on a microplate reader (SpectraMax M2, USA). The maximum excitation and emission band was set at 490/585 nm. The fluorescence variation of similar catalytic system without RGD-BSA-CuCs was set as control.
| (2) |
Measurement of O2 production
For each experiment, reaction buffer at pH 6.5 or pH 7.4 containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 was pipetted into the chamber of oxytherm electrode unit (Hansatech Instrument Ltd., Britain), respectively, which uses a Clark-type electrode to monitor the dissolved oxygen production in the sealed chamber. Samples of catalytic mixture premixed at 37°C for 24 hours were also evaluated generation of O2 in the initial 600 s. The aqueous solution of 100 μM H2O2 dispersed in pH 6.5 or pH 7.4 buffer was set as control groups. The temperature was maintained at 37°C during the measurement. Relative O2 production was recorded by subtracting background value. The O2 content promotion is the O2 production catalyzed by the metalloenzyme in the sealed chamber during the initial 600 s.
Characterization of RGD-BSA-CuCs before and after catalysis by HRTEM
The buffer solution containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 in pH 6.5 or pH 7.4 was first mixed for 24 hours at 37°C. Then, a drop of the sample mentioned above was dropped on the ultrathin carbon-copper grid. The samples of 12 μM RGD-BSA-CuCs dispersed in pH 6.5 or pH 7.4 buffer solution were set as control groups. The HRTEM images were collected using a JEM-2100F microscope (JEOL, Japan) working at 200 kV. The average size analysis was evaluated with image software (ImageJ 1.43) by randomly measuring 50 nanoparticle’s diameters.
Characterization of RGD-BSA-CuCs before and after catalysis by DSL and fluorescence test
The mean hydration diameter and size distribution of 12 μM RGD-BSA-CuCs incubated with or without 100 μM H2O2 in pH 6.5 or pH 7.4 buffer solution at 37°C for 24 hours were documented by a phase analysis light-scattering technique (Zetasizer Nano, Malvern). For fluorescence analysis, aqueous solution containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 at pH 6.5 or pH 7.4 was kept at 37°C water bath. Then, fluorescence emission intensity of catalyst at indicated incubation time (Ft) was evaluated every 2 hours by setting the optimal excitation and emission band at 323/400 nm. The fluorescence emission intensity of aqueous solution containing 12 μM RGD-BSA-CuCs was set as control (F0).
X-ray absorption spectroscopic analysis
Reaction buffer at pH 6.5 or pH 7.4 containing 12 μM RGD-BSA-CuCs and 100 μM H2O2 was premixed for 24 hours before rinsed, concentrated, and freeze-dried into powder. X-ray absorption spectroscopy (XAS) of Cu K edge (8979 eV) was measurements at 1W1B beamline of Beijing Synchrotron Radiation Facility, Beijing, China. RGD-BSA-CuCs’ XAS measurements were performed in fluorescence mode, while standard Cu references (commercial Cu foil, Cu2O, and CuSO4) were measured in transmission mode. Background subtraction, spectra normalization, PCA, and LCF analysis were conducted using Athena software.
Cell viability assay
Human non–small cell lung carcinoma cells (A549 cells) were from Cancer Institute and Hospital, Chinese Academy of Medical Sciences. A549 cells were cultured in 1640 medium and 10% FBS supplemented with 1% penicillin and streptomycin at 37°C in 5% CO2 and 70% humidity environment, reseeded every 3 days to maintain subconfluence. Initially, A549 cells (5 × 104 cells ml−1) were split into 96-well plates and incubated for 24 hours to allow them to attach. Except from controlled cells, the remaining cells were administered with RGD-BSA-CuCs or BSA-CuCs with the final concentrations of Cu at 4.8, 9.6, 19, 29, 39, 58, and 77 μM, respectively. After cells were cultured for 48 hours, the noneffective clusters were removed, and the fresh growth medium was added, respectively. Then, the cells were incubated with fresh medium containing 10% (v/v) CCK-8 reagent for 3 hours at 37°C for cytotoxicity assay. The absorbance of each well at 450 nm was measured using a microplate reader (SpectraMax M2, Sunnyvale, CA). Survival of untreated cells was set as 100%, and that of treated cells were expressed as a percentage of untreated cells. Data are shown as means ± SE from three independent experiments. Cells pretreated with 250 μM mannitol and then 77 μM RGD-BSA-CuCs were set as control, respectively. In addition, A549 cells viability test incubated with 4.8, 9.6, 19, 29, 39, 58, and 77 μM RGD-BSA for 48 hours was also performed. The IC50 was calculated by the software GraphPad Prism 5.0. Clusters’ cytotoxicity was also evaluated on human embryonic lung diploid cell line CCC-HPF-1 by following the same procedure mentioned above.
Imaging intracellular ROS generation
To evaluate intracellular ROS burst by clusters concentration dependent manner, A549 cells were grown in glass-bottomed Petri dishes and then supplemented with culture medium to allow 70% confluence in 24 hours. After exposed with 9.6, 19, and 39 μM RGD-BSA-CuCs at 37°C for 48 hours, cells were rinsed and incubated with 10 μM CM-H2DCFDA for 30 min before observed by confocal laser scanning microscope (CLSM; Olympus, FV1000MPE, Japan). Images of fluorescence emission of CM-H2DCFDA were collected mainly in green channel when excited by 488-nm laser. Cells in the absence of agent, cultured with 39 μM RGD-BSA-CuCs and 250 μM ●OH quencher mannitol, were monitored as control groups. All fluorescent images were collected at the same setting.
Detection of cellular apoptosis and necrosis
Apoptotic cells and necrotic cells were analyzed by double staining with annexin V–FITC/PI. A549 cells cultured in a six-well culture plates with 70% confluence were treated with 9.6, 19, and 39 μM RGD-BSA-CuCs at 37°C for 48 hours. Then, cells were collected, washed with PBS, and resuspended in binding buffer. After that, cells suspension was mixed with annexin V–FITC and incubated for 15 min at room temperature in the dark. At last, they were stained with PI solution and immediately analyzed by flow cytometry. The percentage of apoptotic and necrotic cells were determined using FACS software. A total of 1 × 104 events was counted for analysis.
Plasmid DNA cleavage assays
The ability of RGD-BSA-CuCs to cause DNA double-strand damage in vitro was assessed using agarose gel electrophoresis to separate the cleavage products of pUC19 plasmid DNA. The cleavage efficiency was evaluated by following the conversion of supercoiled DNA (form I) to the open circular DNA (form II) and, lastly, the transformation into smaller fragments. The solution of DNA (20 ng ml−1), a series concentration of RGD-BSA-CuCs, and 100 μM H2O2 were premixed in pH 7.4 buffer to achieve the concentration of clusters at 0, 1.2, 2.4, 4.8, 9.6, 19, 39, and 77 μM. The negative control was set by adding sole DNA to the sample solution. The test groups comprising DNA (20 ng ml−1) and a series concentration of RGD-BSA-CuCs were also prepared. All the reactions were carried out for 12 hours at 37°C. Then, 10 μl of mixture were individually mixed with 2 μl of 6× DNA loading dye and separated over a 1.0% agarose gel stained by GelRed (Biotium, USA). The electrophoresis was carried out at room temperature at 80 to 100 V for about 1 hour. After electrophoresis, the gel was viewed with a charge-coupled device camera by supersensitive multifunctional imager system and photographed (GE Healthcare, AI 600). The time-dependent DNA cutting experiment was performed by following the similar procedure that 9.6 μM RGD-BSA-CuCs was mixed with 100 μM H2O2 and DNA (20 ng ml−1) for 0, 3, 6, and 12 hours. The test that DNA incubated with 100 μM H2O2 at pH 7.4 for 12 hours was set as control.
Single cell gel electrophoresis assays
DNA damage was evaluated using the reagent kit for single cell gel electrophoresis assay/alkaline comet assay (Trevigen, USA), according to the manufacturer’s instructions. Briefly, A549 cells were treated with RGD-BSA-CuCs at different concentrations (0, 9.6, 19, 39, 58, and 77 μM) for 24 hours at 37°C in the dark. Following treatment, the cells were harvested in cold PBS at a density of 1 × 105 cells/ml. An aliquot of this cell suspension was mixed with prewarmed low melting agarose (37°C) at a 1:10 ratio (v/v) and was immediately pipetted onto a comet slide. Comet slides were immersed in cold lysing solution overnight at 4°C, followed by an incubation in alkaline unwinding solution for 20 min at room temperature in the dark. Electrophoresis was then carried out for 20 min at 21 V (~1 V cm−1) at 4°C. Then, DNA was stained by 1:10,000 dilution of SYBR Gold (Molecular Probes, USA). Gel was then photographed by CLSM (Olympus, FV1000MPE, Japan), and DNA content in the comet tail was evaluated through CASP (Comet Assay Software Project) software by analyzing 50 cells randomly on each slide.
Animal experiments
Under the guidance of the items in Ministry of Science and Technology of the People’s Republic China, which concerns use and care of laboratory animals, animal experiments were performed. Moreover, Animal Study Committee of Ministry of Science and Technology of the People’s Republic of China has given approval of the experiments. Female BABL/c athymic nude mice with an average age of 4 weeks (18 to 20 g) were obtained from Beijing HFK Bioscience Co. Ltd.
In vivo anticancer efficacy
BALB/c nude mice were subcutaneously injected in the right flank with 1 × 107 A549 cells suspended in 200 μl of serum-free Dulbecco’s modified Eagle medium and Matrigel mixture, and further fed until tumor volume grew up to 50 mm3. It took 8 days to prepare subcutaneous A549 xenografted tumor model. BALB/c nude mice were then randomly sorted into three groups (n = 5 for each group) before they were administrated intraperitoneally with 200 μl of RGD-BSA-CuCs (Cu dose at 1.0 and 2.5 mg kg−1) every 2 days. Equivoluminal saline was injected as the control group. To evaluate the therapeutic efficacy and toxicity, tumor growth and body weight evolution were monitored every 2 days. Tumor size was measured with a digital caliper. With the length (L) and width (W), the tumor volume (V) was calculated as V = L × W2/2. The relative tumor volume (V/V0) was normalized to tumor initial volume (V0).
Histopathological analysis and TUNEL staining
At the end of therapeutic period, the mice were euthanized. Their tumors were then collected for pathological studies, respectively. The tumors were fixed in 10% formalin, embedded in paraffin, sectioned, and then stained by H&E. The stained tissue sections were then observed using NanoZoomer digital pathology system (Hamamatsu, Japan) to monitor the possible histological changes. For assessment of cell apoptosis, TUNEL assays were performed by following the manufacturer’s protocol (Roche, Germany). The stained tissue sections were counterstained with 4, 6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, USA) and captured by CLSM (Olympus, FV1000MPE, Japan).
Wavelength, dose, and time-dependent chemiluminescence imaging in test tube
The wavelength, dose, and time-dependent chemiluminescence properties of L-012, a kind of in vitro/in vivo ROS probe were recorded by the microplate reader and PerkinElmer IVIS Spectrum (CAS Key Laboratory for Biomedical Effects of Nanomaterial and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China). First, the wavelength-dependent chemiluminescence of 200 μl of reaction mixture containing 77 μM RGD-BSA-CuCs and 100 μM H2O2 together with 1.0 mM L-012 at pH 6.5 was recorded by the microplate reader. Next, 200 μl of pH 6.5 buffer solution containing 100 μM H2O2, 1.0 mM L-012, and a series concentration of RGD-BSA-CuCs (0, 9.6, 19, 39, 58, 77 μM) was placed in the wells of 96-well solid plates, respectively. Then, the chemiluminescence in each well was measured by IVIS imaging system. Imaging exposure time was set at 1 min. In another test, dispersion of 12 μM cluster, 1.0 mM L-012, and a series concentration of H2O2 (0, 20, 40, 60, 80, and 100 μM) were mixed to study the chemiluminescence intensity variation by a H2O2-dependent manner. What’s more, kinetic measurements were conducted by monitoring the chemiluminescence intensity of 1.0 mM L-012 catalyzed by 9.6 μM RGD-BSA-CuCs in the presence of 100 μM H2O2 by a time scan mode for 48 hours. Half-life of chemiluminescence was evaluated through GraphPad Prism 5.0. Luminescence intensity from the region of interest was quantified as photons per second per square centimeter per steradian using the imaging system software package. Data are shown as means ± SE from three independent experiments.
ROS generation through catalysis in tumor microenvironment
BALB/c nude mice were subcutaneously injected in the right flank with 1 × 107 A549 cells and tumors were allowed to grow to 100 mm3. Mice were randomly assigned into six groups (n = 3 for each group). After intraperitoneal injection 200 μl of RGD-BSA-CuCs (2.5 mg kg−1) for 1, 2, 4, 6, 12, and 24 hours, the mice were intratumorally injection 25 μl of L-012 (25 mg kg−1). The mice were first anesthetized by 2% isoflurane in oxygen, and then the images were collected in given intervals after injection to monitor the change of chemiluminescence signal in vivo recorded by the PerkinElmer IVIS Spectrum. Imaging exposure time was set at 2 min. Luminescence intensity from the region of interest was quantified as photons per second per square centimeter per steradian using the imaging system software package. At 1 hour after injection of RGD-BSA-CuCs and L-012, the mice were euthanized, and organs of interest and tumors were harvested. Then, ex vivo chemiluminescence images of organs and tumors were captured. Mice administered with only saline (intraperitoneally) or L-012 (intratumorally) were set as control groups.
Chemiluminescence imaging in vivo to trace the process of therapeutic intervention
In a separate experiment, 18 BALB/c nude mice were subcutaneously injected in the right flank with 1 × 107 A549 cells and further fed until tumor volume grew up to 50 mm3. Then, mice were randomly assigned into two groups (n = 9 for each group). After one group of mice were intraperitoneally injected with 200 μl of RGD-BSA-CuCs (2.5 mg kg−1) for 7, 14, and 21 days every other day (n = 3 for each therapeutic end point), 25 μl of L-012 (25 mg kg−1) was intratumorally injected to the mice by following the same procedure mentioned above. Another group of mice without clusters intervention was injected with the same dosage of L-012 for imaging the intrinsic oxidative pressure in situ (n = 3 for each therapeutic end point). Group of mice without tumor xenograft, clusters, or L-012 treatment was set as control (n = 3 for each therapeutic end point). Chemiluminescence imaging was performed as mentioned above. Then, mice were euthanized, and organs of interest and tumors were harvested and captured. The quantitative analysis was conducted as aforementioned.
Statistical analysis
Quantitative data are expressed as means ± SD. Statistical comparisons were conducted using Student’s two-tailed t test for two group comparison or one-way analysis of variance (ANOVA) or multiple groups’ comparison. P < 0.05 was defined as statistically significant difference.
Supplementary Material
Acknowledgments
We thank H. Yu and Y. Fu (Shanghai Institute of Materia Medica, CAS) for pharmacokinetics data analysis. We appreciate X. Gao (Jiangxi Normal University) for helpful discussions on computational simulation. Funding: This study was supported by the Natural Science Foundation of China (nos. 21425522, 21727818, 11621505, 31971311, 31500815, 31771082, 31971269, and 31700874). We also acknowledge the financial support from the Beijing Municipal High Level Innovative Team Building Program (IDHT20180504), Beijing Municipal Natural Science Foundation (KZ202010005005), and the “Talented People Project” from Beijing University of Technology. Author contributions: X.G., L.Z., and L.G. conceived and designed the experiments. X.G., L.Z., H.J., and X.W. supervised the project and commented on the project. L.G. synthesized and characterized the clusters, conducted the catalytic experiments, and performed in vitro experiments. L.Z. and W.N. performed the computational simulation. L.G., Y.Z., Y.T., F.G., and P.C. contributed to in vivo experiments. Q.Y. and Y.T. contributed to in vitro experiments. L.G., L.Z., X.G., and W.N. analyzed the data and wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or in the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eabb1421/DC1
REFERENCES AND NOTES
- 1.Wang D., Lippard S. J., Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4, 307–320 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Furgason J. M., Bahassi E. M., Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 137, 298–308 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Pan L., Liu J., Shi J., Cancer cell nucleus-targeting nanocomposites for advanced tumor therapeutics. Chem. Soc. Rev. 47, 6930–6946 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Rebelein J. G., Ward T. R., In vivo catalyzed new-to-nature reactions. Curr. Opin. Biotechnol. 53, 106–114 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Lin H., Chen Y., Shi J., Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy. Chem. Soc. Rev. 47, 1938–1958 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Li X., Kim J., Yoon J., Chen X., Cancer-associated, stimuli-driven, turn on theranostics for multimodality imaging and therapy. Adv. Mater. 29, 1606857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J., Pu K., Development of organic semiconducting materials for deep-tissue optical imaging, phototherapy and photoactivation. Chem. Soc. Rev. 48, 38–71 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Mao D., Wu W., Ji S., Chen C., Hu F., Kong D., Ding D., Liu B., Chemiluminescence-guided cancer therapy using a chemiexcited photosensitizer. Chem 3, 991–1007 (2017). [Google Scholar]
- 9.Xu X., An H., Zhang D., Tao H., Dou Y., Li X., Huang J., Zhang J., A self-illuminating nanoparticle for inflammation imaging and cancer therapy. Sci. Adv. 5, eaat2953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J., Chakraborty S., Hosseinzadeh P., Yu Y., Tian S., Petrik I., Bhagi A., Lu Y., Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens P. J., Jollie D. R., Warshel A., Protein control of redox potentials of iron-sulfur proteins. Chem. Rev. 96, 2491–2514 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Zhang C., Chen C., Dong H., Shen J.-R., Dau H., Zhao J., A synthetic Mn4Ca-cluster mimicking the oxygen-evolving center of photosynthesis. Science 348, 690–693 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Yamazoe S., Koyasu K., Tsukuda T., Nonscalable oxidation catalysis of gold clusters. Acc. Chem. Res. 47, 816–824 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Jin R., Zeng C., Zhou M., Chen Y., Atomically precise colloidal metal nanoclusters and nanoparticles: Fundamentals and opportunities. Chem. Rev. 116, 10346–10413 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Fang J., Zhang B., Yao Q., Yang Y., Xie J., Yan N., Recent advances in the synthesis and catalytic applications of ligand-protected, atomically precise metal nanoclusters. Coordin. Chem. Rev. 322, 1–29 (2016). [Google Scholar]
- 16.Oliver-Messeguer J., Liu L., García-García S., Canós-Giménez C., Domínguez I., Gavara R., Doménech-Carbó A., Concepción P., Leyva-Pérez A., Corma A., Stabilized naked sub-nanometric Cu clusters within a polymeric film catalyze C–N, C–C, C–O, C–S, and C–P bond-forming reactions. J. Am. Chem. Soc. 137, 3894–3900 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Maity P., Yamazoe S., Tsukuda T., Dendrimer-encapsulated copper cluster as a chemoselective and regenerable hydrogenation catalyst. ACS Catal. 3, 182–185 (2013). [Google Scholar]
- 18.Vilar-Vidal N., Rey J. R., Lopez Quintela M. A., Green emitter copper clusters as highly efficient and reusable visible degradation photocatalysts. Small 10, 3632–3636 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Tang Q., Lee Y., Li D.-Y., Choi W., Liu C. W., Lee D., Jiang D.-E., Lattice-hydride mechanism in electrocatalytic CO2 reduction by structurally precise copper-hydride nanoclusters. J. Am. Chem. Soc. 139, 9728–9736 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Raj L., Ide T., Gurkar A. U., Foley M., Schenone M., Li X., Tolliday N. J., Golub T. R., Carr S. A., Shamji A. F., Stern A. M., Mandinov A., Schreiber S. L., Lee S. W., Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Weinstain R., Savariar E. N., Felsen C. N., Tsien R. Y., In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. J. Am. Chem. Soc. 136, 874–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Q., Liang C., Sun X., Chen J., Yang Z., Zhao H., Feng L., Liu Z., H2O2-responsive liposomal nanoprobe for photoacoustic inflammation imaging and tumor theranostics via in vivo chromogenic assay. Proc. Natl. Acad. Sci. U.S.A. 114, 5343–5348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coverdale J. P. C., Romero-Canelón I., Sanchez-Cano C., Clarkson G. J., Habtemariam A., Wills M., Sadler P. J., Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 10, 347–354 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Ali M. R., Panikkanvalappil S. R., El-Sayed M. A., Enhancing the efficiency of gold nanoparticles treatment of cancer by increasing their rate of endocytosis and cell accumulation using rifampicin. J. Am. Chem. Soc. 136, 4464–4467 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Danhier F., Le Breton A., Préat V., RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 9, 2961–2973 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Vilar-Vidal N., Rivas J., López-Quintela M. A., Size dependent catalytic activity of reusable subnanometer copper(0) clusters. ACS Catal. 2, 1693–1697 (2012). [Google Scholar]
- 27.Liu X., Astruc D., Atomically precise copper nanoclusters and their applications. Coord. Chem. Rev. 359, 112–126 (2018). [Google Scholar]
- 28.Chakrahari K. K., Liao J.-H., Kahlal S., Liu Y.-C., Chiang M.-H., Saillard J.-Y., Liu C. W., [Cu13{S2CNnB}6(acetylide)4]+: A two-electron superatom. Angew. Chem. Int. Ed. 128, 14924–14928 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Sanner M. F., Python: A programming language for software integration and development. J. Mol. Graph. Model. 17, 57–61 (1999). [PubMed] [Google Scholar]
- 30.Trott O., Olson A. J., AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kogo A., Sakai N., Tatsuma T., Photoelectrochemical analysis of size-dependent electronic structures of gold clusters supported on TiO2. Nanoscale 4, 4217–4221 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Zhang H., Ji Z., Xia T., Meng H., Low-Kam C., Liu R., Pokhre S., Lin S., Wang X., Liao Y.-P., Wang M. Y., Li L. J., Rallo R., Damoiseaux R., Telesca D., Mädler L., Cohen Y., Zink J. I., Nel A. E., Use of metal oxide nanoparticle band gap to develop a predictive paradigm for oxidative stress and acute pulmonary inflammation. ACS Nano 6, 4349–4368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W., Hu S., Yin J.-J., He W., Lu W., Ma M., Gu N., Zhang Y., Prussian blue nanoparticles as multienzyme mimetics and reactive oxygen species scavengers. J. Am. Chem. Soc. 138, 5860–5865 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Yang Y.-C., Wang Y.-T., Tseng W.-L., Amplified peroxidase-like activity in iron oxide nanoparticles using adenosine monophosphate: Application to urinary protein sensing. ACS Appl. Mater. Interfaces. 9, 10069–10077 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Dong J., Song L., Yin J.-J., He W., Wu Y., Gu N., Zhang Y., Co3O4 nanoparticles with multi-enzyme activities and their application in immunohistochemical assay. ACS Appl. Mater. Interfaces. 6, 1959–1970 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Jachimska B., Wasilewska M., Adamczyk Z., Characterization of globular protein solutions by dynamic light scattering, electrophoretic mobility, and viscosity measurements. Langmuir 24, 6866–6872 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Jia X., Yang X., Li J., Li D., Wang E., Stable Cu nanoclusters: From an aggregation-induced emission mechanism to biosensing and catalytic applications. Chem. Commun. 50, 237–239 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Peng Y., Shang L., Cao Y., Waterhouse G. I. N., Zhou C., Bian T., Wu L.-Z., Tung C.-H., Zhang T., Copper(I) cysteine complexes: Efficient earth-abundant oxidation co-catalysts for visible light-driven photocatalytic H2 production. Chem. Commun. 51, 12556–12559 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Malta G., Kondrat S. A., Freakley S. J., Davies C. J., Lu L., Dawson S., Thetford A., Gibson E. K., Morgan D. J., Jones W., Wells P. P., Johnston P., Catlow C. R. A., Kiely C. J., Hutchings G. J., Identification of single-site gold catalysis in acetylene hydrochlorination. Science 355, 1399–1403 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Yin C., Zheng F., Lee S., Guo J., Wang W.-C., Kwon G., Vajda V., Wang H.-H., Lee B., DeBartolo J., Seifert S., Winans R. E., Vajda S., Size- and support-dependent evolution of the oxidation state and structure by oxidation of subnanometer cobalt clusters. J. Phys. Chem. A 118, 8477–8484 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Oyanagi H., Orimoto Y., Hayakawa K., Hatada K., Sun Z., Zhang L., Yamashita K., Nakamura H., Uehara M., Fukano A., Maeda H., Nanoclusters synthesized by synchrotron radiolysis in concert with wet chemistry. Sci. Rep. 4, 7199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C., Wang C., Xu L., Cheng H., Lin Q., Zhang C., Protein-directed synthesis of pH-responsive red fluorescent copper nanoclusters and their applications in cellular imaging and catalysis. Nanoscale 6, 1775–1781 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Fu P. P., Xia Q., Hwang H.-M., Ray P. C., Yu H., Mechanisms of nanotoxicity: Generation of reactive oxygen species. J. Food. Drug Anal. 22, 64–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Z., Zhang H., Liu Y., Ni D., Zhang H., Zhang J., Yao Z., He M., Shi J., Bu W., Antiferromagnetic pyrite as the tumor microenvironment-mediated nanoplatform for self-enhanced tumor imaging and therapy. Adv. Mater. 29, 1701683 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Gao L., Zhuang J., Nie L., Zhang J., Zhang Y., Gu N., Wang T., Feng J., Yang D., Perrett S., Yan X., Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nanotechnol. 2, 577–585 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Wu J., Gao G., Sun P., Long X., Li F., Synergetic catalysis of bimetallic CuCo nanocomposites for selective hydrogenation of bioderived esters. ACS Catal. 7, 7890–7901 (2017). [Google Scholar]
- 47.Isegawa M., Sameera W. C., Sharma A. K., Kitanosono T., Kato M., Kobayashi S., Morokuma K., Copper-catalyzed enantioselective boron conjugate addition: DFT and AFIR study on different selectivities of Cu(I) and Cu(II) catalysts. ACS Catal. 7, 5370–5380 (2017). [Google Scholar]
- 48.Deng Q., Zhao L., Gao X., Zhang M., Luo Y., Zhao Y., Single layer of polymeric cobalt phthalocyanine: Promising low-cost and high-activity nanocatalysts for CO oxidation. Small 9, 3506–3513 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Kang Y.-H., Lee E., Choi M.-K., Ku J.-L., Kim S. H., Park Y.-G., Lim S.-J., Role of reactive oxygen species in the induction of apoptosis by α-tocopheryl succinate. Int. J. Cancer 112, 385–392 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Murakami T., Nakatsuji H., Inada M., Matoba Y., Umeyama T., Tsujimoto M., Isoda S., Hashida M., Imahori H., Photodynamic and photothermal effects of semiconducting and metallic-enriched single-walled carbon nanotubes. J. Am. Chem. Soc. 134, 17862–17865 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Zhu Z., Wang Z., Hao Y., Zhu C., Jiao Y., Chen H., Wang Y.-M., Yan J., Guo Z., Wang X., Glutathione boosting the cytotoxicity of a magnetic platinum(IV) nano-prodrug in tumor cells. Chem. Sci. 7, 2864–2869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pagoria D., Lee A., Geurtsen W., The effect of camphorquinone (CQ) and CQ-related photosensitizers on the generation of reactive oxygen species and the production of oxidative DNA damage. Biomaterials 26, 409–4099 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Sun M., Xu L., Qu A., Zhao P., Hao T., Ma W., Hao C., Wen X., Colombari F. M., de Moura A. F., Kotov N. A., Xu C., Kuang H., Site-selective photoinduced cleavage and profiling of DNA by chiral semiconductor nanoparticles. Nat. Chem. 10, 821–830 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Wang Y., Van Ness B., Site-specific cleavage of supercoiled DNA by ascorbate/Cu(II). Nucleic Acids Res. 17, 6915–6926 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melvin M., Tomlinson J., Saluta G., Kucera G., Lindquist N., Manderville R., Double-strand DNA cleavage by copper•prodigiosin. J. Am. Chem. Soc. 122, 6333–6334 (2000). [Google Scholar]
- 56.Higo T., Naito A. T., Sumida T., Shibamoto M., Okada K., Nomura S., Nakagawa A., Yamaguchi T., Sakai T., Hashimoto A., Kuramoto Y., Ito M., Hikoso S., Akazawa H., Lee J.-K., Shiojima I., McKinnon P. J., Sakata Y., Komuro I., DNA single-strand break-induced DNA damage response causes heart failure. Nat. Commun. 8, 15104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashimoto T., Horikawa D., Saito Y., Kuwahara H., Kozuka-Hata H., Shin-I T., Minakuchi Y., Ohishi K., Motoyama A., Aizu T., Enomoto A., Kondo K., Tanaka S., Hara Y., Koshikawa S., Sagara H., Miura T., Yokobori S.-I., Miyagawa K., Suzuki Y., Kubo T., Oyama M., Kohara Y., Fujiyama A., Arakawa K., Katayama T., Toyoda A., Kunieda T., Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nat. Commun. 7, 12808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kielland A., Blom T., Nandakumar K. S., Holmdahl R., Blomhoff R., Carlsen H., In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic. Biol. Med. 47, 760–766 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Zielonka J., Lambeth J. D., Kalyanaraman B., On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: A reevaluation. Free Radic. Biol. Med. 65, 1310–1314 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santini C., Pellei M., Gandin V., Porchia M., Tisato F., Marzano C., Advances in copper complexes as anticancer agents. Chem. Rev. 114, 815–862 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eabb1421/DC1